Defining the Role of GLI/Hedgehog Signaling in Chemoresistance: Implications in Therapeutic Approaches

 , and
, and 
Abstract
Simple Summary
Abstract
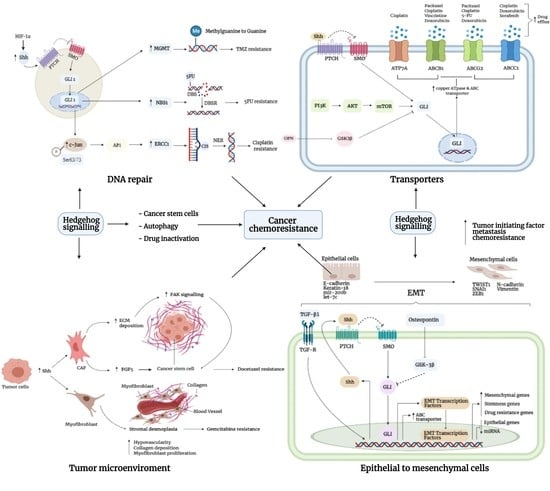
1. Introduction
2. Hedgehog Signaling
3. Mechanism of Chemoresistance
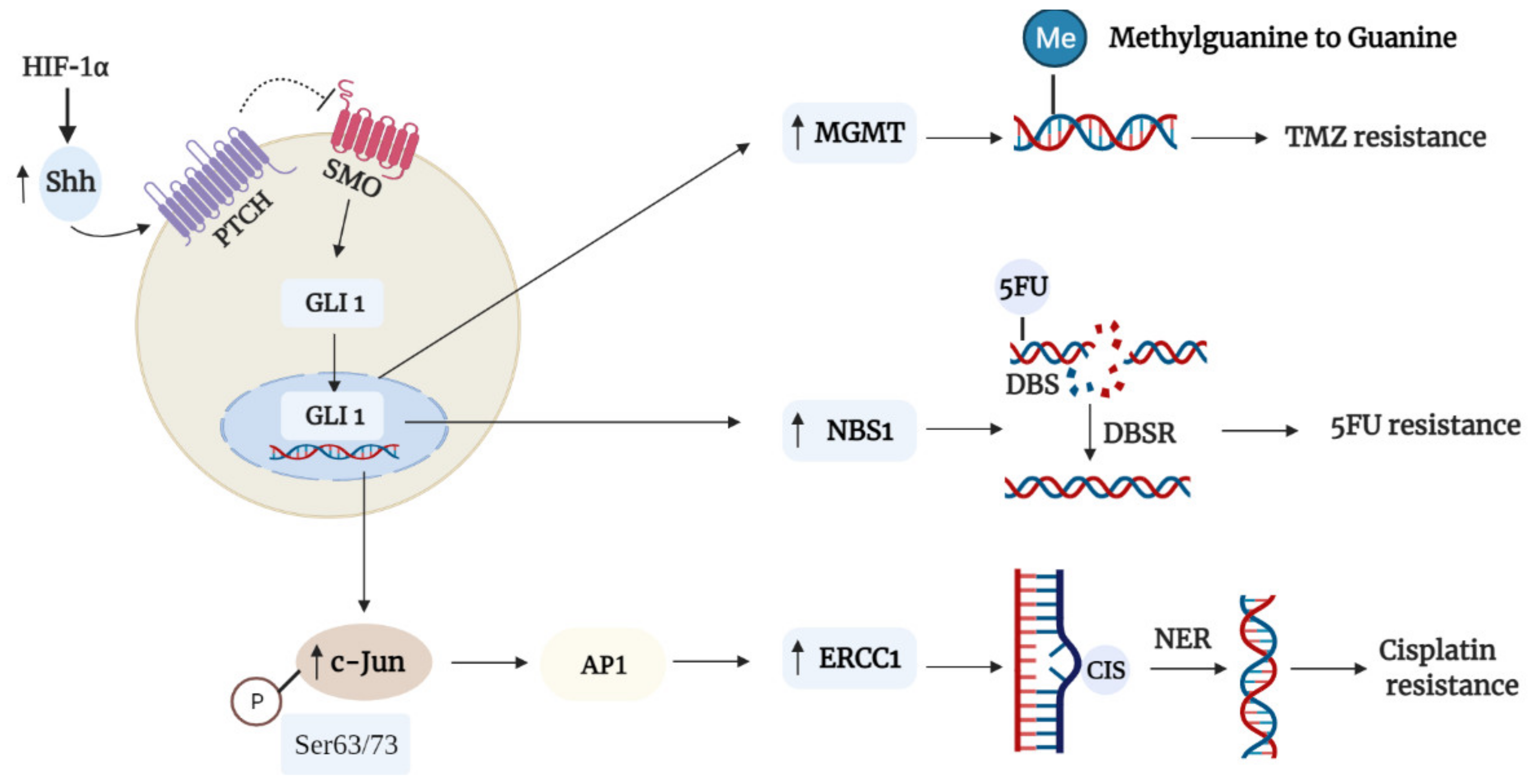
3.1. DNA Repair
3.2. Autophagy
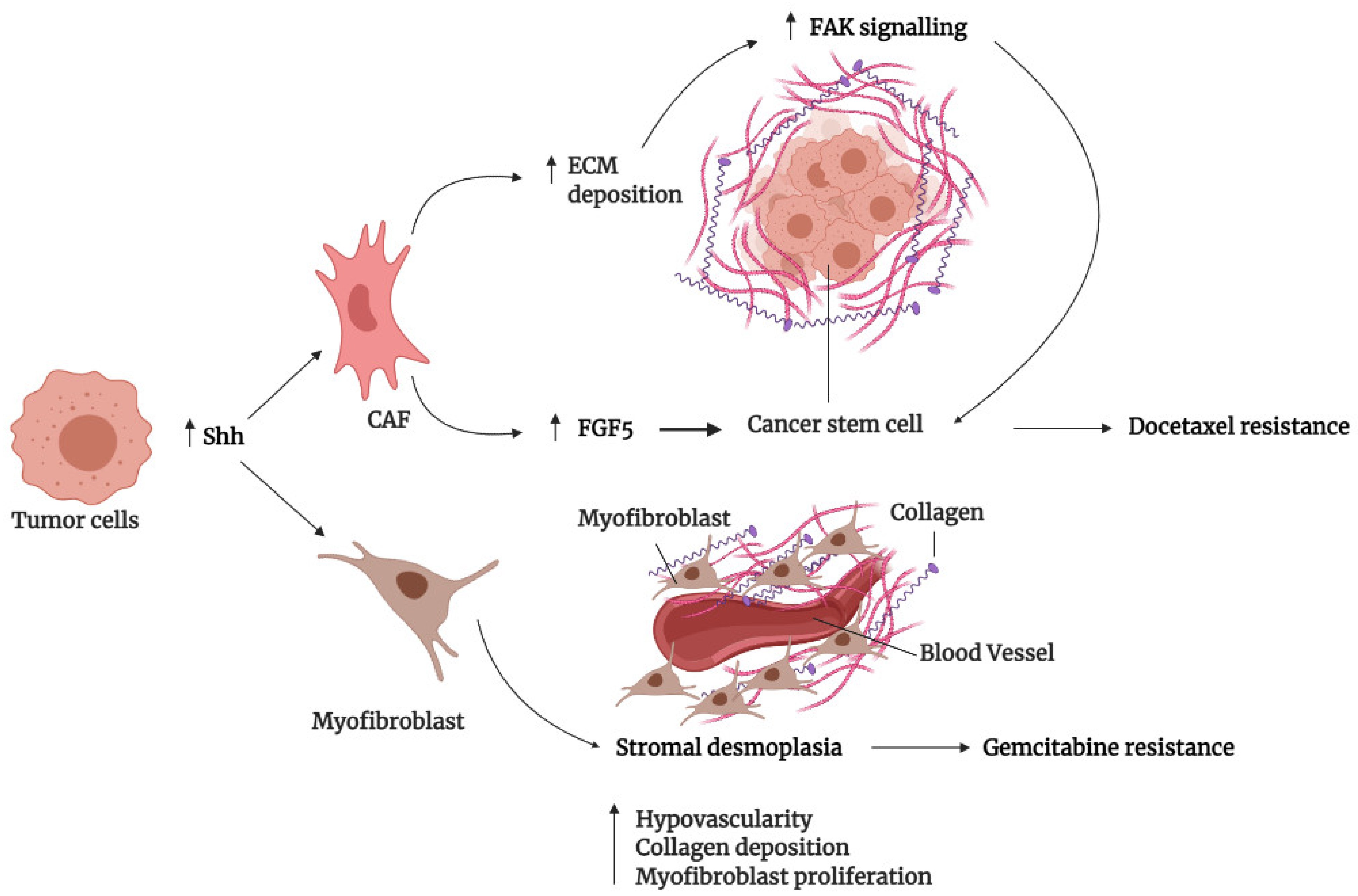
3.3. Tumor Microenvironment
3.4. Drug Inactivation
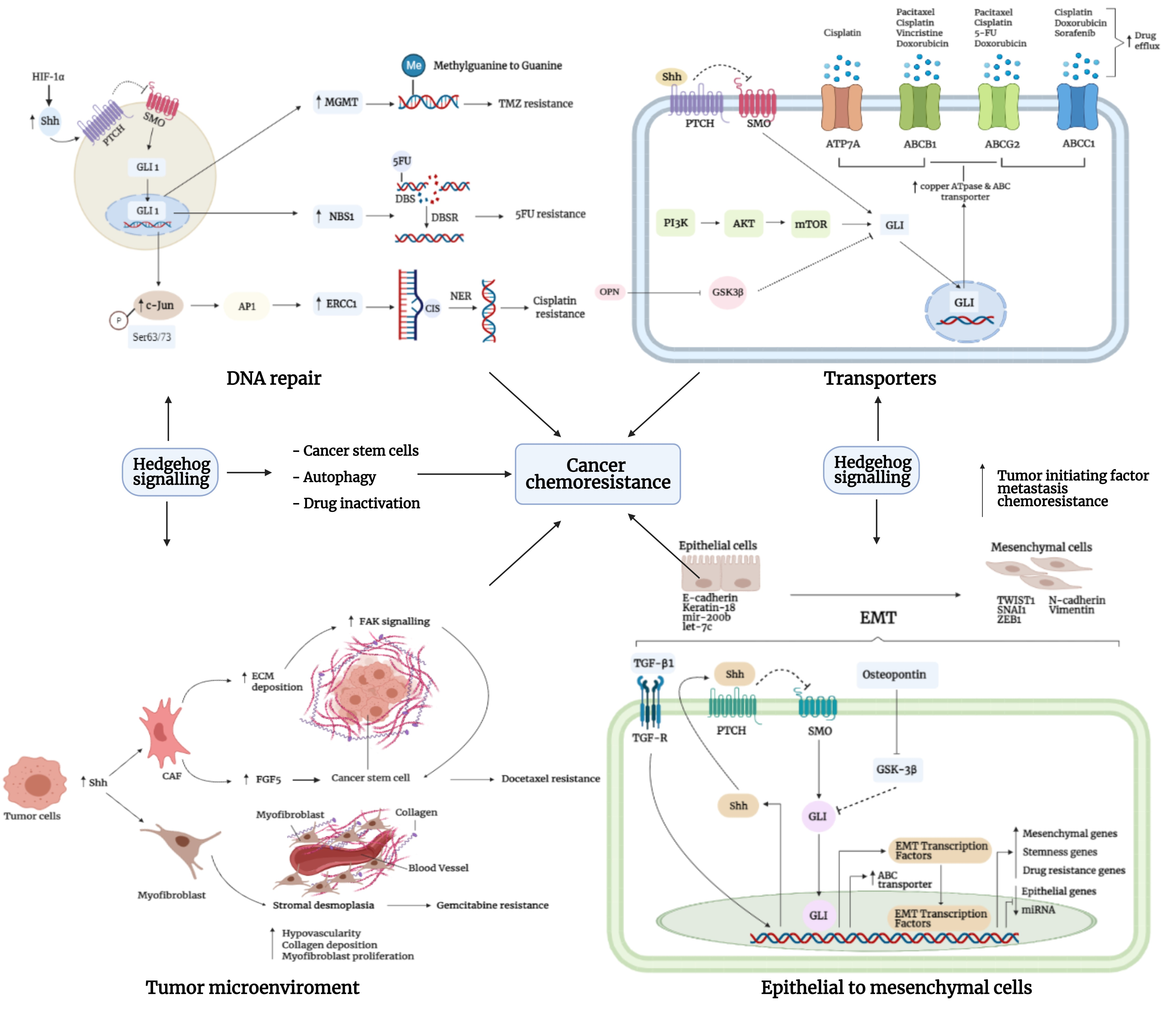
3.5. Transporters
3.6. Epithelial-to-Mesenchymal Interaction
3.7. Cancer Stem Cells
3.8. Section Summary
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global Cancer Statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- André, T.; Boni, C.; Mounedji-Boudiaf, L.; Navarro, M.; Tabernero, J.; Hickish, T.; Topham, C.; Zaninelli, M.; Clingan, P.; Bridgewater, J.; et al. Oxaliplatin, fluorouracil, and leucovorin as adjuvant treatment for colon cancer. N. Engl. J. Med. 2004, 350, 2343–2351. [Google Scholar] [CrossRef] [PubMed]
- Thompson, A.M.; Moulder-Thompson, S.L. Neoadjuvant treatment of breast cancer. Ann. Oncol. 2012, 23, 231. [Google Scholar] [CrossRef] [PubMed]
- D’Alterio, C.; Scala, S.; Sozzi, G.; Roz, L.; Bertolini, G. Paradoxical effects of chemotherapy on tumor relapse and metastasis promotion. Semin. Cancer Biol. 2020, 60, 351–361. [Google Scholar] [CrossRef]
- Madden, E.C.; Gorman, A.M.; Logue, S.E.; Samali, A. Tumour cell secretome in chemoresistance and tumour recurrence. Trends Cancer 2020, 6, 489–505. [Google Scholar] [CrossRef] [PubMed]
- Bailey, A.M.; Mao, Y.; Zeng, J.; Holla, V.; Johnson, A.; Brusco, L.; Chen, K.; Mendelsohn, J.; Routbort, M.J.; Mills, G.B.; et al. Implementation of biomarker-driven cancer therapy: Existing tools and remaining gaps. Discov. Med. 2014, 17, 101–114. [Google Scholar]
- Liu, D. Cancer biomarkers for targeted therapy. Biomark. Res. 2019, 7, 101–114. [Google Scholar] [CrossRef]
- Mokhtari, R.B.; Homayouni, T.S.; Baluch, N.; Morgatskaya, E.; Kumar, S.; Das, B.; Yeger, H. Combination therapy in combating cancer. Oncotarget 2017, 8, 38022–38043. [Google Scholar] [CrossRef]
- Kukcinaviciute, E.; Jonusiene, V.; Sasnauskiene, A.; Dabkeviciene, D.; Eidenaite, E.; Laurinavicius, A. Significance of notch and Wnt signaling for chemoresistance of colorectal cancer cells HCT. J. Cell. Biochem. 2018, 119, 5913–5920. [Google Scholar] [CrossRef] [PubMed]
- Yuan, S.; Tao, F.; Zhang, X.; Zhang, Y.; Sun, X.; Wu, D. Role of Wnt/ β-Catenin signaling in the chemoresistance modulation of colorectal cancer. Biomed Res. Int. 2020, 2020, 9390878. [Google Scholar] [CrossRef] [PubMed]
- Lv, Y.; Cang, W.; Li, Q.; Liao, X.; Zhan, M.; Deng, H.; Li, S.; Jin, W.; Pang, Z.; Qiu, X.; et al. Erlotinib overcomes paclitaxel-resistant cancer stem cells by blocking the EGFR-CREB/GRβ-IL-6 axis in MUC1-positive cervical cancer. Oncogenesis 2019, 8, 70. [Google Scholar] [CrossRef] [PubMed]
- Hopper-Borge, E.A.; Nasto, R.E.; Ratushny, V.; Weiner, L.M.; Golemis, E.A.; Astsaturov, I. Mechanisms of tumor resistance to EGFR-targeted therapies. Expert Opin. Ther. Targets 2009, 13, 339–362. [Google Scholar] [CrossRef] [PubMed]
- West, K.A.; Castillo, S.S.; Dennis, P.A. Activation of the PI3K/Akt pathway and chemotherapeutic resistance. Drug Resist. Updat. 2002, 5, 234–248. [Google Scholar] [CrossRef]
- Deng, J.; Bai, X.; Feng, X.; Ni, J.; Beretov, J.; Graham, P.; Li, Y. Inhibition of PI3K/Akt/MTOR signaling pathway alleviates ovarian cancer chemoresistance through reversing epithelial-mesenchymal transition and decreasing cancer stem cell marker expression. BMC Cancer 2019, 19, 618. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Shen, S.; Guo, J.; Chen, H.; Yu Greenblatt, D.; Kleeff, J.; Liao, Q.; Chen, G.; Friess, H.; Sing Leung, P. Mitogen-activated protein kinases and chemoresistance in pancreatic cancer cells. J. Surg. Res. 2006, 136, 325–335. [Google Scholar] [CrossRef] [PubMed]
- Salaroglio, I.C.; Mungo, E.; Gazzano, E.; Kopecka, J.; Riganti, C. ERK is a pivotal player of chemo-immune-resistance in cancer. Int. J. Mol. Sci. 2019, 20, 2505. [Google Scholar] [CrossRef]
- Godwin, P.; Baird, A.M.; Heavey, S.; Barr, M.P.; O’Byrne, K.J.; Gately, K. Targeting nuclear factor-kappa B to overcome resistance to chemotherapy. Front. Oncol. 2013, 3, 120. [Google Scholar] [CrossRef]
- Li, Q.; Yang, G.; Feng, M.; Zheng, S.; Cao, Z.; Qiu, J.; You, L.; Zheng, L.; Hu, Y.; Zhang, T.; et al. NF-ΚB in pancreatic cancer: Its key role in chemoresistance. Cancer Lett. 2018, 421, 127–134. [Google Scholar] [CrossRef]
- Skoda, A.M.; Simovic, D.; Karin, V.; Kardum, V.; Vranic, S.; Serman, L. The role of the hedgehog signaling pathway in cancer: A comprehensive review. Bosn. J. Basic Med. Sci. 2018, 18, 8–20. [Google Scholar] [CrossRef]
- Sari, I.N.; Phi, L.T.H.; Jun, N.; Wijaya, Y.T.; Lee, S.; Kwon, H.Y. Hedgehog signaling in cancer: A prospective therapeutic target for eradicating cancer stem cells. Cells 2018, 7, 208. [Google Scholar] [CrossRef]
- Lupi, O. Correlations between the sonic hedgehog pathway and basal cell carcinoma. Int. J. Dermatol. 2007, 46, 1113–1117. [Google Scholar] [CrossRef] [PubMed]
- Otsuka, A.; Levesque, M.P.; Dummer, R.; Kabashima, K. Hedgehog signaling in basal cell carcinoma. J. Dermatol. Sci. 2015, 78, 95–100. [Google Scholar] [CrossRef] [PubMed]
- Bakshi, A.; Chaudhary, S.C.; Rana, M.; Elmets, C.A.; Athar, M. Basal cell carcinoma pathogenesis and therapy involving hedgehog signaling and beyond. Mol. Carcinog. 2017, 56, 2543–2557. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.Y.; Yang, J.Y. Targeting the hedgehog pathway in pediatric medulloblastoma. Cancers 2015, 7, 2110–2123. [Google Scholar] [CrossRef]
- Yoon, J.W.; Gilbertson, R.; Iannaccone, S.; Iannaccone, P.; Walterhouse, D. Defining a role for sonic hedgehog pathway activation in desmoplastic medulloblastoma by identifying GLI1 target genes. Int. J. Cancer 2009, 124, 109–119. [Google Scholar] [CrossRef] [PubMed]
- Brancaccio, G.; Pea, F.; Moscarella, E.; Argenziano, G. Sonidegib for the treatment of advanced basal cell carcinoma. Front. Oncol. 2020, 10, 582866. [Google Scholar] [CrossRef]
