
Analysis of Telomere Maintenance Related Genes Reveals NOP10 as a New Metastatic-Risk Marker in Pheochromocytoma/Paraganglioma

 , ,
, ,  , , , ,
, , , ,  ,
,  , , ,
, , , 
 , and
, and
Abstract
:Simple Summary
Abstract
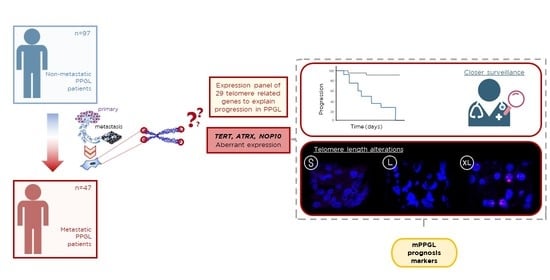
1. Introduction
2. Materials and Methods
2.1. PPGL Cohort and Genetic Characterization
2.2. Tumor DNA Extraction
2.3. Tumor RNA Extraction and Quality Test
2.4. TREx RNA Sequencing
2.5. ATRX Mutations
2.6. Identification of TERT PROMOTER Mutations (TPMs)
2.7. Analysis of TERT Promoter Methylation Levels
2.8. TERT Copy Number Alterations (CNAs)
2.9. Telomerome Significant Genes and Metastasis Prediction Risk Model
2.10. Time to Progression and Validated Telomerome Genes
2.11. Telomere Length Q-FISH, High-Throughput Quantification
2.12. Promelocytic Leukaemia (PML) Bodies and Telomere Co-Localization by Immuno-Q-FISH
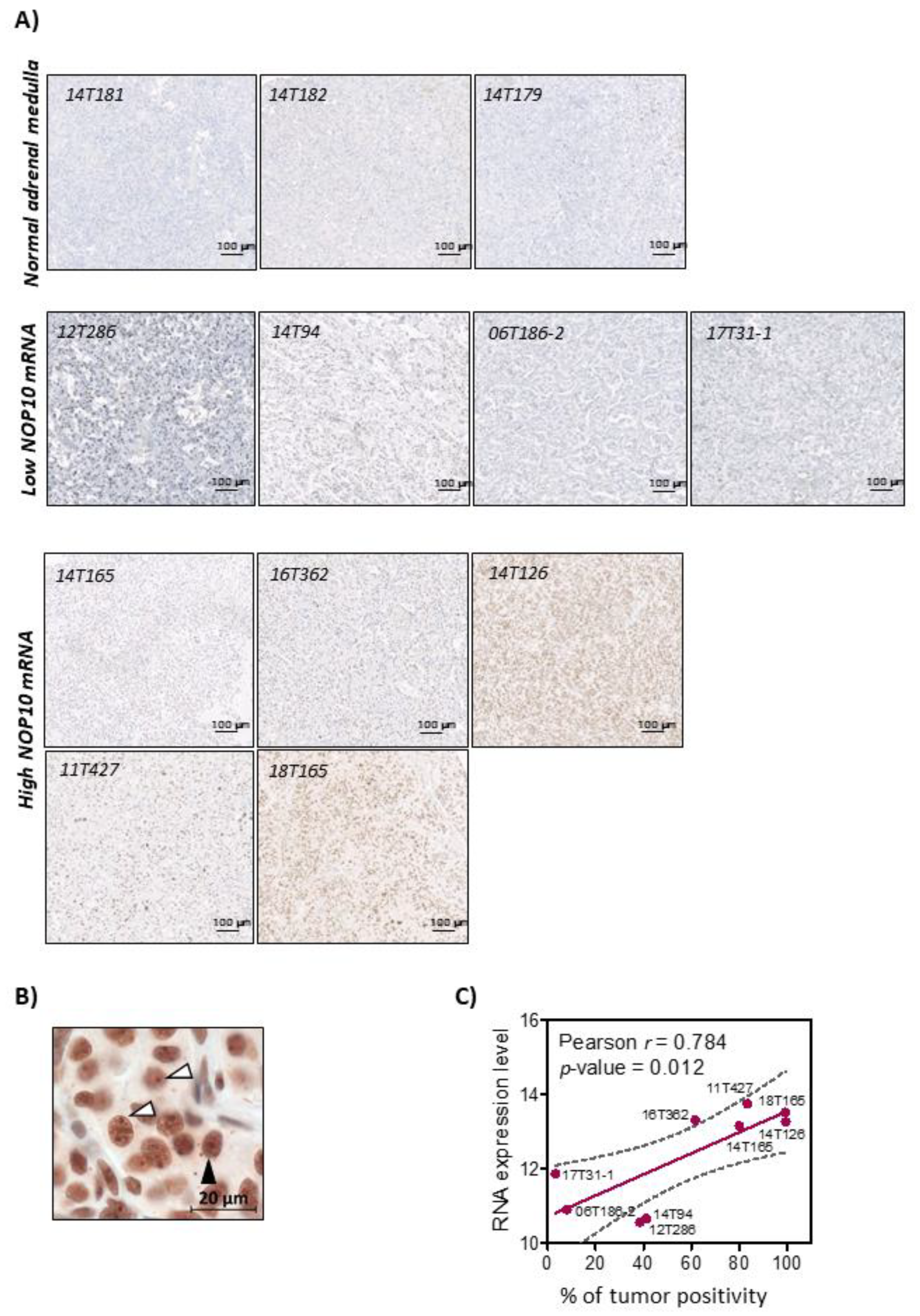
2.13. Characterization of NOP10 Expression Mechanisms
2.14. Cell Culture and Generation of TERT and NOP10 Overexpression Models
- pLV-TERT-IRES-puro: to generate TERT overexpressing cells, lentiviral plasmid pLV-TERT-IRES-hygro was acquired from Addgene repository (Addgene Plasmid #85140 [48]). Selection antibiotic was changed from hygromycin to puromycin.
- pLV-NOP10-IRES-hygro: NOP10 expression plasmid (NM_018648) was acquired from OriGene (CAT#: RC209038). Using pLV-TERT-IRES-hygro backbone, TERT gene was replaced by NOP10 ORF sequence from the aforementioned plasmid, generating a new pLV-NOP10-IRES-hygro vector.
