
Overcoming Immunotherapy Resistance by Targeting the Tumor-Intrinsic NLRP3-HSP70 Signaling Axis

 , , and
, , and
Abstract
:Simple Summary
Abstract
1. Introduction
2. NLRP3 in Cancer
3. Tumor-Intrinsic NLRP3 and Its Regulation
4. NLRP3 and Anti-Tumor Immunity
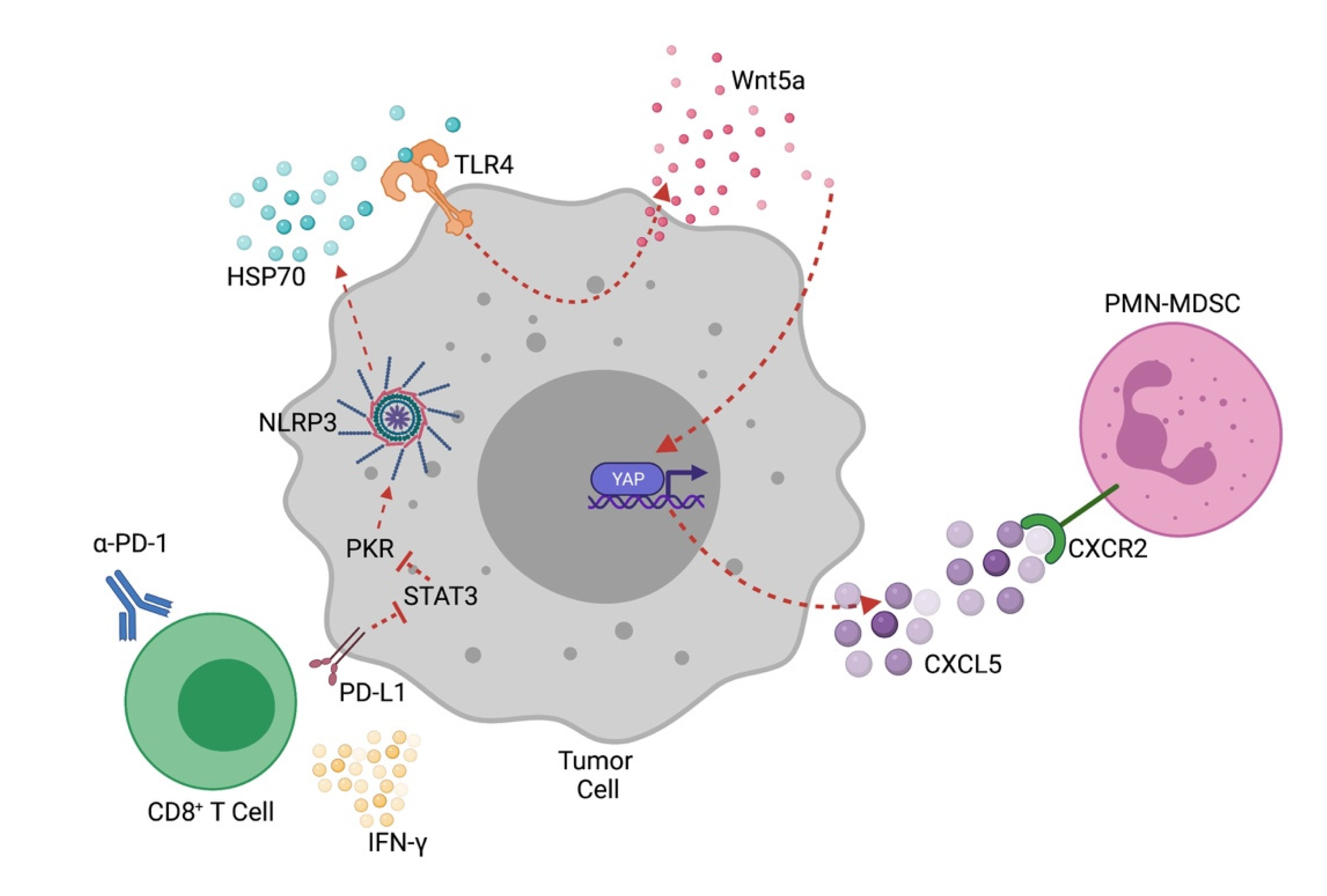
5. Tumor-Intrinsic NLRP3 and Adaptive Immunotherapy Resistance
6. Monitoring Tumor-Intrinsic NLRP3
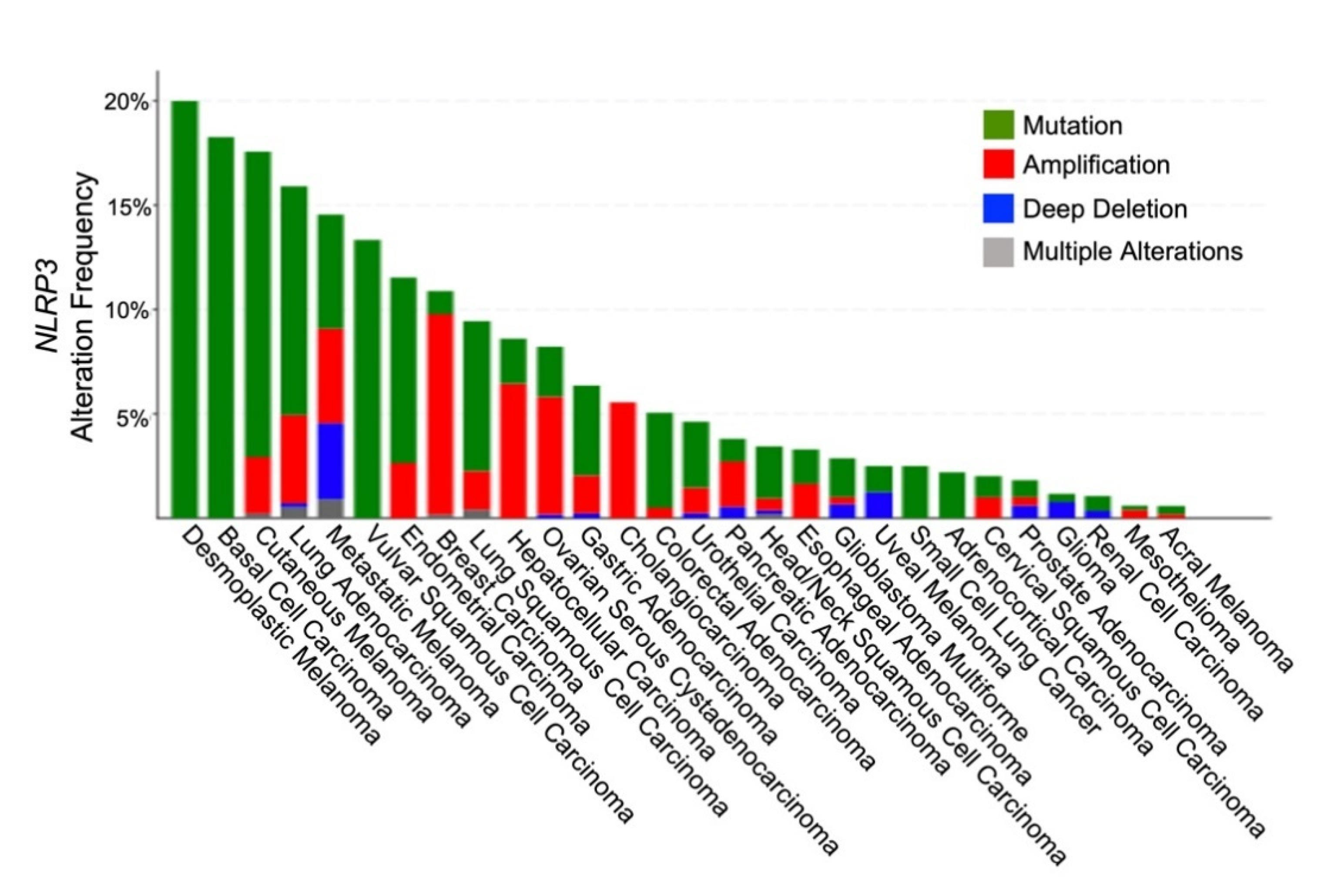
7. Genetics of Tumor-Intrinsic NLRP3
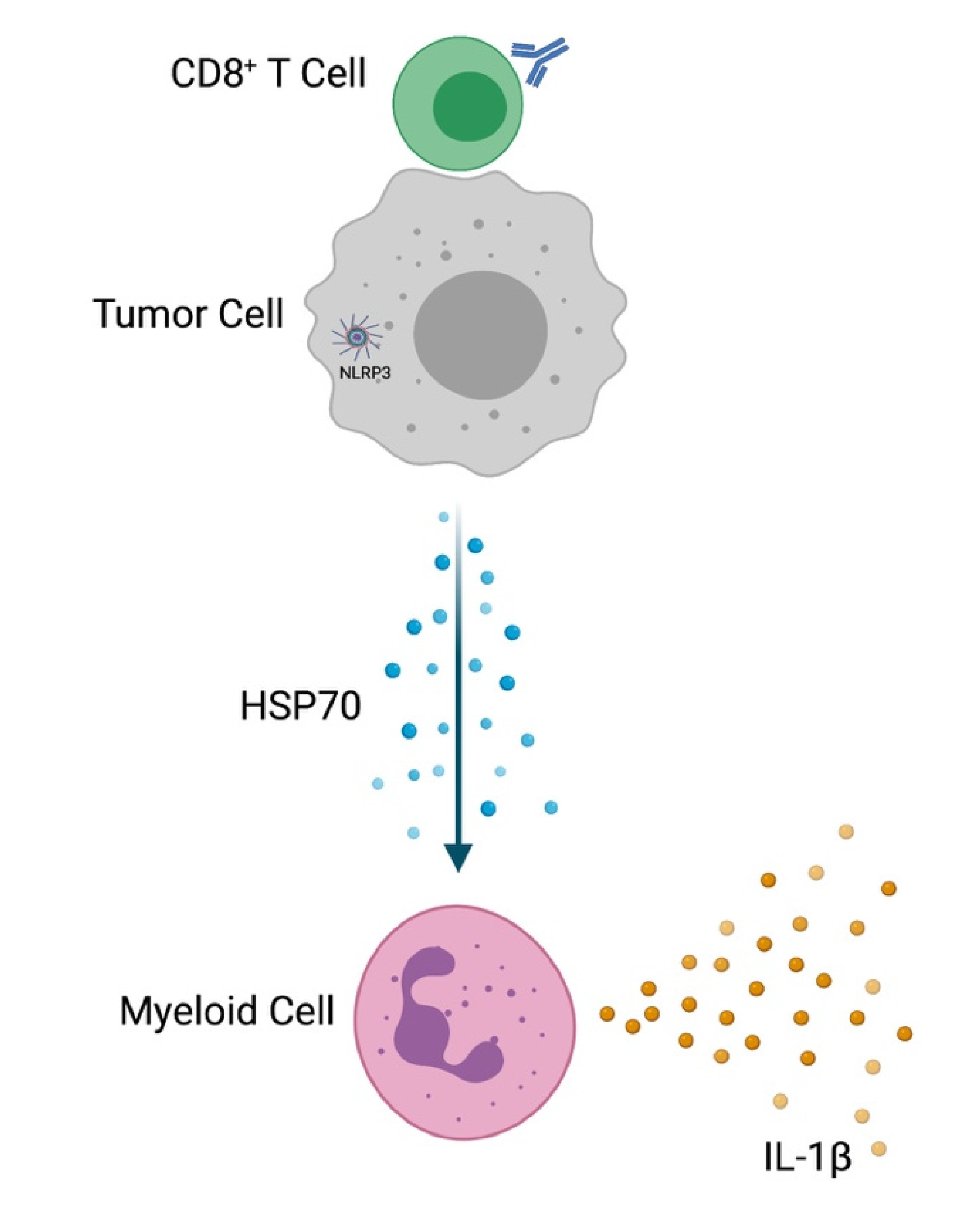
8. Role of HSP70 as a Mediator of Tumor-Intrinsic NLRP3
9. NLRP3 and HSP70 Inhibitors in Development
10. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| CAF | cancer-associated fibroblast |
| CANTOS | Canakinumab Anti-Inflammatory Thrombosis Outcome Study |
| DAMPs | danger-associated molecular patterns |
| EMT | epithelial-mesenchymal transition |
| HSP70 | heat shock protein-70 |
| IL-1β | interleukin-1 beta |
| IL-18 | interleukin-18 |
| NACHT | nucleotide-binding and oligomerization |
| NEK7 | never in mitosis A (NIMA)-related kinase 7 |
| NLRP3 | NOD-like receptor family, pyrin-domain-containing-3 |
| PD-L1 | programmed death-ligand-1 |
| PKR | RNA-dependent protein kinase |
| PMN-MDSC | granulocytic myeloid-derived suppressor cell |
| ROS | reactive oxygen species |
| TCGA | The cancer genome atlas |
| TLR4 | toll-like receptor-4 |
| TME | tumor microenvironment |
| YAP | Yes-associated protein |
References
- Booshehri, L.M.; Hoffman, H.M. CAPS and NLRP3. J. Clin. Immunol. 2019, 39, 277–286. [Google Scholar] [CrossRef]
- Menu, P.; Vince, J.E. The NLRP3 inflammasome in health and disease: The good, the bad and the ugly. Clin. Exp. Immunol. 2011, 166, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhivaki, D.; Kagan, J.C. NLRP3 inflammasomes that induce antitumor immunity. Trends Immunol. 2021, 42, 575–589. [Google Scholar] [CrossRef] [PubMed]
- Swanson, K.V.; Deng, M.; Ting, J.P. The NLRP3 inflammasome: Molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 2019, 19, 477–489. [Google Scholar] [CrossRef]
- Willingham, S.B.; Allen, I.C.; Bergstralh, D.T.; Brickey, W.J.; Huang, M.T.; Taxman, D.J.; Duncan, J.A.; Ting, J.P. NLRP3 (NALP3, Cryopyrin) facilitates in vivo caspase-1 activation, necrosis, and HMGB1 release via inflammasome-dependent and -independent pathways. J. Immunol. 2009, 183, 2008–2015. [Google Scholar] [CrossRef] [PubMed]
- Sharma, B.R.; Kanneganti, T.D. NLRP3 inflammasome in cancer and metabolic diseases. Nat. Immunol. 2021, 22, 550–559. [Google Scholar] [CrossRef]
- He, Y.; Hara, H.; Nunez, G. Mechanism and Regulation of NLRP3 Inflammasome Activation. Trends Biochem. Sci. 2016, 41, 1012–1021. [Google Scholar] [CrossRef] [Green Version]
- Touitou, I.; Lesage, S.; McDermott, M.; Cuisset, L.; Hoffman, H.; Dode, C.; Shoham, N.; Aganna, E.; Hugot, J.P.; Wise, C.; et al. Infevers: An evolving mutation database for auto-inflammatory syndromes. Hum. Mutat. 2004, 24, 194–198. [Google Scholar] [CrossRef]
- Caseley, E.A.; Immunome Project Consortium for Autoinflammatory Disorders (ImmunAID); Poulter, J.A.; Rodrigues, F.; McDermott, M.F. Inflammasome inhibition under physiological and pharmacological conditions. Genes Immun. 2020, 21, 211–223. [Google Scholar] [CrossRef]
- Marchetti, C.; Swartzwelter, B.; Koenders, M.I.; Azam, T.; Tengesdal, I.W.; Powers, N.; de Graaf, D.M.; Dinarello, C.A.; Joosten, L.A.B. NLRP3 inflammasome inhibitor OLT1177 suppresses joint inflammation in murine models of acute arthritis. Arthritis Res. Ther. 2018, 20, 169. [Google Scholar] [CrossRef] [Green Version]