- Sandhiya, S.; Melvin, G.; Kumar, S.S.; Dkhar, S.A. The dawn of hedgehog inhibitors: Vismodegib. J. Pharmacol. Pharmacother. 2013, 4, 4–7. [Google Scholar] [CrossRef]
- Catenacci, D.V.T.; Junttila, M.R.; Karrison, T.; Bahary, N.; Horiba, M.N.; Nattam, S.R.; Marsh, R.; Wallace, J.; Kozloff, M.; Rajdev, L.; et al. Randomized phase Ib/II study of gemcitabine plus placebo or vismodegib, a hedgehog pathway inhibitor, in patients with metastatic pancreatic cancer. J. Clin. Oncol. 2015, 33, 4284–4292. [Google Scholar] [CrossRef]
- Xie, H.; Paradise, B.D.; Ma, W.W.; Fernandez-Zapico, M.E. Recent advances in the clinical targeting of hedgehog/GLI signaling in cancer. Cells 2019, 8, 394. [Google Scholar] [CrossRef]
- Li, Y.; Song, Q.; Day, B.W. Phase I and phase II Sonidegib and Vismodegib clinical trials for the treatment of paediatric and adult MB patients: A systemic review and meta-analysis. Acta Neuropathol. Commun. 2019, 7, 123. [Google Scholar] [CrossRef]
- Gould, S.E.; Low, J.A.; Marsters, J.C.; Robarge, K.; Rubin, L.L.; De Sauvage, F.J.; Sutherlin, D.P.; Wong, H.; Yauch, R.L. Discovery and preclinical development of Vismodegib. Expert Opin. Drug Discov. 2014, 9, 969–984. [Google Scholar] [CrossRef] [PubMed]
- Queiroz, K.; Ruela-de-Sousa, R.; Fuhler, G.; Aberson, H.; Ferreira, C.; Peppelenbosch, M.; Spek, C. Hedgehog signaling maintains chemoresistance in myeloid leukemic cells. Oncogene 2010, 29, 6314–6322. [Google Scholar] [CrossRef] [PubMed]
- Nüsslein-volhard, C.; Wieschaus, E. Mutations affecting segment number and polarity in drosophila. Nature 1980, 287, 795–801. [Google Scholar] [CrossRef] [PubMed]
- Le, H.; Kleinerman, R.; Lerman, O.Z.; Brown, D.; Galiano, R.; Gurtner, G.C.; Warren, S.M.; Levine, J.P.; Saadeh, P.B. Hedgehog signaling is essential for normal wound healing. Wound Repair Regen. 2008, 16, 768–773. [Google Scholar] [CrossRef]
- Carballo, G.B.; Honorato, J.R.; De Lopes, G.P.F.; Spohr, T.C.L.D.S.E. A highlight on sonic hedgehog pathway. Cell Commun. Signal. 2018, 16, 11. [Google Scholar] [CrossRef]
- Merchant, J.L. Hedgehog signalling in gut development, physiology and cancer. J. Physiol. 2012, 590, 421–432. [Google Scholar] [CrossRef]
- Perler, F.B. Protein splicing of inteins and hedgehog autoproteolysis: Structure, function, and evolution. Cell 1998, 92, 1–4. [Google Scholar] [CrossRef]
- Chen, X.; Tukachinsky, H.; Huang, C.H.; Jao, C.; Chu, Y.R.; Tang, H.Y.; Mueller, B.; Schulman, S.; Rapoport, T.A.; Salic, A. Processing and turnover of the hedgehog protein in the endoplasmic reticulum. J. Cell Biol. 2011, 192, 825–838. [Google Scholar] [CrossRef]
- Buglino, J.A.; Resh, M.D. Palmitoylation of hedgehog proteins. Vitam. Horm. 2012, 88, 229–252. [Google Scholar]
- Tukachinsky, H.; Kuzmickas, R.P.; Jao, C.Y.; Liu, J.; Salic, A. Dispatched and scube mediate the efficient secretion of the cholesterol-modified hedgehog ligand. Cell Rep. 2012, 2, 308–320. [Google Scholar] [CrossRef]
- Chan, J.A.; Balasubramanian, S.; Witt, R.M.; Nazemi, K.J.; Choi, Y.; Pazyra-Murphy, M.F.; Walsh, C.O.; Thompson, M.; Segal, R.A. Proteoglycan interactions with sonic hedgehog specify mitogenic responses. Nat. Neurosci. 2009, 12, 409–417. [Google Scholar] [CrossRef] [PubMed]
- Christ, A.; Herzog, K.; Willnow, T.E. LRP2, an auxiliary receptor that controls sonic hedgehog signaling in development and disease. Dev. Dyn. 2016, 245, 569–579. [Google Scholar] [CrossRef]
- Petrov, K.; Wierbowski, B.M.; Salic, A. Sending and receiving hedgehog signals. Annu. Rev. Cell Dev. Biol. 2017, 33, 145–168. [Google Scholar] [CrossRef] [PubMed]
- Marigo, V.; Davey, R.A.; Zuo, Y.; Cunningham, J.M.; Tabin, C.J. Biochemical evidence that patched is the hedgehog receptor. Nature 1996, 384, 176–179. [Google Scholar] [CrossRef]
- Mastronardi, F.G.; Dimitroulakos, J.; Kamel-Reid, S.; Manoukian, A.S. Co-localization of patched and activated sonic hedgehog to lysosomes in neurons. Neuroreport 2000, 11, 581–585. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, J.J.; Whalen, E.J.; Liu, R.; Xiao, K.; Kim, J.; Chen, M.; Wang, J.; Chen, W.; Lefkowitz, R.J. β-arrestin-mediated localization of smoothened to the primary cilium. Science 2008, 320, 1777–1781. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Ren, X.R.; Nelson, C.D.; Barak, L.S.; Chen, J.K.; Beachy, P.A.; De Sauvage, F.; Lefkowitz, R.J. Activity-dependent internalization of smoothened mediated by β-arrestin 2 and GRK. Science 2004, 306, 2257–2260. [Google Scholar] [CrossRef] [PubMed]
- Bangs, F.; Anderson, K.V. Primary cilia and mammalian hedgehog signaling. Cold Spring Harb. Perspect. Biol. 2017, 9, a028175. [Google Scholar] [CrossRef] [PubMed]
- Jia, J.; Tong, C.; Wang, B.; Luo, L.; Jiang, J. Hedgehog signalling activity of smoothened requires phosphorylation by protein kinase A and casein kinase I. Nature 2004, 432, 1045–1050. [Google Scholar] [CrossRef]
- Chen, Y.; Jiang, J. Decoding the phosphorylation code in hedgehog signal transduction. Cell Res. 2013, 23, 186–200. [Google Scholar] [CrossRef]
- Ma, G.; Li, S.; Han, Y.; Li, S.; Yue, T.; Wang, B.; Correspondence, J.J. Regulation of smoothened trafficking and hedgehog signaling by the SUMO pathway. Dev. Cell 2016, 39, 438–451. [Google Scholar] [CrossRef]
- Han, Y.; Shi, Q.; Jiang, J. Multisite interaction with sufu regulates Ci/Gli activity through distinct mechanisms in Hh signal transduction. Proc. Natl. Acad. Sci. USA 2015, 112, 6383–6388. [Google Scholar] [CrossRef]
- Shafique, S.; Rashid, S. Structural basis of ΒTrCP1-associated GLI3 processing. Sci. Rep. 2019, 9, 6865. [Google Scholar] [CrossRef] [PubMed]
- Jia, J.; Zhang, L.; Zhang, Q.; Tong, C.; Wang, B.; Hou, F.; Amanai, K.; Jiang, J. Phosphorylation by double-time/CKIε and CKIα targets cubitus interruptus for slimb/β-TRCP-mediated proteolytic processing. Dev. Cell 2005, 9, 819–830. [Google Scholar] [CrossRef] [PubMed]
- Tukachinsky, H.; Lopez, L.V.; Salic, A. A mechanism for vertebrate hedgehog signaling: Recruitment to cilia and dissociation of SuFu-Gli protein complexes. J. Cell Biol. 2010, 191, 415–428. [Google Scholar] [CrossRef]
- Humke, E.W.; Dorn, K.V.; Milenkovic, L.; Scott, M.P.; Rohatgi, R. The output of hedgehog signaling is controlled by the dynamic association between suppressor of fused and the gli proteins. Genes Dev. 2010, 24, 670–682. [Google Scholar] [CrossRef]