2.15. Telomere Length Q-FISH on Cell Spreads
3. Results
3.1. Description of the PPGL-Telomerome Series
3.2. Study of the Telomerome Expression Profile and Outlier Selection
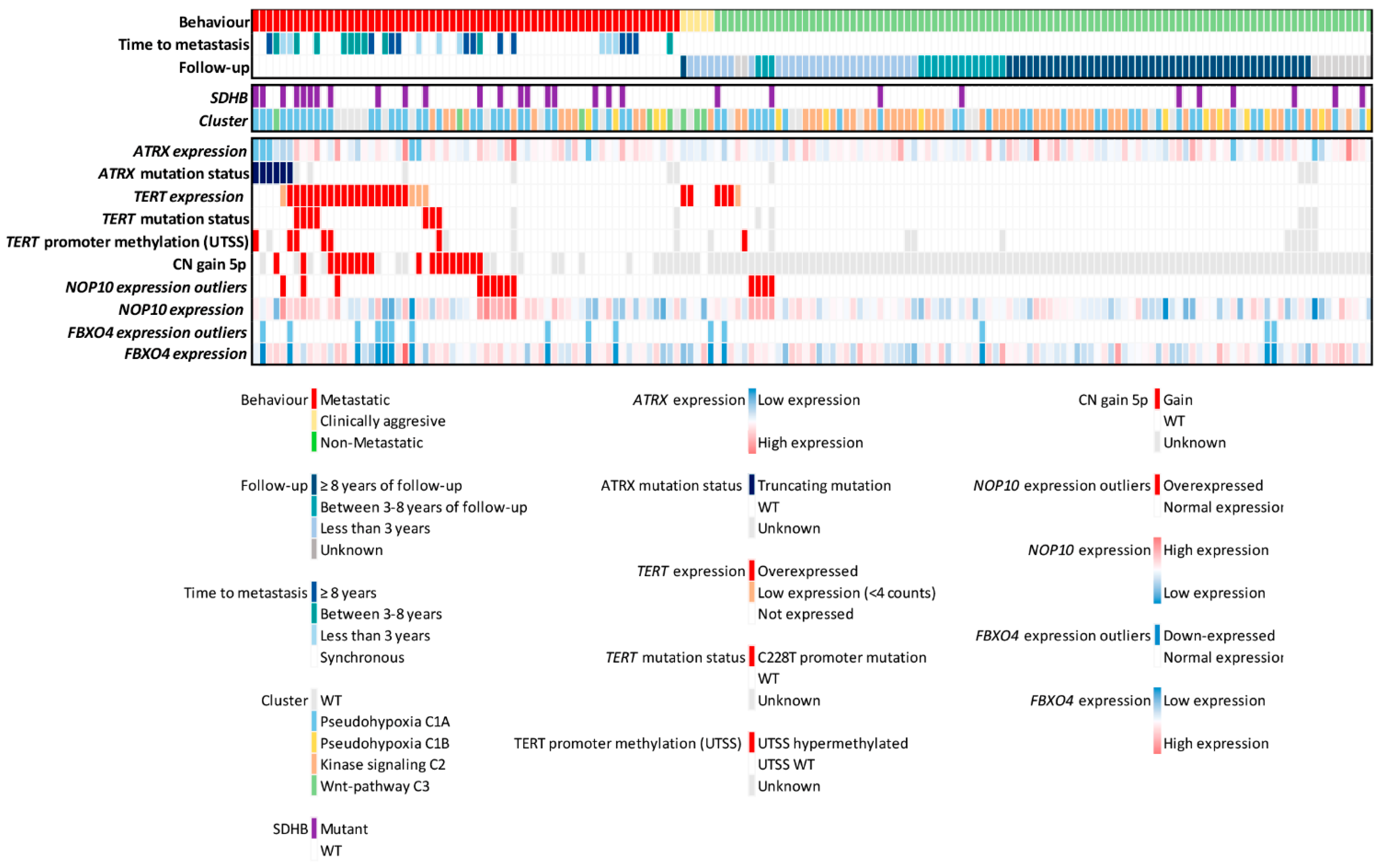
3.3. Mechanisms That Trigger Aberrant Expression of Telomerome Genes in mPPGL
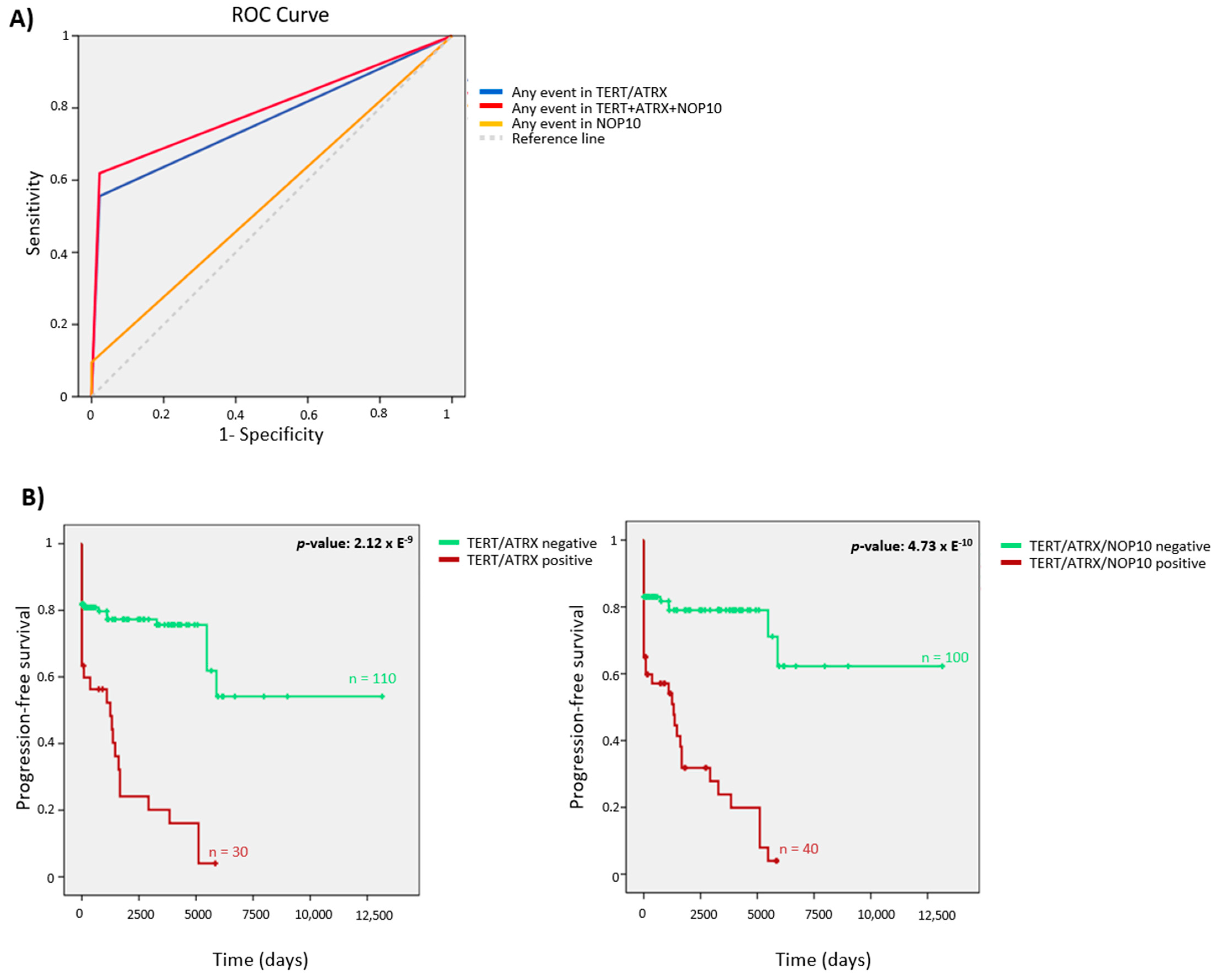
3.4. Telomerome Significant Genes Identification and Predictive Value
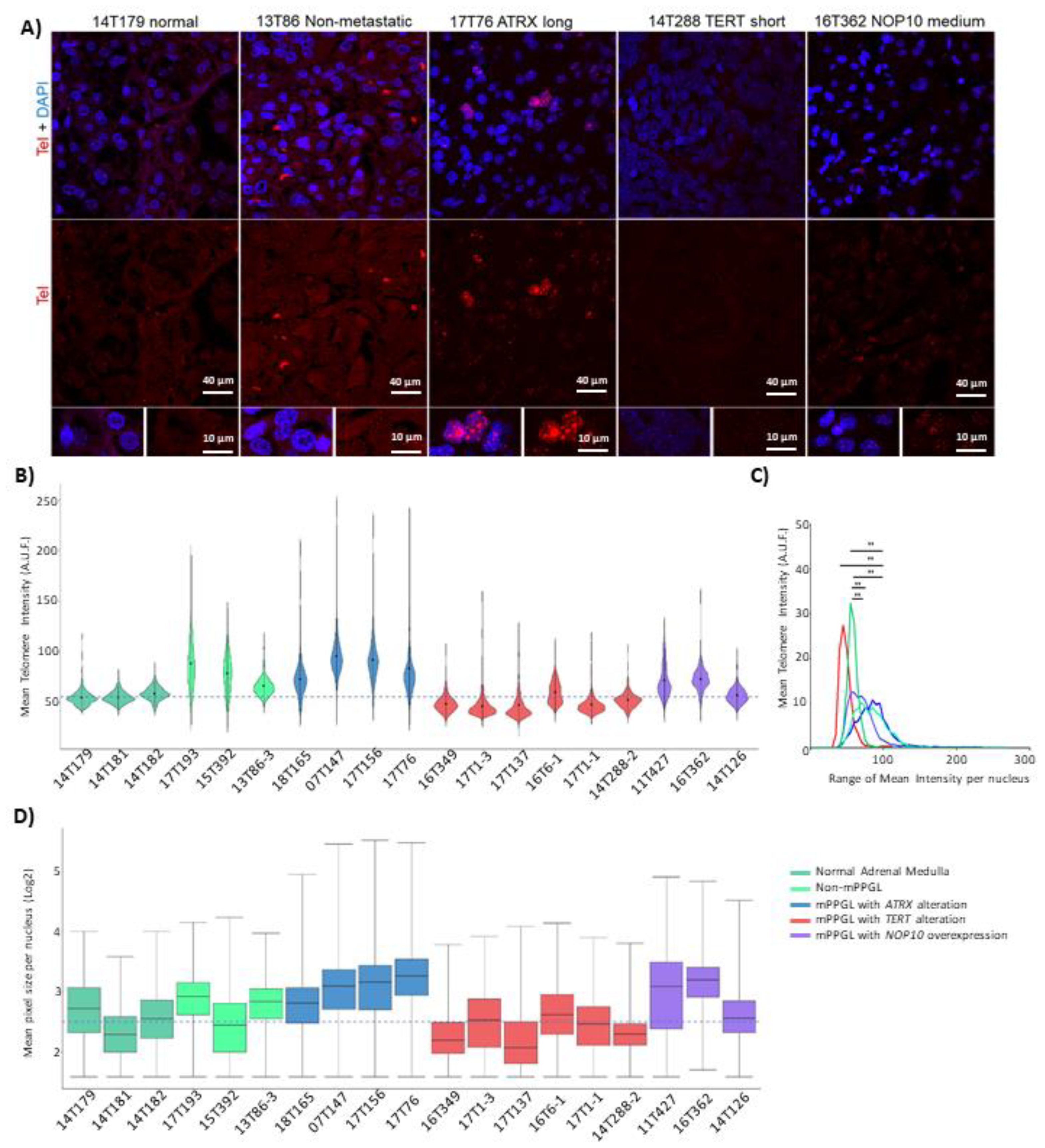
3.5. TERT, ATRX and NOP10 Events Affect Telomere Length
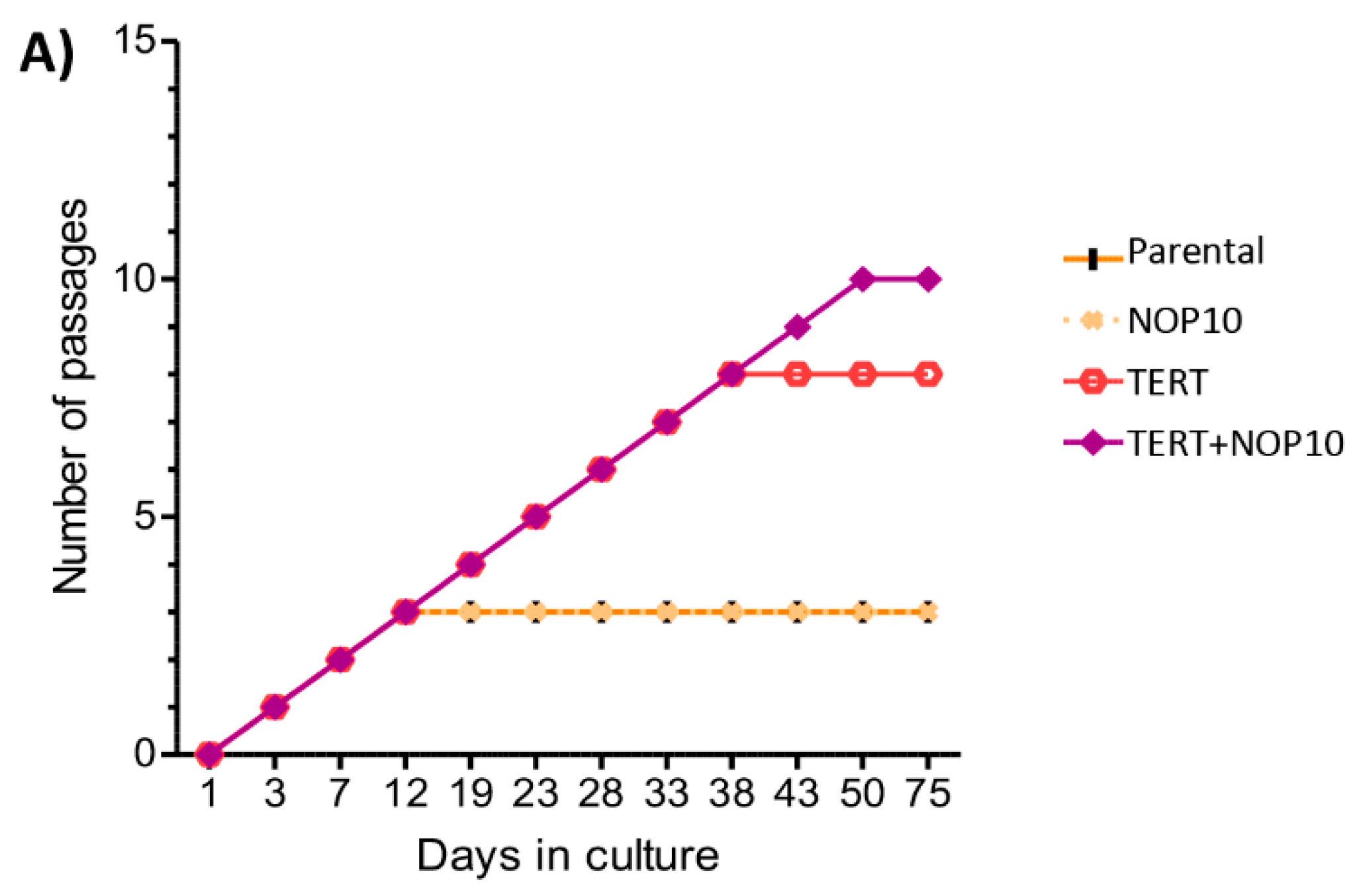
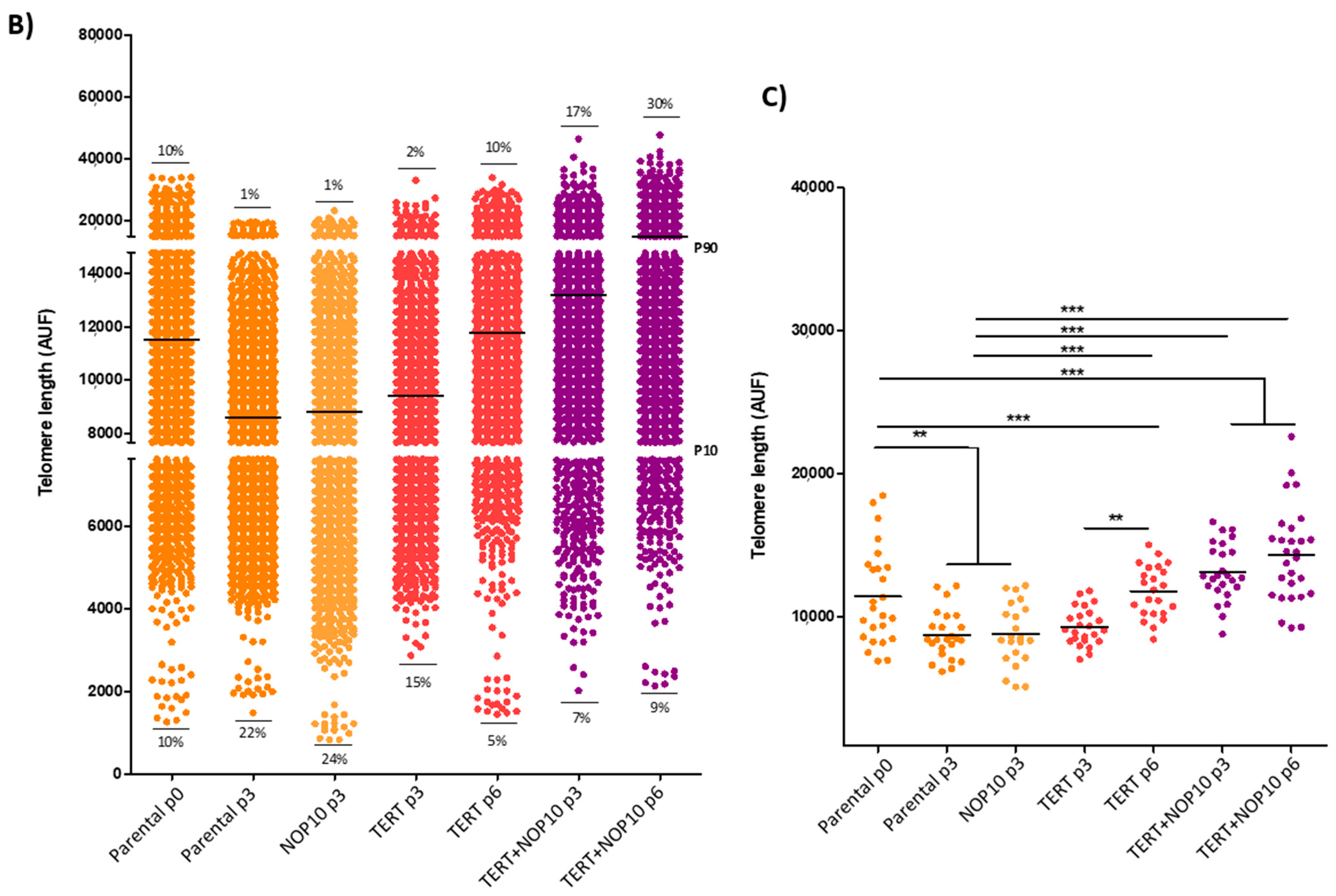
3.6. NOP10 and TERT Expression in Primary Cultures Affect Telomere Length Maintenance
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Favier, J.; Amar, L.; Gimenez-Roqueplo, A.P. Paraganglioma and phaeochromocytoma: From genetics to personalized medicine. Nat. Rev. Endocrinol. 2015, 11, 101–111. [Google Scholar] [CrossRef]
- Cascón, A.; Remacha, L.; Calsina, B.; Robledo, M. Pheochromocytomas and paragangliomas: Bypassing cellular respiration. Cancers 2019, 11, 683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fishbein, L.; Leshchiner, I.; Walter, V.; Danilova, L.; Robertson, A.G.; Johnson, A.R.; Lichtenberg, T.M.; Murray, B.A.; Ghayee, H.K.; Else, T.; et al. Comprehensive molecular characterization of pheochromocytoma and paraganglioma. Cancer Cell 2017, 31, 181–193. [Google Scholar] [CrossRef] [Green Version]
- Baudin, E.; Habra, M.A.; Deschamps, F.; Cote, G.; Dumont, F.; Cabanillas, M.; Arfi-Roufe, J.; Berdelou, A.; Moon, B.; Ghuzlan, A.A.; et al. Therapy of endocrine disease: Treatment of malignant pheochromocytoma and paraganglioma. Eur. J. Endocrinol. 2014, 171, R111–R122. [Google Scholar] [CrossRef] [PubMed]
- Jimenez, C. Treatment for patients with malignant pheochromocytomas and paragangliomas: A perspective from the hallmarks of cancer. Front. Endocrinol. 2018, 9, 277. [Google Scholar] [CrossRef] [PubMed]
- De Filpo, G.; Maggi, M.; Mannelli, M.; Canu, L. Management and outcome of metastatic pheochromocytomas/paragangliomas: An overview. J. Endocrinol. Investig. 2021, 44, 15–25. [Google Scholar] [CrossRef]
- Jochmanova, I.; Pacak, K. Pheochromocytoma: The first metabolic endocrine cancer. Clin. Cancer Res. 2016, 22, 5001–5011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plouin, P.F.; Amar, L.; Dekkers, O.M.; Fassnach, M.; Gimenez-Roqueplo, A.P.; Lenders, J.W.M.; Lussey-Lepoutre, C.; Steichen, O. European society of endocrinology clinical practice guideline for long-term follow-up of patients operated on for a phaeochromocytoma or a paraganglioma. Eur. J. Endocrinol. 2016, 174, G1–G10. [Google Scholar] [CrossRef] [Green Version]
- Plouin, P.F.; Fitzgerald, P.; Rich, T.; Ayala-Ramirez, M.; Perrier, N.D.; Baudin, E.; Jimenez, C. Metastatic pheochromocytoma and paraganglioma: Focus on therapeutics. Horm. Metab. Res. 2012, 44, 390–399. [Google Scholar] [CrossRef]
- John, H.; Ziegler, W.H.; Hauri, D.; Jaeger, P. Pheochromocytomas: Can malignant potential be predicted? Urology 1999, 53, 679–683. [Google Scholar] [CrossRef]
