
VKNG-1 Antagonizes ABCG2-Mediated Multidrug Resistance via p-AKT and Bcl-2 Pathway in Colon Cancer: In Vitro and In Vivo Study


 ,
, 
Abstract
:Simple Summary
Abstract
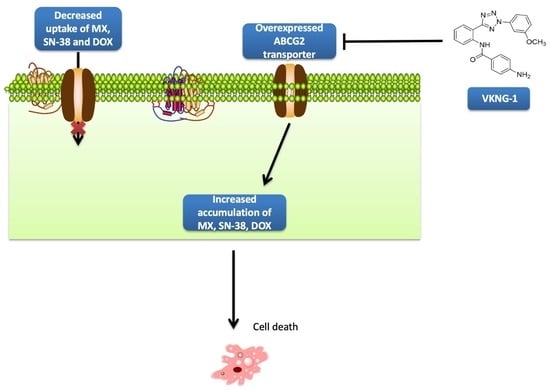
1. Introduction
2. Results
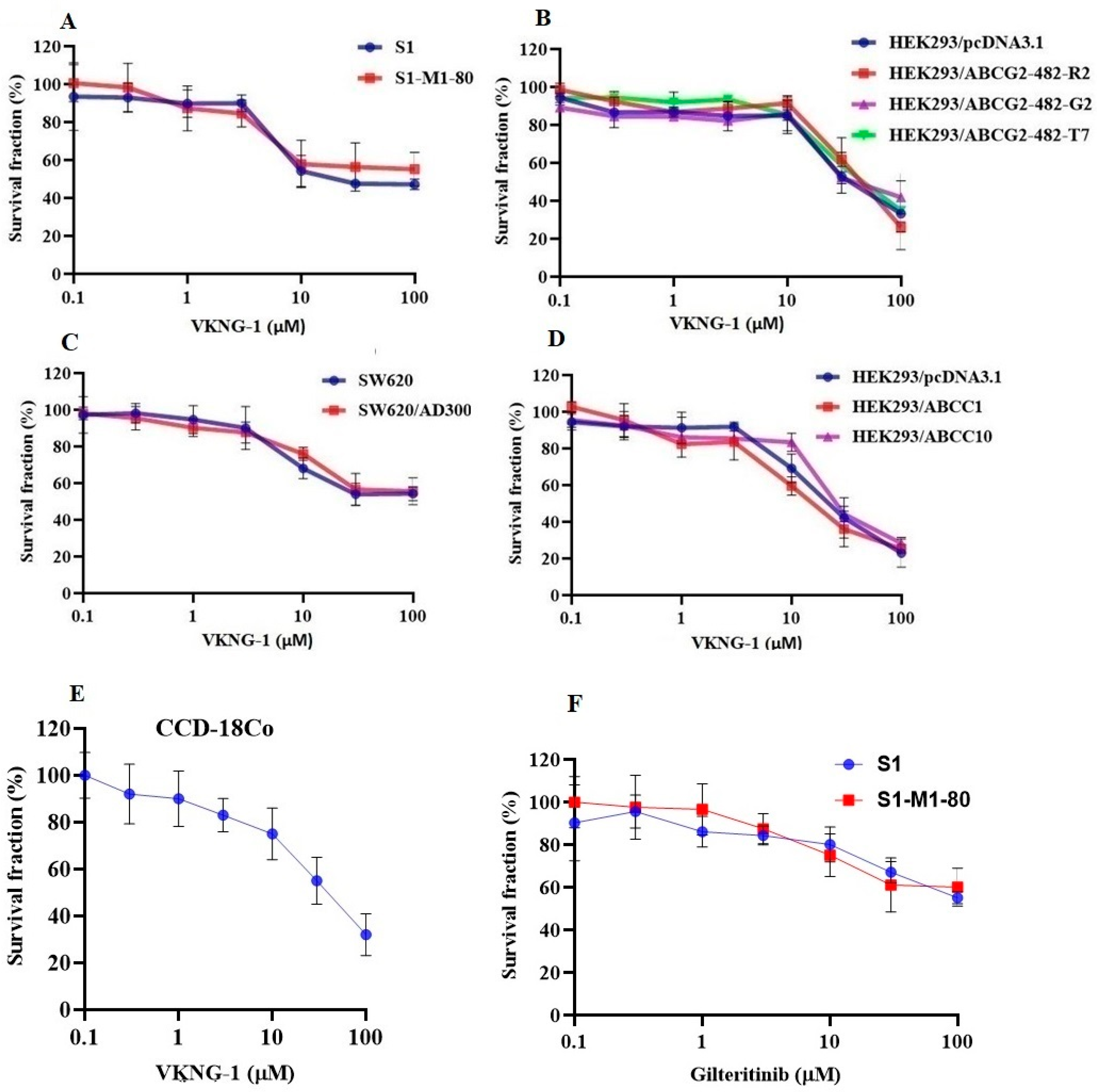
2.1. VKNG-1 Increases the Sensitivity of ABCG2-Ooverexpressing Drug-Resistant Cells to Anticancer Drugs
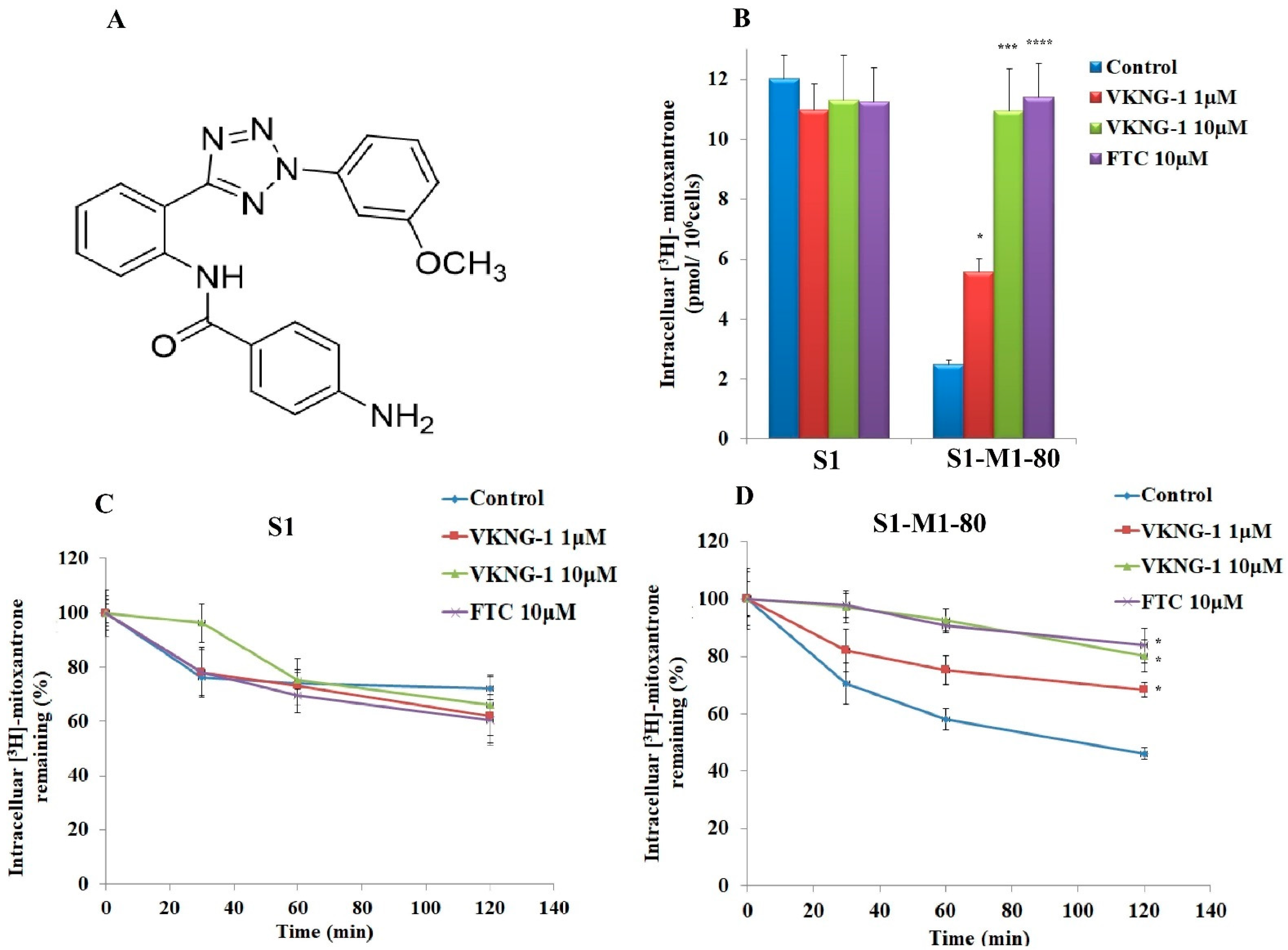
2.2. VKNG-1 Significantly Enhances the Accumulation and Diminishes the Efflux of (3H)-Mitoxantrone in Cells Overexpressing the ABCG2 Transporter
2.3. Effect of VKNG-1 and Gilteritinib on the Expression of ABCG2
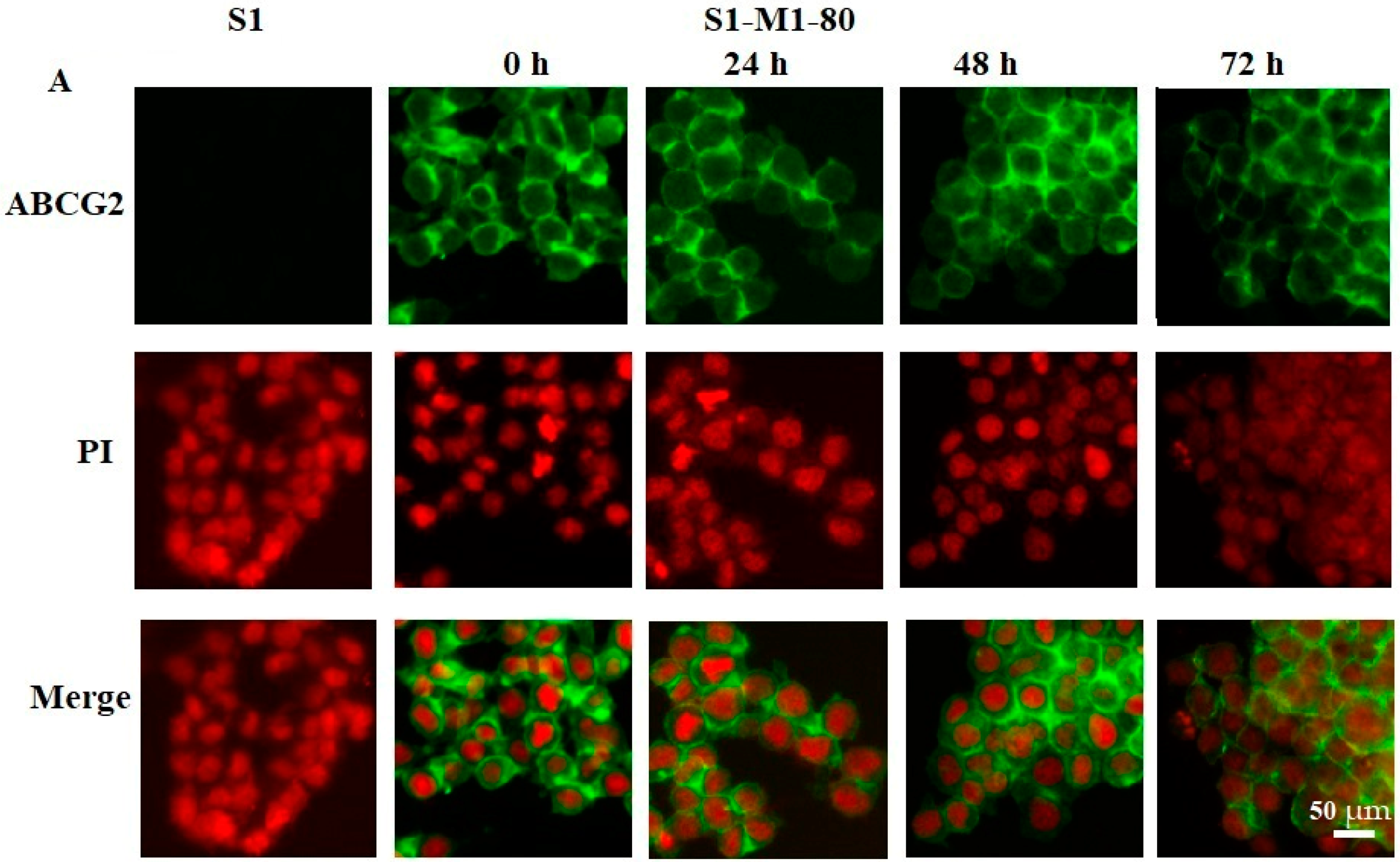
2.4. Effect of VKNG-1 on the Membrane Localization of ABCG2 Transporter
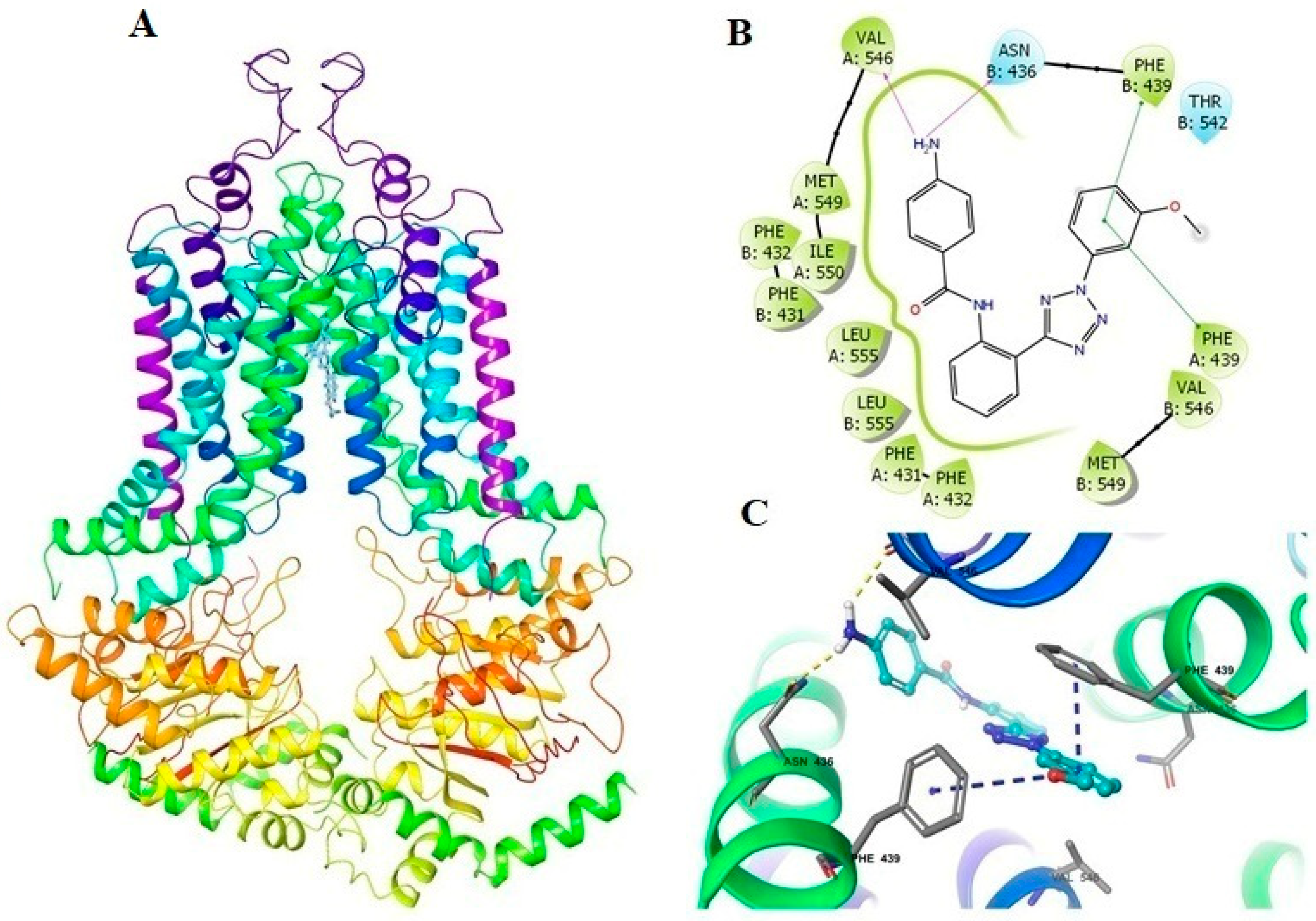
2.5. Docking Analysis of VKNG-1 and the Human ABCG2 Model
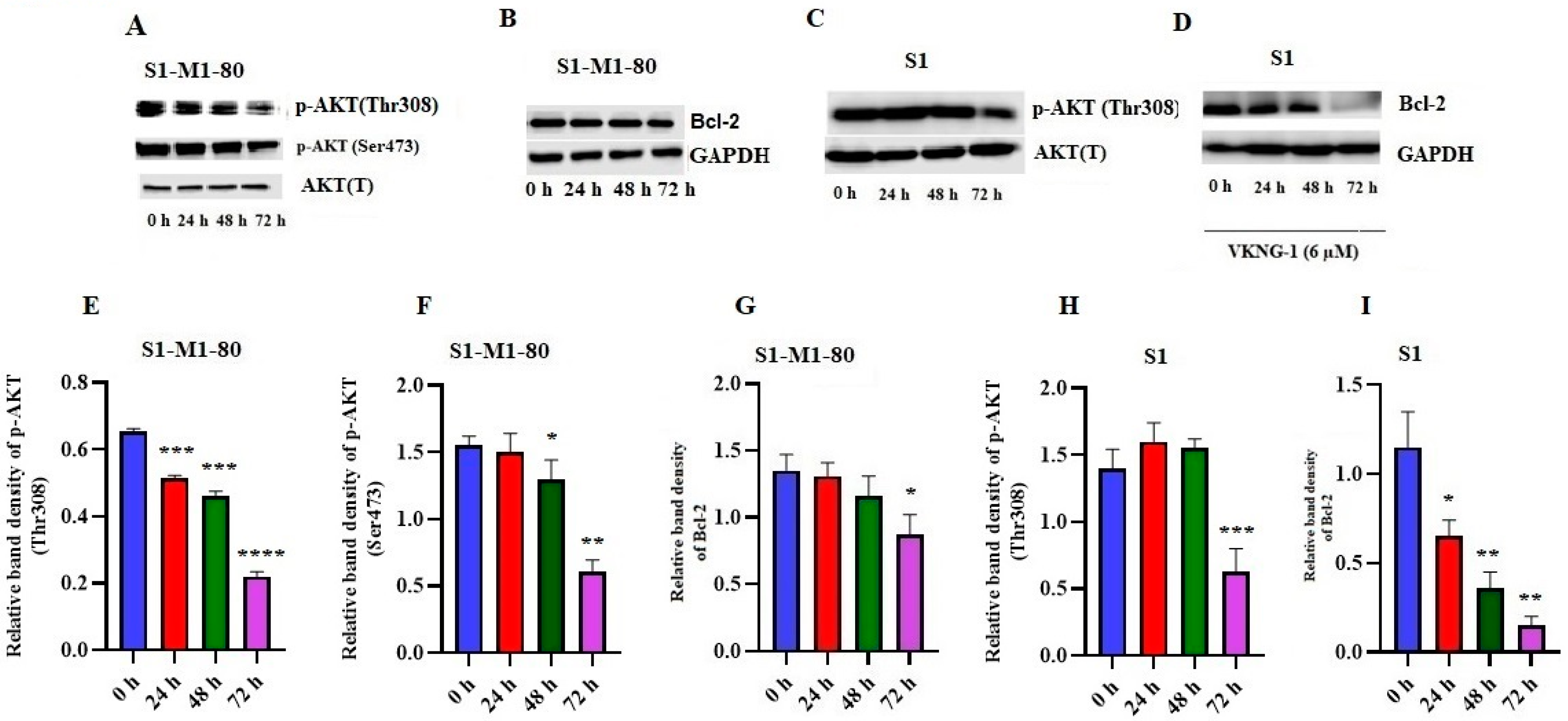
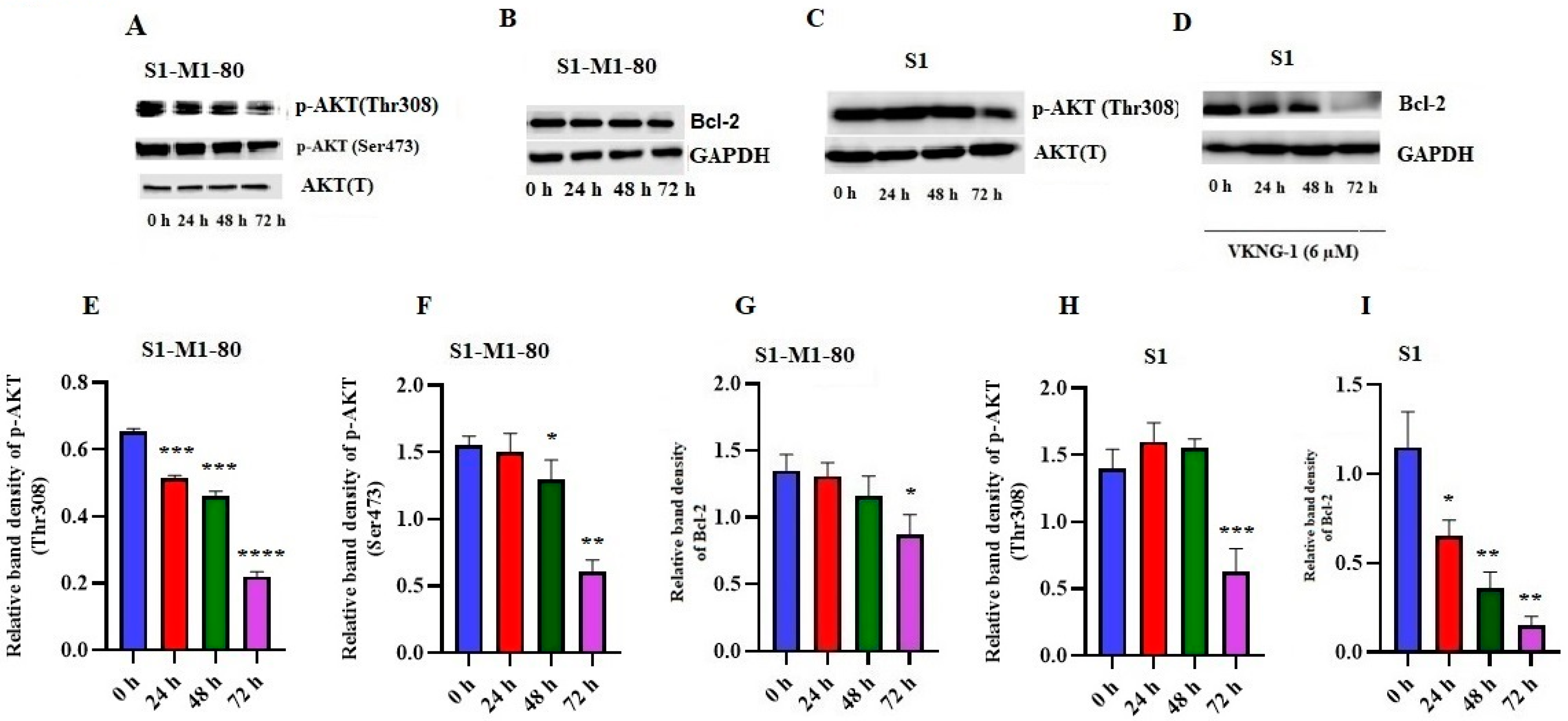
2.6. Effect of VKNG-1 on p-AKT and B-cell lymphoma-2 (Bcl-2) Expression
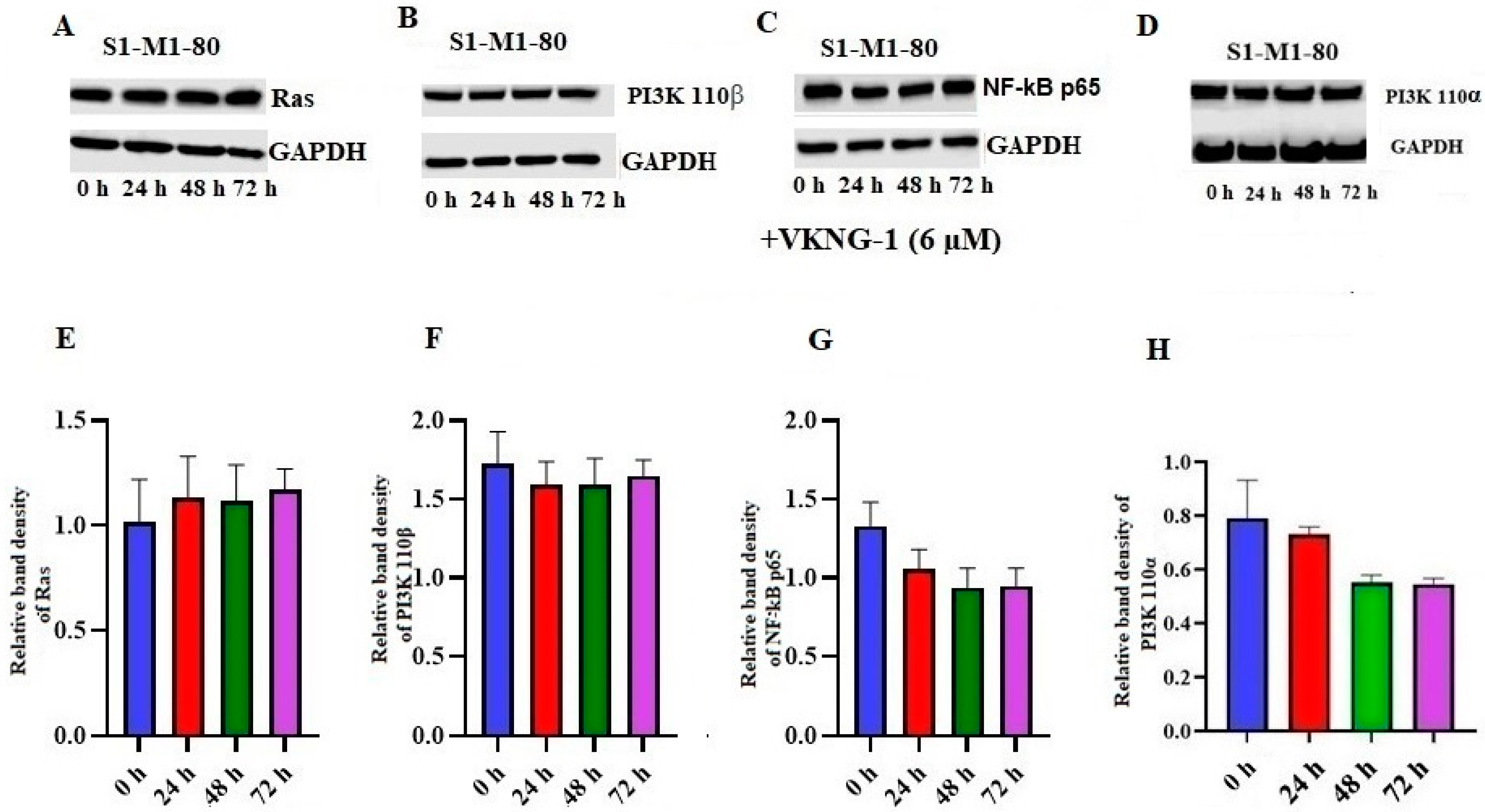
2.7. Effect of VKNG-1 on Ras, NF-kB p65, PI3K p110α and 110β Expression
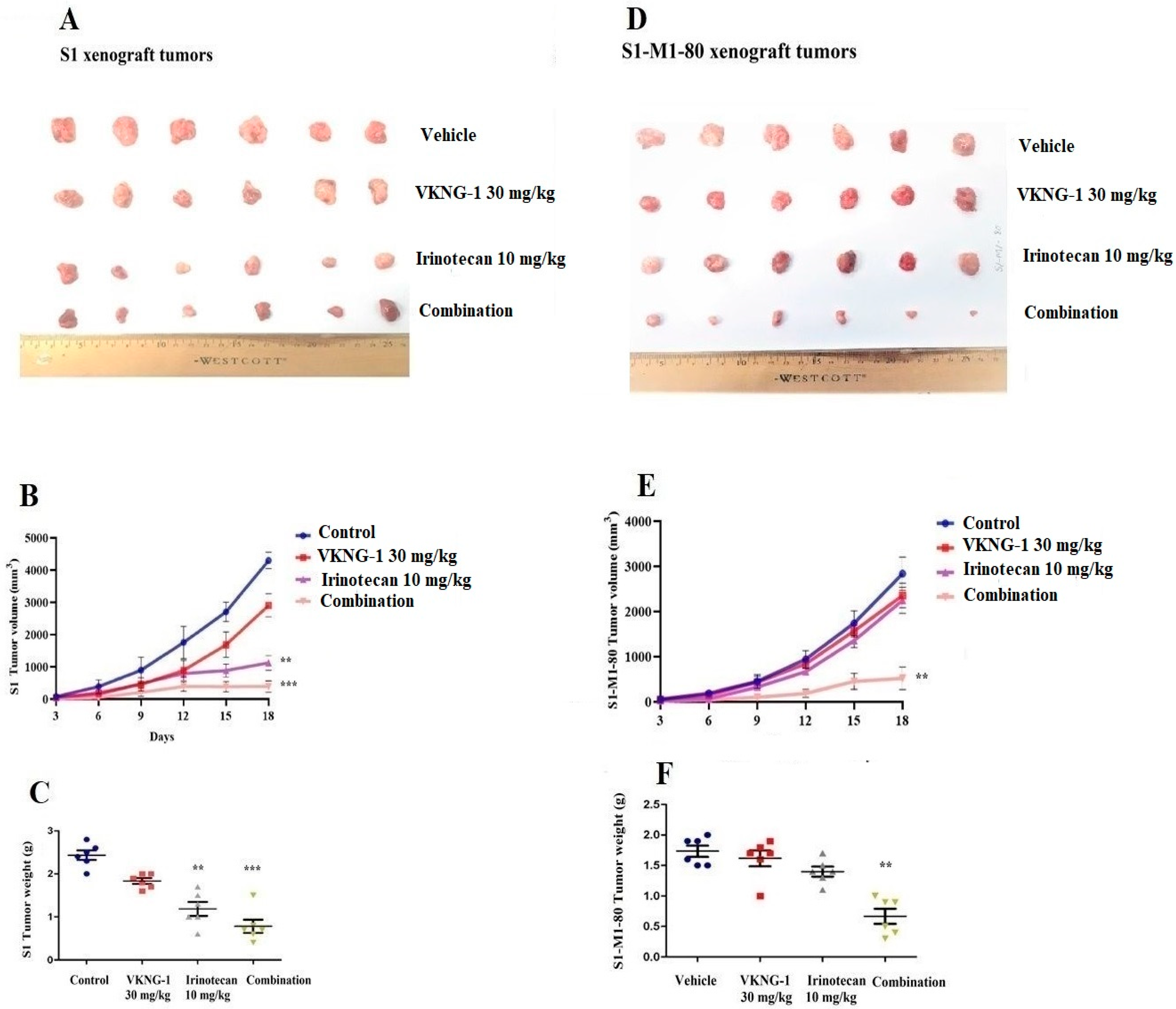
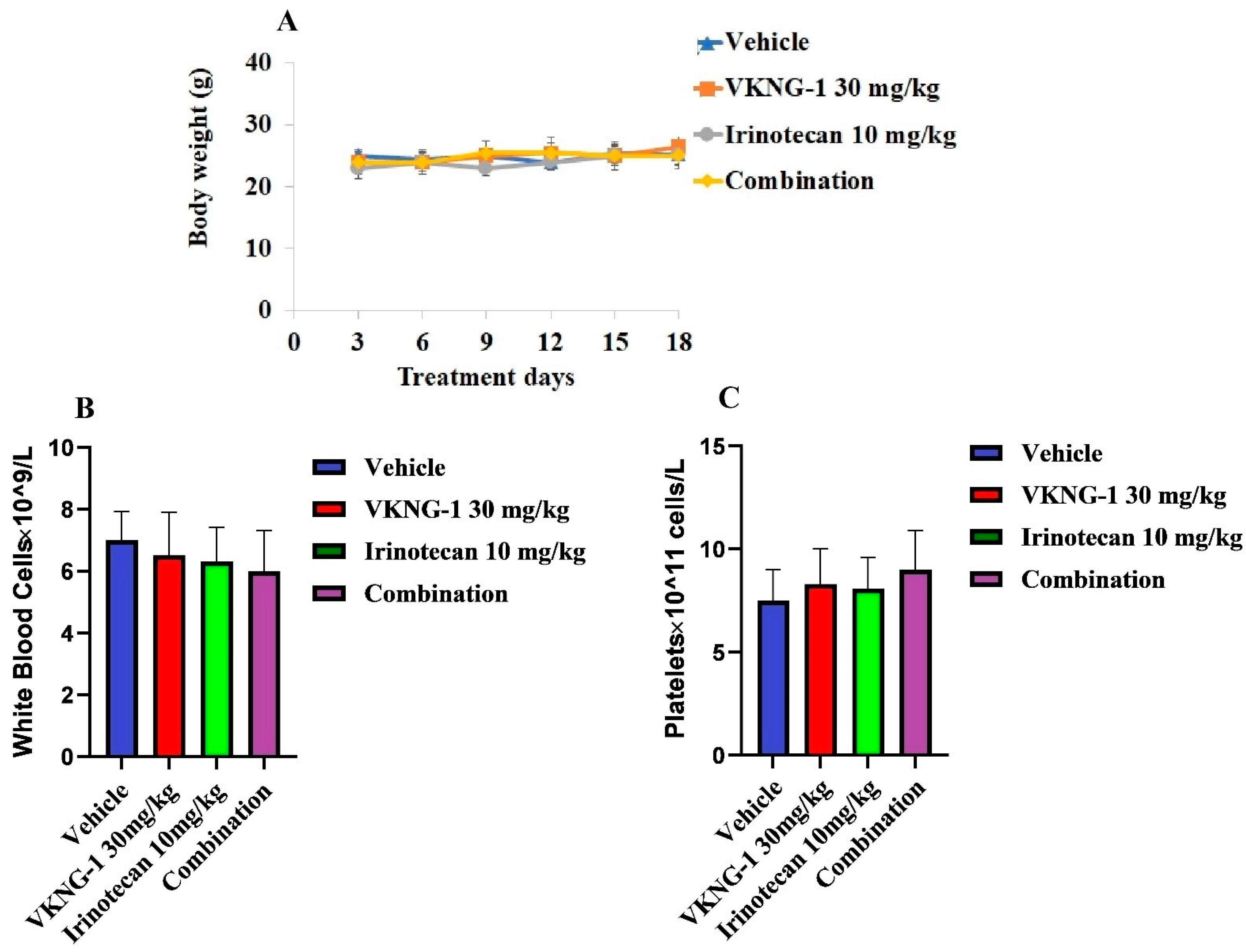
2.8. VKNG-1 Reverses ABCG2-Mediated MDR in a Nude Mouse Xenograft Model
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Cell Lines
4.3. Experimental Animals
4.4. Determination of Cytotoxicity by MTT Assay
4.5. Screening of VKNG-1 for ABC Transporters Reversal Activity
4.6. (3H)-Mitoxantrone Accumulation and Efflux Assay
4.7. Western Blot Analysis
4.8. mRNA Expression
4.9. Immunofluorescence
4.10. Docking Analysis
4.11. MDR Mouse Xenograft Models
4.12. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2016. CA. Cancer J. Clin. 2016, 66, 7–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narayanan, S.; Cai, C.-Y.; Assaraf, Y.G.; Guo, H.-Q.; Cui, Q.; Wei, L.; Huang, J.-J.; Ashby, C.R.; Chen, Z.-S. Targeting the ubiquitin-proteasome pathway to overcome anti-cancer drug resistance. Drug Resist. Updat. 2020, 48, 100663. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA. Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, T.; Narayanan, S.; Brown, D.P.; Chen, Z.-S. Synthesis and Cytotoxicity Studies of Stilbene Long-Chain Fatty Acid Conjugates. J. Nat. Prod. 2020, 83, 1563–1570. [Google Scholar] [CrossRef] [PubMed]
- Gupta, P.; Narayanan, S.; Yang, D.-H. Chapter 9—CDK inhibitors as sensitizing agents for cancer chemotherapy. In Cancer Sensitizing Agents for Chemotherapy; Chen, Z.-S., Yang, D.-H., Eds.; Academic Press: Cambridge, MA, USA, 2019; Volume 4, pp. 125–149. ISBN 24683183. [Google Scholar]
- De Vera, A.A.; Gupta, P.; Lei, Z.; Liao, D.; Narayanan, S.; Teng, Q.; Reznik, S.E.; Chen, Z.-S. Immuno-oncology agent IPI-549 is a modulator of P-glycoprotein (P-gp, MDR1, ABCB1)-mediated multidrug resistance (MDR) in cancer: In vitro and in vivo. Cancer Lett. 2019, 442, 91–103. [Google Scholar] [CrossRef] [PubMed]
