
ALK-Negative Anaplastic Large Cell Lymphoma: Current Concepts and Molecular Pathogenesis of a Heterogeneous Group of Large T-Cell Lymphomas

 ,
,  ,
,
Simple Summary
Abstract
1. Introduction
2. Systemic ALK- ALCL
2.1. Definition
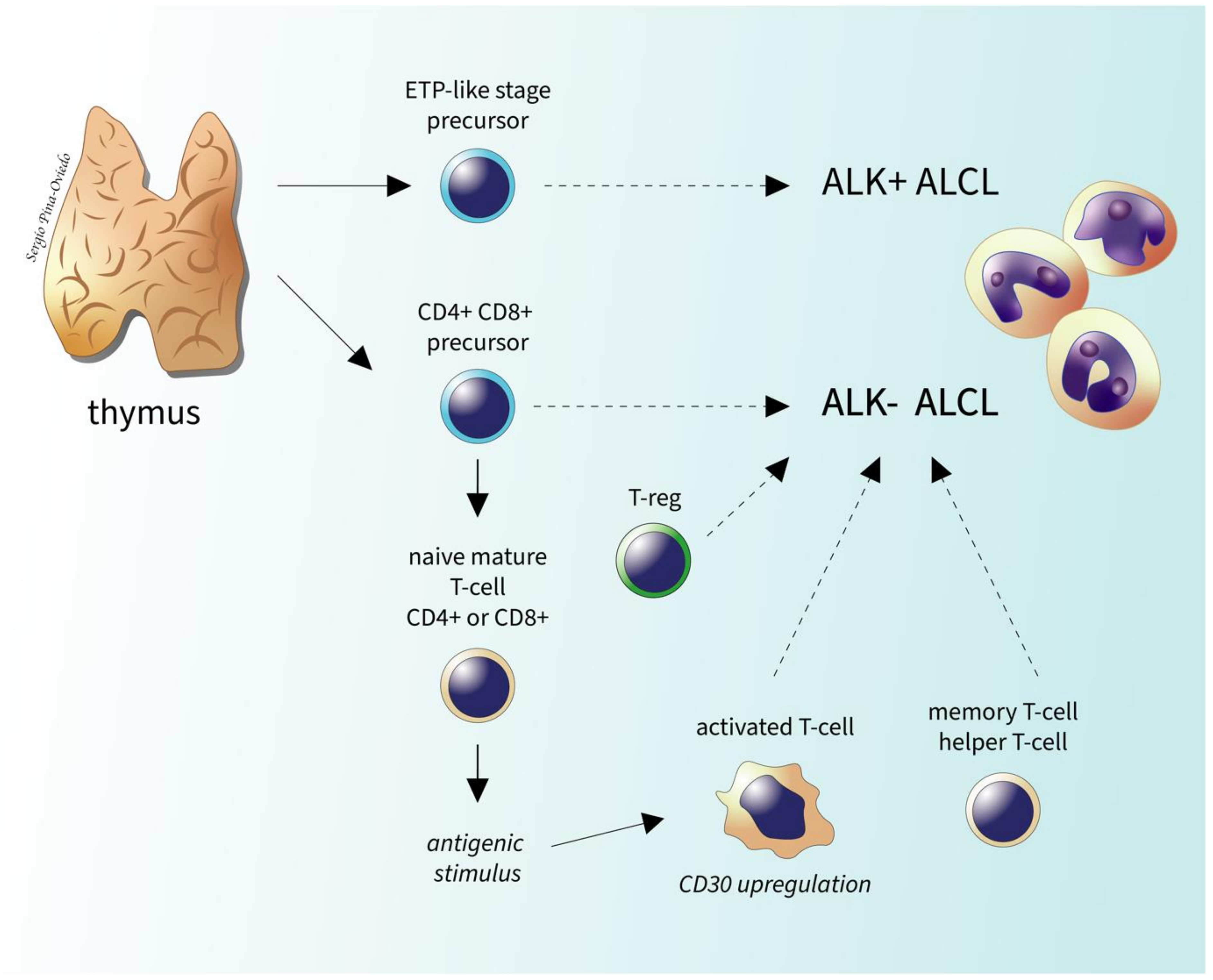
2.2. Historical Aspects