- Katoh, Y.; Katoh, M. Hedgehog target genes: Mechanisms of carcinogenesis induced by aberrant hedgehog signaling activation. Curr. Mol. Med. 2009, 9, 873–886. [Google Scholar] [CrossRef] [PubMed]
- Cochrane, C.R.; Szczepny, A.; Watkins, D.N.; Cain, J.E. Hedgehog signaling in the maintenance of cancer stem cells. Cancers 2015, 7, 1554–1585. [Google Scholar] [CrossRef]
- Niewiadomski, P.; Niedziółka, S.M.; Markiewicz, Ł.; Uśpieński, T.; Baran, B.; Chojnowska, K. Gli proteins: Regulation in development and cancer. Cells 2019, 8, 147. [Google Scholar] [CrossRef]
- De Reyniès, A.; Javelaud, D.; Elarouci, N.; Marsaud, V.; Gilbert, C.; Mauviel, A. Large-scale pan-cancer analysis reveals broad prognostic association between TGF-β ligands, not hedgehog, and GLI1/2 expression in tumors. Sci. Rep. 2020, 10, 14491. [Google Scholar] [CrossRef]
- Nolan-Stevaux, O.; Lau, J.; Truitt, M.L.; Chu, G.C.; Hebrok, M.; Fernández-Zapico, M.E.; Hanahan, D. GLI1 Is regulated through smoothened-independent mechanisms in neoplastic pancreatic ducts and mediates PDAC cell survival and transformation. Genes Dev. 2009, 23, 24–36. [Google Scholar] [CrossRef]
- Eberl, M.; Klingler, S.; Mangelberger, D.; Loipetzberger, A.; Damhofer, H.; Zoidl, K.; Schnidar, H.; Hache, H.; Bauer, H.C.; Solca, F.; et al. Hedgehog-EGFR cooperation response genes determine the oncogenic phenotype of basal cell carcinoma and tumour-initiating pancreatic cancer cells. EMBO Mol. Med. 2012, 4, 218–233. [Google Scholar] [CrossRef]
- Po, A.; Silvano, M.; Miele, E.; Capalbo, C.; Eramo, A.; Salvati, V.; Todaro, M.; Besharat, Z.M.; Catanzaro, G.; Cucchi, D.; et al. Noncanonical GLI1 signaling promotes stemness features and in vivo growth in lung adenocarcinoma. Oncogene 2017, 36, 4641–4652. [Google Scholar] [CrossRef]
- Varnat, F.; Siegl-Cachedenier, I.; Malerba, M.; Gervaz, P.; Ruiz, I.; Altaba, A. Loss of WNT-TCF Addiction and enhancement of HH-GLI1 signalling define the metastatic transition of human colon carcinomas. EMBO Mol. Med. 2010, 2, 440–457. [Google Scholar] [CrossRef]
- Noubissi, F.K.; Goswami, S.; Sanek, N.A.; Kawakami, K.; Minamoto, T.; Moser, A.; Grinblat, Y.; Spiegelman, V.S. Wnt signaling stimulates transcriptional outcome of the hedgehog pathway by stabilizing GLI1 MRNA. Cancer Res. 2009, 69, 8572–8578. [Google Scholar] [CrossRef]
- Zubčić, V.; Rinčić, N.; Kurtović, M.; Trnski, D.; Musani, V.; Ozretić, P.; Levanat, S.; Leović, D.; Sabol, M. GANT61 and lithium chloride inhibit the growth of head and neck cancer cell lines through the regulation of GLI3 processing by GSK3β. Int. J. Mol. Sci. 2020, 21, 6410. [Google Scholar] [CrossRef]
- Ozretić, P.; Trnski, D.; Musani, V.; Maurac, I.; Kalafatić, D.; Orešković, S.; Levanat, S.; Sabol, M. Non-canonical hedgehog signaling activation in ovarian borderline tumors and ovarian carcinomas. Int. J. Oncol. 2017, 51, 1869–1877. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, Y.; Matsubara, S.; Ding, Q.; Tsukasa, K.; Yoshimitsu, M.; Kosai, K.; Takao, S. Efficient elimination of pancreatic cancer stem cells by hedgehog/GLI Inhibitor GANT61 in combination with MTOR inhibition. Mol. Cancer 2016, 15, 49. [Google Scholar] [CrossRef]
- Chatterjee, N.; Walker, G.C. Mechanisms of DNA damage, repair, and mutagenesis. Environ. Mol. Mutagen. 2017, 58, 235–263. [Google Scholar] [CrossRef] [PubMed]
- An, Q.; Robins, P.; Lindahl, T.; Barnes, D.E. 5-Fluorouracil incorporated into DNA is excised by the Smug1 DNA glycosylase to reduce drug cytotoxicity. Cancer Res. 2007, 67, 940–945. [Google Scholar] [CrossRef] [PubMed]
- Meng, E.; Hanna, A.; Samant, R.S.; Shevde, L.A. The impact of hedgehog signaling pathway on DNA repair mechanisms in human cancer. Cancers 2015, 7, 1333–1348. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.Y.; Che, J.; Sun, K.K.; Shen, X.J.; Yang, D.; Zhong, N.; Zhao, H. Cyclopamine increases the radiosensitivity of human pancreatic cancer cells by regulating the DNA repair signal pathway through an epidermal growth factor receptor-dependent pathway. Mol. Med. Rep. 2013, 8, 979–983. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Cai, J.; Zhao, S.; Yao, K.; Sun, Y.; Li, Y.; Chen, L.; Li, R.; Zhai, X.; Zhang, J.; et al. GANT61, a GLI inhibitor, sensitizes glioma cells to the temozolomide treatment. J. Exp. Clin. Cancer Res. 2016, 35, 184. [Google Scholar] [CrossRef]
- Wang, K.; Chen, D.; Qian, Z.; Cui, D.; Gao, L.; Lou, M. Hedgehog/Gli1 Signaling pathway regulates MGMT expression and chemoresistance to temozolomide in human glioblastoma. Cancer Cell Int. 2017, 17, 117. [Google Scholar] [CrossRef]
- Wei, M.; Ma, R.; Huang, S.; Liao, Y.; Ding, Y.; Li, Z.; Guo, Q.; Tan, R.; Zhang, L.; Zhao, L. Oroxylin A increases the sensitivity of temozolomide on glioma cells by hypoxia-inducible factor 1α/hedgehog pathway under hypoxia. J. Cell. Physiol. 2019, 234, 17392–17404. [Google Scholar] [CrossRef]
- Lee, S.Y. Temozolomide resistance in glioblastoma multiforme. Genes Dis. 2016, 3, 198–210. [Google Scholar] [CrossRef]
- Zhu, H.; Lo, H.-W. The human glioma-associated oncogene homolog 1 (GLI1) family of transcription factors in gene regulation and diseases. Curr. Genom. 2010, 11, 238–245. [Google Scholar] [CrossRef]
- Alnaim, L. Individualization of 5-fluorouracil in the treatment of colorectal cancer. SRX Pharmacol. 2010, 2010, 1–12. [Google Scholar] [CrossRef][Green Version]
- Ho, V.; Chung, L.; Singh, A.; Lea, V.; Abubakar, A.; Lim, S.H.; Ng, W.; Lee, M.; de Souza, P.; Shin, J.S.; et al. Overexpression of the MRE11-RAD50-NBS1 (MRN) complex in rectal cancer correlates with poor response to neoadjuvant radiotherapy and prognosis. BMC Cancer 2018, 18, 869. [Google Scholar] [CrossRef]
- Zhang, L.; Song, R.; Gu, D.; Zhang, X.; Yu, B.; Liu, B.; Xie, J. The role of GLI1 for 5-Fu resistance in colorectal cancer. Cell Biosci. 2017, 7, 17. [Google Scholar] [CrossRef]