- Narayanan, S.; Gupta, P.; Nazim, U.; Ali, M.; Karadkhelkar, N.; Ahmad, M.; Chen, Z.-S. Anti-cancer effect of Indanone-based thiazolyl hydrazone derivative on colon cancer cell lines. Int. J. Biochem. Cell Biol. 2019, 110, 21–28. [Google Scholar] [CrossRef]
- Barbuti, A.M.; Zhang, G.-N.; Gupta, P.; Narayanan, S.; Chen, Z.-S. Chapter 1—EGFR and HER2 inhibitors as sensitizing agents for cancer chemotherapy. In Cancer Sensitizing Agents for Chemotherapy; Chen, Z.-S., Yang, D.-H., Eds.; Academic Press: Cambridge, MA, USA, 2019; Volume 4, pp. 1–11. ISBN 24683183. [Google Scholar]
- Narayanan, S.; Koya, J.; Wang, J.; Assaraf, Y.G.; Ashby, C.R., Jr.; Chen, Z.-S. Poly (ADP-ribose) polymerase (PARP) inhibitors as chemosensitizing compounds for the treatment of drug resistant cancers. J. Mol. Clin. Med. 2019, 2, 55–67. [Google Scholar]
- Gupta, P.; Xie, M.; Narayanan, S.; Wang, Y.-J.; Wang, X.-Q.; Yuan, T.; Wang, Z.; Yang, D.-H.; Chen, Z.-S. GSK1904529A, a Potent IGF-IR inhibitor, reverses MRP1-Mediated multidrug resistance. J. Cell. Biochem. 2017, 118, 3260–3267. [Google Scholar] [CrossRef]
- Alfarouk, K.O.; Stock, C.-M.; Taylor, S.; Walsh, M.; Muddathir, A.K.; Verduzco, D.; Bashir, A.H.H.; Mohammed, O.Y.; Elhassan, G.O.; Harguindey, S.; et al. Resistance to cancer chemotherapy: Failure in drug response from ADME to P-gp. Cancer Cell Int. 2015, 15, 71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Z.-X.; Yang, Y.; Wang, J.-Q.; Narayanan, S.; Lei, Z.-N.; Teng, Q.-X.; Zeng, L.; Chen, Z.-S. Overexpression of ABCG2 confers resistance to MLN7243, a Ubiquitin-Activating enzyme (UAE) inhibitor. Front. Cell Dev. Biol. 2021, 9, 1727. [Google Scholar] [CrossRef]
- Favoriti, P.; Carbone, G.; Greco, M.; Pirozzi, F.; Pirozzi, R.; Corcione, F. Worldwide burden of colorectal cancer: A review. Updates Surg. 2016, 68. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.-H.; Chen, Y.-X.; Fang, J.-Y. Comprehensive review of targeted therapy for colorectal cancer. Signal Transduct. Target. Ther. 2020, 5, 22. [Google Scholar] [CrossRef]
- Nielsen, D.L.; Palshof, J.A.; Brünner, N.; Stenvang, J.; Viuff, B.M. Implications of ABCG2 expression on irinotecan treatment of colorectal cancer patients: A review. Int. J. Mol. Sci. 2017, 18, 1926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, J.; Bebawy, M. Proteins regulating microvesicle biogenesis and multidrug resistance in cancer. Proteomics 2019, 19, 1800165. [Google Scholar] [CrossRef] [Green Version]
- Hu, J.; Zhang, X.; Wang, F.; Wang, X.; Yang, K.; Xu, M.; To, K.K.W.; Li, Q.; Fu, L. Effect of ceritinib (LDK378) on enhancement of chemotherapeutic agents in ABCB1 and ABCG2 overexpressing cells in vitro and in vivo. Oncotarget 2015, 6, 44643–44659. [Google Scholar] [CrossRef] [Green Version]
- El-Awady, R.; Saleh, E.; Hashim, A.; Soliman, N.; Dallah, A.; Elrasheed, A.; Elakraa, G. The role of eukaryotic and prokaryotic ABC transporter family in failure of chemotherapy. Front. Pharmacol. 2017, 7, 535. [Google Scholar] [CrossRef] [Green Version]
