
Determinants of Homologous Recombination Deficiency in Pancreatic Cancer

Abstract
:Simple Summary
Abstract
1. Introduction
2. Tumor Intrinsic and Extrinsic Determinants of HRD
2.1. Homologous Recombination and Sensitivity to Platinum Chemotherapy and Poly (ADP-Ribose) Polymerase Inhibitors
2.1.1. Platinum Chemotherapy in Pancreatic Cancer
2.1.2. Mutations in BRCA Genes and Sensitivity to Platinum Chemotherapy
2.1.3. Mutations in Non-BRCA HR Genes and Sensitivity to Platinum Chemotherapy
2.1.4. Poly (ADP-Ribose) Polymerase Inhibitors in Cancer
2.1.5. Mutations in BRCA Genes and Sensitivity to Poly (ADP-Ribose) Polymerase Inhibitors
2.1.6. Mutations in Non-BRCA Genes and Sensitivity to Poly (ADP-Ribose) Polymerase Inhibitors
2.2. Additional Intrinsic Determinants of HRD Phenotype
2.3. Extrinsic Determinants of HRD
2.3.1. Interplay of the Tumor Immune Microenvironment and HRD
2.3.2. Interplay among PARP Inhibition and the Immune System
3. Identifying HRD in Pancreatic Cancer
3.1. Clinical Biomarkers
3.1.1. Family History of Cancer
3.1.2. Platinum Sensitivity
3.2. Laboratory Biomarkers
3.2.1. Stereotyped Genomic Features
DNA Scars
Mutational Patterns
3.2.2. Functional Assays
3.3. Current Limitations and Next-Generation Strategies in Testing for HRD
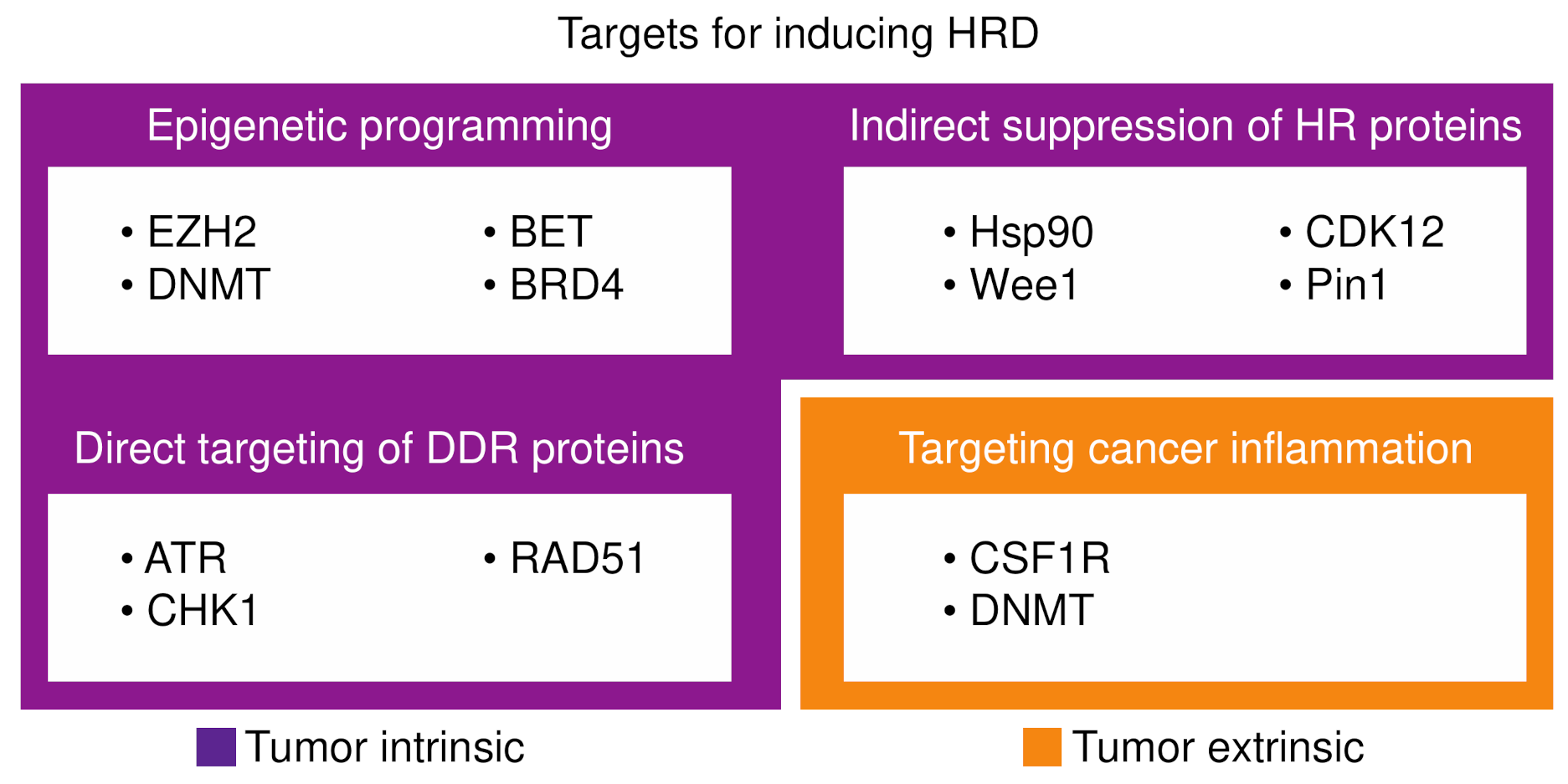
4. Inducing HRD
4.1. Epigenetic Programming of HRD
4.2. Targeting DNA Damage Repair Proteins
4.3. Indirect Suppression of Homologous Recombination Proteins
5. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef]
- Von Hoff, D.D.; Ervin, T.; Arena, F.P.; Chiorean, E.G.; Infante, J.; Moore, M.; Seay, T.; Tjulandin, S.A.; Ma, W.W.; Saleh, M.N.; et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N. Engl. J. Med. 2013, 369, 1691–1703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conroy, T.; Desseigne, F.; Ychou, M.; Bouché, O.; Guimbaud, R.; Bécouarn, Y.; Adenis, A.; Raoul, J.L.; Gourgou-Bourgade, S.; de la Fouchardière, C.; et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N. Engl. J. Med. 2011, 364, 1817–1825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turner, N.; Tutt, A.; Ashworth, A. Hallmarks of ‘BRCAness’ in sporadic cancers. Nat. Rev. Cancer 2004, 4, 814–819. [Google Scholar] [CrossRef]
- Lord, C.J.; Ashworth, A. BRCAness revisited. Nat. Rev. Cancer 2016, 16, 110–120. [Google Scholar] [CrossRef]
- Thompson, D.; Easton, D.F.; Consortium, B.C.L. Cancer Incidence in BRCA1 mutation carriers. J. Natl. Cancer Inst. 2002, 94, 1358–1365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Consortium, B.C.L. Cancer risks in BRCA2 mutation carriers. J. Natl. Cancer Inst. 1999, 91, 1310–1316. [Google Scholar] [CrossRef]
- Toh, M.; Ngeow, J. Homologous Recombination Deficiency: Cancer Predispositions and Treatment Implications. Oncologist 2021, 26, e1526–e1537. [Google Scholar] [CrossRef]
- Golan, T.; O’Kane, G.M.; Denroche, R.E.; Raitses-Gurevich, M.; Grant, R.C.; Holter, S.; Wang, Y.; Zhang, A.; Jang, G.H.; Stossel, C.; et al. Genomic Features and Classification of Homologous Recombination Deficient Pancreatic Ductal Adenocarcinoma. Gastroenterology 2021, 160, 2119–2132.e2119. [Google Scholar] [CrossRef]
- Casolino, R.; Paiella, S.; Azzolina, D.; Beer, P.A.; Corbo, V.; Lorenzoni, G.; Gregori, D.; Golan, T.; Braconi, C.; Froeling, F.E.M.; et al. Homologous Recombination Deficiency in Pancreatic Cancer: A Systematic Review and Prevalence Meta-Analysis. J. Clin. Oncol. 2021, 39, 2617–2631. [Google Scholar] [CrossRef]
- Jasin, M.; Rothstein, R. Repair of strand breaks by homologous recombination. Cold Spring Harb. Perspect. Biol. 2013, 5, a012740. [Google Scholar] [CrossRef]
- Stover, E.H.; Konstantinopoulos, P.A.; Matulonis, U.A.; Swisher, E.M. Biomarkers of Response and Resistance to DNA Repair Targeted Therapies. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2016, 22, 5651–5660. [Google Scholar] [CrossRef] [Green Version]
- Sung, P.; Klein, H. Mechanism of homologous recombination: Mediators and helicases take on regulatory functions. Nat. Rev. Mol. Cell Biol. 2006, 7, 739–750. [Google Scholar] [CrossRef]
- Wright, W.D.; Shah, S.S.; Heyer, W.D. Homologous recombination and the repair of DNA double-strand breaks. J. Biol. Chem. 2018, 293, 10524–10535. [Google Scholar] [CrossRef] [Green Version]
- Symington, L.S. End resection at double-strand breaks: Mechanism and regulation. Cold Spring Harb. Perspect. Biol. 2014, 6, a016436. [Google Scholar] [CrossRef] [Green Version]
- Lavin, M.F. ATM and the Mre11 complex combine to recognize and signal DNA double-strand breaks. Oncogene 2007, 26, 7749–7758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cortez, D.; Wang, Y.; Qin, J.; Elledge, S.J. Requirement of ATM-dependent phosphorylation of brca1 in the DNA damage response to double-strand breaks. Science 1999, 286, 1162–1166. [Google Scholar] [CrossRef] [PubMed]
- Christou, C.M.; Kyriacou, K. BRCA1 and Its Network of Interacting Partners. Biology 2013, 2, 40–63. [Google Scholar] [CrossRef] [Green Version]
- Prakash, R.; Zhang, Y.; Feng, W.; Jasin, M. Homologous recombination and human health: The roles of BRCA1, BRCA2, and associated proteins. Cold Spring Harb. Perspect. Biol. 2015, 7, a016600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, Q.; Chen, C.F.; Li, S.; Chen, Y.; Wang, C.C.; Xiao, J.; Chen, P.L.; Sharp, Z.D.; Lee, W.H. Association of BRCA1 with the hRad50-hMre11-p95 complex and the DNA damage response. Science 1999, 285, 747–750. [Google Scholar] [CrossRef] [PubMed]
- Sy, S.M.; Huen, M.S.; Chen, J. PALB2 is an integral component of the BRCA complex required for homologous recombination repair. Proc. Natl. Acad. Sci. USA 2009, 106, 7155–7160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lord, C.J.; Ashworth, A. RAD51, BRCA2 and DNA repair: A partial resolution. Nat. Struct. Mol. Biol. 2007, 14, 461–462. [Google Scholar] [CrossRef] [PubMed]
- Daley, J.M.; Gaines, W.A.; Kwon, Y.; Sung, P. Regulation of DNA pairing in homologous recombination. Cold Spring Harb. Perspect. Biol. 2014, 6, a017954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colucci, G.; Labianca, R.; Di Costanzo, F.; Gebbia, V.; Cartenì, G.; Massidda, B.; Dapretto, E.; Manzione, L.; Piazza, E.; Sannicolò, M.; et al. Randomized phase III trial of gemcitabine plus cisplatin compared with single-agent gemcitabine as first-line treatment of patients with advanced pancreatic cancer: The GIP-1 study. J. Clin. Oncol. 2010, 28, 1645–1651. [Google Scholar] [CrossRef] [PubMed]
- Lowery, M.A.; Wong, W.; Jordan, E.J.; Lee, J.W.; Kemel, Y.; Vijai, J.; Mandelker, D.; Zehir, A.; Capanu, M.; Salo-Mullen, E.; et al. Prospective Evaluation of Germline Alterations in Patients with Exocrine Pancreatic Neoplasms. J. Natl. Cancer Inst. 2018, 110, 1067–1074. [Google Scholar] [CrossRef] [Green Version]
- O’Reilly, E.M.; Lee, J.W.; Zalupski, M.; Capanu, M.; Park, J.; Golan, T.; Tahover, E.; Lowery, M.A.; Chou, J.F.; Sahai, V.; et al. Randomized, Multicenter, Phase II Trial of Gemcitabine and Cisplatin with or without Veliparib in Patients with Pancreas Adenocarcinoma and a Germline. J. Clin. Oncol. 2020, 38, 1378–1388. [Google Scholar] [CrossRef]
- Wattenberg, M.M.; Asch, D.; Yu, S.; O’Dwyer, P.J.; Domchek, S.M.; Nathanson, K.L.; Rosen, M.A.; Beatty, G.L.; Siegelman, E.S.; Reiss, K.A. Platinum response characteristics of patients with pancreatic ductal adenocarcinoma and a germline BRCA1, BRCA2 or PALB2 mutation. Br. J. Cancer 2020, 122, 333–339. [Google Scholar] [CrossRef]