- Missiroli, S.; Perrone, M.; Boncompagni, C.; Borghi, C.; Campagnaro, A.; Marchetti, F.; Anania, G.; Greco, P.; Fiorica, F.; Pinton, P.; et al. Targeting the NLRP3 Inflammasome as a New Therapeutic Option for Overcoming Cancer. Cancers 2021, 13, 2297. [Google Scholar] [CrossRef]
- Hu, H.; Wang, Y.; Ding, X.; He, Y.; Lu, Z.; Wu, P.; Tian, L.; Yuan, H.; Liu, D.; Shi, G.; et al. Long non-coding RNA XLOC_000647 suppresses progression of pancreatic cancer and decreases epithelial-mesenchymal transition-induced cell invasion by down-regulating NLRP3. Mol. Cancer 2018, 17, 18. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Kong, H.; Zeng, X.; Liu, W.; Wang, Z.; Yan, X.; Wang, H.; Xie, W. Activation of NLRP3 inflammasome enhances the proliferation and migration of A549 lung cancer cells. Oncol. Rep. 2016, 35, 2053–2064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Luo, Q.; Feng, X.; Zhang, R.; Li, J.; Chen, F. NLRP3 promotes tumor growth and metastasis in human oral squamous cell carcinoma. BMC Cancer 2018, 18, 500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, F.; Wei, B.; Lan, T.; Xiao, Y.; Quan, X.; Chen, J.; Zhao, C.; Gao, J. Low NLRP3 expression predicts a better prognosis of colorectal cancer. Biosci. Rep. 2021, 41. [Google Scholar] [CrossRef] [PubMed]
- Zou, J.; Yang, Y.; Yang, Y.; Liu, X. Polydatin suppresses proliferation and metastasis of non-small cell lung cancer cells by inhibiting NLRP3 inflammasome activation via NF-kappaB pathway. Biomed. Pharmacother. 2018, 108, 130–136. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.; Zhang, S.; Zhang, Q.; Kang, L.; Ma, C.; Feng, L.; Li, S.; Li, J.; Yang, L.; Liu, J.; et al. Resveratrol inhibits tumor progression by down-regulation of NLRP3 in renal cell carcinoma. J. Nutr. Biochem. 2020, 85, 108489. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Yin, J.J.; Miao, J.X.; Li, S.G.; Huang, C.Z.; Huang, N.; Fan, T.L.; Li, X.N.; Wang, Y.H.; Han, S.N.; et al. Activation of NLRP3 inflammasome promotes the proliferation and migration of esophageal squamous cell carcinoma. Oncol. Rep. 2020, 43, 1113–1124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Zhang, H.; Xu, Y.; Peng, T.; Meng, X.; Zou, F. NLRP3 induces the autocrine secretion of IL-1beta to promote epithelial-mesenchymal transition and metastasis in breast cancer. Biochem. Biophys. Res. Commun. 2021, 560, 72–79. [Google Scholar] [CrossRef]
- Yang, D.; Cao, X.; Wang, F.; Jiang, H.; Feng, D.; Guo, H.; Du, L.; Jin, Y.; Chen, Y.; Yin, X.; et al. LFG-500, a novel synthetic flavonoid, suppresses epithelial-mesenchymal transition in human lung adenocarcinoma cells by inhibiting NLRP3 in inflammatory microenvironment. Cancer Lett. 2017, 400, 137–148. [Google Scholar] [CrossRef]
- Hamarsheh, S.; Zeiser, R. NLRP3 Inflammasome Activation in Cancer: A Double-Edged Sword. Front. Immunol. 2020, 11, 1444. [Google Scholar] [CrossRef]
- Wang, B.; Li, H.; Wang, X.; Zhu, X. The association of aberrant expression of NLRP3 and p-S6K1 in colorectal cancer. Pathol. Res. Pract. 2020, 216, 152737. [Google Scholar] [CrossRef]
- Xue, Y.; Du, H.D.; Tang, D.; Zhang, D.; Zhou, J.; Zhai, C.W.; Yuan, C.C.; Hsueh, C.Y.; Li, S.J.; Heng, Y.; et al. Correlation Between the NLRP3 Inflammasome and the Prognosis of Patients with LSCC. Front. Oncol. 2019, 9, 588. [Google Scholar] [CrossRef] [PubMed]