- Eisenhofer, G.; Lenders, J.W.M.; Siegert, G.; Bornstein, S.R.; Friberg, P.; Milosevic, D.; Mannelli, M.; Linehan, W.M.; Adams, K.; Timmers, H.J.; et al. Plasma methoxytyramine: A novel biomarker of metastatic pheochromocytoma and paraganglioma in relation to established risk factors of tumour size, location and SDHB mutation status. Eur. J. Cancer 2012, 48, 1739–1749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gimenez-Roqueplo, A.P.; Favier, J.; Rustin, P.; Rieubland, C.; Crespin, M.; Nau, V.; van Kien, P.K.; Corvol, P.; Plouin, P.F.; Jeunemaitre, X. Mutations in the SDHB gene are associated with extra-adrenal and/or malignant phaeochromocytomas. Cancer Res. 2003, 63, 5615–5621. [Google Scholar] [PubMed]
- Hescot, S.; Curras-Freixes, M.; Deutschbein, T.; van Berkel, A.; Vezzosi, D.; Amar, L.; de La Fouchardiere, C.; Valdes, N.; Riccardi, F.; Do Cao, C.; et al. Prognosis of malignant pheochromocytoma and paraganglioma (MAPP-PronO Study): A European network for the study of adrenal tumors retrospective study. J. Clin. Endocrinol. Metab. 2019, 104, 2367–2374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Job, S.; Draskovic, I.; Burnichon, N.; Buffet, A.; Cros, J.; Lepine, C.; Venisse, A.; Robidel, E.; Verkarre, V.; Meatchi, T.; et al. Telomerase Activation and ATRX Mutations Are Independent Risk Factors for Metastatic Pheochromocytoma and Paraganglioma. Clin. Cancer Res. 2019, 25, 760–770. [Google Scholar] [CrossRef] [Green Version]
- Dwight, T.; Flynn, A.; Amarasinghe, K.; Benn, D.E.; Lupat, R.; Li, J.; Cameron, D.L.; Hogg, A.; Balachander, S.; Candiloro, I.L.M.; et al. TERT Structural Rearrangements in Metastatic Pheochromocytomas. Endocr. Relat. Cancer 2018, 25, 1–9. [Google Scholar] [CrossRef] [Green Version]
- De Lange, T. Shelterin: The protein complex that shapes and safeguards human telomeres. Genes Dev. 2005, 19, 2100–2110. [Google Scholar] [CrossRef] [Green Version]
- Chan, S.W.-L.; Blackburn, E.H. New ways not to make ends meet: Telomerase, DNA damage proteins and heterochromatin. Oncogene 2002, 21, 553–563. [Google Scholar] [CrossRef]
- Funk, W.D.; Wang, C.K.; Shelton, D.N.; Harley, C.B.; Pagon, G.D.; Hoeffler, W.K. Telomerase expression restores dermal integrity to in vitro-aged fibroblasts in a reconstituted skin model. Exp. Cell Res. 2000, 258, 270–278. [Google Scholar] [CrossRef] [PubMed]
- Blasco, M.A.; Funk, W.; Villeponteau, B.; Greider, C.W. Functional characterization and developmental regulation of mouse telomerase RNA. Science 1995, 269, 1267–1270. [Google Scholar] [CrossRef] [Green Version]
- Shay, J.W.; Wright, W.E. Role of telomeres and telomerase in cancer. Semin. Cancer Biol. 2011, 21, 349–353. [Google Scholar] [CrossRef] [Green Version]
- Maciejowski, J.; de Lange, T. Telomeres in cancer: Tumour suppression and genome instability. Nat. Rev. Mol. Cell Biol. 2017, 18, 175–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wright, W.E.; Piatyszek, M.A.; Rainey, W.E.; Shgy, J.W.; Byrd, W. Telomerase activity in human germline embryonic tissues and cells. Dev. Genet. 1996, 18, 18173–18179. [Google Scholar] [CrossRef]
- Martínez, P.; Blasco, M.A. Telomeric and extra-telomeric roles for telomerase and the telomere-binding proteins. Nat. Rev. Cancer 2011, 11, 161–176. [Google Scholar] [CrossRef] [PubMed]
- Greider, C.W.; Blackburn, E.H. Identification of a specific telomere terminal transferase activity in tetrahymena extracts. Cell 1985, 43, 405–413. [Google Scholar] [CrossRef]