- Pishvaian, M.J.; Blais, E.M.; Brody, J.R.; Rahib, L.; Lyons, E.; Arbeloa, P.D.; Hendifar, A.; Mikhail, S.; Chung, V.; Sohal, D.P.S.; et al. Outcomes in Patients with Pancreatic Adenocarcinoma with Genetic Mutations in DNA Damage Response Pathways: Results From the Know Your Tumor Program. JCO Precis. Oncol. 2019, 3, 1–10. [Google Scholar] [CrossRef]
- Kelland, L. The resurgence of platinum-based cancer chemotherapy. Nat. Rev. Cancer 2007, 7, 573–584. [Google Scholar] [CrossRef]
- Raymond, E.; Faivre, S.; Chaney, S.; Woynarowski, J.; Cvitkovic, E. Cellular and molecular pharmacology of oxaliplatin. Mol. Cancer Ther. 2002, 1, 227–235. [Google Scholar]
- Bruno, P.M.; Liu, Y.; Park, G.Y.; Murai, J.; Koch, C.E.; Eisen, T.J.; Pritchard, J.R.; Pommier, Y.; Lippard, S.J.; Hemann, M.T. A subset of platinum-containing chemotherapeutic agents kills cells by inducing ribosome biogenesis stress. Nat. Med. 2017, 23, 461–471. [Google Scholar] [CrossRef]
- Kondo, T.; Kanai, M.; Kou, T.; Sakuma, T.; Mochizuki, H.; Kamada, M.; Nakatsui, M.; Uza, N.; Kodama, Y.; Masui, T.; et al. Association between homologous recombination repair gene mutations and response to oxaliplatin in pancreatic cancer. Oncotarget 2018, 9, 19817–19825. [Google Scholar] [CrossRef] [Green Version]
- Golan, T.; Kanji, Z.S.; Epelbaum, R.; Devaud, N.; Dagan, E.; Holter, S.; Aderka, D.; Paluch-Shimon, S.; Kaufman, B.; Gershoni-Baruch, R.; et al. Overall survival and clinical characteristics of pancreatic cancer in BRCA mutation carriers. Br. J. Cancer 2014, 111, 1132–1138. [Google Scholar] [CrossRef]
- Reiss, K.A.; Yu, S.; Judy, R.; Symecko, H.; Nathanson, K.L.; Domchek, S.M. Retrospective Survival Analysis of Patients with Advanced Pancreatic Ductal Adenocarcinoma and Germline BRCA or PALB2 Mutations. JCO Precis. Oncol. 2018, 2, 1–9. [Google Scholar] [CrossRef]
- Jonsson, P.; Bandlamudi, C.; Cheng, M.L.; Srinivasan, P.; Chavan, S.S.; Friedman, N.D.; Rosen, E.Y.; Richards, A.L.; Bouvier, N.; Selcuklu, S.D.; et al. Tumour lineage shapes BRCA-mediated phenotypes. Nature 2019, 571, 576–579. [Google Scholar] [CrossRef]
- Ashworth, A. Drug resistance caused by reversion mutation. Cancer Res. 2008, 68, 10021–10023. [Google Scholar] [CrossRef] [Green Version]
- Tobalina, L.; Armenia, J.; Irving, E.; O’Connor, M.J.; Forment, J.V. A meta-analysis of reversion mutations in BRCA genes identifies signatures of DNA end-joining repair mechanisms driving therapy resistance. Ann. Oncol. 2021, 32, 103–112. [Google Scholar] [CrossRef]
- Lin, K.K.; Harrell, M.I.; Oza, A.M.; Oaknin, A.; Ray-Coquard, I.; Tinker, A.V.; Helman, E.; Radke, M.R.; Say, C.; Vo, L.T.; et al. Reversion Mutations in Circulating Tumor DNA Predict Primary and Acquired Resistance to the PARP Inhibitor Rucaparib in High-Grade Ovarian Carcinoma. Cancer Discov. 2019, 9, 210–219. [Google Scholar] [CrossRef] [Green Version]
- Kondo, T.; Kanai, M.; Matsubara, J.; Quy, P.N.; Fukuyama, K.; Yamamoto, Y.; Yamada, T.; Nishigaki, M.; Minamiguchi, S.; Takeda, M.; et al. BRCA2 Reversion Mutation Identified by Liquid Biopsy After Durable Response to FOLFIRINOX in BRCA2-Associated Pancreatic Cancer. Pancreas 2020, 49, e101–e103. [Google Scholar] [CrossRef] [PubMed]
- Tao, H.; Liu, S.; Huang, D.; Han, X.; Wu, X.; Shao, Y.W.; Hu, Y. Acquired multiple secondary BRCA2 mutations upon PARPi resistance in a metastatic pancreatic cancer patient harboring a BRCA2 germline mutation. Am. J. Transl. Res. 2020, 12, 612–617. [Google Scholar] [PubMed]
- Pishvaian, M.J.; Biankin, A.V.; Bailey, P.; Chang, D.K.; Laheru, D.; Wolfgang, C.L.; Brody, J.R. BRCA2 secondary mutation-mediated resistance to platinum and PARP inhibitor-based therapy in pancreatic cancer. Br. J. Cancer 2017, 116, 1021–1026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aguirre, A.J.; Nowak, J.A.; Camarda, N.D.; Moffitt, R.A.; Ghazani, A.A.; Hazar-Rethinam, M.; Raghavan, S.; Kim, J.; Brais, L.K.; Ragon, D.; et al. Real-time Genomic Characterization of Advanced Pancreatic Cancer to Enable Precision Medicine. Cancer Discov. 2018, 8, 1096–1111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, W.; Chen, J.; Chou, J.F.; Varghese, A.M.; Yu, K.H.; Wong, W.; Capanu, M.; Balachandran, V.; McIntyre, C.A.; El Dika, I.; et al. Genomic Methods Identify Homologous Recombination Deficiency in Pancreas Adenocarcinoma and Optimize Treatment Selection. Clin. Cancer Res. 2020, 26, 3239–3247. [Google Scholar] [CrossRef] [PubMed]
- Vyas, S.; Chesarone-Cataldo, M.; Todorova, T.; Huang, Y.H.; Chang, P. A systematic analysis of the PARP protein family identifies new functions critical for cell physiology. Nat. Commun. 2013, 4, 2240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ray Chaudhuri, A.; Nussenzweig, A. The multifaceted roles of PARP1 in DNA repair and chromatin remodelling. Nat. Rev. Mol. Cell Biol. 2017, 18, 610–621. [Google Scholar] [CrossRef]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef]
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005, 434, 913–917. [Google Scholar] [CrossRef]
- Dias, M.P.; Moser, S.C.; Ganesan, S.; Jonkers, J. Understanding and overcoming resistance to PARP inhibitors in cancer therapy. Nat. Rev. Clin. Oncol. 2021. Epub ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Ashworth, A. A synthetic lethal therapeutic approach: Poly(ADP) ribose polymerase inhibitors for the treatment of cancers deficient in DNA double-strand break repair. J. Clin. Oncol. 2008, 26, 3785–3790. [Google Scholar] [CrossRef]
- Hoeijmakers, J.H. Genome maintenance mechanisms for preventing cancer. Nature 2001, 411, 366–374. [Google Scholar] [CrossRef] [PubMed]
- Helleday, T. The underlying mechanism for the PARP and BRCA synthetic lethality: Clearing up the misunderstandings. Mol. Oncol. 2011, 5, 387–393. [Google Scholar] [CrossRef] [Green Version]
- Patel, A.G.; Sarkaria, J.N.; Kaufmann, S.H. Nonhomologous end joining drives poly(ADP-ribose) polymerase (PARP) inhibitor lethality in homologous recombination-deficient cells. Proc. Natl. Acad. Sci. USA 2011, 108, 3406–3411. [Google Scholar] [CrossRef] [Green Version]
- Pommier, Y.; O’Connor, M.J.; de Bono, J. Laying a trap to kill cancer cells: PARP inhibitors and their mechanisms of action. Sci. Transl. Med. 2016, 8, 362ps317. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Zhao, W.; Ju, Z.; Wang, L.; Peng, Y.; Labrie, M.; Yap, T.A.; Mills, G.B.; Peng, G. PARPi Triggers the STING-Dependent Immune Response and Enhances the Therapeutic Efficacy of Immune Checkpoint Blockade Independent of BRCAness. Cancer Res. 2019, 79, 311–319. [Google Scholar] [CrossRef] [Green Version]
- Kaufman, B.; Shapira-Frommer, R.; Schmutzler, R.K.; Audeh, M.W.; Friedlander, M.; Balmaña, J.; Mitchell, G.; Fried, G.; Stemmer, S.M.; Hubert, A.; et al. Olaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutation. J. Clin. Oncol. 2015, 33, 244–250. [Google Scholar] [CrossRef]
- Shroff, R.T.; Hendifar, A.; McWilliams, R.R.; Geva, R.; Epelbaum, R.; Rolfe, L.; Goble, S.; Lin, K.K.; Biankin, A.V.; Giordano, H.; et al. Rucaparib Monotherapy in Patients with Pancreatic Cancer and a Known Deleterious BRCA Mutation. JCO Precis. Oncol. 2018, 2, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Lowery, M.A.; Kelsen, D.P.; Capanu, M.; Smith, S.C.; Lee, J.W.; Stadler, Z.K.; Moore, M.J.; Kindler, H.L.; Golan, T.; Segal, A.; et al. Phase II trial of veliparib in patients with previously treated BRCA-mutated pancreas ductal adenocarcinoma. Eur. J. Cancer 2018, 89, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Golan, T.; Hammel, P.; Reni, M.; Van Cutsem, E.; Macarulla, T.; Hall, M.J.; Park, J.O.; Hochhauser, D.; Arnold, D.; Oh, D.Y.; et al. Maintenance Olaparib for Germline BRCA-Mutated Metastatic Pancreatic Cancer. N. Engl. J. Med. 2019, 381, 317–327. [Google Scholar] [CrossRef]