- Hasanabady, M.H.; Kalalinia, F. ABCG2 inhibition as a therapeutic approach for overcoming multidrug resistance in cancer. J. Biosci. 2016, 41, 313–324. [Google Scholar] [CrossRef] [PubMed]
- Mo, W.; Zhang, J.-T. Human ABCG2: Structure, function, and its role in multidrug resistance. Int. J. Biochem. Mol. Biol. 2012, 3, 1–27. [Google Scholar]
- Mao, Q.; Unadkat, J.D. Role of the breast cancer resistance protein (ABCG2) in drug transport. Aaps J. 2005, 7, E118–E133. [Google Scholar] [CrossRef] [Green Version]
- Fletcher, J.I.; Williams, R.T.; Henderson, M.J.; Norris, M.D.; Haber, M. ABC transporters as mediators of drug resistance and contributors to cancer cell biology. Drug Resist. Update Rev. Comment. Antimicrob. Anticancer Chemother. 2016, 26, 1–9. [Google Scholar] [CrossRef]
- Dei, S.; Braconi, L.; Romanelli, M.; Teodori, E. Recent advances in the search of BCRP- and dual P-gp/BCRP-based multidrug resistance modulators. Cancer Drug Resist. 2019. [Google Scholar] [CrossRef] [Green Version]
- Lei, Z.-N.; Teng, Q.-X.; Gupta, P.; Zhang, W.; Narayanan, S.; Yang, D.-H.; Wurpel, J.N.D.; Fan, Y.-F.; Chen, Z.-S. Cabozantinib reverses topotecan resistance in human non-small cell lung cancer NCI-H460/TPT10 cell line and tumor xenograft model. Front. Cell Dev. Biol. 2021, 9, 643. [Google Scholar] [CrossRef]
- Gujarati, N.A.; Zeng, L.; Gupta, P.; Chen, Z.-S.; Korlipara, V.L. Design, synthesis and biological evaluation of benzamide and phenyltetrazole derivatives with amide and urea linkers as BCRP inhibitors. Bioorg. Med. Chem. Lett. 2017, 27, 4698–4704. [Google Scholar] [CrossRef] [PubMed]
- Narayanan, S.; Gujarati, N.A.; Wang, J.-Q.; Wu, Z.-X.; Koya, J.; Cui, Q.; Korlipara, V.L.; Ashby, C.R., Jr.; Chen, Z.-S. The novel benzamide derivative, VKNG-2, restores the efficacy of chemotherapeutic drugs in colon cancer cell lines by inhibiting the ABCG2 transporter. Int. J. Mol. Sci. 2021, 22, 2463. [Google Scholar] [CrossRef]
- Bhardwaj, B.; Baidya, A.T.K.; Amin, S.A.; Adhikari, N.; Jha, T.; Gayen, S. Insight into structural features of phenyltetrazole derivatives as ABCG2 inhibitors for the treatment of multidrug resistance in cancer. SAR QSAR Environ. Res. 2019, 30, 457–475. [Google Scholar] [CrossRef]
- Pampaloni, F.; Reynaud, E.G.; Stelzer, E.H.K. The third dimension bridges the gap between cell culture and live tissue. Nat. Rev. Mol. Cell Biol. 2007, 8, 839–845. [Google Scholar] [CrossRef] [PubMed]
- Sodani, K.; Patel, A.; Anreddy, N.; Singh, S.; Yang, D.-H.; Kathawala, R.J.; Kumar, P.; Talele, T.T.; Chen, Z.-S. Telatinib reverses chemotherapeutic multidrug resistance mediated by ABCG2 efflux transporter in vitro and in vivo. Biochem. Pharmacol. 2014, 89, 52–61. [Google Scholar] [CrossRef] [Green Version]
- Vesel, M.; Rapp, J.; Feller, D.; Kiss, E.; Jaromi, L.; Meggyes, M.; Miskei, G.; Duga, B.; Smuk, G.; Laszlo, T.; et al. ABCB1 and ABCG2 drug transporters are differentially expressed in non-small cell lung cancers (NSCLC) and expression is modified by cisplatin treatment via altered Wnt signaling. Respir. Res. 2017, 18, 52. [Google Scholar] [CrossRef] [Green Version]