- Ulaner, G.A.; Hu, J.-F.; Vu, T.H.; Giudice, L.C.; Hoffman2, A.R. Telomerase activity in human development is regulated by human telomerase reverse transcriptase (HTERT) transcription and by alternate splicing of HTERT Transcripts1. CANCER Res. 1998, 58, 4168–4172. [Google Scholar] [PubMed]
- Dunham, M.A.; Neumann, A.A.; Fasching, C.L.; Reddel, R.R. Telomere maintenance by recombination in human cells. Nat. Genet. 2000, 26, 447–450. [Google Scholar] [CrossRef]
- Shay, J.W.; Bacchetti, S. A survey of telomerase activity in human cancer. Eur. J. Cancer Part A 1997, 33, 787–791. [Google Scholar] [CrossRef]
- Dilley, R.L.; Greenberg, R.A. Alternative telomere maintenance and cancer. Trends Cancer 2015, 1, 145–156. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, J.C.; Cech, T.R. Human telomerase: Biogenesis, trafficking, recruitment, and activation. Genes Dev. 2015, 29, 1095–1105. [Google Scholar] [CrossRef] [Green Version]
- Venteicher, A.S.; Abreu, E.B.; Meng, Z.; McCann, K.E.; Terns, R.M.; Veenstra, T.D.; Terns, M.P.; Artandi, S.E. A human telomerase holoenzyme protein required for cajal body localization and telomere synthesis. Science 2009, 323, 644–648. [Google Scholar] [CrossRef] [Green Version]
- Nykamp, K.; Anderson, M.; Powers, M.; Garcia, J.; Herrera, B.; Ho, Y.-Y.; Kobayashi, Y.; Patil, N.; Thusberg, J.; Westbrook, M.; et al. Sherloc: A comprehensive refinement of the ACMG–AMP variant classification criteria. Genet. Med. 2017, 19, 1105–1117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toledo, R.A.; Burnichon, N.; Cascon, A.; Benn, D.E.; Bayley, J.P.; Welander, J.; Tops, C.M.; Firth, H.; Dwight, T.; Ercolino, T.; et al. Consensus statement on next-generation-sequencing-based diagnostic testing of hereditary phaeochromocytomas and paragangliomas. Nat. Rev. Endocrinol. 2017, 13, 233–247. [Google Scholar] [CrossRef]
- Trapnell, C.; Pachter, L.; Salzberg, S.L. TopHat: Discovering splice junctions with RNA-Seq. Bioinformatics 2009, 25, 1105–1111. [Google Scholar] [CrossRef] [PubMed]
- Graña, O.; Rubio-Camarillo, M.; Fdez-Riverola, F.; Pisano, D.G.; Glez-Peña, D. Nextpresso: Next generation sequencing expression analysis pipeline. Curr. Bioinform. 2017, 13, 583–591. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Rubio-Camarillo, M.; López, G.G.; Fernández, J.M.; Valencia, A.; Pisano, D. RUbioSeq: A suite of parallelized pipelines to automate exome variation and bisulfite-seq analyses. Bioinformatics 2013, 29, 1687–1689. [Google Scholar] [CrossRef] [Green Version]
- Poplin, R.; Ruano-Rubio, V.; DePristo, M.; Fennell, T.; Carneiro, M.; van der Auwera, G.; Kling, D.; Gauthier, L.; Levy-Moonshine, A.; Roazen, D.; et al. Scaling accurate genetic variant discovery to tens of thousands of samples. bioRxiv 2017. [Google Scholar] [CrossRef] [Green Version]
- Cibulskis, K.; Lawrence, M.S.; Carter, S.L.; Sivachenko, A.; Jaffe, D.; Sougnez, C.; Gabriel, S.; Meyerson, M.; Lander, E.S.; Getz, G. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat. Biotechnol. 2013, 31, 213–219. [Google Scholar] [CrossRef]