- Kan, Z.; Jaiswal, B.S.; Stinson, J.; Janakiraman, V.; Bhatt, D.; Stern, H.M.; Yue, P.; Haverty, P.M.; Bourgon, R.; Zheng, J.; et al. Diverse somatic mutation patterns and pathway alterations in human cancers. Nature 2010, 466, 869–873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.S.; Mendiratta, S.; Kim, J.; Pecot, C.V.; Larsen, J.E.; Zubovych, I.; Seo, B.Y.; Kim, J.; Eskiocak, B.; Chung, H.; et al. Systematic identification of molecular subtype-selective vulnerabilities in non-small-cell lung cancer. Cell 2013, 155, 552–566. [Google Scholar] [CrossRef] [Green Version]
- Basiorka, A.A.; McGraw, K.L.; Eksioglu, E.A.; Chen, X.; Johnson, J.; Zhang, L.; Zhang, Q.; Irvine, B.A.; Cluzeau, T.; Sallman, D.A.; et al. The NLRP3 inflammasome functions as a driver of the myelodysplastic syndrome phenotype. Blood 2016, 128, 2960–2975. [Google Scholar] [CrossRef]
- Xia, X.; Wang, X.; Cheng, Z.; Qin, W.; Lei, L.; Jiang, J.; Hu, J. The role of pyroptosis in cancer: Pro-cancer or pro-“host”? Cell Death Dis. 2019, 10, 650. [Google Scholar] [CrossRef] [Green Version]
- Ershaid, N.; Sharon, Y.; Doron, H.; Raz, Y.; Shani, O.; Cohen, N.; Monteran, L.; Leider-Trejo, L.; Ben-Shmuel, A.; Yassin, M.; et al. NLRP3 inflammasome in fibroblasts links tissue damage with inflammation in breast cancer progression and metastasis. Nat. Commun. 2019, 10, 4375. [Google Scholar] [CrossRef] [Green Version]
- van Deventer, H.W.; Burgents, J.E.; Wu, Q.P.; Woodford, R.M.; Brickey, W.J.; Allen, I.C.; McElvania-Tekippe, E.; Serody, J.S.; Ting, J.P. The inflammasome component NLRP3 impairs antitumor vaccine by enhancing the accumulation of tumor-associated myeloid-derived suppressor cells. Cancer Res. 2010, 70, 10161–10169. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Jia, Y.; Wen, L.; Mu, W.; Wu, X.; Liu, T.; Liu, X.; Fang, J.; Luan, Y.; Chen, P.; et al. Porphyromonas gingivalis Promotes Colorectal Carcinoma by Activating the Hematopoietic NLRP3 Inflammasome. Cancer Res. 2021, 81, 2745–2759. [Google Scholar] [CrossRef] [PubMed]
- Hamarsheh, S.; Osswald, L.; Saller, B.S.; Unger, S.; De Feo, D.; Vinnakota, J.M.; Konantz, M.; Uhl, F.M.; Becker, H.; Lubbert, M.; et al. Oncogenic Kras(G12D) causes myeloproliferation via NLRP3 inflammasome activation. Nat. Commun. 2020, 11, 1659. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; An, Y.; Wang, Y.; Shen, K.; Wang, X.; Luan, W.; Ma, F.; Ni, L.; Liu, M.; Yu, L. Insulin-like growth factor-I activates NFkappaB and NLRP3 inflammatory signalling via ROS in cancer cells. Mol. Cell. Probes 2020, 52, 101583. [Google Scholar] [CrossRef]
- Chen, W.; Wei, T.; Chen, Y.; Yang, L.; Wu, X. Downregulation of IRAK1 Prevents the Malignant Behavior of Hepatocellular Carcinoma Cells by Blocking Activation of the MAPKs/NLRP3/IL-1beta Pathway. OncoTargets Ther. 2020, 13, 12787–12796. [Google Scholar] [CrossRef]
- Liu, S.G.; Wu, X.X.; Hua, T.; Xin, X.Y.; Feng, D.L.; Chi, S.Q.; Wang, X.X.; Wang, H.B. NLRP3 inflammasome activation by estrogen promotes the progression of human endometrial cancer. OncoTargets Ther. 2019, 12, 6927–6936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raut, P.K.; Kim, S.H.; Choi, D.Y.; Jeong, G.S.; Park, P.H. Growth of breast cancer cells by leptin is mediated via activation of the inflammasome: Critical roles of estrogen receptor signaling and reactive oxygen species production. Biochem. Pharmacol. 2019, 161, 73–88. [Google Scholar] [CrossRef]