- Danielsen, S.A.; Eide, P.W.; Nesbakken, A.; Guren, T.; Leithe, E.; Lothe, R.A. Portrait of the PI3K/AKT pathway in colorectal cancer. Biochim. Biophys. Acta 2015, 1855, 104–121. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-J.; Zhang, Y.-K.; Kathawala, R.J.; Chen, Z.-S. Repositioning of tyrosine kinase inhibitors as antagonists of ATP-Binding cassette transporters in anticancer drug resistance. Cancers 2014, 6, 1925–1952. [Google Scholar] [CrossRef]
- Zhang, W.; Li, J.; Allen, S.M.; Weiskircher, E.A.; Huang, Y.; George, R.A.; Fong, R.G.; Owen, A.; Hidalgo, I.J. Silencing the breast cancer resistance protein expression and function in caco-2 cells using lentiviral vector-based short hairpin RNA. Drug Metab. Dispos. 2009, 37, 737–744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Li, Y.; Wang, Q.; Chen, Z.; Li, X.; Wu, Z.; Hu, C.; Liao, D.; Zhang, W.; Chen, Z.-S. The PI3K subunits, P110α and P110β are potential targets for overcoming P-gp and BCRP-mediated MDR in cancer. Mol. Cancer 2020, 19, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daenen, S.; van der Holt, B.; Verhoef, G.E.G.; Löwenberg, B.; Wijermans, P.W.; Huijgens, P.C.; van Marwijk Kooy, R.; Schouten, H.C.; Kramer, M.H.H.; Ferrant, A.; et al. Addition of cyclosporin A to the combination of mitoxantrone and etoposide to overcome resistance to chemotherapy in refractory or relapsing acute myeloid leukaemia: A randomised phase II trial from HOVON, the Dutch-Belgian Haemato-Oncology Working Group. Leuk. Res. 2004, 28, 1057–1067. [Google Scholar] [CrossRef]
- Mlejnek, P.; Kosztyu, P.; Dolezel, P.; Bates, S.E.; Ruzickova, E. Reversal of ABCB1 mediated efflux by imatinib and nilotinib in cells expressing various transporter levels. Chem. Biol. Interact. 2017, 273, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Kosztyu, P.; Bukvova, R.; Dolezel, P.; Mlejnek, P. Resistance to daunorubicin, imatinib, or nilotinib depends on expression levels of ABCB1 and ABCG2 in human leukemia cells. Chem. Biol. Interact. 2014, 219, 203–210. [Google Scholar] [CrossRef] [PubMed]
- Kosztyu, P.; Dolezel, P.; Vaclavikova, R.; Mlejnek, P. Can the assessment of ABCB1 gene expression predict its function in vitro? Eur. J. Haematol. 2015, 95, 150–159. [Google Scholar] [CrossRef]
- Fojo, A.T.; Ueda, K.; Slamon, D.J.; Poplack, D.G.; Gottesman, M.M.; Pastan, I. Expression of a multidrug-resistance gene in human tumors and tissues. Proc. Natl. Acad. Sci. USA 1987, 84, 265–269. [Google Scholar] [CrossRef] [Green Version]
- Goldstein, L.J.; Galski, H.; Fojo, A.; Willingham, M.; Lai, S.-L.; Gazdar, A.; Pirker, R.; Green, A.; Crist, W.; Brodeur, G.M.; et al. Expression of multidrug resistance gene in human cancers. JNCI J. Natl. Cancer Inst. 1989, 81, 116–124. [Google Scholar] [CrossRef]