- Zhang, R.; Ma, J.; Avery, J.T.; Sambandam, V.; Nguyen, T.H.; Xu, B.; Suto, M.J.; Boohaker, R.J. GLI1 Inhibitor SRI-38832 attenuates chemotherapeutic resistance by downregulating NBS1 transcription in BRAFV600E colorectal cancer. Front. Oncol. 2020, 10, 241. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Zhang, L.; Tsang, B.; Gardner, K.; Bostick-Bruton, F.; Reed, E. Phorbol ester exposure activates an AP-1-mediated increase in ERCC-1 messenger RNA expression in human ovarian tumor cells. Cell. Mol. Life Sci. 1999, 55, 456–466. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Gardner, K.; Zhang, L.; Tsang, B.; Bostick-Brutont, F.; Reed, E. Cisplatin induction of ERCC-1 MRNA expression in A2780/CP70 human ovarian cancer cells. J. Biol. Chem. 1998, 273, 23419–23425. [Google Scholar] [CrossRef] [PubMed]
- Kudo, K.; Gavin, E.; Das, S.; Amable, L.; Shevde, L.A.; Reed, E. Inhibition of Gli1 results in altered C-jun activation, inhibition of cisplatin-induced upregulation of ERCC1, XPD and XRCC1, and inhibition of platinum-DNA adduct repair. Oncogene 2012, 31, 4718–4724. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Zhao, Z.; Yang, Y.; O’Connell, D.; Zhang, X.; Oh, S.; Ma, B.; Lee, J.H.; Zhang, T.; Varghese, B.; et al. Truncating mutation in the autophagy gene UVRAG confers oncogenic properties and chemosensitivity in colorectal cancers. Nat. Commun. 2015, 6, 7839. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Jeong, E.G.; Ahn, C.H.; Kim, S.S.; Lee, S.H.; Yoo, N.J. Frameshift mutation of UVRAG, an autophagy-related gene, in gastric carcinomas with microsatellite instability. Hum. Pathol. 2008, 39, 1059–1063. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, Y.; Coppola, D.; Matsushita, N.; Cualing, H.D.; Sun, M.; Sato, Y.; Liang, C.; Jung, J.U.; Cheng, J.Q.; Mulé, J.J.; et al. Bif-1 interacts with beclin 1 through UVRAG and regulates autophagy and tumorigenesis. Nat. Cell Biol. 2007, 9, 1142–1151. [Google Scholar] [CrossRef] [PubMed]
- Yun, C.W.; Lee, S.H. The roles of autophagy in cancer. Int. J. Mol. Sci. 2018, 19, 3466. [Google Scholar] [CrossRef]
- Degenhardt, K.; Mathew, R.; Beaudoin, B.; Bray, K.; Anderson, D.; Chen, G.; Mukherjee, C.; Shi, Y.; Gélinas, C.; Fan, Y.; et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell 2006, 10, 51–64. [Google Scholar] [CrossRef]
- Sun, T.; Liu, H.; Ming, L. Multiple roles of autophagy in the sorafenib resistance of hepatocellular carcinoma. Cell. Physiol. Biochem. 2017, 44, 716–727. [Google Scholar] [CrossRef]
- Saxena, R.; Klochkova, A.; Murray, M.G.; Kabir, M.F.; Samad, S.; Beccari, T.; Gang, J.; Patel, K.; Hamilton, K.E.; Whelan, K.A. Roles for autophagy in esophageal carcinogenesis: Implications for improving patient outcomes. Cancers 2019, 11, 1697. [Google Scholar] [CrossRef]
- Wang, J.; Wu, G.S. Role of autophagy in cisplatin resistance in ovarian cancer cells. J. Biol. Chem. 2014, 289, 17163–17173. [Google Scholar] [CrossRef] [PubMed]
- Drullion, C.; Trégoat, C.; Lagarde, V.; Tan, S.; Gioia, R.; Priault, M.; Djavaheri-Mergny, M.; Brisson, A.; Auberger, P.; Mahon, F.X.; et al. Apoptosis and autophagy have opposite roles on imatinib-induced K562 leukemia cell senescence. Cell Death Dis. 2012, 3, e373. [Google Scholar] [CrossRef] [PubMed]
- Palumbo, S.; Pirtoli, L.; Tini, P.; Cevenini, G.; Calderaro, F.; Toscano, M.; Miracco, C.; Comincini, S. Different involvement of autophagy in human malignant glioma cell lines undergoing irradiation and temozolomide combined treatments. J. Cell. Biochem. 2012, 113, 2308–2318. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Deng, L.; Chen, Q.; Wang, Y.; Xu, R.; Shi, C.; Shao, J.; Hu, G.; Gao, M.; Rao, H.; et al. Inhibition of hedgehog signaling pathway impedes cancer cell proliferation by promotion of autophagy. Eur. J. Cell Biol. 2015, 94, 223–233. [Google Scholar] [CrossRef] [PubMed]
- Lo Ré, A.E.; Fernández-Barrena, M.G.; Almada, L.L.; Mills, L.D.; Elsawa, S.F.; Lund, G.; Ropolo, A.; Molejon, M.I.; Vaccaro, M.I.; Fernandez-Zapico, M.E. Novel AKT1-GLI3-VMP1 pathway mediates KRAS oncogene-induced autophagy in cancer cells. J. Biol. Chem. 2012, 287, 25325–25334. [Google Scholar] [CrossRef]
- Amantini, C.; Morelli, M.B.; Nabissi, M.; Cardinali, C.; Santoni, M.; Gismondi, A.; Santoni, G. Capsaicin triggers autophagic cell survival which drives epithelial mesenchymal transition and chemoresistance in bladder cancer cells in an hedgehog-dependent manner. Oncotarget 2016, 7, 50180–50194. [Google Scholar] [CrossRef]
- Honorato, J.R.; Hauser-Davis, R.A.; Saggioro, E.M.; Correia, F.V.; Sales-Junior, S.F.; Soares, L.O.S.; Lima, L.D.R.; Moura-Neto, V.; Lopes, G.P.D.F.; Spohr, T.C.L.D.S. Role of sonic hedgehog signaling in cell cycle, oxidative stress, and autophagy of temozolomide resistant glioblastoma. J. Cell. Physiol. 2020, 235, 3798–3814. [Google Scholar] [CrossRef]
- Jin, M.Z.; Jin, W.L. The updated landscape of tumor microenvironment and drug repurposing. Signal Transduct. Target. Ther. 2020, 5, 166. [Google Scholar] [CrossRef]
- Lei, J.; Ma, J.; Ma, Q.; Li, X.; Liu, H.; Xu, Q.; Duan, W.; Sun, Q.; Xu, J.; Wu, Z.; et al. Hedgehog signaling regulates hypoxia induced epithelial to mesenchymal transition and invasion in pancreatic cancer cells via a ligand-independent manner. Mol. Cancer 2013, 12, 66. [Google Scholar] [CrossRef]
- Fischer, K.; Hoffmann, P.; Voelkl, S.; Meidenbauer, N.; Ammer, J.; Edinger, M.; Gottfried, E.; Schwarz, S.; Rothe, G.; Hoves, S.; et al. Inhibitory effect of tumor cell-derived lactic acid on human T cells. Blood 2007, 109, 3812–3819. [Google Scholar] [CrossRef]
- Ward, C.; Meehan, J.; Gray, M.E.; Murray, A.F.; Argyle, D.J.; Kunkler, I.H.; Langdon, S.P. The impact of tumour PH on cancer progression: Strategies for clinical intervention. Explor. Target. Antitumor Ther. 2020, 1, 71–100. [Google Scholar] [CrossRef]
- Senthebane, D.A.; Rowe, A.; Thomford, N.E.; Shipanga, H.; Munro, D.; Al Mazeedi, M.A.M.; Almazyadi, H.A.M.; Kallmeyer, K.; Dandara, C.; Pepper, M.S.; et al. The role of tumor microenvironment in chemoresistance: To survive, keep your enemies closer. Int. J. Mol. Sci. 2017, 18, 1586. [Google Scholar] [CrossRef] [PubMed]
- Hui, M.; Cazet, A.; Jessica, Y.; Cooper, C.; McFarland, A.; Nair, R.; O’Toole, S.; Swarbrick, A. Targeting the hedgehog signalling pathway in triple negative breast cancer. Ann. Oncol. 2015, 26, iii31. [Google Scholar] [CrossRef]