- Feng, X.; Luo, Q.; Wang, H.; Zhang, H.; Chen, F. MicroRNA-22 suppresses cell proliferation, migration and invasion in oral squamous cell carcinoma by targeting NLRP3. J. Cell. Physiol. 2018, 233, 6705–6713. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Cao, G.; Zhao, G.; Wang, C.; Qin, Q. LncRNA differentiation antagonizing non-protein coding RNA promotes proliferation and invasion through regulating miR-135a/NLRP37 axis in pancreatic cancer. Investig. New Drugs 2020, 38, 714–721. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Li, H.; Zang, Y.; Wang, F. NLRP3 inflammasome inactivation driven by miR2233p reduces tumor growth and increases anticancer immunity in breast cancer. Mol. Med. Rep. 2019, 19, 2180–2188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rameshbabu, S.; Labadie, B.W.; Argulian, A.; Patnaik, A. Targeting Innate Immunity in Cancer Therapy. Vaccines 2021, 9, 138. [Google Scholar] [CrossRef]
- Lu, F.; Zhao, Y.; Pang, Y.; Ji, M.; Sun, Y.; Wang, H.; Zou, J.; Wang, Y.; Li, G.; Sun, T.; et al. NLRP3 inflammasome upregulates PD-L1 expression and contributes to immune suppression in lymphoma. Cancer Lett. 2021, 497, 178–189. [Google Scholar] [CrossRef] [PubMed]
- Chow, M.T.; Sceneay, J.; Paget, C.; Wong, C.S.; Duret, H.; Tschopp, J.; Moller, A.; Smyth, M.J. NLRP3 suppresses NK cell-mediated responses to carcinogen-induced tumors and metastases. Cancer Res. 2012, 72, 5721–5732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Huang, C.F.; Li, Y.C.; Deng, W.W.; Mao, L.; Wu, L.; Zhang, W.F.; Zhang, L.; Sun, Z.J. Blockage of the NLRP3 inflammasome by MCC950 improves anti-tumor immune responses in head and neck squamous cell carcinoma. Cell. Mol. Life Sci. 2018, 75, 2045–2058. [Google Scholar] [CrossRef] [PubMed]
- Daley, D.; Mani, V.R.; Mohan, N.; Akkad, N.; Pandian, G.; Savadkar, S.; Lee, K.B.; Torres-Hernandez, A.; Aykut, B.; Diskin, B.; et al. NLRP3 signaling drives macrophage-induced adaptive immune suppression in pancreatic carcinoma. J. Exp. Med. 2017, 214, 1711–1724. [Google Scholar] [CrossRef] [Green Version]
- Deng, Q.; Geng, Y.; Zhao, L.; Li, R.; Zhang, Z.; Li, K.; Liang, R.; Shao, X.; Huang, M.; Zuo, D.; et al. NLRP3 inflammasomes in macrophages drive colorectal cancer metastasis to the liver. Cancer Lett. 2019, 442, 21–30. [Google Scholar] [CrossRef]
- Lee, H.E.; Lee, J.Y.; Yang, G.; Kang, H.C.; Cho, Y.Y.; Lee, H.S.; Lee, J.Y. Inhibition of NLRP3 inflammasome in tumor microenvironment leads to suppression of metastatic potential of cancer cells. Sci. Rep. 2019, 9, 12277. [Google Scholar] [CrossRef] [Green Version]
- Bruchard, M.; Mignot, G.; Derangere, V.; Chalmin, F.; Chevriaux, A.; Vegran, F.; Boireau, W.; Simon, B.; Ryffel, B.; Connat, J.L.; et al. Chemotherapy-triggered cathepsin B release in myeloid-derived suppressor cells activates the Nlrp3 inflammasome and promotes tumor growth. Nat. Med. 2013, 19, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Guo, B.; Fu, S.; Zhang, J.; Liu, B.; Li, Z. Targeting inflammasome/IL-1 pathways for cancer immunotherapy. Sci. Rep. 2016, 6, 36107. [Google Scholar] [CrossRef]
- Voigt, C.; May, P.; Gottschlich, A.; Markota, A.; Wenk, D.; Gerlach, I.; Voigt, S.; Stathopoulos, G.T.; Arendt, K.A.M.; Heise, C.; et al. Cancer cells induce interleukin-22 production from memory CD4(+) T cells via interleukin-1 to promote tumor growth. Proc. Natl. Acad. Sci. USA 2017, 114, 12994–12999. [Google Scholar] [CrossRef] [Green Version]