- Reiss, K.A.; Mick, R.; O’Hara, M.H.; Teitelbaum, U.; Karasic, T.B.; Schneider, C.; Cowden, S.; Southwell, T.; Romeo, J.; Izgur, N.; et al. Phase II Study of Maintenance Rucaparib in Patients with Platinum-Sensitive Advanced Pancreatic Cancer and a Pathogenic Germline or Somatic Variant in. J. Clin. Oncol. 2021, 39, 2497–2505. [Google Scholar] [CrossRef] [PubMed]
- Javle, M.; Shacham-Shmueli, E.; Xiao, L.; Varadhachary, G.; Halpern, N.; Fogelman, D.; Boursi, B.; Uruba, S.; Margalit, O.; Wolff, R.A.; et al. Olaparib Monotherapy for Previously Treated Pancreatic Cancer with DNA Damage Repair Genetic Alterations Other Than Germline BRCA Variants: Findings From 2 Phase 2 Nonrandomized Clinical Trials. JAMA Oncol. 2021, 7, 693–699. [Google Scholar] [CrossRef]
- Hannan, Z.; Yu, S.; Domchek, S.; Mamtani, R.; Reiss, K.A. Clinical Characteristics of Patients with Pancreatic Cancer and Pathogenic ATM Alterations. JNCI Cancer Spectr. 2021, 5, pkaa121. [Google Scholar] [CrossRef]
- Abida, W.; Campbell, D.; Patnaik, A.; Shapiro, J.D.; Sautois, B.; Vogelzang, N.J.; Voog, E.G.; Bryce, A.H.; McDermott, R.; Ricci, F.; et al. Non-BRCA DNA Damage Repair Gene Alterations and Response to the PARP Inhibitor Rucaparib in Metastatic Castration-Resistant Prostate Cancer: Analysis From the Phase II TRITON2 Study. Clin. Cancer Res. 2020, 26, 2487–2496. [Google Scholar] [CrossRef] [Green Version]
- de Bono, J.; Mateo, J.; Fizazi, K.; Saad, F.; Shore, N.; Sandhu, S.; Chi, K.N.; Sartor, O.; Agarwal, N.; Olmos, D.; et al. Olaparib for Metastatic Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2020, 382, 2091–2102. [Google Scholar] [CrossRef] [PubMed]
- Tung, N.M.; Robson, M.E.; Ventz, S.; Santa-Maria, C.A.; Nanda, R.; Marcom, P.K.; Shah, P.D.; Ballinger, T.J.; Yang, E.S.; Vinayak, S.; et al. TBCRC 048: Phase II Study of Olaparib for Metastatic Breast Cancer and Mutations in Homologous Recombination-Related Genes. J. Clin. Oncol. 2020, 38, 4274–4282. [Google Scholar] [CrossRef]
- Tacheci, I.; Kopacova, M.; Bures, J. Peutz-Jeghers syndrome. Curr. Opin. Gastroenterol. 2021, 37, 245–254. [Google Scholar] [CrossRef]
- Wang, Y.S.; Chen, J.; Cui, F.; Wang, H.; Wang, S.; Hang, W.; Zeng, Q.; Quan, C.S.; Zhai, Y.X.; Wang, J.W.; et al. LKB1 is a DNA damage response protein that regulates cellular sensitivity to PARP inhibitors. Oncotarget 2016, 7, 73389–73401. [Google Scholar] [CrossRef] [Green Version]
- Su, G.H.; Hruban, R.H.; Bansal, R.K.; Bova, G.S.; Tang, D.J.; Shekher, M.C.; Westerman, A.M.; Entius, M.M.; Goggins, M.; Yeo, C.J.; et al. Germline and somatic mutations of the STK11/LKB1 Peutz-Jeghers gene in pancreatic and biliary cancers. Am. J. Pathol. 1999, 154, 1835–1840. [Google Scholar] [CrossRef] [Green Version]
- Deng, J.; Thennavan, A.; Dolgalev, I.; Chen, T.; Li, J.; Marzio, A.; Poirier, J.T.; Peng, D.; Bulatovic, M.; Mukhopadhyay, S.; et al. ULK1 inhibition overcomes compromised antigen presentation and restores antitumor immunity in LKB1 mutant lung cancer. Nat. Cancer 2021, 2, 503–514. [Google Scholar] [CrossRef]
- Armstrong, S.A.; Schultz, C.W.; Azimi-Sadjadi, A.; Brody, J.R.; Pishvaian, M.J. ATM Dysfunction in Pancreatic Adenocarcinoma and Associated Therapeutic Implications. Mol. Cancer Ther. 2019, 18, 1899–1908. [Google Scholar] [CrossRef] [Green Version]
- Lloyd, R.L.; Wijnhoven, P.W.G.; Ramos-Montoya, A.; Wilson, Z.; Illuzzi, G.; Falenta, K.; Jones, G.N.; James, N.; Chabbert, C.D.; Stott, J.; et al. Combined PARP and ATR inhibition potentiates genome instability and cell death in ATM-deficient cancer cells. Oncogene 2020, 39, 4869–4883. [Google Scholar] [CrossRef] [PubMed]
- Clements, K.E.; Schleicher, E.M.; Thakar, T.; Hale, A.; Dhoonmoon, A.; Tolman, N.J.; Sharma, A.; Liang, X.; Imamura Kawasawa, Y.; Nicolae, C.M.; et al. Identification of regulators of poly-ADP-ribose polymerase inhibitor response through complementary CRISPR knockout and activation screens. Nat. Commun. 2020, 11, 6118. [Google Scholar] [CrossRef] [PubMed]
- Schlacher, K.; Wu, H.; Jasin, M. A distinct replication fork protection pathway connects Fanconi anemia tumor suppressors to RAD51-BRCA1/2. Cancer Cell 2012, 22, 106–116. [Google Scholar] [CrossRef] [Green Version]
- Ying, S.; Hamdy, F.C.; Helleday, T. Mre11-dependent degradation of stalled DNA replication forks is prevented by BRCA2 and PARP1. Cancer Res. 2012, 72, 2814–2821. [Google Scholar] [CrossRef] [Green Version]
- Daza-Martin, M.; Starowicz, K.; Jamshad, M.; Tye, S.; Ronson, G.E.; MacKay, H.L.; Chauhan, A.S.; Walker, A.K.; Stone, H.R.; Beesley, J.F.J.; et al. Isomerization of BRCA1-BARD1 promotes replication fork protection. Nature 2019, 571, 521–527. [Google Scholar] [CrossRef] [PubMed]
- Schlacher, K.; Christ, N.; Siaud, N.; Egashira, A.; Wu, H.; Jasin, M. Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell 2011, 145, 529–542. [Google Scholar] [CrossRef] [Green Version]
- Ray Chaudhuri, A.; Callen, E.; Ding, X.; Gogola, E.; Duarte, A.A.; Lee, J.E.; Wong, N.; Lafarga, V.; Calvo, J.A.; Panzarino, N.J.; et al. Replication fork stability confers chemoresistance in BRCA-deficient cells. Nature 2016, 535, 382–387. [Google Scholar] [CrossRef]
- Guillemette, S.; Serra, R.W.; Peng, M.; Hayes, J.A.; Konstantinopoulos, P.A.; Green, M.R.; Cantor, S.B. Resistance to therapy in BRCA2 mutant cells due to loss of the nucleosome remodeling factor CHD4. Genes Dev. 2015, 29, 489–494. [Google Scholar] [CrossRef] [PubMed]
- Rondinelli, B.; Gogola, E.; Yücel, H.; Duarte, A.A.; van de Ven, M.; van der Sluijs, R.; Konstantinopoulos, P.A.; Jonkers, J.; Ceccaldi, R.; Rottenberg, S.; et al. EZH2 promotes degradation of stalled replication forks by recruiting MUS81 through histone H3 trimethylation. Nat. Cell Biol. 2017, 19, 1371–1378. [Google Scholar] [CrossRef]
- Taglialatela, A.; Alvarez, S.; Leuzzi, G.; Sannino, V.; Ranjha, L.; Huang, J.W.; Madubata, C.; Anand, R.; Levy, B.; Rabadan, R.; et al. Restoration of Replication Fork Stability in BRCA1- and BRCA2-Deficient Cells by Inactivation of SNF2-Family Fork Remodelers. Mol. Cell 2017, 68, 414–430.e418. [Google Scholar] [CrossRef] [Green Version]
- Bouwman, P.; Aly, A.; Escandell, J.M.; Pieterse, M.; Bartkova, J.; van der Gulden, H.; Hiddingh, S.; Thanasoula, M.; Kulkarni, A.; Yang, Q.; et al. 53BP1 loss rescues BRCA1 deficiency and is associated with triple-negative and BRCA-mutated breast cancers. Nat. Struct. Mol. Biol. 2010, 17, 688–695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bunting, S.F.; Callén, E.; Wong, N.; Chen, H.T.; Polato, F.; Gunn, A.; Bothmer, A.; Feldhahn, N.; Fernandez-Capetillo, O.; Cao, L.; et al. 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell 2010, 141, 243–254. [Google Scholar] [CrossRef] [Green Version]
- Jaspers, J.E.; Kersbergen, A.; Boon, U.; Sol, W.; van Deemter, L.; Zander, S.A.; Drost, R.; Wientjens, E.; Ji, J.; Aly, A.; et al. Loss of 53BP1 causes PARP inhibitor resistance in Brca1-mutated mouse mammary tumors. Cancer Discov. 2013, 3, 68–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Virgilio, M.; Callen, E.; Yamane, A.; Zhang, W.; Jankovic, M.; Gitlin, A.D.; Feldhahn, N.; Resch, W.; Oliveira, T.Y.; Chait, B.T.; et al. Rif1 prevents resection of DNA breaks and promotes immunoglobulin class switching. Science 2013, 339, 711–715. [Google Scholar] [CrossRef] [Green Version]