2.3. Epidemiology, Risk Factors, and Clinical Features
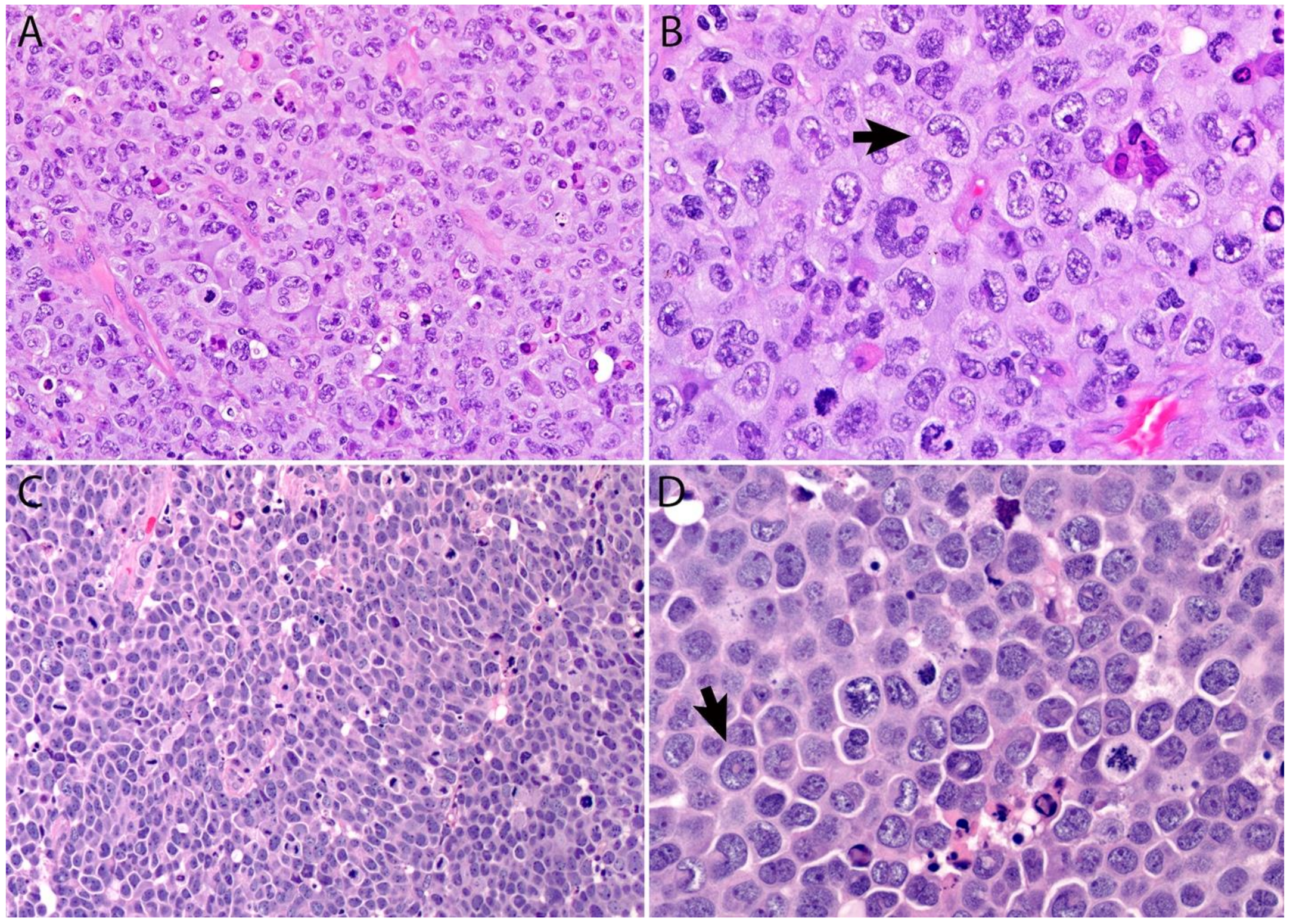