- Ridker, P.M.; MacFadyen, J.G.; Thuren, T.; Everett, B.M.; Libby, P.; Glynn, R.J.; Group, C.T. Effect of interleukin-1beta inhibition with canakinumab on incident lung cancer in patients with atherosclerosis: Exploratory results from a randomised, double-blind, placebo-controlled trial. Lancet 2017, 390, 1833–1842. [Google Scholar] [CrossRef]
- Lee, P.H.; Yamamoto, T.N.; Gurusamy, D.; Sukumar, M.; Yu, Z.; Hu-Li, J.; Kawabe, T.; Gangaplara, A.; Kishton, R.J.; Henning, A.N.; et al. Host conditioning with IL-1beta improves the antitumor function of adoptively transferred T cells. J. Exp. Med. 2019, 216, 2619–2634. [Google Scholar] [CrossRef] [Green Version]
- Le, T.T.; Skak, K.; Schroder, K.; Schroder, W.A.; Boyle, G.M.; Pierce, C.J.; Suhrbier, A. IL-1 Contributes to the Anti-Cancer Efficacy of Ingenol Mebutate. PLoS ONE 2016, 11, e0153975. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.E.; Phan, T.X.; Nguyen, V.H.; Dinh-Vu, H.V.; Zheng, J.H.; Yun, M.; Park, S.G.; Hong, Y.; Choy, H.E.; Szardenings, M.; et al. Salmonella typhimurium Suppresses Tumor Growth via the Pro-Inflammatory Cytokine Interleukin-1beta. Theranostics 2015, 5, 1328–1342. [Google Scholar] [CrossRef] [Green Version]
- Han, C.; Godfrey, V.; Liu, Z.; Han, Y.; Liu, L.; Peng, H.; Weichselbaum, R.R.; Zaki, H.; Fu, Y.X. The AIM2 and NLRP3 inflammasomes trigger IL-1-mediated antitumor effects during radiation. Sci. Immunol. 2021, 6. [Google Scholar] [CrossRef] [PubMed]
- Theivanthiran, B.; Evans, K.S.; DeVito, N.C.; Plebanek, M.; Sturdivant, M.; Wachsmuth, L.P.; Salama, A.K.; Kang, Y.; Hsu, D.; Balko, J.M.; et al. A tumor-intrinsic PD-L1/NLRP3 inflammasome signaling pathway drives resistance to anti-PD-1 immunotherapy. J. Clin. Investig. 2020, 130, 2570–2586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, H.W.; Kim, Y.C.; Yu, B.; Moroishi, T.; Mo, J.S.; Plouffe, S.W.; Meng, Z.; Lin, K.C.; Yu, F.X.; Alexander, C.M.; et al. Alternative Wnt Signaling Activates YAP/TAZ. Cell 2015, 162, 780–794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, G.; Lu, X.; Dey, P.; Deng, P.; Wu, C.C.; Jiang, S.; Fang, Z.; Zhao, K.; Konaparthi, R.; Hua, S.; et al. Targeting YAP-Dependent MDSC Infiltration Impairs Tumor Progression. Cancer Discov. 2016, 6, 80–95. [Google Scholar] [CrossRef] [Green Version]
- Asea, A.; Rehli, M.; Kabingu, E.; Boch, J.A.; Bare, O.; Auron, P.E.; Stevenson, M.A.; Calderwood, S.K. Novel signal transduction pathway utilized by extracellular HSP70: Role of toll-like receptor (TLR) 2 and TLR4. J. Biol. Chem. 2002, 277, 15028–15034. [Google Scholar] [CrossRef] [Green Version]
- Blumenthal, A.; Ehlers, S.; Lauber, J.; Buer, J.; Lange, C.; Goldmann, T.; Heine, H.; Brandt, E.; Reiling, N. The Wingless homolog WNT5A and its receptor Frizzled-5 regulate inflammatory responses of human mononuclear cells induced by microbial stimulation. Blood 2006, 108, 965–973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mambula, S.S.; Calderwood, S.K. Heat shock protein 70 is secreted from tumor cells by a nonclassical pathway involving lysosomal endosomes. J. Immunol. 2006, 177, 7849–7857. [Google Scholar] [CrossRef] [Green Version]
- Gato-Canas, M.; Zuazo, M.; Arasanz, H.; Ibanez-Vea, M.; Lorenzo, L.; Fernandez-Hinojal, G.; Vera, R.; Smerdou, C.; Martisova, E.; Arozarena, I.; et al. PDL1 Signals through Conserved Sequence Motifs to Overcome Interferon-Mediated Cytotoxicity. Cell Rep. 2017, 20, 1818–1829. [Google Scholar] [CrossRef] [Green Version]