- Hui, M.N.; Cazet, A.; Elsworth, B.; Roden, D.; Cox, T.; Yang, J.; McFarland, A.; Deng, N.; Chan, C.-L.; O’Toole, S.; et al. Targeting the hedgehog signalling pathway in triple negative breast cancer. J. Clin. Oncol. 2018, 36, e24216. [Google Scholar] [CrossRef]
- Cazet, A.S.; Hui, M.N.; Elsworth, B.L.; Wu, S.Z.; Roden, D.; Chan, C.L.; Skhinas, J.N.; Collot, R.; Yang, J.; Harvey, K.; et al. Targeting stromal remodeling and cancer stem cell plasticity overcomes chemoresistance in triple negative breast cancer. Nat. Commun. 2018, 9, 2897. [Google Scholar] [CrossRef] [PubMed]
- Feig, C.; Gopinathan, A.; Neesse, A.; Chan, D.S.; Cook, N.; Tuveson, D.A. The pancreas cancer microenvironment. Clin. Cancer Res. 2012, 18, 4266–4276. [Google Scholar] [CrossRef]
- Thayer, S.P.; Di Magliano, M.P.; Heiser, P.W.; Nielsen, C.M.; Roberts, D.J.; Lauwers, G.Y.; Qi, Y.P.; Gysin, S.; Fernández-del Castillo, C.; Yajnik, V.; et al. Hedgehog is an early and late mediator of pancreatic cancer tumorigenesis. Nature 2003, 425, 851–856. [Google Scholar] [CrossRef]
- Hingorani, S.R.; Wang, L.; Multani, A.S.; Combs, C.; Deramaudt, T.B.; Hruban, R.H.; Rustgi, A.K.; Chang, S.; Tuveson, D.A. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell 2005, 7, 469–483. [Google Scholar] [CrossRef]
- Tian, H.; Callahan, C.A.; Dupree, K.J.; Darbonne, W.C.; Ahn, C.P.; Scales, S.J.; De Sauvage, F.J. Hedgehog signaling is restricted to the stromal compartment during pancreatic carcinogenesis. Proc. Natl. Acad. Sci. USA 2009, 106, 4254–4259. [Google Scholar] [CrossRef]
- Olive, K.P.; Jacobetz, M.A.; Davidson, C.J.; Gopinathan, A.; McIntyre, D.; Honess, D.; Madhu, B.; Goldgraben, M.A.; Caldwell, M.E.; Allard, D.; et al. Inhibition of hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science 2009, 324, 1457–1461. [Google Scholar] [CrossRef] [PubMed]
- Ge, S.; Tu, Y.; Hu, M. Challenges and opportunities with predicting in vivo phase II metabolism via glucuronidation from in vitro data. Curr. Pharmacol. Rep. 2016, 2, 326–338. [Google Scholar] [CrossRef] [PubMed]
- Allain, E.P.; Rouleau, M.; Lévesque, E.; Guillemette, C. Emerging roles for UDP-glucuronosyltransferases in drug resistance and cancer progression. Br. J. Cancer 2020, 122, 1277–1287. [Google Scholar] [CrossRef] [PubMed]
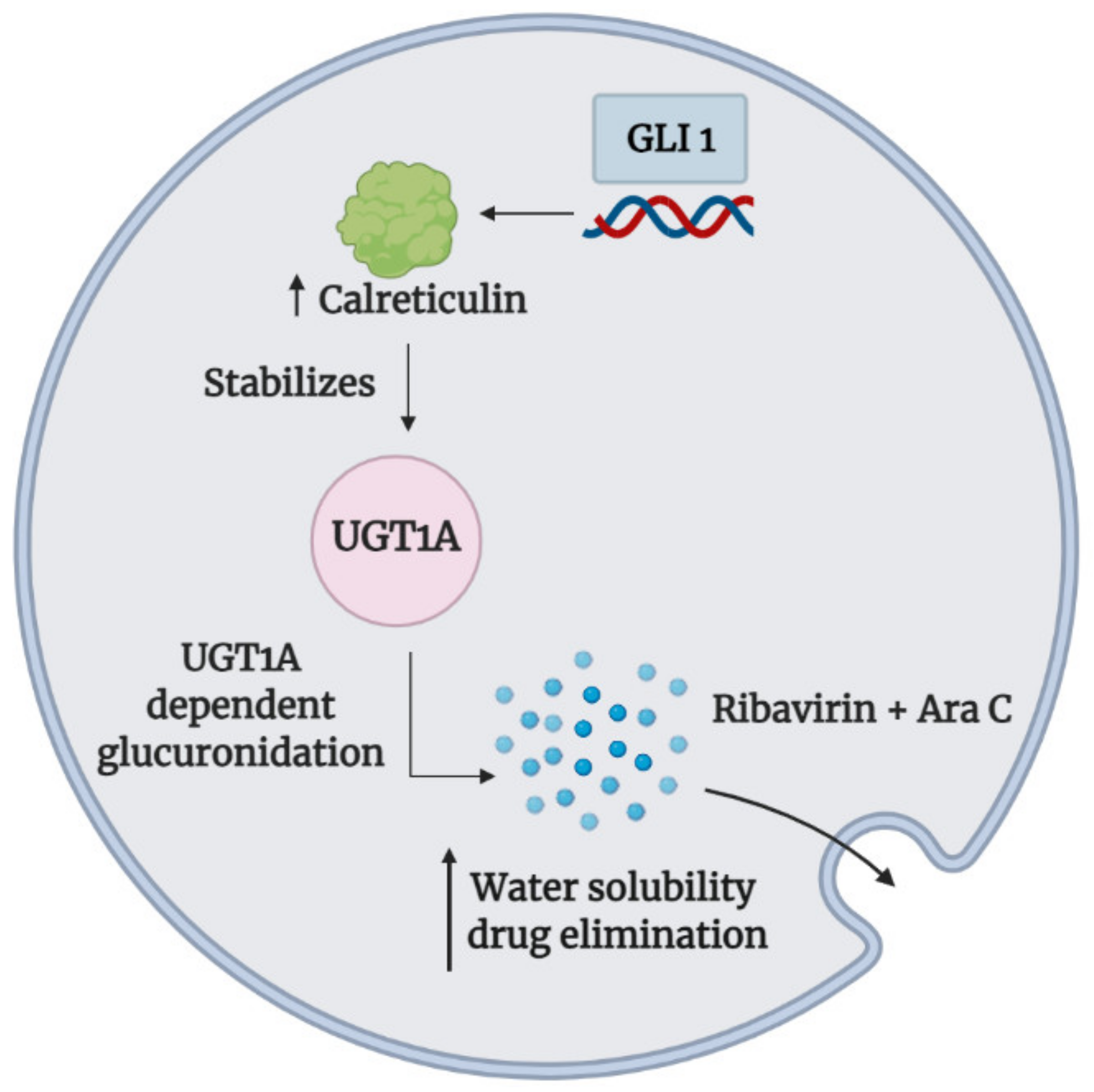
- Zahreddine, H.A.; Culjkovic-Kraljacic, B.; Assouline, S.; Gendron, P.; Romeo, A.A.; Morris, S.J.; Cormack, G.; Jaquith, J.B.; Cerchietti, L.; Cocolakis, E.; et al. The sonic hedgehog factor GLI1 imparts drug resistance through inducible glucuronidation. Nature 2014, 511, 90–93. [Google Scholar] [CrossRef]
- Kentsis, A.; Volpon, L.; Topisirovic, I.; Soll, C.E.; Culjkovic, B.; Shao, L.; Borden, K.L.B. Further evidence that ribavirin interacts with EIF4E. RNA 2005, 11, 1762–1766. [Google Scholar] [CrossRef] [PubMed]
- Kentsis, A.; Topisirovic, I.; Culjkovic, B.; Shao, L.; Borden, K.L.B. Ribavirin suppresses ElF4E-mediated oncogenic transformation by physical mimicry of the 7-Methyl Guanosine MRNA Cap. Proc. Natl. Acad. Sci. USA 2004, 101, 18105–18110. [Google Scholar] [CrossRef] [PubMed]
- Zahreddine, H.A.; Culjkovic-Kraljacic, B.; Gasiorek, J.; Duchaine, J.; Borden, K.L.B. GLI1-Inducible glucuronidation targets a broad spectrum of drugs. ACS Chem. Biol. 2019, 14, 348–355. [Google Scholar] [CrossRef]
- Ford, R.C.; Beis, K. Learning the ABCs one at a time: Structure and mechanism of ABC transporters. Biochem. Soc. Trans. 2019, 47, 23–36. [Google Scholar] [CrossRef]
- Pan, S.-T.; Li, Z.-L.; He, Z.-X.; Qiu, J.-X.; Zhou, S.-F. Molecular mechanisms for tumour resistance to chemotherapy. Clin. Exp. Pharmacol. Physiol. 2016, 43, 723–737. [Google Scholar] [CrossRef]
- Begicevic, R.R.; Falasca, M. ABC Transporters in cancer stem cells: Beyond chemoresistance. Int. J. Mol. Sci. 2017, 18, 2362. [Google Scholar] [CrossRef]
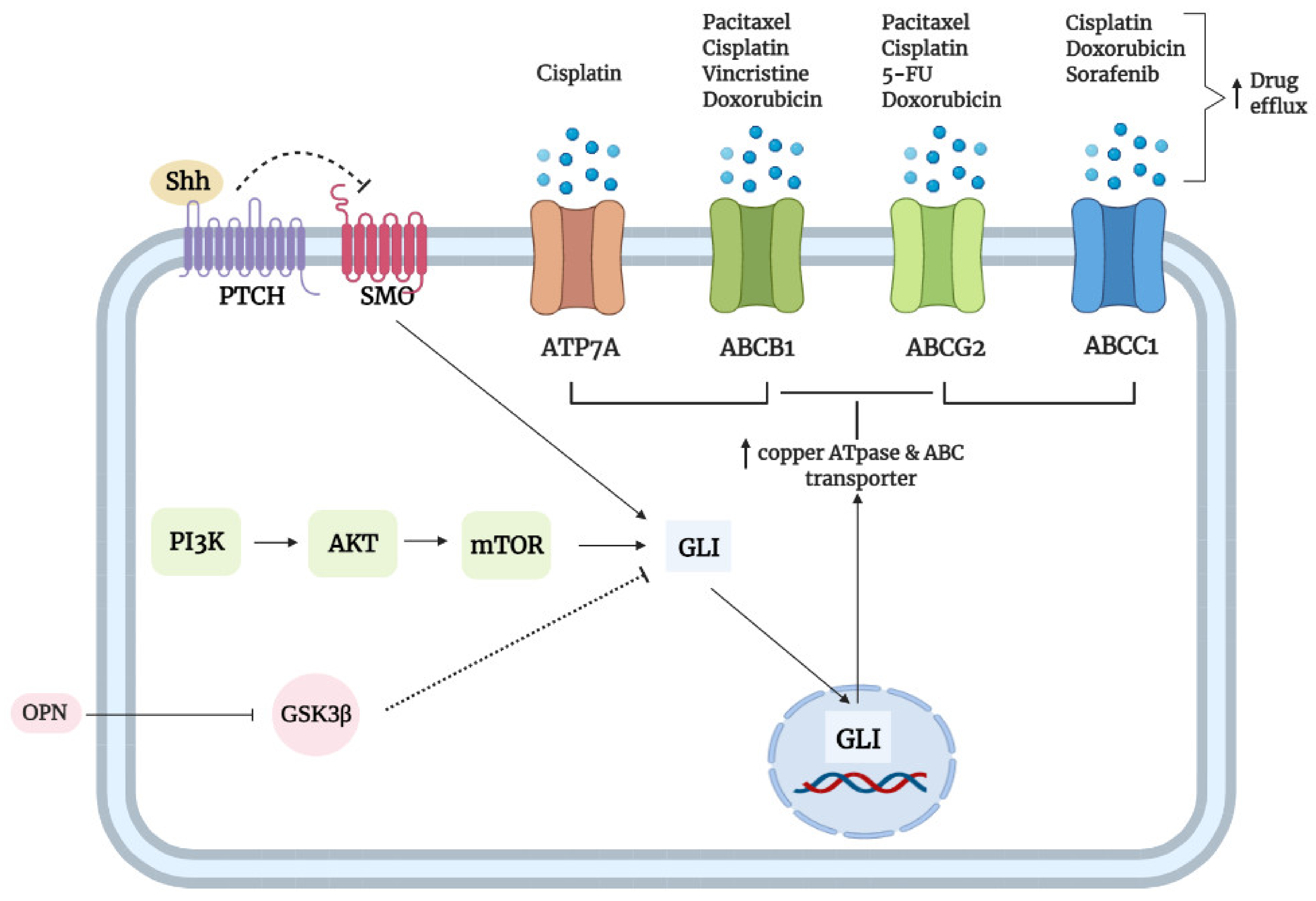
- Santisteban, M. ABC Transporters as molecular effectors of pancreatic oncogenic pathways: The hedgehog-GLI model. J. Gastrointest. Cancer 2010, 41, 153–158. [Google Scholar] [CrossRef]
- Steg, A.D.; Katre, A.A.; Bevis, K.S.; Ziebarth, A.; Dobbin, Z.C.; Shah, M.M.; Alvarez, R.D.; Landen, C.N. Smoothened antagonists reverse taxane resistance in ovarian cancer. Mol. Cancer Ther. 2012, 11, 1587–1597. [Google Scholar] [CrossRef]
- Zhang, H.; Hu, L.; Cheng, M.; Wang, Q.; Hu, X.; Chen, Q. The hedgehog signaling pathway promotes chemotherapy resistance via multidrug resistance protein 1 in ovarian cancer. Oncol. Rep. 2020, 44, 2610–2620. [Google Scholar] [CrossRef]
- Chen, Y.; Bieber, M.M.; Teng, N.N.H. Hedgehog signaling regulates drug sensitivity by targeting ABC transporters ABCB1 and ABCG2 in epithelial ovarian cancer. Mol. Carcinog. 2014, 53, 625–634. [Google Scholar] [CrossRef] [PubMed]
- Cui, W.; Fang, T.; Duan, Z.; Xiang, D.; Wang, Y.; Zhang, M.; Zhai, F.; Cui, X.; Yang, L. Dihydroartemisinin sensitizes esophageal squamous cell carcinoma to cisplatin by inhibiting sonic hedgehog signaling. Front. Cell Dev. Biol. 2020, 8596788. [Google Scholar] [CrossRef]
- Das, S.; Samant, R.S.; Shevde, L.A. Nonclassical activation of hedgehog signaling enhances multidrug resistance and makes cancer cells refractory to smoothened-targeting hedgehog inhibition. J. Biol. Chem. 2013, 288, 11824–11833. [Google Scholar] [CrossRef] [PubMed]
- Yu, B.; Gu, D.; Zhang, X.; Liu, B.; Xie, J. The role of GLI2-ABCG2 signaling axis for 5Fu resistance in gastric cancer. J. Genet. Genomics 2017, 44, 375–383. [Google Scholar] [CrossRef] [PubMed]
- Yu, B.; Gu, D.; Zhang, X.; Li, J.; Liu, B.; Xie, J. GLI1-mediated regulation of side population is responsible for drug resistance in gastric cancer. Oncotarget 2017, 8, 27412–27427. [Google Scholar] [CrossRef]
- Yao, Y.; Zhou, D.; Shi, D.; Zhang, H.; Zhan, S.; Shao, X.; Sun, K.; Sun, L.; Wu, G.; Tian, K.; et al. GLI1 Overexpression promotes gastric cancer cell proliferation and migration and induces drug resistance by combining with the AKT-MTOR pathway. Biomed. Pharmacother. 2019, 111, 993–1004. [Google Scholar] [CrossRef]
- Xu, M.; Gong, A.; Yang, H.; George, S.K.; Jiao, Z.; Huang, H.; Jiang, X.; Zhang, Y. Sonic hedgehog-glioma associated oncogene homolog 1 signaling enhances drug resistance in CD44+/Musashi-1+ gastric cancer stem cells. Cancer Lett. 2015, 369, 124–133. [Google Scholar] [CrossRef]