- Tazzari, P.L.; Tabellini, G.; Ricci, F.; Papa, V.; Bortul, R.; Chiarini, F.; Evangelisti, C.; Martinelli, G.; Bontadini, A.; Cocco, L.; et al. Synergistic proapoptotic activity of recombinant TRAIL plus the Akt inhibitor perifosine in acute myelogenous leukemia cells. Cancer Res. 2008, 68, 9394. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Lin, N.; Li, Y. The PI3K/AKT signaling pathway regulates ABCG2 expression and confers resistance to chemotherapy in human multiple myeloma. Oncol. Rep. 2019, 41, 1678–1690. [Google Scholar] [CrossRef]
- Liu, R.; Chen, Y.; Liu, G.; Li, C.; Song, Y.; Cao, Z.; Li, W.; Hu, J.; Lu, C.; Liu, Y. PI3K/AKT pathway as a key link modulates the multidrug resistance of cancers. Cell Death Dis. 2020, 11, 797. [Google Scholar] [CrossRef]
- Bleau, A.-M.; Hambardzumyan, D.; Ozawa, T.; Fomchenko, E.I.; Huse, J.T.; Brennan, C.W.; Holland, E.C. PTEN/PI3K/Akt pathway regulates the side population phenotype and ABCG2 activity in glioma tumor stem-like cells. Cell Stem Cell 2009, 4, 226–235. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Lin, L.; Li, M.; Li, W. Gilteritinib induces PUMA-dependent apoptotic cell death via AKT/GSK-3β/NF-κB pathway in colorectal cancer cells. J. Cell. Mol. Med. 2020, 24, 2308–2318. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, E.; Brattain, M.G.; Chowdhury, S. Cell survival and metastasis regulation by Akt signaling in colorectal cancer. Cell. Signal. 2013, 25, 1711–1719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, C.; Wu, J.; Chen, Y.; Nie, J.; Chen, C. Activation of PI3K/AKT/mTOR pathway causes drug resistance in breast cancer. Front. Pharmacol. 2021, 12, 628690. [Google Scholar] [CrossRef]
- Zhang, Y.-K.; Zhang, G.-N.; Wang, Y.-J.; Patel, B.A.; Talele, T.T.; Yang, D.-H.; Chen, Z.-S. Bafetinib (INNO-406) reverses multidrug resistance by inhibiting the efflux function of ABCB1 and ABCG2 transporters. Sci. Rep. 2016, 6, 25694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anreddy, N.; Patel, A.; Zhang, Y.-K.; Wang, Y.-J.; Shukla, S.; Kathawala, R.J.; Kumar, P.; Gupta, P.; Ambudkar, S.V.; Wurpel, J.N.D.; et al. A-803467, a tetrodotoxin-resistant sodium channel blocker, modulates ABCG2-mediated MDR in vitro and in vivo. Oncotarget 2015, 6, 39276–39291. [Google Scholar] [CrossRef]
- Miyake, K.; Mickley, L.; Litman, T.; Zhan, Z.; Robey, R.; Cristensen, B.; Brangi, M.; Greenberger, L.; Dean, M.; Fojo, T.; et al. Molecular cloning of cDNAs which are highly overexpressed in mitoxantrone-resistant cells. Cancer Res. 1999, 59, 8. [Google Scholar]
- Huang, X.-C.; Xiao, X.; Zhang, Y.-K.; Talele, T.T.; Salim, A.A.; Chen, Z.-S.; Capon, R.J. Lamellarin O, a pyrrole alkaloid from an Australian marine sponge, Ianthella sp., reverses BCRP mediated drug resistance in cancer cells. Mar. Drugs 2014, 12, 3818–3837. [Google Scholar] [CrossRef] [Green Version]
- Robey, R.W.; Honjo, Y.; Morisaki, K.; Nadjem, T.A.; Runge, S.; Risbood, M.; Poruchynsky, M.S.; Bates, S.E. Mutations at amino-acid 482 in the ABCG 2 gene affect substrate and antagonist specificity. Br. J. Cancer 2003, 89, 1971–1978. [Google Scholar] [CrossRef]