- Shen, S.; Niso-Santano, M.; Adjemian, S.; Takehara, T.; Malik, S.A.; Minoux, H.; Souquere, S.; Marino, G.; Lachkar, S.; Senovilla, L.; et al. Cytoplasmic STAT3 represses autophagy by inhibiting PKR activity. Mol. Cell 2012, 48, 667–680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ju, M.; Bi, J.; Wei, Q.; Jiang, L.; Guan, Q.; Zhang, M.; Song, X.; Chen, T.; Fan, J.; Li, X.; et al. Pan-cancer analysis of NLRP3 inflammasome with potential implications in prognosis and immunotherapy in human cancer. Brief. Bioinform. 2021, 22. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Network. Genomic Classification of Cutaneous Melanoma. Cell 2015, 161, 1681–1696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rayamajhi, M.; Miao, E.A. Just say NO to NLRP3. Nat. Immunol. 2013, 14, 12–14. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, Z.F.; Jin, X.R.; Lin, J.J.; Zhang, X.; Liu, Y.; Xu, H.L.; Xie, D.Y. NALP3 orchestrates cellular bioenergetics to facilitate non-small cell lung cancer cell growth. Life Sci. 2020, 241, 117165. [Google Scholar] [CrossRef]
- Verma, D.; Bivik, C.; Farahani, E.; Synnerstad, I.; Fredrikson, M.; Enerback, C.; Rosdahl, I.; Soderkvist, P. Inflammasome polymorphisms confer susceptibility to sporadic malignant melanoma. Pigment. Cell Melanoma Res. 2012, 25, 506–513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ungerback, J.; Belenki, D.; Jawad ul-Hassan, A.; Fredrikson, M.; Fransen, K.; Elander, N.; Verma, D.; Soderkvist, P. Genetic variation and alterations of genes involved in NFkappaB/TNFAIP3- and NLRP3-inflammasome signaling affect susceptibility and outcome of colorectal cancer. Carcinogenesis 2012, 33, 2126–2134. [Google Scholar] [CrossRef] [Green Version]
- Vande Walle, L.; Stowe, I.B.; Sacha, P.; Lee, B.L.; Demon, D.; Fossoul, A.; Van Hauwermeiren, F.; Saavedra, P.H.V.; Simon, P.; Subrt, V.; et al. MCC950/CRID3 potently targets the NACHT domain of wild-type NLRP3 but not disease-associated mutants for inflammasome inhibition. PLoS Biol. 2019, 17, e3000354. [Google Scholar] [CrossRef] [Green Version]
- Radons, J. The human HSP70 family of chaperones: Where do we stand? Cell Stress Chaperones 2016, 21, 379–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tengesdal, I.W.; Menon, D.R.; Osborne, D.G.; Neff, C.P.; Powers, N.E.; Gamboni, F.; Mauro, A.G.; D’Alessandro, A.; Stefanoni, D.; Henen, M.A.; et al. Targeting tumor-derived NLRP3 reduces melanoma progression by limiting MDSCs expansion. Proc. Natl. Acad. Sci. USA 2021, 118. [Google Scholar] [CrossRef]
- Okamoto, M.; Liu, W.; Luo, Y.; Tanaka, A.; Cai, X.; Norris, D.A.; Dinarello, C.A.; Fujita, M. Constitutively active inflammasome in human melanoma cells mediating autoinflammation via caspase-1 processing and secretion of interleukin-1beta. J. Biol. Chem. 2010, 285, 6477–6488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwaid, A.G.; Spencer, K.B. Strategies for Targeting the NLRP3 Inflammasome in the Clinical and Preclinical Space. J. Med. Chem. 2021, 64, 101–122. [Google Scholar] [CrossRef]
- Vong, C.T.; Tseng, H.H.L.; Yao, P.; Yu, H.; Wang, S.; Zhong, Z.; Wang, Y. Specific NLRP3 inflammasome inhibitors: Promising therapeutic agents for inflammatory diseases. Drug Discov. Today 2021, 26, 1394–1408. [Google Scholar] [CrossRef] [PubMed]
- Coll, R.C.; Robertson, A.A.; Chae, J.J.; Higgins, S.C.; Munoz-Planillo, R.; Inserra, M.C.; Vetter, I.; Dungan, L.S.; Monks, B.G.; Stutz, A.; et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat. Med. 2015, 21, 248–255. [Google Scholar] [CrossRef] [Green Version]