- Huang, F.T.; Zhuan-Sun, Y.X.; Zhuang, Y.Y.; Wei, S.L.; Tang, J.; Chen, W.B.; Zhang, S.N. Inhibition of hedgehog signaling depresses self-renewal of pancreatic cancer stem cells and reverses chemoresistance. Int. J. Oncol. 2012, 41, 1707–1714. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.; Zhou, X.T.; Zou, H.Y.; Wu, J. Hedgehog signaling pathway affects the sensitivity of hepatoma cells to drug therapy through the ABCC1 transporter. Lab. Investig. 2017, 97, 819–832. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Lingala, S.; Khoobyari, S.; Nolta, J.; Zern, M.A.; Wu, J. Epithelial mesenchymal transition and hedgehog signaling activation are associated with chemoresistance and invasion of hepatoma subpopulations. J. Hepatol. 2011, 55, 838–845. [Google Scholar] [CrossRef]
- Zhou, X.T.; Ding, J.; Li, H.Y.; Zuo, J.L.; Ge, S.Y.; Jia, H.L.; Wu, J. Hedgehog signalling mediates drug resistance through targeting TAP1 in hepatocellular carcinoma. J. Cell. Mol. Med. 2020, 24, 4298–4311. [Google Scholar] [CrossRef]
- Amable, L.; Fain, J.; Gavin, E.; Reed, E. Gli1 Contributes to cellular resistance to cisplatin through altered cellular accumulation of the drug. Oncol. Rep. 2014, 32, 469–474. [Google Scholar] [CrossRef] [PubMed]
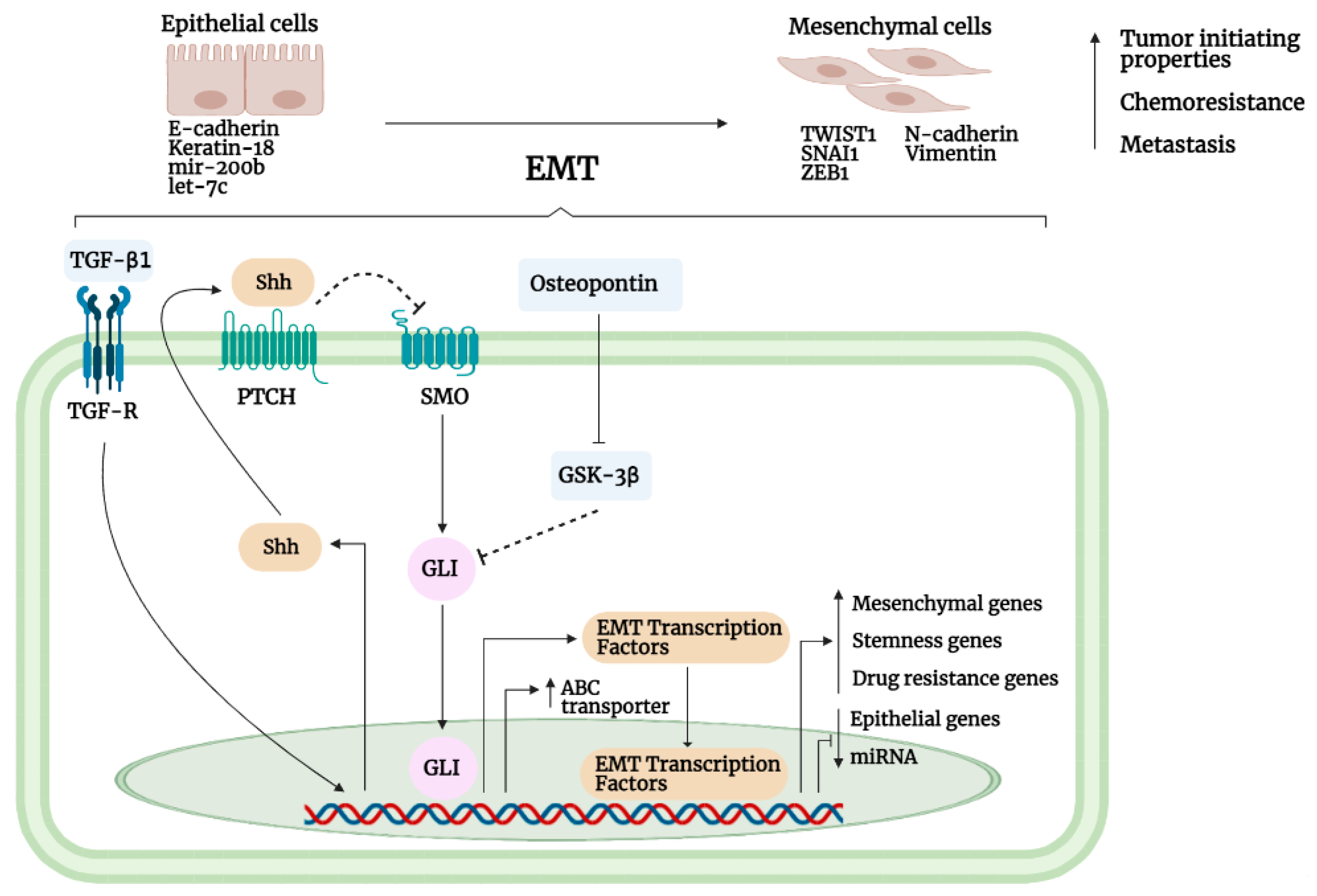
- Dongre, A.; Weinberg, R.A. New insights into the mechanisms of epithelial–mesenchymal transition and implications for cancer. Nat. Rev. Mol. Cell Biol. 2019, 20, 69–84. [Google Scholar] [CrossRef]
- Kim, A.Y.; Kwak, J.H.; Je, N.K.; Lee, Y.; Jung, Y.S. Epithelial-mesenchymal transition is associated with acquired resistance to 5-fluorocuracil in HT-29 colon cancer cells. Toxicol. Res. 2015, 31, 151–156. [Google Scholar] [CrossRef]
- Ashrafizadeh, M.; Zarrabi, A.; Hushmandi, K.; Kalantari, M.; Mohammadinejad, R.; Javaheri, T.; Sethi, G. Association of the Epithelial–Mesenchymal Transition (EMT) with cisplatin resistance. Int. J. Mol. Sci. 2020, 21, 4002. [Google Scholar] [CrossRef]
- Liu, Y.-R.; Liang, L.; Zhao, J.M.; Zhang, Y.; Zhang, M.; Zhong, W.-L.; Zhang, Q.; Wei, J.-J.; Li, M.; Yuan, J.; et al. Twist1 confers multidrug resistance in colon cancer through upregulation of ATP-binding cassette transporters. Oncotarget 2017, 8, 52901–52912. [Google Scholar] [CrossRef]
- Jiang, Z.S.; Sun, Y.Z.; Wang, S.M.; Ruan, J.S. Epithelial-mesenchymal transition: Potential regulator of ABC transporters in tumor progression. J. Cancer 2017, 8, 2319–2327. [Google Scholar] [CrossRef]
- Liu, Y.; Du, F.; Zhao, Q.; Jin, J.; Ma, X.; Li, H. Acquisition of 5-fluorouracil resistance induces epithelial-mesenchymal transitions through the hedgehog signaling pathway in HCT-8 colon cancer cells. Oncol. Lett. 2015, 9, 2675–2679. [Google Scholar] [CrossRef]
- Maitah, M.Y.; Ali, S.; Ahmad, A.; Gadgeel, S.; Sarkar, F.H. Up-regulation of sonic hedgehog contributes to TGF-Β1-induced epithelial to mesenchymal transition in NSCLC Cells. PLoS ONE 2011, 6, e16068. [Google Scholar] [CrossRef]
- Wellner, U.; Schubert, J.; Burk, U.C.; Schmalhofer, O.; Zhu, F.; Sonntag, A.; Waldvogel, B.; Vannier, C.; Darling, D.; Hausen, A.Z.; et al. The EMT-activator ZEB1 promotes tumorigenicity by repressing stemness-inhibiting MicroRNAs. Nat. Cell Biol. 2009, 11, 1487–1495. [Google Scholar] [CrossRef]
- Hojo, N.; Huisken, A.L.; Wang, H.; Chirshev, E.; Kim, N.S.; Nguyen, S.M.; Campos, H.; Glackin, C.A.; Ioffe, Y.J.; Unternaehrer, J.J. Snail knockdown reverses stemness and inhibits tumour growth in ovarian cancer. Sci. Rep. 2018, 8, 8704. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.H.; Lan, H.Y.; Huang, C.H.; Tai, S.K.; Tzeng, C.H.; Kao, S.Y.; Wu, K.J.; Hung, M.C.; Yang, M.H. RAC1 Activation mediates twist1-induced cancer cell migration. Nat. Cell Biol. 2012, 14, 366–374. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, A.; Maitah, M.Y.; Ginnebaugh, K.R.; Li, Y.; Bao, B.; Gadgeel, S.M.; Sarkar, F.H. Inhibition of hedgehog signaling sensitizes NSCLC cells to standard therapies through modulation of EMT-regulating MiRNAs. J. Hematol. Oncol. 2013, 6, 77. [Google Scholar] [CrossRef]
- Fischer, K.R.; Durrans, A.; Lee, S.; Sheng, J.; Li, F.; Wong, S.T.C.; Choi, H.; El Rayes, T.; Ryu, S.; Troeger, J.; et al. Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature 2015, 527, 472–476. [Google Scholar] [CrossRef]
- Singh, A.; Settleman, J. EMT, Cancer stem cells and drug resistance: An emerging axis of evil in the war on cancer. Oncogene 2010, 29, 4741–4751. [Google Scholar] [CrossRef] [PubMed]
- Zhan, X.; Wang, J.; Liu, Y.; Peng, Y.; Tan, W. GPCR-like signaling mediated by smoothened contributes to acquired chemoresistance through activating gli. Mol. Cancer 2014, 13, 4. [Google Scholar] [CrossRef] [PubMed]
- Kong, Y.; Peng, Y.; Liu, Y.; Xin, H.; Zhan, X.; Tan, W. Twist1 and snail link hedgehog signaling to tumor-initiating cell-like properties and acquired chemoresistance independently of ABC transporters. Stem Cells 2015, 33, 1063–1074. [Google Scholar] [CrossRef]
- Mani, S.A.; Guo, W.; Liao, M.J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef]
- Izadpanah, M.H.; Abbaszadegan, M.R.; Fahim, Y.; Forghanifard, M.M. Ectopic expression of TWIST1 upregulates the stemness marker OCT4 in the esophageal squamous cell carcinoma cell line KYSE. Cell. Mol. Biol. Lett. 2017, 22, 33. [Google Scholar] [CrossRef]
- Zhou, W.; Lv, R.; Qi, W.; Wu, D.; Xu, Y.; Liu, W.; Mou, Y.; Wang, L. Snail contributes to the maintenance of stem cell-like phenotype cells in human pancreatic cancer. PLoS ONE 2014, 9, 87409. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Ma, L.; Zhang, Z.; Liu, X.; Gao, H.; Zhuang, Y.; Yang, P.; Kornmann, M.; Tian, X.; Yang, Y. hedgehog signaling regulates epithelial-mesenchymal transition in pancreatic cancer stem-like cells. J. Cancer 2016, 7, 408–417. [Google Scholar] [CrossRef] [PubMed]
- Atashzar, M.R.; Baharlou, R.; Karami, J.; Abdollahi, H.; Rezaei, R.; Pourramezan, F.; Zoljalali Moghaddam, S.H. Cancer stem cells: A review from origin to therapeutic implications. J. Cell. Physiol. 2020, 235, 790–803. [Google Scholar] [CrossRef]
- Kim, W.T.; Ryu, C.J. Cancer stem cell surface markers on normal stem cells. BMB Rep. 2017, 50, 285–298. [Google Scholar] [CrossRef] [PubMed]