- Yang, Y.; Liu, Y.; Dong, Z.; Xu, J.; Peng, H.; Liu, Z.; Zhang, J.-T. Regulation of function by dimerization through the amino-terminal membrane-spanning domain of human ABCC1/MRP1. J. Biol. Chem. 2007, 282, 8821–8830. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.-S.; Hopper-Borge, E.; Belinsky, M.G.; Shchaveleva, I.; Kotova, E.; Kruh, G.D. Characterization of the Transport Properties of Human Multidrug Resistance Protein 7 (MRP7, ABCC10). Mol. Pharmacol. 2003, 63, 351. [Google Scholar] [CrossRef]
- Bahuguna, A.; Khan, I.; Bajpai, V.K.; Chul, S. MTT assay to evaluate the cytotoxic potential of a drug. Bangladesh J. Pharmacol. 2017, 12, 8. [Google Scholar] [CrossRef]
- Tiwari, A.K.; Sodani, K.; Dai, C.-L.; Abuznait, A.H.; Singh, S.; Xiao, Z.-J.; Patel, A.; Talele, T.T.; Fu, L.; Kaddoumi, A.; et al. Nilotinib potentiates anticancer drug sensitivity in murine ABCB1-, ABCG2-, and ABCC10-multidrug resistance xenograft models. Cancer Lett. 2013, 328, 307–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shukla, S.; Jhamtani, R.; Dahiya, M.; Agarwal, D.R. A novel method to achieve high yield of total RNA from zebrafish for expression studies. Int. J. Bioassays 2017, 6, 5383. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.-J.; Zhang, Y.-K.; Zhang, G.-N.; Al Rihani, S.B.; Wei, M.-N.; Gupta, P.; Zhang, X.-Y.; Shukla, S.; Ambudkar, S.V.; Kaddoumi, A.; et al. Regorafenib overcomes chemotherapeutic multidrug resistance mediated by ABCB1 transporter in colorectal cancer: In vitro and in vivo study. Cancer Lett. 2017, 396, 145–154. [Google Scholar] [CrossRef] [PubMed]
- Cai, C.-Y.; Zhai, H.; Lei, Z.-N.; Tan, C.-P.; Chen, B.-L.; Du, Z.-Y.; Wang, J.-Q.; Zhang, Y.-K.; Wang, Y.-J.; Gupta, P.; et al. Benzoyl indoles with metabolic stability as reversal agents for ABCG2-mediated multidrug resistance. Eur. J. Med. Chem. 2019, 179, 849–862. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.M.; Manolaridis, I.; Kowal, J.; Zechner, M.; Taylor, N.M.I.; Bause, M.; Bauer, S.; Bartholomaeus, R.; Bernhardt, G.; Koenig, B.; et al. Structural basis of small-molecule inhibition of human multidrug transporter ABCG2. Nat. Struct. Mol. Biol. 2018, 25, 333–340. [Google Scholar] [CrossRef]
- Bunting, K.D. ABC Transporters as phenotypic markers and functional regulators of stem cells. Stem Cells 2002, 20, 274. [Google Scholar] [CrossRef]
- Milczarek, M.; Rosińska, S.; Psurski, M.; Maciejewska, M.; Kutner, A.; Wietrzyk, J. Combined Colonic cancer treatment with vitamin D analogs and irinotecan or oxaliplatin. Anticancer Res. 2013, 33, 433–444. [Google Scholar]
- Wang, Y.-J.; Huang, Y.; Anreddy, N.; Zhang, G.-N.; Zhang, Y.-K.; Xie, M.; Lin, D.; Yang, D.-H.; Zhang, M.; Chen, Z.-S. Tea nanoparticle, a safe and biocompatible nanocarrier, greatly potentiates the anticancer activity of doxorubicin. Oncotarget 2016, 7, 5877–5891. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Lines | S1 | S1-M1-80 | ||