- He, H.; Jiang, H.; Chen, Y.; Ye, J.; Wang, A.; Wang, C.; Liu, Q.; Liang, G.; Deng, X.; Jiang, W.; et al. Oridonin is a covalent NLRP3 inhibitor with strong anti-inflammasome activity. Nat. Commun. 2018, 9, 2550. [Google Scholar] [CrossRef] [Green Version]
- Jin, H.; Tan, X.; Liu, X.; Ding, Y. Downregulation of AP-1 gene expression is an initial event in the oridonin-mediated inhibition of colorectal cancer: Studies in vitro and in vivo. J. Gastroenterol. Hepatol. 2011, 26, 706–715. [Google Scholar] [CrossRef]
- Platten, M.; Wild-Bode, C.; Wick, W.; Leitlein, J.; Dichgans, J.; Weller, M. N-[3,4-dimethoxycinnamoyl]-anthranilic acid (tranilast) inhibits transforming growth factor-beta relesase and reduces migration and invasiveness of human malignant glioma cells. Int. J. Cancer 2001, 93, 53–61. [Google Scholar] [CrossRef]
- Kluck, V.; Jansen, T.; Janssen, M.; Comarniceanu, A.; Efde, M.; Tengesdal, I.W.; Schraa, K.; Cleophas, M.C.P.; Scribner, C.L.; Skouras, D.B.; et al. Dapansutrile, an oral selective NLRP3 inflammasome inhibitor, for treatment of gout flares: An open-label, dose-adaptive, proof-of-concept, phase 2a trial. Lancet Rheumatol. 2020, 2, e270–e280. [Google Scholar] [CrossRef]
- Marchetti, C.; Swartzwelter, B.; Gamboni, F.; Neff, C.P.; Richter, K.; Azam, T.; Carta, S.; Tengesdal, I.; Nemkov, T.; D’Alessandro, A.; et al. OLT1177, a beta-sulfonyl nitrile compound, safe in humans, inhibits the NLRP3 inflammasome and reverses the metabolic cost of inflammation. Proc. Natl. Acad. Sci. USA 2018, 115, E1530–E1539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brunnert, D.; Langer, C.; Zimmermann, L.; Bargou, R.C.; Burchardt, M.; Chatterjee, M.; Stope, M.B. The heat shock protein 70 inhibitor VER155008 suppresses the expression of HSP27, HOP and HSP90beta and the androgen receptor, induces apoptosis, and attenuates prostate cancer cell growth. J. Cell. Biochem. 2020, 121, 407–417. [Google Scholar] [CrossRef] [PubMed]
- Wen, W.; Liu, W.; Shao, Y.; Chen, L. VER-155008, a small molecule inhibitor of HSP70 with potent anti-cancer activity on lung cancer cell lines. Exp. Biol. Med. 2014, 239, 638–645. [Google Scholar] [CrossRef] [PubMed]
- Ko, S.K.; Kim, J.; Na, D.C.; Park, S.; Park, S.H.; Hyun, J.Y.; Baek, K.H.; Kim, N.D.; Kim, N.K.; Park, Y.N.; et al. A small molecule inhibitor of ATPase activity of HSP70 induces apoptosis and has antitumor activities. Chem. Biol. 2015, 22, 391–403. [Google Scholar] [CrossRef] [Green Version]
- Adam, C.; Baeurle, A.; Brodsky, J.L.; Wipf, P.; Schrama, D.; Becker, J.C.; Houben, R. The HSP70 modulator MAL3-101 inhibits Merkel cell carcinoma. PLoS ONE 2014, 9, e92041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barnoud, T.; Leung, J.C.; Leu, J.I.; Basu, S.; Poli, A.N.R.; Parris, J.L.D.; Indeglia, A.; Martynyuk, T.; Good, M.; Gnanapradeepan, K.; et al. A Novel Inhibitor of HSP70 Induces Mitochondrial Toxicity and Immune Cell Recruitment in Tumors. Cancer Res. 2020, 80, 5270–5281. [Google Scholar] [CrossRef]
- Arora, N.; Alsaied, O.; Dauer, P.; Majumder, K.; Modi, S.; Giri, B.; Dudeja, V.; Banerjee, S.; Von Hoff, D.; Saluja, A. Downregulation of Sp1 by Minnelide leads to decrease in HSP70 and decrease in tumor burden of gastric cancer. PLoS ONE 2017, 12, e0171827. [Google Scholar] [CrossRef]