- Gao, J.; Wan, C.; Zhang, H.; Li, A.; Zang, Q.; Ban, R.; Ali, A.; Yu, Z.; Shi, Q.; Jiang, X.; et al. Anaconda: An automated pipeline for somatic copy number variation detection and annotation from tumor exome sequencing data. BMC Bioinform. 2017, 18, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Mermel, C.H.; Schumacher, S.E.; Hill, B.; Meyerson, M.L.; Beroukhim, R.; Getz, G. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol. 2011, 12, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Cascón, A.; Comino-Méndez, I.; Currás-Freixes, M.; de Cubas, A.A.; Contreras, L.; Richter, S.; Peitzsch, M.; Mancikova, V.; Inglada-Pérez, L.; Pérez-Barrios, A.; et al. Whole-exome sequencing identifies MDH2 as a new familial paraganglioma gene. J. Natl. Cancer Inst. 2015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Cubas, A.A.; Korpershoek, E.; Inglada-Pérez, L.; Letouzé, E.; Currás-Freixes, M.; Fernández, A.F.; Comino-Méndez, I.; Schiavi, F.; Mancikova, V.; Eisenhofer, G.; et al. DNA methylation profiling in pheochromocytoma and paraganglioma reveals diagnostic and prognostic markers. Clin. Cancer Res. 2015, 21, 3020–3030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Remacha, L.; Currás-Freixes, M.; Torres-Ruiz, R.; Schiavi, F.; Torres-Pérez, R.; Calsina, B.; Letón, R.; Comino-Méndez, I.; Roldán-Romero, J.M.; Montero-Conde, C.; et al. Gain-of-function mutations in DNMT3A in patients with paraganglioma. Genet. Med. 2018, 20, 1644–1651. [Google Scholar] [CrossRef] [PubMed]
- Remacha, L.; Pirman, D.; Mahoney, C.E.; Coloma, J.; Calsina, B.; Currás-Freixes, M.; Letón, R.; Torres-Pérez, R.; Richter, S.; Pita, G.; et al. Recurrent germline DLST mutations in individuals with multiple pheochromocytomas and paragangliomas. Am. J. Hum. Genet. 2019, 104, 651–664. [Google Scholar] [CrossRef] [Green Version]
- Calsina, B.; Castro-Vega, L.J.; Torres-Pérez, R.; Inglada-Pérez, L.; Currás-Freixes, M.; Roldán-Romero, J.M.; Mancikova, V.; Letón, R.; Remacha, L.; Santos, M.; et al. Integrative multi-omics analysis identifies a prognostic MiRNA signature and a targetable MiR-21-3p/TSC2/MTOR axis in metastatic pheochromocytoma/paraganglioma. Theranostics 2019, 9, 4946–4958. [Google Scholar] [CrossRef]
- Bankhead, P.; Loughrey, M.B.; Fernández, J.A.; Dombrowski, Y.; McArt, D.G.; Dunne, P.D.; McQuaid, S.; Gray, R.T.; Murray, L.J.; Coleman, H.G.; et al. QuPath: Open source software for digital pathology image analysis. Sci. Rep. 2017, 7, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Torres-Ruiz, R.; Martinez-Lage, M.; Martin, M.C.; Garcia, A.; Bueno, C.; Castaño, J.; Ramirez, J.C.; Menendez, P.; Cigudosa, J.C.; Rodriguez-Perales, S. Efficient recreation of t(11;22) EWSR1-FLI1+ in human stem cells using CRISPR/Cas9. Stem Cell Rep. 2017, 8, 1408–1420. [Google Scholar] [CrossRef] [Green Version]
- Hayer, A.; Shao, L.; Chung, M.; Joubert, L.M.; Yang, H.W.; Tsai, F.C.; Bisaria, A.; Betzig, E.; Meyer, T. Engulfed cadherin fingers are polarized junctional structures between collectively migrating endothelial cells. Nat. Cell Biol. 2016, 18, 1311–1323. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Samper, E.; Flores, J.M.; Blasco, M.A. Restoration of telomerase activity rescues chromosomal instability and premature aging in Terc−/− mice with short telomeres. EMBO Rep. 2001, 2, 800–807. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.D.; Leão, R.; Komosa, M.; Gallo, M.; Zhang, C.H.; Lipman, T.; Remke, M.; Heidari, A.; Nunes, N.M.; Apolónio, J.D.; et al. DNA hypermethylation within TERT promoter upregulates TERT expression in cancer. J. Clin. Investig. 2019, 129, 223–229. [Google Scholar] [CrossRef]