- Zimmermann, M.; Lottersberger, F.; Buonomo, S.B.; Sfeir, A.; de Lange, T. 53BP1 regulates DSB repair using Rif1 to control 5’ end resection. Science 2013, 339, 700–704. [Google Scholar] [CrossRef] [Green Version]
- Xu, G.; Chapman, J.R.; Brandsma, I.; Yuan, J.; Mistrik, M.; Bouwman, P.; Bartkova, J.; Gogola, E.; Warmerdam, D.; Barazas, M.; et al. REV7 counteracts DNA double-strand break resection and affects PARP inhibition. Nature 2015, 521, 541–544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghezraoui, H.; Oliveira, C.; Becker, J.R.; Bilham, K.; Moralli, D.; Anzilotti, C.; Fischer, R.; Deobagkar-Lele, M.; Sanchiz-Calvo, M.; Fueyo-Marcos, E.; et al. 53BP1 cooperation with the REV7-shieldin complex underpins DNA structure-specific NHEJ. Nature 2018, 560, 122–127. [Google Scholar] [CrossRef]
- Dev, H.; Chiang, T.W.; Lescale, C.; de Krijger, I.; Martin, A.G.; Pilger, D.; Coates, J.; Sczaniecka-Clift, M.; Wei, W.; Ostermaier, M.; et al. Shieldin complex promotes DNA end-joining and counters homologous recombination in BRCA1-null cells. Nat. Cell Biol. 2018, 20, 954–965. [Google Scholar] [CrossRef] [PubMed]
- He, Y.J.; Meghani, K.; Caron, M.C.; Yang, C.; Ronato, D.A.; Bian, J.; Sharma, A.; Moore, J.; Niraj, J.; Detappe, A.; et al. DYNLL1 binds to MRE11 to limit DNA end resection in BRCA1-deficient cells. Nature 2018, 563, 522–526. [Google Scholar] [CrossRef] [PubMed]
- Nagathihalli, N.S.; Nagaraju, G. RAD51 as a potential biomarker and therapeutic target for pancreatic cancer. Biochim. Biophys. Acta 2011, 1816, 209–218. [Google Scholar] [CrossRef]
- Klein, H.L. The consequences of Rad51 overexpression for normal and tumor cells. DNA Repair 2008, 7, 686–693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clements, K.E.; Thakar, T.; Nicolae, C.M.; Liang, X.; Wang, H.G.; Moldovan, G.L. Loss of E2F7 confers resistance to poly-ADP-ribose polymerase (PARP) inhibitors in BRCA2-deficient cells. Nucleic Acids Res. 2018, 46, 8898–8907. [Google Scholar] [CrossRef] [PubMed]
- Vaidyanathan, A.; Sawers, L.; Gannon, A.L.; Chakravarty, P.; Scott, A.L.; Bray, S.E.; Ferguson, M.J.; Smith, G. ABCB1 (MDR1) induction defines a common resistance mechanism in paclitaxel- and olaparib-resistant ovarian cancer cells. Br. J. Cancer 2016, 115, 431–441. [Google Scholar] [CrossRef] [Green Version]
- O’Driscoll, L.; Walsh, N.; Larkin, A.; Ballot, J.; Ooi, W.S.; Gullo, G.; O’Connor, R.; Clynes, M.; Crown, J.; Kennedy, S. MDR1/P-glycoprotein and MRP-1 drug efflux pumps in pancreatic carcinoma. Anticancer Res. 2007, 27, 2115–2120. [Google Scholar] [PubMed]
- Lu, Z.; Kleeff, J.; Shrikhande, S.; Zimmermann, T.; Korc, M.; Friess, H.; Büchler, M.W. Expression of the multidrug-resistance 1 (MDR1) gene and prognosis in human pancreatic cancer. Pancreas 2000, 21, 240–247. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Kang, Y.; Chen, L.; Wang, H.; Liu, J.; Zeng, S.; Yu, L. The Drug-Resistance Mechanisms of Five Platinum-Based Antitumor Agents. Front. Pharmacol. 2020, 11, 343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gogola, E.; Duarte, A.A.; de Ruiter, J.R.; Wiegant, W.W.; Schmid, J.A.; de Bruijn, R.; James, D.I.; Guerrero Llobet, S.; Vis, D.J.; Annunziato, S.; et al. Selective Loss of PARG Restores PARylation and Counteracts PARP Inhibitor-Mediated Synthetic Lethality. Cancer Cell 2018, 33, 1078–1093.e1012. [Google Scholar] [CrossRef] [Green Version]
- Pettitt, S.J.; Krastev, D.B.; Brandsma, I.; Dréan, A.; Song, F.; Aleksandrov, R.; Harrell, M.I.; Menon, M.; Brough, R.; Campbell, J.; et al. Genome-wide and high-density CRISPR-Cas9 screens identify point mutations in PARP1 causing PARP inhibitor resistance. Nat. Commun. 2018, 9, 1849. [Google Scholar] [CrossRef] [PubMed]
- Ho, W.J.; Jaffee, E.M.; Zheng, L. The tumour microenvironment in pancreatic cancer—Clinical challenges and opportunities. Nat. Rev. Clin. Oncol. 2020, 17, 527–540. [Google Scholar] [CrossRef]
- Beatty, G.L.; Werba, G.; Lyssiotis, C.A.; Simeone, D.M. The biological underpinnings of therapeutic resistance in pancreatic cancer. Genes Dev. 2021, 35, 940–962. [Google Scholar] [CrossRef] [PubMed]
- Dauer, P.; Nomura, A.; Saluja, A.; Banerjee, S. Microenvironment in determining chemo-resistance in pancreatic cancer: Neighborhood matters. Pancreatology 2017, 17, 7–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liudahl, S.M.; Betts, C.B.; Sivagnanam, S.; Morales-Oyarvide, V.; da Silva, A.; Yuan, C.; Hwang, S.; Grossblatt-Wait, A.; Leis, K.R.; Larson, W.; et al. Leukocyte Heterogeneity in Pancreatic Ductal Adenocarcinoma: Phenotypic and Spatial Features Associated with Clinical Outcome. Cancer Discov. 2021, 11, 2014–2031. [Google Scholar] [CrossRef]
- Väyrynen, S.A.; Zhang, J.; Yuan, C.; Väyrynen, J.P.; Dias Costa, A.; Williams, H.; Morales-Oyarvide, V.; Lau, M.C.; Rubinson, D.A.; Dunne, R.F.; et al. Composition, Spatial Characteristics, and Prognostic Significance of Myeloid Cell Infiltration in Pancreatic Cancer. Clin. Cancer Res. 2021, 27, 1069–1081. [Google Scholar] [CrossRef] [PubMed]
- Eikesdal, H.P.; Yndestad, S.; Elzawahry, A.; Llop-Guevara, A.; Gilje, B.; Blix, E.S.; Espelid, H.; Lundgren, S.; Geisler, J.; Vagstad, G.; et al. Olaparib monotherapy as primary treatment in unselected triple negative breast cancer. Ann. Oncol. 2021, 32, 240–249. [Google Scholar] [CrossRef]
- Lama-Sherpa, T.D.; Shevde, L.A. An Emerging Regulatory Role for the Tumor Microenvironment in the DNA Damage Response to Double-Strand Breaks. Mol. Cancer Res. 2020, 18, 185–193. [Google Scholar] [CrossRef] [Green Version]
- Chan, N.; Koritzinsky, M.; Zhao, H.; Bindra, R.; Glazer, P.M.; Powell, S.; Belmaaza, A.; Wouters, B.; Bristow, R.G. Chronic hypoxia decreases synthesis of homologous recombination proteins to offset chemoresistance and radioresistance. Cancer Res. 2008, 68, 605–614. [Google Scholar] [CrossRef] [Green Version]
- Mehibel, M.; Xu, Y.; Li, C.G.; Moon, E.J.; Thakkar, K.N.; Diep, A.N.; Kim, R.K.; Bloomstein, J.D.; Xiao, Y.; Bacal, J.; et al. Eliminating hypoxic tumor cells improves response to PARP inhibitors in homologous recombination-deficient cancer models. J. Clin. Investig. 2021, 131, e146256. [Google Scholar] [CrossRef]
- Wattenberg, M.M.; Beatty, G.L. Overcoming immunotherapeutic resistance by targeting the cancer inflammation cycle. Semin. Cancer Biol. 2020, 65, 38–50. [Google Scholar] [CrossRef]
- Zhu, Y.; Herndon, J.M.; Sojka, D.K.; Kim, K.W.; Knolhoff, B.L.; Zuo, C.; Cullinan, D.R.; Luo, J.; Bearden, A.R.; Lavine, K.J.; et al. Tissue-Resident Macrophages in Pancreatic Ductal Adenocarcinoma Originate from Embryonic Hematopoiesis and Promote Tumor Progression. Immunity 2017, 47, 323–338.e326. [Google Scholar] [CrossRef] [PubMed]
- Hamarsheh, S.; Groß, O.; Brummer, T.; Zeiser, R. Immune modulatory effects of oncogenic KRAS in cancer. Nat. Commun. 2020, 11, 5439. [Google Scholar] [CrossRef] [PubMed]
- Park, D.; Shakya, R.; Koivisto, C.; Pitarresi, J.R.; Szabolcs, M.; Kladney, R.; Hadjis, A.; Mace, T.A.; Ludwig, T. Murine models for familial pancreatic cancer: Histopathology, latency and drug sensitivity among cancers of Palb2, Brca1 and Brca2 mutant mouse strains. PLoS ONE 2019, 14, e0226714. [Google Scholar] [CrossRef] [Green Version]
- Samstein, R.M.; Krishna, C.; Ma, X.; Pei, X.; Lee, K.W.; Makarov, V.; Kuo, F.; Chung, J.; Srivastava, R.M.; Purohit, T.A.; et al. Mutations in BRCA1 and BRCA2 differentially affect the tumor microenvironment and response to checkpoint blockade immunotherapy. Nat. Cancer 2021, 1, 1188–1203. [Google Scholar] [CrossRef]