|---|---|---|---|---|
| IC50 ± SD (µM) | FR | IC50 ± SD (µM) | FR | |
| Mitoxantrone | 0.147 ± 0.013 | (1.0) | 23.321 ± 2.602 | (151.7) |
| +VKNG-1 (1 µM) | 0.117 ± 0.018 | (0.8) | 0.771 ± 0.149 | (4.7) |
| +VKNG-1 (6 µM) | 0.115 ± 0.019 | (0.8) | 0.268 ± 0.055 | (1.6) |
| +FTC (6 µM) | 0.129 ± 0.013 | (0.9) | 0.175 ± 0.033 | (1.2) |
| SN-38 | 0.113 ± 0.071 | (1.0) | 25.867 ± 2.884 | (228.2) |
| +VKNG-1 (1 µM) | 0.153 ± 0.053 | (1.3) | 0.889 ± 0.127 | (5.6) |
| +VKNG-1 (6 µM) | 0.117 ± 0.024 | (1.0) | 0.245 ± 0.092 | (2.2) |
| +FTC (6 µM) | 0.121 ± 0.020 | (1.0) | 0.197 ± 0.025 | (1.7) |
| Doxorubicin | 0.139 ± 0.022 | (1.0) | 9.579 ± 1.534 | (73.1) |
| +VKNG-1 (1 µM) | 0.083 ± 0.011 | (0.6) | 0.581 ± 0.097 | (4.5) |
| +VKNG-1 (6 µM) | 0.165 ± 0.015 | (1.2) | 0.063 ± 0.013 | (0.5) |
| +FTC (6 µM) | 0.125 ± 0.019 | (0.9) | 0.193 ± 0.032 | (1.5) |
| Cisplatin | 154.959 ± 22.426 | (1.0) | 194.845 ± 24.119 | (1.3) |
| +VKNG-1 (1 µM) | 150.539 ± 24.745 | (0.9) | 180.931 ± 32.527 | (1.2) |
| +VKNG-1 (6 µM) | 159.259 ± 18.131 | (1.1) | 173.985 ± 30.406 | (1.1) |
| +FTC (6 µM) | 153.51 ± 34.951 | (1.2) | 195.785 ± 23.335 | (1.3) |
| Cell Lines | HEK293/pcDNA3.1 | HEK293/R2 | HEK293/G2 | HEK293/T7 | ||||
|---|---|---|---|---|---|---|---|---|
| IC50 ± SD (µM) | FR | IC50 ± SD (µM) | FR | IC50 ± SD (µM) | FR | IC50 ± SD (µM) | FR | |
| Mitoxantrone | 0.0180 ± 0.0001 | (1.0) | 0.7449 ± 0.0214 | (41.3) | 0.7111 ± 0.0536 | (39.5) | 0.6065 ± 0.0394 | (33.6) |
| +VKNG-1 (1 µM) | 0.0187 ± 0.0006 | (1.0) | 0.6827 ± 0.0572 | (37.9) | 0.3974 ± 0.0051 | (22.1) | 0.5542 ± 0.0306 | (30.7) |
| +VKNG-1 (6 µM) | 0.0182 ± 0.0008 | (1.0) | 0.1008 ± 0.0347 | (5.5) | 0.0600 ± 0.0172 | (3.3) | 0.1372 ± 0.0265 | (7.6) |
| +FTC (6 µM) | 0.0167 ± 0.0005 | (0.9) | 0.1428 ± 0.0377 | (7.9) | 0.0772 ± 0.0189 | (4.3) | 0.1482 ± 0.0206 | (8.3) |
| SN-38 | 0.0129 ± 0.0007 | (1.0) | 0.7319 ± 0.0023 | (56.7) | 0.4932 ± 0.0012 | (38.2) | 0.4430 ± 0.0010 | (34.3) |
| +VKNG-1 (1 µM) | 0.0157 ± 0.0028 | (1.2) | 0.3090 ± 0.0377 | (23.9) | 0.1033 ± 0.0082 | (8.0) | 0.1314 ± 0.0169 | (10.1) |
| +VKNG-1 (6 µM) | 0.0113 ± 0.0007 | (0.8) | 0.0309 ± 0.0065 | (2.3) | 0.0534 ± 0.0047 | (4.1) | 0.0218 ± 0.0015 | (1.7) |
| +FTC (6 µM) | 0.0129 ± 0.0012 | (1.0) | 0.0355 ± 0.0050 | (2.8) | 0.0577 ± 0.0007 | (4.5) | 0.0233 ± 0.0030 | (1.8) |
| Doxorubicin | 0.0152 ± 0.0001 | (1.0) | 0.6225 ± 0.0332 | (40.9) | 0.5994 ± 0.0054 | (39.3) | 0.6858 ± 0.0367 | (44.9) |
| +VKNG-1 (1 µM) | 0.0154 ± 0.0001 | (1.0) | 0.2395 ± 0.0257 | (15.7) | 0.2345 ± 0.0213 | (15.3) | 0.2222 ± 0.0407 | (14.5) |
| +VKNG-1 (6 µM) | 0.0157 ± 0.0001 | (1.0) | 0.0662 ± 0.0004 | (4.3) | 0.0597 ± 0.0073 | (3.9) | 0.0752 ± 0.0052 | (4.9) |
| +FTC (6 µM) | 0.0180 ± 0.0001 | (1.2) | 0.0771 ± 0.0105 | (5.1) | 0.0694 ± 0.0024 | (4.6) | 0.0817 ± 0.0097 | (5.3) |
| Cisplatin | 1.8455 ± 0.2609 | (1.0) | 1.8567 ± 0.0598 | (1.0) | 1.7348 ± 0.0379 | (0.9) | 1.9224 ± 0.0543 | (1.0) |