- de Nonneville, A.; Reddel, R.R. Alternative lengthening of telomeres is not synonymous with mutations in ATRX/DAXX. Nat. Commun. 2021, 12, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Amorim, J.P.; Santos, G.; Vinagre, J.; Soares, P. The role of ATRX in the alternative lengthening of telomeres (ALT) phenotype. Genes 2016, 7, 66. [Google Scholar] [CrossRef] [PubMed]
- Cesare, A.J.; Reddel, R.R. Alternative lengthening of telomeres: Models, mechanisms and implications. Nat. Rev. Genet. 2010, 11, 319–330. [Google Scholar] [CrossRef]
- Klöppel, G.; Couvelard, A.; Hruban, R.H.; Klimstra, D.S.; Komminoth, P.; Osamura, R.Y.; Perren, A.; Rindi, G. Neoplasms of the neuroendocrine pancreas. In WHO Classification of Tumours of the Endocrine Organs; WHO/IARC Classification of Tumours; Lioyd, R.V., Osamura, R.Y., Kloppel, G., Eds.; IARC Press: Lyon, France, 2017; Volume 10, pp. 210–239. ISBN 978-92-832-4493-6. [Google Scholar]
- Barthel, F.P.; Wei, W.; Tang, M.; Martinez-Ledesma, E.; Hu, X.; Amin, S.B.; Akdemir, K.C.; Seth, S.; Song, X.; Wang, Q.; et al. Systematic analysis of telomere length and somatic alterations in 31 cancer types. Nat. Genet. 2017, 49, 349–357. [Google Scholar] [CrossRef] [PubMed]
- Stern, J.L.; Theodorescu, D.; Vogelstein, B.; Papadopoulos, N.; Cech, T.R. Mutation of the TERT promoter, switch to active chromatin, and monoallelic TERT expression in multiple cancers. Genes Dev. 2015, 29, 2219. [Google Scholar] [CrossRef] [Green Version]
- Bell, R.J.A.; Rube, H.T.; Xavier-Magalhães, A.; Costa, B.M.; Mancini, A.; Song, J.S.; Costello, J.F. Understanding TERT promoter mutations: A common path to immortality. Mol. Cancer Res. 2016, 14, 315–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, T.; Brown, T.C.; Juhlin, C.C.; Andreasson, A.; Wang, N.; Bäckdahl, M.; Healy, J.M.; Prasad, M.L.; Korah, R.; Carling, T.; et al. The activating TERT promoter mutation C228T is recurrent in subsets of adrenal tumors. Endocr. Relat. Cancer 2014, 21, 427–434. [Google Scholar] [CrossRef]
- Gupta, S.; Vanderbilt, C.M.; Lin, Y.-T.; Benhamida, J.K.; Jungbluth, A.A.; Rana, S.; Momeni-Boroujeni, A.; Chang, J.C.; Mcfarlane, T.; Salazar, P.; et al. A pan-cancer study of somatic TERT promoter mutations and amplification in 30,773 tumors profiled by clinical genomic sequencing. J. Mol. Diagnostics 2021, 23, 253–263. [Google Scholar] [CrossRef]
- Zhang, A.; Zheng, C.; Hou, M.; Lindvall, C.; Wallin, K.-L.; Ångström, T.; Yang, X.; Hellström, A.-C.; Blennow, E.; Björkholm, M.; et al. Amplification of the telomerase reverse transcriptase (HTERT) gene in cervical carcinomas. Genes Chromosom. Cancer 2002, 34, 269–275. [Google Scholar] [CrossRef]
- Dagg, R.A.; Pickett, H.A.; Neumann, A.A.; Napier, C.E.; Henson, J.D.; Teber, E.T.; Arthur, J.W.; Reynolds, C.P.; Murray, J.; Haber, M.; et al. Extensive proliferation of human cancer cells with ever-shorter telomeres. Cell Rep. 2017, 19, 2544–2556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viceconte, N.; Dheur, M.S.; Majerova, E.; Pierreux, C.E.; Baurain, J.F.; van Baren, N.; Decottignies, A. Highly aggressive metastatic melanoma cells unable to maintain telomere length. Cell Rep. 2017, 19, 2529–2543. [Google Scholar] [CrossRef] [Green Version]