- Mehta, A.K.; Cheney, E.M.; Hartl, C.A.; Pantelidou, C.; Oliwa, M.; Castrillon, J.A.; Lin, J.R.; Hurst, K.E.; de Oliveira Taveira, M.; Johnson, N.T.; et al. Targeting immunosuppressive macrophages overcomes PARP inhibitor resistance in BRCA1-associated triple-negative breast cancer. Nat. Cancer 2021, 2, 66–82. [Google Scholar] [CrossRef]
- Sanford, D.E.; Belt, B.A.; Panni, R.Z.; Mayer, A.; Deshpande, A.D.; Carpenter, D.; Mitchem, J.B.; Plambeck-Suess, S.M.; Worley, L.A.; Goetz, B.D.; et al. Inflammatory monocyte mobilization decreases patient survival in pancreatic cancer: A role for targeting the CCL2/CCR2 axis. Clin. Cancer Res. 2013, 19, 3404–3415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nywening, T.M.; Belt, B.A.; Cullinan, D.R.; Panni, R.Z.; Han, B.J.; Sanford, D.E.; Jacobs, R.C.; Ye, J.; Patel, A.A.; Gillanders, W.E.; et al. Targeting both tumour-associated CXCR2+ neutrophils and CCR2+ macrophages disrupts myeloid recruitment and improves chemotherapeutic responses in pancreatic ductal adenocarcinoma. Gut 2018, 67, 1112–1123. [Google Scholar] [CrossRef] [Green Version]
- Martí, J.M.; Fernández-Cortés, M.; Serrano-Sáenz, S.; Zamudio-Martinez, E.; Delgado-Bellido, D.; Garcia-Diaz, A.; Oliver, F.J. The Multifactorial Role of PARP-1 in Tumor Microenvironment. Cancers 2020, 12, 739. [Google Scholar] [CrossRef] [Green Version]
- Borcoman, E.; Kanjanapan, Y.; Champiat, S.; Kato, S.; Servois, V.; Kurzrock, R.; Goel, S.; Bedard, P.; Le Tourneau, C. Novel patterns of response under immunotherapy. Ann. Oncol. 2019, 30, 385–396. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Kim, H.J.; Wang, Q.; Kearns, M.; Jiang, T.; Ohlson, C.E.; Li, B.B.; Xie, S.; Liu, J.F.; Stover, E.H.; et al. PARP Inhibition Elicits STING-Dependent Antitumor Immunity in Brca1-Deficient Ovarian Cancer. Cell Rep. 2018, 25, 2972–2980.e2975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pantelidou, C.; Sonzogni, O.; de Oliveira Taveira, M.; Mehta, A.K.; Kothari, A.; Wang, D.; Visal, T.; Li, M.K.; Pinto, J.; Castrillon, J.A.; et al. PARP inhibitor efficacy depends on CD8+ T cell recruitment via intratumoral STING pathway activation in BRCA-deficient models of triple-negative breast cancer. Cancer Discov. 2019, 9, 722–737. [Google Scholar] [CrossRef] [Green Version]
- Motwani, M.; Pesiridis, S.; Fitzgerald, K.A. DNA sensing by the cGAS-STING pathway in health and disease. Nat. Rev. Genet. 2019, 20, 657–674. [Google Scholar] [CrossRef]
- Li, J.; Duran, M.A.; Dhanota, N.; Chatila, W.K.; Bettigole, S.E.; Kwon, J.; Sriram, R.K.; Humphries, M.P.; Salto-Tellez, M.; James, J.A.; et al. Metastasis and Immune Evasion from Extracellular cGAMP Hydrolysis. Cancer Discov. 2021, 11, 1212–1227. [Google Scholar] [CrossRef]
- Jiao, S.; Xia, W.; Yamaguchi, H.; Wei, Y.; Chen, M.K.; Hsu, J.M.; Hsu, J.L.; Yu, W.H.; Du, Y.; Lee, H.H.; et al. PARP Inhibitor Upregulates PD-L1 Expression and Enhances Cancer-Associated Immunosuppression. Clin. Cancer Res. 2017, 23, 3711–3720. [Google Scholar] [CrossRef] [Green Version]
- Mersch, J.; Jackson, M.A.; Park, M.; Nebgen, D.; Peterson, S.K.; Singletary, C.; Arun, B.K.; Litton, J.K. Cancers associated with BRCA1 and BRCA2 mutations other than breast and ovarian. Cancer 2015, 121, 269–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fogelman, D.; Sugar, E.A.; Oliver, G.; Shah, N.; Klein, A.; Alewine, C.; Wang, H.; Javle, M.; Shroff, R.; Wolff, R.A.; et al. Family history as a marker of platinum sensitivity in pancreatic adenocarcinoma. Cancer Chemother. Pharmacol. 2015, 76, 489–498. [Google Scholar] [CrossRef] [Green Version]
- Okano, N.; Morizane, C.; Nomura, S.; Takahashi, H.; Tsumura, H.; Satake, H.; Mizuno, N.; Tsuji, K.; Shioji, K.; Asagi, A.; et al. Phase II clinical trial of gemcitabine plus oxaliplatin in patients with metastatic pancreatic adenocarcinoma with a family history of pancreatic/breast/ovarian/prostate cancer or personal history of breast/ovarian/prostate cancer (FABRIC study). Int. J. Clin. Oncol. 2020, 25, 1835–1843. [Google Scholar] [CrossRef] [PubMed]
- Hoppe, M.M.; Sundar, R.; Tan, D.S.P.; Jeyasekharan, A.D. Biomarkers for Homologous Recombination Deficiency in Cancer. J. Natl. Cancer Inst. 2018, 110, 704–713. [Google Scholar] [CrossRef] [Green Version]
- Fong, P.C.; Yap, T.A.; Boss, D.S.; Carden, C.P.; Mergui-Roelvink, M.; Gourley, C.; De Greve, J.; Lubinski, J.; Shanley, S.; Messiou, C.; et al. Poly(ADP)-ribose polymerase inhibition: Frequent durable responses in BRCA carrier ovarian cancer correlating with platinum-free interval. J. Clin. Oncol. 2010, 28, 2512–2519. [Google Scholar] [CrossRef] [PubMed]
- Gelmon, K.A.; Tischkowitz, M.; Mackay, H.; Swenerton, K.; Robidoux, A.; Tonkin, K.; Hirte, H.; Huntsman, D.; Clemons, M.; Gilks, B.; et al. Olaparib in patients with recurrent high-grade serous or poorly differentiated ovarian carcinoma or triple-negative breast cancer: A phase 2, multicentre, open-label, non-randomised study. Lancet Oncol. 2011, 12, 852–861. [Google Scholar] [CrossRef]
- Coleman, R.L.; Oza, A.M.; Lorusso, D.; Aghajanian, C.; Oaknin, A.; Dean, A.; Colombo, N.; Weberpals, J.I.; Clamp, A.; Scambia, G.; et al. Rucaparib maintenance treatment for recurrent ovarian carcinoma after response to platinum therapy (ARIEL3): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017, 390, 1949–1961. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.H.; Zhu, J.Q.; Yin, R.T.; Yang, J.X.; Liu, J.H.; Wang, J.; Wu, L.Y.; Liu, Z.L.; Gao, Y.N.; Wang, D.B.; et al. Niraparib maintenance therapy in patients with platinum-sensitive recurrent ovarian cancer using an individualized starting dose (NORA): A randomized, double-blind, placebo-controlled phase III trial. Ann. Oncol. 2021, 32, 512–521. [Google Scholar] [CrossRef]
- Birkbak, N.J.; Wang, Z.C.; Kim, J.Y.; Eklund, A.C.; Li, Q.; Tian, R.; Bowman-Colin, C.; Li, Y.; Greene-Colozzi, A.; Iglehart, J.D.; et al. Telomeric allelic imbalance indicates defective DNA repair and sensitivity to DNA-damaging agents. Cancer Discov. 2012, 2, 366–375. [Google Scholar] [CrossRef] [Green Version]
- Abkevich, V.; Timms, K.M.; Hennessy, B.T.; Potter, J.; Carey, M.S.; Meyer, L.A.; Smith-McCune, K.; Broaddus, R.; Lu, K.H.; Chen, J.; et al. Patterns of genomic loss of heterozygosity predict homologous recombination repair defects in epithelial ovarian cancer. Br. J. Cancer 2012, 107, 1776–1782. [Google Scholar] [CrossRef] [Green Version]
- Popova, T.; Manié, E.; Rieunier, G.; Caux-Moncoutier, V.; Tirapo, C.; Dubois, T.; Delattre, O.; Sigal-Zafrani, B.; Bollet, M.; Longy, M.; et al. Ploidy and large-scale genomic instability consistently identify basal-like breast carcinomas with BRCA1/2 inactivation. Cancer Res. 2012, 72, 5454–5462. [Google Scholar] [CrossRef] [Green Version]
- Timms, K.M.; Abkevich, V.; Hughes, E.; Neff, C.; Reid, J.; Morris, B.; Kalva, S.; Potter, J.; Tran, T.V.; Chen, J.; et al. Association of BRCA1/2 defects with genomic scores predictive of DNA damage repair deficiency among breast cancer subtypes. Breast Cancer Res. 2014, 16, 475. [Google Scholar] [CrossRef] [Green Version]