- Yoon, C.; Park, D.J.; Schmidt, B.; Thomas, N.J.; Lee, H.J.; Kim, T.S.; Janjigian, Y.Y.; Cohen, D.J.; Yoon, S.S. CD44 Expression denotes a subpopulation of gastric cancer cells in which hedgehog signaling promotes chemotherapy resistance. Clin. Cancer Res. 2014, 20, 3974–3988. [Google Scholar] [CrossRef]
- Song, Z.; Yue, W.; Wei, B.; Wang, N.; Li, T.; Guan, L.; Shi, S.; Zeng, Q.; Pei, X.; Chen, L. Sonic hedgehog pathway is essential for maintenance of cancer stem-like cells in human gastric cancer. PLoS ONE 2011, 6, e17687. [Google Scholar] [CrossRef]
- Wang, D.; Nagle, P.W.; Wang, H.H.; Smit, J.K.; Faber, H.; Baanstra, M.; Karrenbeld, A.; Chiu, R.K.; Plukker, J.T.M.; Coppes, R.P. Hedgehog pathway as a potential intervention target in esophageal cancer. Cancers 2019, 11, 821. [Google Scholar] [CrossRef]
- Kobune, M.; Ito, Y.; Kawano, Y.; Sasaki, K.; Uchida, H.; Nakamura, K.; Dehari, H.; Chiba, H.; Takimoto, R.; Matsunaga, T.; et al. Indian hedgehog gene transfer augments hematopoietic support of human stromal cells including NOD/SCID-B2m-/- repopulating cells. Blood 2004, 104, 1002–1009. [Google Scholar] [CrossRef]
- Kobune, M.; Takimoto, R.; Murase, K.; Iyama, S.; Sato, T.; Kikuchi, S.; Kawano, Y.; Miyanishi, K.; Sato, Y.; Niitsu, Y.; et al. Drug resistance is dramatically restored by hedgehog inhibitors in CD34+ leukemic cells. Cancer Sci. 2009, 100, 948–955. [Google Scholar] [CrossRef] [PubMed]
- Long, B.; Wang, L.-X.; Zheng, F.-M.; Lai, S.-P.; Xu, D.-R.; Hu, Y.; Lin, D.-J.; Zhang, X.-Z.; Dong, L.; Long, Z.-J.; et al. Targeting GLI1 suppresses cell growth and enhances chemosensitivity in CD34+ enriched acute myeloid leukemia progenitor cells. Cell. Physiol. Biochem. 2016, 38, 1288–1302. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.; An, Y.; Wei, J.S.; Ji, Z.L.; Lu, Z.P.; Wu, J.L.; Jiang, K.R.; Chen, P.; Xu, Z.K.; Miao, Y. Cyclopamine reverts acquired chemoresistance and down-regulates cancer stem cell markers in pancreatic cancer cell lines. Swiss Med. Wkly. 2011, 141, w13208. [Google Scholar] [CrossRef]
- Ren, Y.; Deng, R.; Cai, R.; Lu, X.; Luo, Y.; Wang, Z.; Zhu, Y.; Yin, M.; Ding, Y.; Lin, J. TUSC3 Induces drug resistance and cellular stemness via hedgehog signaling pathway in colorectal cancer. Carcinogenesis 2020, 41, 1755–1766. [Google Scholar] [CrossRef]
- Gu, Y.; Wang, Q.; Guo, K.; Qin, W.; Liao, W.; Wang, S.; Ding, Y.; Lin, J. TUSC3 promotes colorectal cancer progression and Epithelial-Mesenchymal Transition (EMT) through WNT/β-catenin and MAPK signalling. J. Pathol. 2016, 239, 60–71. [Google Scholar] [CrossRef]
- Varnat, F.; Duquet, A.; Malerba, M.; Zbinden, M.; Mas, C.; Gervaz, P.; Ruiz, I.; Altaba, A. human colon cancer epithelial cells harbour active HEDGEHOG-GLI signalling that is essential for tumour growth, recurrence, metastasis and stem cell survival and expansion. EMBO Mol. Med. 2009, 1, 338–351. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.A.; Chen, Y.F.; Bao, Y.; Mahara, S.; Yatim, S.M.J.M.; Oguz, G.; Lee, P.L.; Feng, M.; Cai, Y.; Tan, E.Y.; et al. Hypoxic tumor microenvironment activates GLI2 via HIF-1α and TGF-Β2 to promote chemoresistance in colorectal cancer. Proc. Natl. Acad. Sci. USA 2018, 115, E5990–E5999. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecular Target | Drug | Status | Cancer Target | Drug Combination | Mechanism of Chemoresistance | Treatment Outcome | Citation |
|---|---|---|---|---|---|---|---|
| Shh/Ihh | 5E1 | Preclinical study | Gastric | Oxaliplatin and Mitomycin | Hh signaling enhanced self-renewing capacity of tumorspheres, which promotes oxaliplatin and mitomycin resistance | Hh signaling inhibition enhanced cell death of CD44+ HGC-7 tumorsphere cells and primary gastric tumor samples when co-treated with oxaliplatin and mitomycin | [158] |
| SMO | Sonidegib/Erismodegib (NVP-LDE-225) | Preclinical study and phase I clinical trail EDALINE | Breast | Docetaxel | M6-Hh cells activate CAFs via paracrine Hh signaling to induce ECM remodeling and consequently chemoresistant CSCs phenotype in adjacent epithelial cells | SMO inhibition reduced collagen fibrillar deposition and consequently reversed the CSC phenotype of M6-Hh cells, which sensitizes M6-Hh tumors, patient-derived xenografts, and metastatic TNBC patients to docetaxel | [105,106] |
| Preclinical study | Ovarian | Paclitaxel | The upregulation of SMO, GLI1, and GLI2 modulate ABCB1 expression to promote paclitaxel resistance | SMO inhibition sensitized taxane resistant ALDH-negative and positive A2780 and SKOV3 cell lines to paclitaxel by downregulating ABCB1 expression | [122] | ||
| Vismodegib (GDC-0449) | Preclinical study | Myeloid Leukemia | Ribavirin, cytarabine, methotrexate, venetoclax, 5-fluorouracil, sunitinib, and idarubicin | High levels of GLI1 mediated the UGT1A-dependent glucuronidation of several cancer therapeutics via calreticulin-dependent UGT1A protein stabilization to enhance drug elimination | Inhibition of GLI1 reduced UGT1A dependent glucuronidation of cancer therapeutics to promote drug accumulation (e.g., eIF4E-ribavirin complex) in primary AML specimens and acquired resistant cell lines | [114,117] | |
| Colon | 5-fluorouracil | Elevated GLI1 levels induced EMT phenotype, which was associated with 5-fluorouracil resistance | Inhibition of SMO reversed EMT signals and 5-fluorouracil resistance in colon cancer HCT-8 cell line | [141] | |||
| Lung | Cisplatin | Elevated Hh signaling contributed to TGF-β1 induced EMT and cisplatin resistance via the downregulation of miR-200b and let-7c | The inhibition of Hh signaling sensitized A549 EMT cells to cisplatin through the restoration of miR-200b and let-7c expression and reversal of EMT genes | [146] | |||
| Esophageal | Carboplatin | High levels of Shh and PTCH1 were associated with increased CSC traits and therapeutic resistance | Inhibition of Hh pathway via SMO decreased CD44+ CSC phenotype and sphere-forming potential in esophageal cancer OE21 cell line, which restored carboplatin sensitivity | [159] | |||