- Papathomas, T.G.; Oudijk, L.; Zwarthoff, E.C.; Post, E.; Duijkers, F.A.; van Noesel, M.M.; Hofland, L.J.; Pollard, P.J.; Maher, E.R.; Restuccia, D.F.; et al. Telomerase reverse transcriptase promoter mutations in tumors originating from the adrenal gland and extra-adrenal paraganglia. Endocr. Relat. Cancer 2014, 21, 653–661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vinagre, J.; Almeida, A.; Pópulo, H.; Batista, R.; Lyra, J.; Pinto, V.; Coelho, R.; Celestino, R.; Prazeres, H.; Lima, L.; et al. Frequency of TERT promoter mutations in human cancers. Nat. Commun. 2013, 4, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Letouzé, E.; Martinelli, C.; Loriot, C.; Burnichon, N.; Abermil, N.; Ottolenghi, C.; Janin, M.; Menara, M.; Nguyen, A.T.; Benit, P.; et al. SDH mutations establish a hypermethylator phenotype in paraganglioma. Cancer Cell 2013, 23, 739–752. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Goldkorn, A. Telomere and telomerase therapeutics in cancer. Genes 2016, 7, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reddel, R. Telomere maintenance mechanisms in cancer: Clinical implications. Curr. Pharm. Des. 2014, 20, 6361–6374. [Google Scholar] [CrossRef] [Green Version]
- Benn, D.E.; Robinson, B.G.; Clifton-Bligh, R.J. 15 years of paraganglioma: Clinical manifestations of paraganglioma syndromes types 1–5. Endocr. Relat. Cancer 2015, 22, T91–T103. [Google Scholar] [CrossRef] [Green Version]
- Heaphy, C.M.; Subhawong, A.P.; Hong, S.M.; Goggins, M.G.; Montgomery, E.A.; Gabrielson, E.; Netto, G.J.; Epstein, J.I.; Lotan, T.L.; Westra, W.H.; et al. Prevalence of the alternative lengthening of telomeres telomere maintenance mechanism in human cancer subtypes. Am. J. Pathol. 2011, 179, 1608–1615. [Google Scholar] [CrossRef]
- Qin, J.; Autexier, C. Regulation of human telomerase RNA biogenesis and localization. RNA Biol. 2021, 18, 305–315. [Google Scholar] [CrossRef]
- Grozdanov, P.N.; Roy, S.; Kittur, N.; Meier, U.T. SHQ1 is required prior to NAF1 for assembly of H/ACA small nucleolar and telomerase RNPs. RNA 2009, 15, 1188–1197. [Google Scholar] [CrossRef] [Green Version]
- Kiss, T. Small nucleolar RNA-guided post-transcriptional modification of cellular RNAs. EMBO J. 2001, 20, 3617–3622. [Google Scholar] [CrossRef]
- Kiss, A.M.; Jády, B.E.; Darzacq, X.; Verheggen, C.; Bertrand, E.; Kiss, T. A cajal body-specific pseudouridylation guide RNA is composed of two box H/ACA SnoRNA-like domains. Nucleic Acids Res. 2002, 30, 4643–4649. [Google Scholar] [CrossRef] [Green Version]
- Mcmahon, M.; Contreras, A.; Ruggero, D. Small RNAs with big implications: New insights into H/ACA SnoRNA function and their role in human disease. Wiley Interdiscip. Rev. RNA 2015, 6, 173–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- von Stedingk, K.; Koster, J.; Piqueras, M.; Noguera, R.; Navarro, S.; Påhlman, S.; Versteeg, R.; Øra, I.; Gisselsson, D.; Lindgren, D.; et al. SnoRNPs regulate telomerase activity in neuroblastoma and are associated with poor prognosis. Transl. Oncol. 2013, 6, 447-IN6. [Google Scholar] [CrossRef] [Green Version]
- Elsharawy, K.A.; Althobiti, M.; Mohammed, O.J.; Aljohani, A.I.; Toss, M.S.; Green, A.R.; Rakha, E.A. Nucleolar protein 10 (NOP10) predicts poor prognosis in invasive breast cancer. Breast Cancer Res. Treat. 2021, 185, 615–627. [Google Scholar] [CrossRef]