- Telli, M.L.; Timms, K.M.; Reid, J.; Hennessy, B.; Mills, G.B.; Jensen, K.C.; Szallasi, Z.; Barry, W.T.; Winer, E.P.; Tung, N.M.; et al. Homologous Recombination Deficiency (HRD) Score Predicts Response to Platinum-Containing Neoadjuvant Chemotherapy in Patients with Triple-Negative Breast Cancer. Clin. Cancer Res. 2016, 22, 3764–3773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- González-Martín, A.; Pothuri, B.; Vergote, I.; DePont Christensen, R.; Graybill, W.; Mirza, M.R.; McCormick, C.; Lorusso, D.; Hoskins, P.; Freyer, G.; et al. Niraparib in Patients with Newly Diagnosed Advanced Ovarian Cancer. N. Engl. J. Med. 2019, 381, 2391–2402. [Google Scholar] [CrossRef] [Green Version]
- Ray-Coquard, I.; Pautier, P.; Pignata, S.; Pérol, D.; González-Martín, A.; Berger, R.; Fujiwara, K.; Vergote, I.; Colombo, N.; Mäenpää, J.; et al. Olaparib plus Bevacizumab as First-Line Maintenance in Ovarian Cancer. N. Engl. J. Med. 2019, 381, 2416–2428. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Park, J.Y.P.; Pacis, A.; Denroche, R.E.; Jang, G.H.; Zhang, A.; Cuggia, A.; Domecq, C.; Monlong, J.; Raitses-Gurevich, M.; et al. A Preclinical Trial and Molecularly Annotated Patient Cohort Identify Predictive Biomarkers in Homologous Recombination-deficient Pancreatic Cancer. Clin. Cancer Res. 2020, 26, 5462–5476. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexandrov, L.B.; Kim, J.; Haradhvala, N.J.; Huang, M.N.; Tian Ng, A.W.; Wu, Y.; Boot, A.; Covington, K.R.; Gordenin, D.A.; Bergstrom, E.N.; et al. The repertoire of mutational signatures in human cancer. Nature 2020, 578, 94–101. [Google Scholar] [CrossRef] [Green Version]
- Zámborszky, J.; Szikriszt, B.; Gervai, J.Z.; Pipek, O.; Póti, Á.; Krzystanek, M.; Ribli, D.; Szalai-Gindl, J.M.; Csabai, I.; Szallasi, Z.; et al. Loss of BRCA1 or BRCA2 markedly increases the rate of base substitution mutagenesis and has distinct effects on genomic deletions. Oncogene 2017, 36, 746–755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Børresen-Dale, A.L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Xu, X. Microhomology-mediated end joining: New players join the team. Cell Biosci. 2017, 7, 6. [Google Scholar] [CrossRef] [Green Version]
- Waddell, N.; Pajic, M.; Patch, A.M.; Chang, D.K.; Kassahn, K.S.; Bailey, P.; Johns, A.L.; Miller, D.; Nones, K.; Quek, K.; et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature 2015, 518, 495–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nik-Zainal, S.; Davies, H.; Staaf, J.; Ramakrishna, M.; Glodzik, D.; Zou, X.; Martincorena, I.; Alexandrov, L.B.; Martin, S.; Wedge, D.C.; et al. Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature 2016, 534, 47–54. [Google Scholar] [CrossRef]
- Davies, H.; Glodzik, D.; Morganella, S.; Yates, L.R.; Staaf, J.; Zou, X.; Ramakrishna, M.; Martin, S.; Boyault, S.; Sieuwerts, A.M.; et al. HRDetect is a predictor of BRCA1 and BRCA2 deficiency based on mutational signatures. Nat. Med. 2017, 23, 517–525. [Google Scholar] [CrossRef] [PubMed]
- Naipal, K.A.; Verkaik, N.S.; Ameziane, N.; van Deurzen, C.H.; Ter Brugge, P.; Meijers, M.; Sieuwerts, A.M.; Martens, J.W.; O’Connor, M.J.; Vrieling, H.; et al. Functional ex vivo assay to select homologous recombination-deficient breast tumors for PARP inhibitor treatment. Clin. Cancer Res. 2014, 20, 4816–4826. [Google Scholar] [CrossRef] [Green Version]
- Graeser, M.; McCarthy, A.; Lord, C.J.; Savage, K.; Hills, M.; Salter, J.; Orr, N.; Parton, M.; Smith, I.E.; Reis-Filho, J.S.; et al. A marker of homologous recombination predicts pathologic complete response to neoadjuvant chemotherapy in primary breast cancer. Clin. Cancer Res. 2010, 16, 6159–6168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cruz, C.; Castroviejo-Bermejo, M.; Gutiérrez-Enríquez, S.; Llop-Guevara, A.; Ibrahim, Y.H.; Gris-Oliver, A.; Bonache, S.; Morancho, B.; Bruna, A.; Rueda, O.M.; et al. RAD51 foci as a functional biomarker of homologous recombination repair and PARP inhibitor resistance in germline BRCA-mutated breast cancer. Ann. Oncol. 2018, 29, 1203–1210. [Google Scholar] [CrossRef]
- Castroviejo-Bermejo, M.; Cruz, C.; Llop-Guevara, A.; Gutiérrez-Enríquez, S.; Ducy, M.; Ibrahim, Y.H.; Gris-Oliver, A.; Pellegrino, B.; Bruna, A.; Guzmán, M.; et al. A RAD51 assay feasible in routine tumor samples calls PARP inhibitor response beyond BRCA mutation. EMBO Mol. Med. 2018, 10, e9172. [Google Scholar] [CrossRef]
- Meijer, T.G.; Verkaik, N.S.; Sieuwerts, A.M.; van Riet, J.; Naipal, K.A.T.; van Deurzen, C.H.M.; den Bakker, M.A.; Sleddens, H.F.B.M.; Dubbink, H.J.; den Toom, T.D.; et al. Functional Ex Vivo Assay Reveals Homologous Recombination Deficiency in Breast Cancer Beyond BRCA Gene Defects. Clin. Cancer Res. 2018, 24, 6277–6287. [Google Scholar] [CrossRef] [Green Version]
- Voabil, P.; de Bruijn, M.; Roelofsen, L.M.; Hendriks, S.H.; Brokamp, S.; van den Braber, M.; Broeks, A.; Sanders, J.; Herzig, P.; Zippelius, A.; et al. An ex vivo tumor fragment platform to dissect response to PD-1 blockade in cancer. Nat. Med. 2021, 27, 1250–1261. [Google Scholar] [CrossRef] [PubMed]
- Echle, A.; Rindtorff, N.T.; Brinker, T.J.; Luedde, T.; Pearson, A.T.; Kather, J.N. Deep learning in cancer pathology: A new generation of clinical biomarkers. Br. J. Cancer 2021, 124, 686–696. [Google Scholar] [CrossRef]
- Lu, M.Y.; Chen, T.Y.; Williamson, D.F.K.; Zhao, M.; Shady, M.; Lipkova, J.; Mahmood, F. AI-based pathology predicts origins for cancers of unknown primary. Nature 2021, 594, 106–110. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zou, C.; Zhang, Y.; Li, X.; Wang, C.; Ke, F.; Chen, J.; Wang, W.; Wang, D.; Xu, X.; et al. Prediction of BRCA Gene Mutation in Breast Cancer Based on Deep Learning and Histopathology Images. Front. Genet. 2021, 12, 661109. [Google Scholar] [CrossRef]
- Schonhoft, J.D.; Zhao, J.L.; Jendrisak, A.; Carbone, E.A.; Barnett, E.S.; Hullings, M.A.; Gill, A.; Sutton, R.; Lee, J.; Dago, A.E.; et al. Morphology-Predicted Large-Scale Transition Number in Circulating Tumor Cells Identifies a Chromosomal Instability Biomarker Associated with Poor Outcome in Castration-Resistant Prostate Cancer. Cancer Res. 2020, 80, 4892–4903. [Google Scholar] [CrossRef] [PubMed]
- Karakashev, S.; Fukumoto, T.; Zhao, B.; Lin, J.; Wu, S.; Fatkhutdinov, N.; Park, P.H.; Semenova, G.; Jean, S.; Cadungog, M.G.; et al. EZH2 Inhibition Sensitizes CARM1-High, Homologous Recombination Proficient Ovarian Cancers to PARP Inhibition. Cancer Cell 2020, 37, 157–167.e156. [Google Scholar] [CrossRef] [PubMed]
- Abbotts, R.; Topper, M.J.; Biondi, C.; Fontaine, D.; Goswami, R.; Stojanovic, L.; Choi, E.Y.; McLaughlin, L.; Kogan, A.A.; Xia, L.; et al. DNA methyltransferase inhibitors induce a BRCAness phenotype that sensitizes NSCLC to PARP inhibitor and ionizing radiation. Proc. Natl. Acad. Sci. USA 2019, 116, 22609–22618. [Google Scholar] [CrossRef]
- McLaughlin, L.J.; Stojanovic, L.; Kogan, A.A.; Rutherford, J.L.; Choi, E.Y.; Yen, R.C.; Xia, L.; Zou, Y.; Lapidus, R.G.; Baylin, S.B.; et al. Pharmacologic induction of innate immune signaling directly drives homologous recombination deficiency. Proc. Natl. Acad. Sci. USA 2020, 117, 17785–17795. [Google Scholar] [CrossRef]
- Pulliam, N.; Fang, F.; Ozes, A.R.; Tang, J.; Adewuyi, A.; Keer, H.; Lyons, J.; Baylin, S.B.; Matei, D.; Nakshatri, H.; et al. An Effective Epigenetic-PARP Inhibitor Combination Therapy for Breast and Ovarian Cancers Independent of BRCA Mutations. Clin. Cancer Res. 2018, 24, 3163–3175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taniguchi, Y. The Bromodomain and Extra-Terminal Domain (BET) Family: Functional Anatomy of BET Paralogous Proteins. Int. J. Mol. Sci. 2016, 17, 1849. [Google Scholar] [CrossRef] [Green Version]