| Preclinical study and phase II clinical trial | Gastric | 5-fluorouracil, cisplatin, oxaliplatin, and leucovorin | Increased Hh signaling enhanced CSC traits in gastric cancer (GC) cells to promote 5-fluorouracil and cisplatin resistance | SMO inhibition sensitized CD44+ AGS, MKN45, and N87 spheroid cells and mice xenografts as well as GC patients with high CD44 median scores to the combination treatment of 5-fluorouracil and cisplatin and 5-fluorouracil, oxaliplatin, and leucovorin, respectively | [157] | ||
| Saridegib (IPI-926) | Preclinical study | Pancreatic | Gemcitabine | Hh pathway activation enhanced stromal desmoplasia and hypovascularity to reduced gemcitabine delivery | SMO inhibition depletes stromal desmoplasia and increases MVD to enhance gemcitabine delivery and consequently intracellular dFdCTP accumulation in KPC mice tissues | [111] | |
| Cyclopamine | Preclinical study | Pancreatic | Gemcitabine and 5-fluorouracil | Hh signaling induced gemcitabine resistance through CSC induction, as well as ABCB1 and ABCG2-mediated drug efflux | Inhibition of SMO restored gemcitabine sensitivity in acquired gemcitabine-resistant SW1990 and CFPAC cell lines expressing CD44 and CD133, as well as restored gemcitabine and 5-fluorouracil sensitivity in PANC-1 tumorspheres by inhibiting their self-renewing capacity and ABCB1 and ABCG2 expression | [131,163] | |
| Glioma | Temozolomide | GLI1 upregulates MGMT at the promoter level to promote TMZ resistance | SMO inhibition restricts Hh/GLI signaling to downregulate MGMT expression and consequently improved TMZ toxicity in GBM U251 and U87 cell lines and xenografts | [75] | |||
| Myeloid Leukemia | Cytarabine | Ihh and its downstream effector (GLI1 and GLI2) were enriched in CD34+ subpopulations, which was associated with cytarabine resistance | Inhibition of Ihh autocrine Hh signaling via SMO induced apoptosis and sensitized cytokine responsive CD34+ Kasumi-1, Kasumi-3, and TF-1 cell lines to cytarabine treatment | [161] | |||
| GLI1/2 | GANT-58 | Preclinical study | Myeloid leukemia and cervical | Doxorubicin and vincristine | GLI1 transcriptionally upregulates TWIST1 and SNAI1 expression to promote tumor-initiating properties and consequently chemoresistance | GLI1 inhibition reduced TWIST1 and SNAI1 levels, which restored the chemosensitivity of multidrug-resistant chronic myelogenous leukemia K562 and human cervical epidermoid carcinoma KB sublines to doxorubicin and vincristine | [150] |
| GANT-61 | Preclinical study | Myeloid Leukemia | Vincristine | High GLI1 levels upregulate ABCB1 to promote vincristine resistance | GLI1 inhibition reversed vincristine resistance of Lucena-1 cell line by interfering with ABCB1 expression | [32] | |
| Cytarabine | GLI1 enrichment in CD34+ cells enhanced colony-forming capacity and cytarabine resistance | GLI1 inhibition induced a significant reduction in cell viability of CD34+ compared to CD34- primary AML cells when treated with cytarabine | [162] | ||||
| Ovarian | Cisplatin | GLI2 transcriptionally upregulates ABCB1 to promotes cisplatin resistance | GLI2 inhibition reduced ABCB1 levels, which enhanced cisplatin-induced DNA damage in cisplatin-resistant OC SK-OV-3 cell line | [123] | |||
| Glioma | Temozolomide | High levels of GLI and MGMT expression in glioblastoma cells lead to TMZ resistance | Inhibition of GLI with GANT-61 reduced the level of MGMT, which restored the sensitivity to temozolomide in GBM U251 and U87 cell lines | [73] | |||
| Hepatoma | Cisplatin, doxorubicin, and sorafenib | The binding of GLI1/2 to the GLI1-binding consensus sequence within the ABCB2 promoter initiated its transcription to confer drug resistance | Inhibition of GLI1/2 downregulated the ABCB2 expression in Huh-7 DN (CD133-/EpCAM-) and trans (CD133−/EpCAM− transwell-selected) hepatoma subpopulations, which enhanced their chemotherapeutic drug sensitivity | [132] | |||
| Colorectal | 5-fluorouracil and cisplatin | Hh signaling has SMO as a mediator of TUSC3-induced CSC phenotype and drug resistance | Inhibition of Hh signaling at the GLI level inhibited the expression of CD133 and ABCC1 and decreased the number of TUSC3-overexpressing CACO2 and RKO2 tumorspheres, which was associated with increased 5-fluorouracil and cisplatin sensitivity | [164] | |||
| SIR-38832 | Preclinical study | Colorectal | 5-fluorouracil | GLI1 transcriptionally upregulates NBS1 and consequently MRN complex function to reduce 5-fluorouracil induced DNA damage | Inhibition of GLI1 activity reduced total NBS1 levels and impaired MRN complex function to increase 5-fluorouracil induced DNA damage in HT29 cell lines and xenografts | [81] | |
| Not specified | Dihydroartemisinin | Preclinical study | Esophageal | Cisplatin | Hh activation upregulates ABCB1 levels to reduce cisplatin enrichment | Hh inhibition reduced ABCB1 levels and consequently enhanced cisplatin accumulation in the TE-1 cell line | [125] |
| HIF-1α | Oroxylin A | Preclinical study | Glioma | TMZ | HIF-1α activates Shh/GLI1/MGMT signaling to promote TMZ resistance | HIF-1α degradation inhibits Hh pathway activation and increases SUFU expression, thus reducing MGMT levels and restoring TMZ sensitivity in glioma U251 and C6 cell lines and xenografts | [75] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chai, J.Y.; Sugumar, V.; Alshanon, A.F.; Wong, W.F.; Fung, S.Y.; Looi, C.Y. Defining the Role of GLI/Hedgehog Signaling in Chemoresistance: Implications in Therapeutic Approaches. Cancers 2021, 13, 4746. https://doi.org/10.3390/cancers13194746
Chai JY, Sugumar V, Alshanon AF, Wong WF, Fung SY, Looi CY. Defining the Role of GLI/Hedgehog Signaling in Chemoresistance: Implications in Therapeutic Approaches. Cancers. 2021; 13(19):4746. https://doi.org/10.3390/cancers13194746
Chicago/Turabian StyleChai, Jian Yi, Vaisnevee Sugumar, Ahmed F. Alshanon, Won Fen Wong, Shin Yee Fung, and Chung Yeng Looi. 2021. "Defining the Role of GLI/Hedgehog Signaling in Chemoresistance: Implications in Therapeutic Approaches" Cancers 13, no. 19: 4746. https://doi.org/10.3390/cancers13194746
APA StyleChai, J. Y., Sugumar, V., Alshanon, A. F., Wong, W. F., Fung, S. Y., & Looi, C. Y. (2021). Defining the Role of GLI/Hedgehog Signaling in Chemoresistance: Implications in Therapeutic Approaches. Cancers, 13(19), 4746. https://doi.org/10.3390/cancers13194746