- Cui, C.; Liu, Y.; Gerloff, D.; Rohde, C.; Pauli, C.; Köhn, M.; Misiak, D.; Oellerich, T.; Schwartz, S.; Schmidt, L.H.; et al. NOP10 predicts lung cancer prognosis and its associated small nucleolar RNAs drive proliferation and migration. Oncogene 2020, 40, 909–921. [Google Scholar] [CrossRef]
- Cristofari, G.; Adolf, E.; Reichenbach, P.; Sikora, K.; Terns, R.M.; Terns, M.P.; Lingner, J. Human telomerase RNA accumulation in cajal bodies facilitates telomerase recruitment to telomeres and telomere elongation. Mol. Cell 2007, 27, 882–889. [Google Scholar] [CrossRef]
- Ghanim, G.E.; Fountain, A.J.; van Roon, A.-M.M.; Rangan, R.; Das, R.; Collins, K.; Nguyen, T.H.D. Structure of human telomerase holoenzyme with bound telomeric DNA. Nature 2021, 593, 449–453. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | Patients |
|---|---|
| CNIO series (n = 149) | |
| Gender | |
| Female | 54.4% (81) |
| Male | 43% (64) |
| Unknown | 2.7% (4) |
| Age at initial diagnosis of PCC/PGL; (range) in years | |
| 45 (9–82) | |
| Cluster | |
| C1A | 40.3% (55) |
| C1B | 9.4% (14) |
| C2 | 36.9% (54) |
| C3 | 4.7% (7) |
| WT | 13.4% (19) |
| Clinical behavior | |
| Metastatic | 34.6% (47) |
| Synchronous | 19.9% (28) |
| Metachronous | 14.7% (19) |
| Clinically aggressive | 3.2% (5) |
| Non-metastatic | 62.2% (97) |
| Driver | |
| SDHB | 21.2% (30) |
| Tumor type | |
| PCC | 54.4% (81) |
| PGL | 29.5% (44) |
| Bilateral PCC | 3.4% (5) |
| Multiple PGL | 3.4% (5) |
| PCC+PGL | 7.4% (11) |
| Unknown | 2% (3) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Monteagudo, M.; Martínez, P.; Leandro-García, L.J.; Martínez-Montes, Á.M.; Calsina, B.; Pulgarín-Alfaro, M.; Díaz-Talavera, A.; Mellid, S.; Letón, R.; Gil, E.; et al. Analysis of Telomere Maintenance Related Genes Reveals NOP10 as a New Metastatic-Risk Marker in Pheochromocytoma/Paraganglioma. Cancers 2021, 13, 4758. https://doi.org/10.3390/cancers13194758
Monteagudo M, Martínez P, Leandro-García LJ, Martínez-Montes ÁM, Calsina B, Pulgarín-Alfaro M, Díaz-Talavera A, Mellid S, Letón R, Gil E, et al. Analysis of Telomere Maintenance Related Genes Reveals NOP10 as a New Metastatic-Risk Marker in Pheochromocytoma/Paraganglioma. Cancers. 2021; 13(19):4758. https://doi.org/10.3390/cancers13194758
Chicago/Turabian StyleMonteagudo, María, Paula Martínez, Luis J. Leandro-García, Ángel M. Martínez-Montes, Bruna Calsina, Marta Pulgarín-Alfaro, Alberto Díaz-Talavera, Sara Mellid, Rocío Letón, Eduardo Gil, and et al. 2021. "Analysis of Telomere Maintenance Related Genes Reveals NOP10 as a New Metastatic-Risk Marker in Pheochromocytoma/Paraganglioma" Cancers 13, no. 19: 4758. https://doi.org/10.3390/cancers13194758
APA StyleMonteagudo, M., Martínez, P., Leandro-García, L. J., Martínez-Montes, Á. M., Calsina, B., Pulgarín-Alfaro, M., Díaz-Talavera, A., Mellid, S., Letón, R., Gil, E., Pérez-Martínez, M., Megías, D., Torres-Ruiz, R., Rodriguez-Perales, S., González, P., Caleiras, E., Jiménez-Villa, S., Roncador, G., Álvarez-Escolá, C., ... Robledo, M. (2021). Analysis of Telomere Maintenance Related Genes Reveals NOP10 as a New Metastatic-Risk Marker in Pheochromocytoma/Paraganglioma. Cancers, 13(19), 4758. https://doi.org/10.3390/cancers13194758