- Miller, A.L.; Fehling, S.C.; Garcia, P.L.; Gamblin, T.L.; Council, L.N.; van Waardenburg, R.C.A.M.; Yang, E.S.; Bradner, J.E.; Yoon, K.J. The BET inhibitor JQ1 attenuates double-strand break repair and sensitizes models of pancreatic ductal adenocarcinoma to PARP inhibitors. EBioMedicine 2019, 44, 419–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Zhang, Y.; Shan, W.; Hu, Z.; Yuan, J.; Pi, J.; Wang, Y.; Fan, L.; Tang, Z.; Li, C.; et al. Repression of BET activity sensitizes homologous recombination-proficient cancers to PARP inhibition. Sci. Transl. Med. 2017, 9, eaal1645. [Google Scholar] [CrossRef] [Green Version]
- Sun, C.; Yin, J.; Fang, Y.; Chen, J.; Jeong, K.J.; Chen, X.; Vellano, C.P.; Ju, Z.; Zhao, W.; Zhang, D.; et al. BRD4 Inhibition Is Synthetic Lethal with PARP Inhibitors through the Induction of Homologous Recombination Deficiency. Cancer Cell 2018, 33, 401–416.e408. [Google Scholar] [CrossRef] [Green Version]
- Parsels, L.A.; Engelke, C.G.; Parsels, J.; Flanagan, S.A.; Zhang, Q.; Tanska, D.; Wahl, D.R.; Canman, C.E.; Lawrence, T.S.; Morgan, M.A. Combinatorial Efficacy of Olaparib with Radiation and ATR Inhibitor Requires PARP1 Protein in Homologous Recombination-Proficient Pancreatic Cancer. Mol. Cancer Ther. 2021, 20, 263–273. [Google Scholar] [CrossRef]
- Yap, T.A.; O’Carrigan, B.; Penney, M.S.; Lim, J.S.; Brown, J.S.; de Miguel Luken, M.J.; Tunariu, N.; Perez-Lopez, R.; Rodrigues, D.N.; Riisnaes, R.; et al. Phase I Trial of First-in-Class ATR Inhibitor M6620 (VX-970) as Monotherapy or in Combination with Carboplatin in Patients with Advanced Solid Tumors. J. Clin. Oncol. 2020, 38, 3195–3204. [Google Scholar] [CrossRef] [PubMed]
- Sørensen, C.S.; Hansen, L.T.; Dziegielewski, J.; Syljuåsen, R.G.; Lundin, C.; Bartek, J.; Helleday, T. The cell-cycle checkpoint kinase Chk1 is required for mammalian homologous recombination repair. Nat. Cell Biol. 2005, 7, 195–201. [Google Scholar] [CrossRef]
- Morgan, M.A.; Parsels, L.A.; Zhao, L.; Parsels, J.D.; Davis, M.A.; Hassan, M.C.; Arumugarajah, S.; Hylander-Gans, L.; Morosini, D.; Simeone, D.M.; et al. Mechanism of radiosensitization by the Chk1/2 inhibitor AZD7762 involves abrogation of the G2 checkpoint and inhibition of homologous recombinational DNA repair. Cancer Res. 2010, 70, 4972–4981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morimoto, Y.; Takada, K.; Takeuchi, O.; Takagi, A.; Watanabe, K.; Hirohara, M.; Hamamoto, T.; Masuda, Y. Prexasertib increases the sensitivity of pancreatic cancer cells to gemcitabine and S-1. Oncol. Rep. 2020, 43, 689–699. [Google Scholar] [CrossRef]
- Engelke, C.G.; Parsels, L.A.; Qian, Y.; Zhang, Q.; Karnak, D.; Robertson, J.R.; Tanska, D.M.; Wei, D.; Davis, M.A.; Parsels, J.D.; et al. Sensitization of pancreatic cancer to chemoradiation by the Chk1 inhibitor MK8776. Clin. Cancer Res. 2013, 19, 4412–4421. [Google Scholar] [CrossRef] [Green Version]
- Do, K.T.; Kochupurakkal, B.; Kelland, S.; de Jonge, A.; Hedglin, J.; Powers, A.; Quinn, N.; Gannon, C.; Vuong, L.; Parmar, K.; et al. Phase 1 Combination Study of the CHK1 Inhibitor Prexasertib and the PARP Inhibitor Olaparib in High-grade Serous Ovarian Cancer and Other Solid Tumors. Clin. Cancer Res. 2021, 27, 4710–7416. [Google Scholar] [CrossRef]
- Laquente, B.; Lopez-Martin, J.; Richards, D.; Illerhaus, G.; Chang, D.Z.; Kim, G.; Stella, P.; Richel, D.; Szcylik, C.; Cascinu, S.; et al. A phase II study to evaluate LY2603618 in combination with gemcitabine in pancreatic cancer patients. BMC Cancer 2017, 17, 137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Infante, J.R.; Hollebecque, A.; Postel-Vinay, S.; Bauer, T.M.; Blackwood, E.M.; Evangelista, M.; Mahrus, S.; Peale, F.V.; Lu, X.; Sahasranaman, S.; et al. Phase I Study of GDC-0425, a Checkpoint Kinase 1 Inhibitor, in Combination with Gemcitabine in Patients with Refractory Solid Tumors. Clin. Cancer Res. 2017, 23, 2423–2432. [Google Scholar] [CrossRef] [Green Version]
- Grundy, M.K.; Buckanovich, R.J.; Bernstein, K.A. Regulation and pharmacological targeting of RAD51 in cancer. NAR Cancer 2020, 2, zcaa024. [Google Scholar] [CrossRef] [PubMed]
- Guy, J.L.; Maclay, T.; Day, M.; Burness, M.L.; Mills, K. Abstract P2-05-05: RAD51 inhibition using CYT-0851, shows anti-cancer activity in cellular models of breast cancer and acts synergistically with PARP inhibitors. Cancer Res. 2020, 80, P2-05-05-P02-05-05. [Google Scholar] [CrossRef]
- Stecklein, S.R.; Kumaraswamy, E.; Behbod, F.; Wang, W.; Chaguturu, V.; Harlan-Williams, L.M.; Jensen, R.A. BRCA1 and HSP90 cooperate in homologous and non-homologous DNA double-strand-break repair and G2/M checkpoint activation. Proc. Natl. Acad. Sci. USA 2012, 109, 13650–13655. [Google Scholar] [CrossRef] [Green Version]
- Gabbasov, R.; Benrubi, I.D.; O’Brien, S.W.; Krais, J.J.; Johnson, N.; Litwin, S.; Connolly, D.C. Targeted blockade of HSP90 impairs DNA-damage response proteins and increases the sensitivity of ovarian carcinoma cells to PARP inhibition. Cancer Biol. Ther. 2019, 20, 1035–1045. [Google Scholar] [CrossRef]
- Ha, K.; Fiskus, W.; Choi, D.S.; Bhaskara, S.; Cerchietti, L.; Devaraj, S.G.; Shah, B.; Sharma, S.; Chang, J.C.; Melnick, A.M.; et al. Histone deacetylase inhibitor treatment induces ‘BRCAness’ and synergistic lethality with PARP inhibitor and cisplatin against human triple negative breast cancer cells. Oncotarget 2014, 5, 5637–5650. [Google Scholar] [CrossRef] [Green Version]
- Shimomura, A.; Yamamoto, N.; Kondo, S.; Fujiwara, Y.; Suzuki, S.; Yanagitani, N.; Horiike, A.; Kitazono, S.; Ohyanagi, F.; Doi, T.; et al. First-in-Human Phase I Study of an Oral HSP90 Inhibitor, TAS-116, in Patients with Advanced Solid Tumors. Mol. Cancer Ther. 2019, 18, 531–540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Renouf, D.J.; Hedley, D.; Krzyzanowska, M.K.; Schmuck, M.; Wang, L.; Moore, M.J. A phase II study of the HSP90 inhibitor AUY922 in chemotherapy refractory advanced pancreatic cancer. Cancer Chemother. Pharmacol. 2016, 78, 541–545. [Google Scholar] [CrossRef]
- Pedersen, K.S.; Kim, G.P.; Foster, N.R.; Wang-Gillam, A.; Erlichman, C.; McWilliams, R.R. Phase II trial of gemcitabine and tanespimycin (17AAG) in metastatic pancreatic cancer: A Mayo Clinic Phase II Consortium study. Investig. New Drugs 2015, 33, 963–968. [Google Scholar] [CrossRef] [Green Version]
- Pillai, R.N.; Fennell, D.A.; Kovcin, V.; Ciuleanu, T.E.; Ramlau, R.; Kowalski, D.; Schenker, M.; Yalcin, I.; Teofilovici, F.; Vukovic, V.M.; et al. Randomized Phase III Study of Ganetespib, a Heat Shock Protein 90 Inhibitor, with Docetaxel Versus Docetaxel in Advanced Non-Small-Cell Lung Cancer (GALAXY-2). J. Clin. Oncol. 2020, 38, 613–622. [Google Scholar] [CrossRef]
- Hubbard, J.; Erlichman, C.; Toft, D.O.; Qin, R.; Stensgard, B.A.; Felten, S.; Ten Eyck, C.; Batzel, G.; Ivy, S.P.; Haluska, P. Phase I study of 17-allylamino-17 demethoxygeldanamycin, gemcitabine and/or cisplatin in patients with refractory solid tumors. Investig. New Drugs 2011, 29, 473–480. [Google Scholar] [CrossRef] [Green Version]
- Sanchez, J.; Carter, T.R.; Cohen, M.S.; Blagg, B.S.J. Old and New Approaches to Target the Hsp90 Chaperone. Curr. Cancer Drug Targets 2020, 20, 253–270. [Google Scholar] [CrossRef] [PubMed]
- Krajewska, M.; Heijink, A.M.; Bisselink, Y.J.; Seinstra, R.I.; Silljé, H.H.; de Vries, E.G.; van Vugt, M.A. Forced activation of Cdk1 via wee1 inhibition impairs homologous recombination. Oncogene 2013, 32, 3001–3008. [Google Scholar] [CrossRef] [PubMed]