| +VKNG-1 (1 µM) | 1.7990 ± 0.2319 | (0.9) | 1.8607 ± 0.0464 | (1.0) | 1.9977 ± 0.0471 | (1.1) | 1.9400 ± 0.1171 | (1.0) |
| +VKNG-1 (6 µM) | 1.8835 ± 0.0955 | (1.0) | 1.9156 ± 0.0896 | (1.0) | 1.7695 ± 0.0213 | (0.9) | 1.9864 ± 0.1278 | (1.1) |
| +FTC (6 µM) | 1.9950 ± 0.1245 | (1.0) | 2.0023 ± 0.1251 | (1.1) | 1.7413 ± 0.0731 | (0.9) | 1.8896 ± 0.0611 | (1.0) |
| Cell Lines | SW620 | SW620/AD300 | ||
|---|---|---|---|---|
| IC50 ± SD (µM) | FR | IC50 ± SD (µM) | FR | |
| Doxorubicin | 0.083 ± 0.012 | (1.0) | 9.371 ± 1.45 | (117.1) |
| +VKNG-1 (1 µM) | 0.101 ± 0.014 | (1.2) | 9.321 ± 1.67 | (112.3) |
| +VKNG-1 (5 µM) | 0.098 ± 0.011 | (1.1) | 8.759 ± 1.89 | (105.5) |
| +Verapamil (5 µM) | 0.094 ± 0.013 | (1.1) | 1.056 ± 0.153 | (13.2) |
| Cell Lines | HEK293/pcDNA3.1 | HEK293/ABCC1 | ||
|---|---|---|---|---|
| IC50 ± SD (µM) | FR | IC50 ± SD (µM) | FR | |
| Vincristine | 0.037 ± 0.007 | (1.0) | 1.169 ± 0.225 | (30.5) |
| +VKNG-1 (1 µM) | 0.035 ± 0.005 | (0.9) | 0.959 ± 0.153 | (24.3) |
| +VKNG-1 (5 µM) | 0.049 ± 0.005 | (1.3) | 0.811 ± 0.147 | (21.9) |
| MK-571 (25 µM) | 0.025 ± 0.002 | (0.7) | 0.225 ± 0.047 | (6.1) |
| Cell Lines | HEK293/pcDNA3.1 | HEK293/ABCC10 | ||
|---|---|---|---|---|
| IC50 ±SD (µM) | FR | IC50 ±SD (µM) | FR | |
| Paclitaxel | 3.523 ± 0.539 | (1.0) | 53.356 ± 4.33 | (15.1) |
| +VKNG-1 (1 µM) | 3.213 ± 0.471 | (0.9) | 50.891 ± 6.67 | (14.3) |
| +VKNG-1 (5 µM) | 3.888 ± 0.613 | (1.3) | 45.323 ± 5.33 | (12.9) |
| Cepharanthine (5 µM) | 3.789 ± 0.469 | (1.1) | 3.588 ± 0.519 | (1.1) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Narayanan, S.; Fan, Y.-F.; Gujarati, N.A.; Teng, Q.-X.; Wang, J.-Q.; Cai, C.-Y.; Yang, Y.; Chintalapati, A.J.; Lei, Y.; Korlipara, V.L.; et al. VKNG-1 Antagonizes ABCG2-Mediated Multidrug Resistance via p-AKT and Bcl-2 Pathway in Colon Cancer: In Vitro and In Vivo Study. Cancers 2021, 13, 4675. https://doi.org/10.3390/cancers13184675
Narayanan S, Fan Y-F, Gujarati NA, Teng Q-X, Wang J-Q, Cai C-Y, Yang Y, Chintalapati AJ, Lei Y, Korlipara VL, et al. VKNG-1 Antagonizes ABCG2-Mediated Multidrug Resistance via p-AKT and Bcl-2 Pathway in Colon Cancer: In Vitro and In Vivo Study. Cancers. 2021; 13(18):4675. https://doi.org/10.3390/cancers13184675
Chicago/Turabian StyleNarayanan, Silpa, Ying-Fang Fan, Nehaben A. Gujarati, Qiu-Xu Teng, Jing-Quan Wang, Chao-Yun Cai, Yuqi Yang, Anirudh J. Chintalapati, Yixiong Lei, Vijaya L. Korlipara, and et al. 2021. "VKNG-1 Antagonizes ABCG2-Mediated Multidrug Resistance via p-AKT and Bcl-2 Pathway in Colon Cancer: In Vitro and In Vivo Study" Cancers 13, no. 18: 4675. https://doi.org/10.3390/cancers13184675
APA StyleNarayanan, S., Fan, Y.-F., Gujarati, N. A., Teng, Q.-X., Wang, J.-Q., Cai, C.-Y., Yang, Y., Chintalapati, A. J., Lei, Y., Korlipara, V. L., & Chen, Z.-S. (2021). VKNG-1 Antagonizes ABCG2-Mediated Multidrug Resistance via p-AKT and Bcl-2 Pathway in Colon Cancer: In Vitro and In Vivo Study. Cancers, 13(18), 4675. https://doi.org/10.3390/cancers13184675