- Jacobson, B.A.; Chen, E.Z.; Tang, S.; Belgum, H.S.; McCauley, J.A.; Evenson, K.A.; Etchison, R.G.; Jay-Dixon, J.; Patel, M.R.; Raza, A.; et al. Triptolide and its prodrug minnelide suppress Hsp70 and inhibit in vivo growth in a xenograft model of mesothelioma. Genes Cancer 2015, 6, 144–152. [Google Scholar] [CrossRef] [Green Version]
- Ghiringhelli, F.; Apetoh, L.; Tesniere, A.; Aymeric, L.; Ma, Y.; Ortiz, C.; Vermaelen, K.; Panaretakis, T.; Mignot, G.; Ullrich, E.; et al. Activation of the NLRP3 inflammasome in dendritic cells induces IL-1beta-dependent adaptive immunity against tumors. Nat. Med. 2009, 15, 1170–1178. [Google Scholar] [CrossRef]
- Allen, I.C.; Scull, M.A.; Moore, C.B.; Holl, E.K.; McElvania-TeKippe, E.; Taxman, D.J.; Guthrie, E.H.; Pickles, R.J.; Ting, J.P. The NLRP3 inflammasome mediates in vivo innate immunity to influenza A virus through recognition of viral RNA. Immunity 2009, 30, 556–565. [Google Scholar] [CrossRef] [Green Version]
- Baylis, R.A.; Gomez, D.; Mallat, Z.; Pasterkamp, G.; Owens, G.K. The CANTOS Trial: One Important Step for Clinical Cardiology but a Giant Leap for Vascular Biology. Arterioscler. Thromb. Vasc. Biol. 2017, 37, e174–e177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruchard, M.; Rebe, C.; Derangere, V.; Togbe, D.; Ryffel, B.; Boidot, R.; Humblin, E.; Hamman, A.; Chalmin, F.; Berger, H.; et al. The receptor NLRP3 is a transcriptional regulator of TH2 differentiation. Nat. Immunol. 2015, 16, 859–870. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Target | Compound Name | Trial Phase | Indication | Status | NCT Identifier |
|---|---|---|---|---|---|
| NLRP3 | Dapansutrile (OLT1177) | I/II | w/ Pembrolizumb in Anti-PD-1-Resistant Melanoma | Not Yet Recruiting | NCT04971499 |
| Resveratrol | I | Colon cancer | Completed | NCT00256334 | |
| Resveratrol | I | Colon cancer | Completed | NCT00433576 | |
| Resveratrol | I | Colon Cancer | Completed | NCT00920803 | |
| HSP70 | Minnelide | I | Advanced GI Tumors | Completed | NCT01927965 |
| Minnelide | II | Refractory Pancreatic Adenocarcinoma | Completed | NCT03117920 | |
| Minnelide | II | Refractory Pancreatic Adenosquamous carcinoma | Recruiting | NCT04896073 | |
| Minnelide | I | Relapsed or Refractory Acute Myeloid Leukemia | Recruiting | NCT03760523 | |
| Minnelide | I | Advanced Solid Tumors | Recruiting | NCT03129139 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Theivanthiran, B.; Haykal, T.; Cao, L.; Holtzhausen, A.; Plebanek, M.; DeVito, N.C.; Hanks, B.A. Overcoming Immunotherapy Resistance by Targeting the Tumor-Intrinsic NLRP3-HSP70 Signaling Axis. Cancers 2021, 13, 4753. https://doi.org/10.3390/cancers13194753
Theivanthiran B, Haykal T, Cao L, Holtzhausen A, Plebanek M, DeVito NC, Hanks BA. Overcoming Immunotherapy Resistance by Targeting the Tumor-Intrinsic NLRP3-HSP70 Signaling Axis. Cancers. 2021; 13(19):4753. https://doi.org/10.3390/cancers13194753
Chicago/Turabian StyleTheivanthiran, Balamayooran, Tarek Haykal, Linda Cao, Alisha Holtzhausen, Michael Plebanek, Nicholas C. DeVito, and Brent A. Hanks. 2021. "Overcoming Immunotherapy Resistance by Targeting the Tumor-Intrinsic NLRP3-HSP70 Signaling Axis" Cancers 13, no. 19: 4753. https://doi.org/10.3390/cancers13194753
APA StyleTheivanthiran, B., Haykal, T., Cao, L., Holtzhausen, A., Plebanek, M., DeVito, N. C., & Hanks, B. A. (2021). Overcoming Immunotherapy Resistance by Targeting the Tumor-Intrinsic NLRP3-HSP70 Signaling Axis. Cancers, 13(19), 4753. https://doi.org/10.3390/cancers13194753