- Hartman, S.J.; Bagby, S.M.; Yacob, B.W.; Simmons, D.M.; MacBeth, M.; Lieu, C.H.; Davis, S.L.; Leal, A.D.; Tentler, J.J.; Diamond, J.R.; et al. WEE1 Inhibition in Combination with Targeted Agents and Standard Chemotherapy in Preclinical Models of Pancreatic Ductal Adenocarcinoma. Front. Oncol. 2021, 11, 642328. [Google Scholar] [CrossRef]
- Karnak, D.; Engelke, C.G.; Parsels, L.A.; Kausar, T.; Wei, D.; Robertson, J.R.; Marsh, K.B.; Davis, M.A.; Zhao, L.; Maybaum, J.; et al. Combined inhibition of Wee1 and PARP1/2 for radiosensitization in pancreatic cancer. Clin. Cancer Res. 2014, 20, 5085–5096. [Google Scholar] [CrossRef] [Green Version]
- Kausar, T.; Schreiber, J.S.; Karnak, D.; Parsels, L.A.; Parsels, J.D.; Davis, M.A.; Zhao, L.; Maybaum, J.; Lawrence, T.S.; Morgan, M.A. Sensitization of Pancreatic Cancers to Gemcitabine Chemoradiation by WEE1 Kinase Inhibition Depends on Homologous Recombination Repair. Neoplasia 2015, 17, 757–766. [Google Scholar] [CrossRef] [Green Version]
- Cuneo, K.C.; Morgan, M.A.; Sahai, V.; Schipper, M.J.; Parsels, L.A.; Parsels, J.D.; Devasia, T.; Al-Hawaray, M.; Cho, C.S.; Nathan, H.; et al. Dose Escalation Trial of the Wee1 Inhibitor Adavosertib (AZD1775) in Combination with Gemcitabine and Radiation for Patients with Locally Advanced Pancreatic Cancer. J. Clin. Oncol. 2019, 37, 2643–2650. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, E.; Falchook, G.S.; Wang, J.S.; Fu, S.; Oza, A.; Karen, S.; Imedio, E.R.; Kumar, S.; Ottesen, L.; Mugundu, G.M.; et al. Abstract CT025: Phase Ib study of adavosertib in combination with olaparib in patients with refractory solid tumors: Dose escalation. Cancer Res. 2019, 79, CT025. [Google Scholar] [CrossRef]
- Quereda, V.; Bayle, S.; Vena, F.; Frydman, S.M.; Monastyrskyi, A.; Roush, W.R.; Duckett, D.R. Therapeutic Targeting of CDK12/CDK13 in Triple-Negative Breast Cancer. Cancer Cell 2019, 36, 545–558.e547. [Google Scholar] [CrossRef]
- Luo, M.L.; Zheng, F.; Chen, W.; Liang, Z.M.; Chandramouly, G.; Tan, J.; Willis, N.A.; Chen, C.H.; Taveira, M.O.; Zhou, X.Z.; et al. Inactivation of the Prolyl Isomerase Pin1 Sensitizes BRCA1-Proficient Breast Cancer to PARP Inhibition. Cancer Res. 2020, 80, 3033–3045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, C. A Novel Mechanism to Induce BRCAness in Cancer Cells. Cancer Res. 2020, 80, 2977–2978. [Google Scholar] [CrossRef] [PubMed]
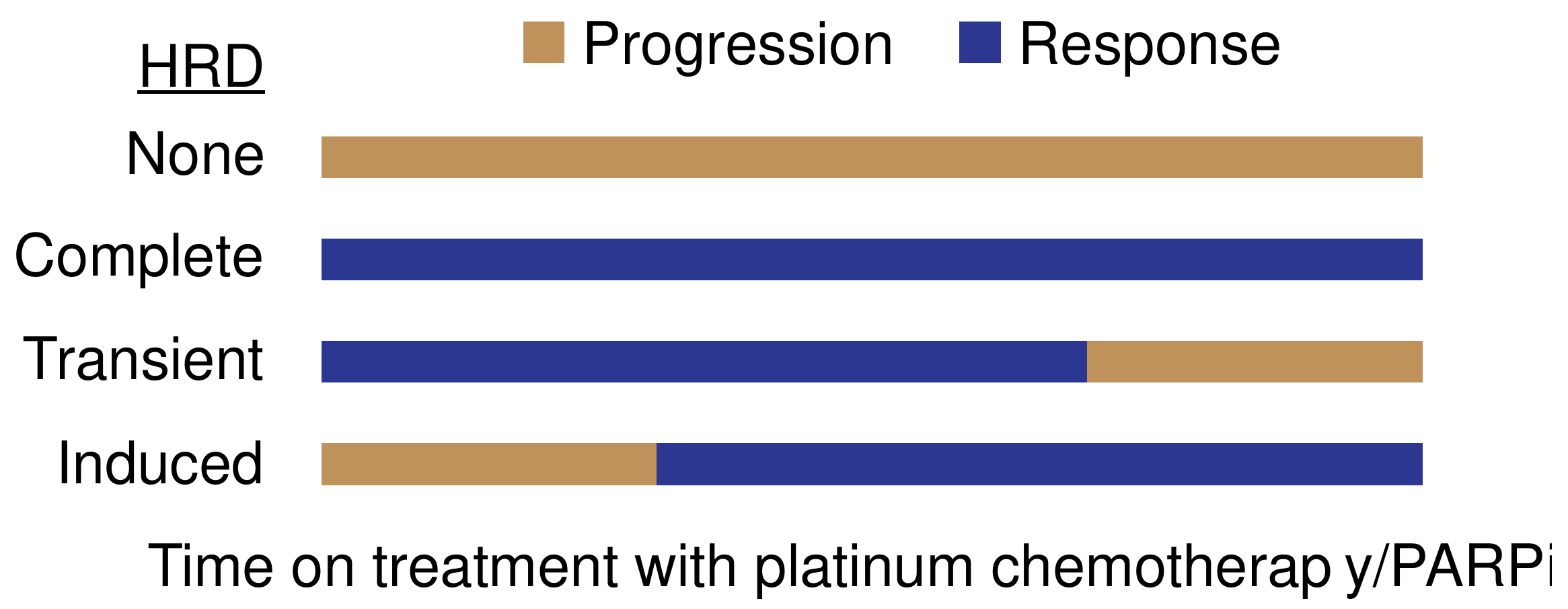

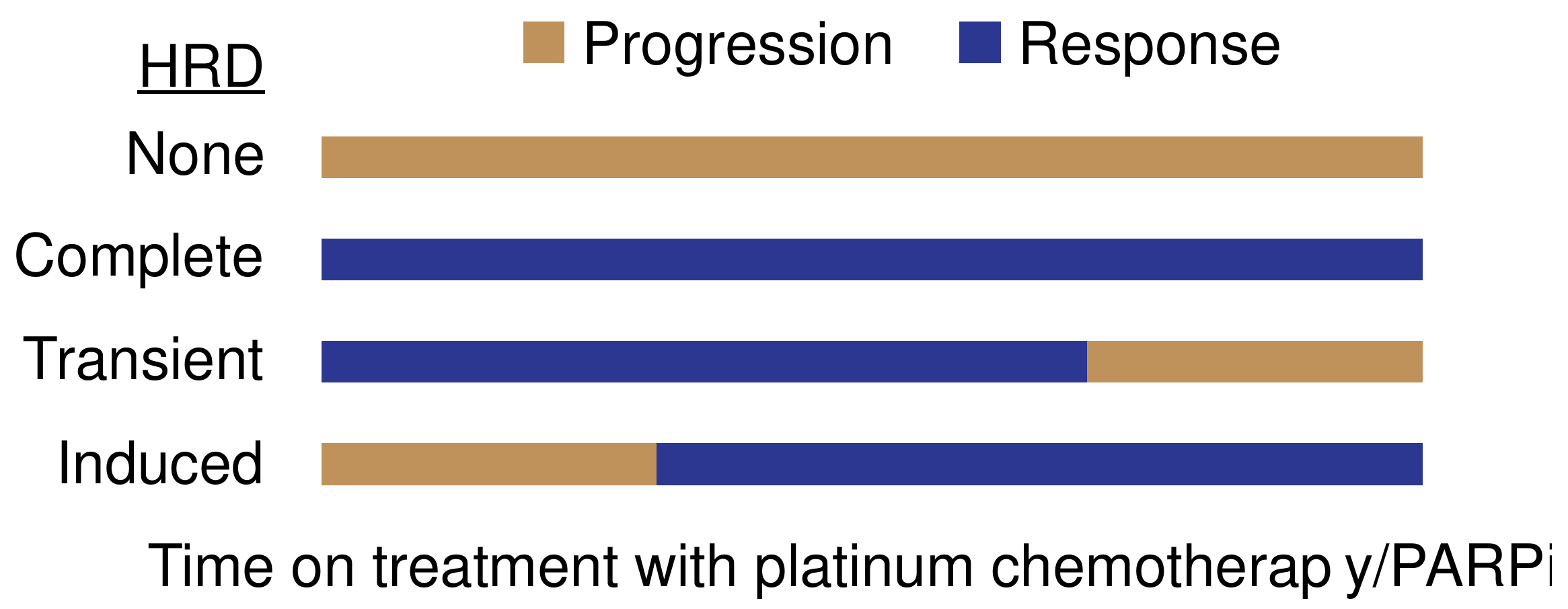{kind=link}
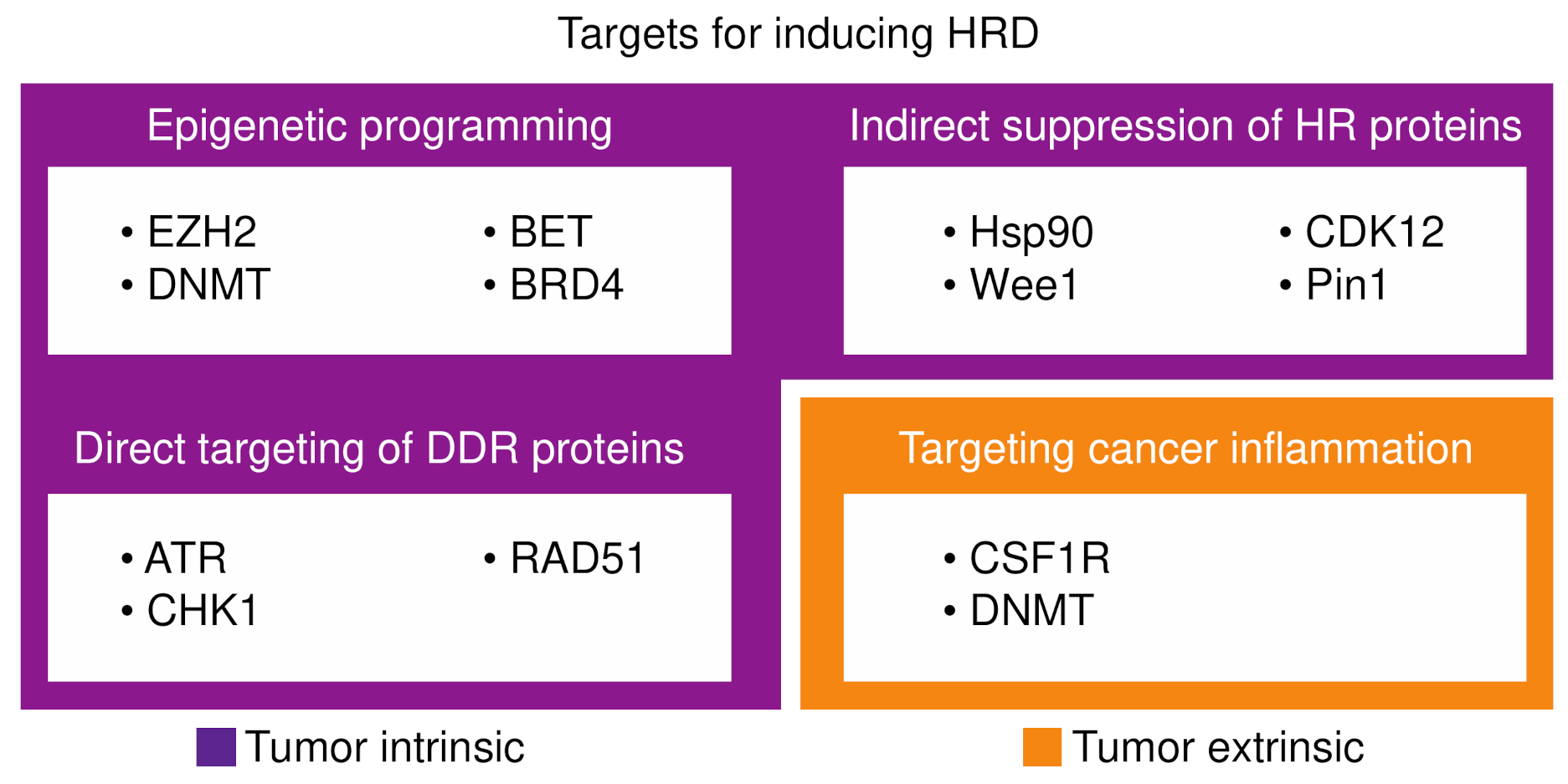{kind=link}
| Determinant | Mechanism | Platinum Sensitivity | PARPi Sensitivity | Notes |
|---|---|---|---|---|
| Primary determinants * | ||||
| HR gene mutations | ||||
| BRCA1 | Abnormal end resection and invasion | ↑ | ↑ | |
| BRCA2 | Abnormal strand invasion | ↑ | ↑ | Germline BRCA2 mutations make up ~70% of BRCA mutations in pancreatic cancer |
| Non-BRCA HR genes | Variable loss of HR | ↑ or ←→ | ↑ or ←→ | RAD51C/D and XRCC2 alterations may drive HRD in pancreatic cancer; Germline ATM and CHEK2 are not associated with signatures of HRD in pancreatic cancer |
| Secondary reversion mutations | Frame-shift mutation allows translation of active truncated protein | ↓ | ↓ | |
| Secondary determinants# | ||||
| Replication fork stability | ||||
| CDH4 | Loss leads to replication fork stabilization via reduced resection | ↓ | ↓ | Clinical relevance in pancreatic cancer remains to be determined |
| EZH2 | ||||
| PTIP | ||||
| SMARCAL1 | ||||
| HR reconstitution/bypass | ||||
| 53BP1-RIF1-REV7-shielden | Loss leads to reduced NHEJ and release of end resection antagonism | ↓ | ↓ | Specific to BRCA1 mutant cancers |
| DYNLL1 | Loss drives reduced inhibition of MRE11-mediated end resection | |||
| RAD51 | Overexpression increases HR proficiency | |||
| Drug specific alterations | ||||
| Drug transport | Excess drug efflux or inefficient drug uptake | ↓ | ↓ | P-glycoprotein/MDR1 is frequently overexpressed in pancreatic cancer |
| PARP1 point-mutations | Reduced DNA binding capacity prevents PARP trapping | |||
| dePARylation | Loss of PARG reconstitutes PARylation activity | |||
| Tumor microenvironment | ||||
| Tumor infiltrating T cells | PARPi drives cGAS-STING activation and T cell anti-tumor immunity | - | ↑ | Pre-treatment T cell infiltration associated with improved response to PARPi in breast cancer |
| Tumor associated macrophages | - | - | ↓ | Anti-CSF1R enhances PARPi activity in BRCA1 mutant breast cancer models |
| Tissue hypoxia | Suppression of RAD51 and BRCA2 translation | ↑↓ | ↑↓ | Severe hypoxia is associated with supression of HR, while moderate hypoxia promotes resistance to PARPi |
| Target | Definition | Diagnostic techniques | Clinical tests | Benefits | Limitations | Notes |
|---|---|---|---|---|---|---|
| Genomic scars | ||||||
| Telomeric allelic imbalance (TAI) | Subchromosomal regions of allelic imbalance extending to the telomere | SNP array; Array-based comparative genomic hybridization; Next-generation sequencing | Genomic instability score (GIS) an unweighted sum of TAI, LOH and LSTs | Can be performed on FFPE tissues | GIS has reduced sensitivity and specificity for identifying HRD as compared to mutational signatures; Scars persist regardless of change in HR proficiency | LST are increased in HRD pancreatic cancer, but may not be associated with response to platinum chemotherapy |
| Loss of heterozygosity (LOH) | Regions of LOH of moderate size (greater than 15 Mb but less than the entire chromosome) | |||||
| Large scale transitions (LST) | Chromosomal break between two regions of at least 10 Mb | |||||
| Mutational signatures | ||||||
| Signature 3 | Large deletions (up to 50 bp) | Whole genome sequencing | HRDetect a weighted model incorporating mutational signatures | Signature 3 and HRDetect have high sensitivity and specificity for detection of HRD in pancreatic cancer | Requires fresh tissue due to FFPE associated artifacts; Signatures persist regardless of change in HR proficiency | RS3 is associated with BRCA1/2 mutations and RS5 is associated with BRCA1 mutations |
| Rearrangement signatures (RS) 3 and 5 | Tandem duplications <10kb (RS3) or deletions <100kb (RS5) | |||||
| HR function | ||||||
| RAD51 foci | RAD51 foci arise during nucleoprotein filament formation | Immunofluorescence (IF) microscopy | Various, including assessment of ex vivo irradiatiated or post-chemotherapy biopsy specimen using IF microscopy | Dynamic marker of HR proficiency | Some assays require on-treatment biopsy due to limited RAD51 staining in pre-treatment FFPE specimens; IF microscopy is challenging to scale for clinical application | Established in breast cancer; Validation in pancreatic cancer needed |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wattenberg, M.M.; Reiss, K.A. Determinants of Homologous Recombination Deficiency in Pancreatic Cancer. Cancers 2021, 13, 4716. https://doi.org/10.3390/cancers13184716
Wattenberg MM, Reiss KA. Determinants of Homologous Recombination Deficiency in Pancreatic Cancer. Cancers. 2021; 13(18):4716. https://doi.org/10.3390/cancers13184716
Chicago/Turabian StyleWattenberg, Max M., and Kim A. Reiss. 2021. "Determinants of Homologous Recombination Deficiency in Pancreatic Cancer" Cancers 13, no. 18: 4716. https://doi.org/10.3390/cancers13184716
APA StyleWattenberg, M. M., & Reiss, K. A. (2021). Determinants of Homologous Recombination Deficiency in Pancreatic Cancer. Cancers, 13(18), 4716. https://doi.org/10.3390/cancers13184716