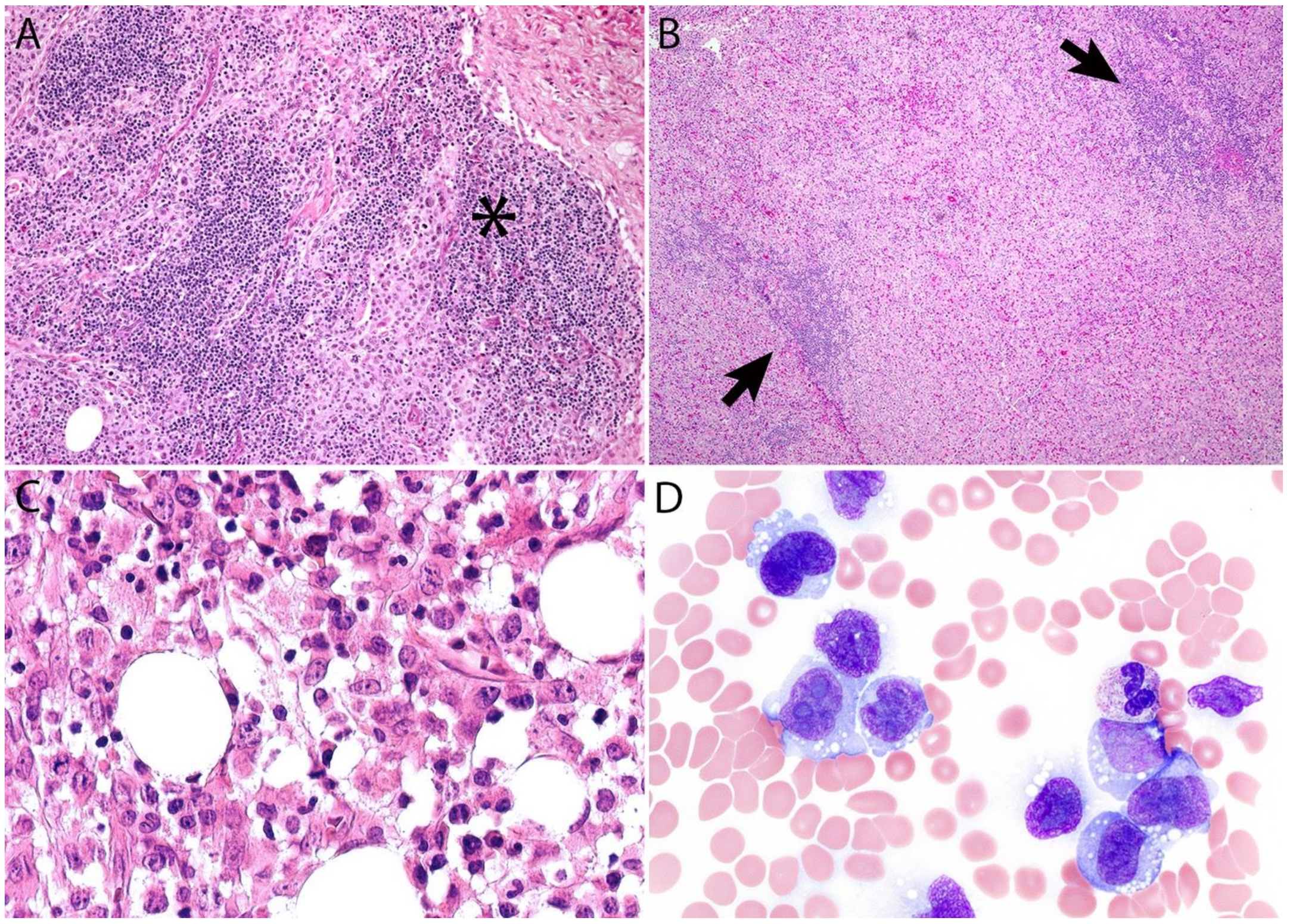
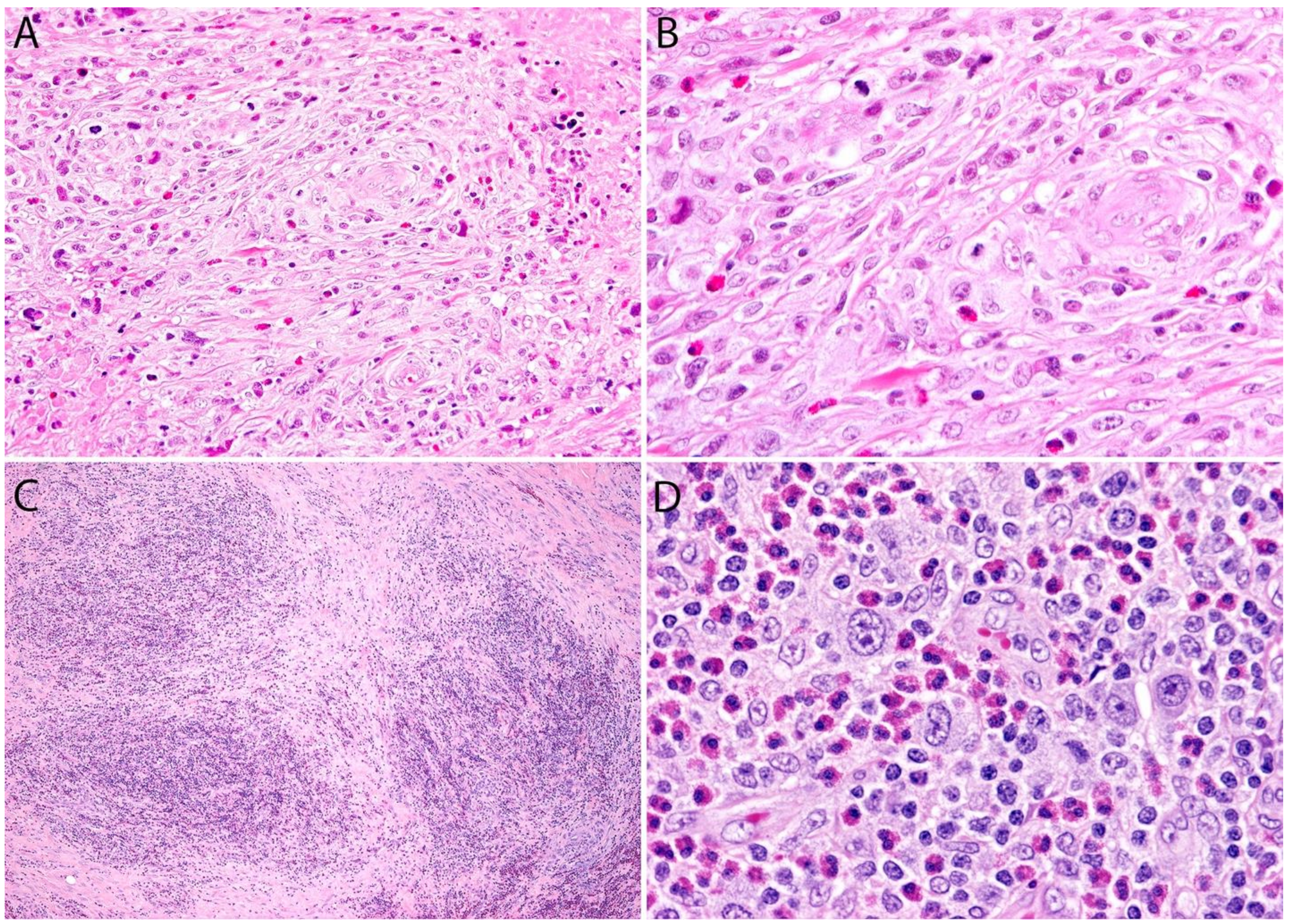
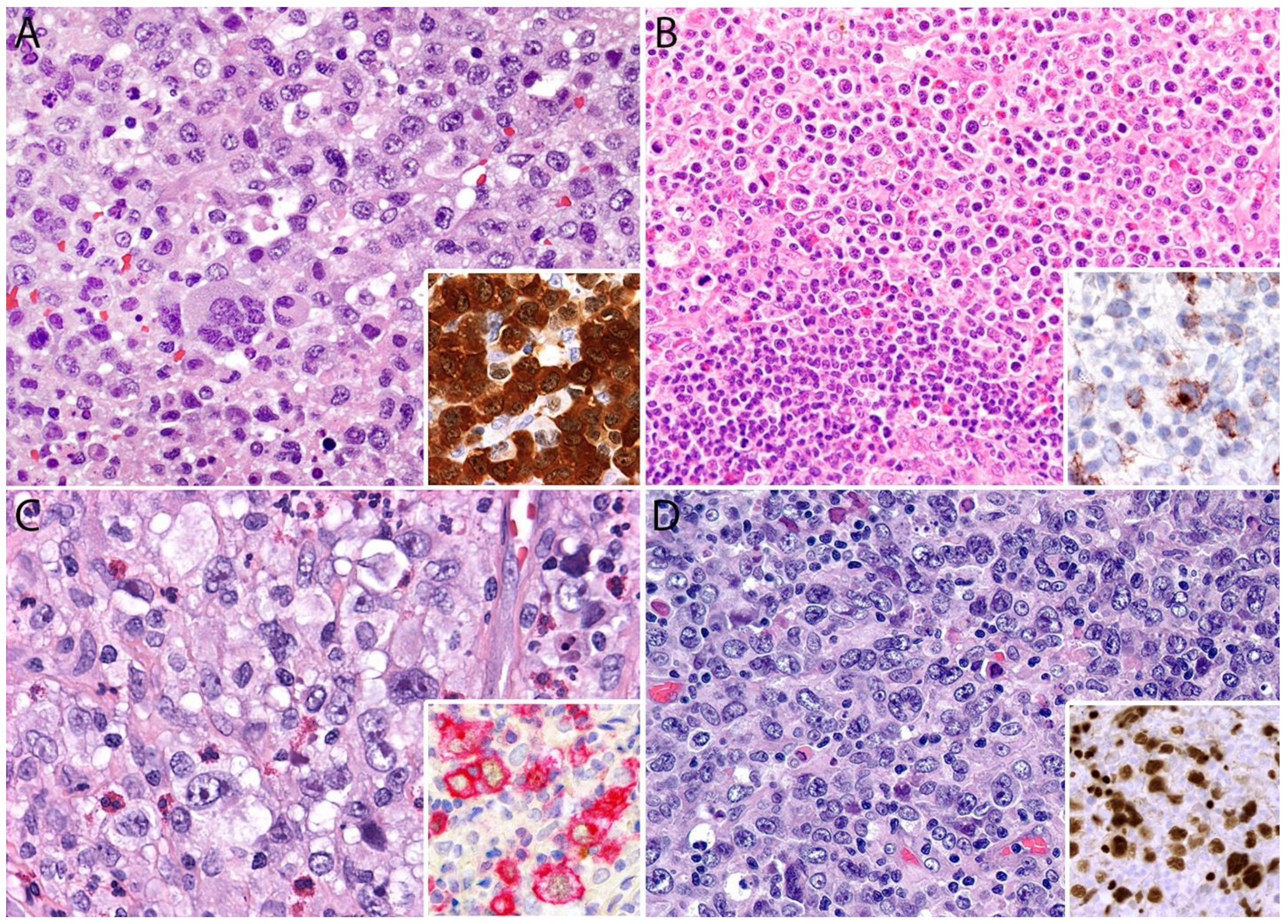
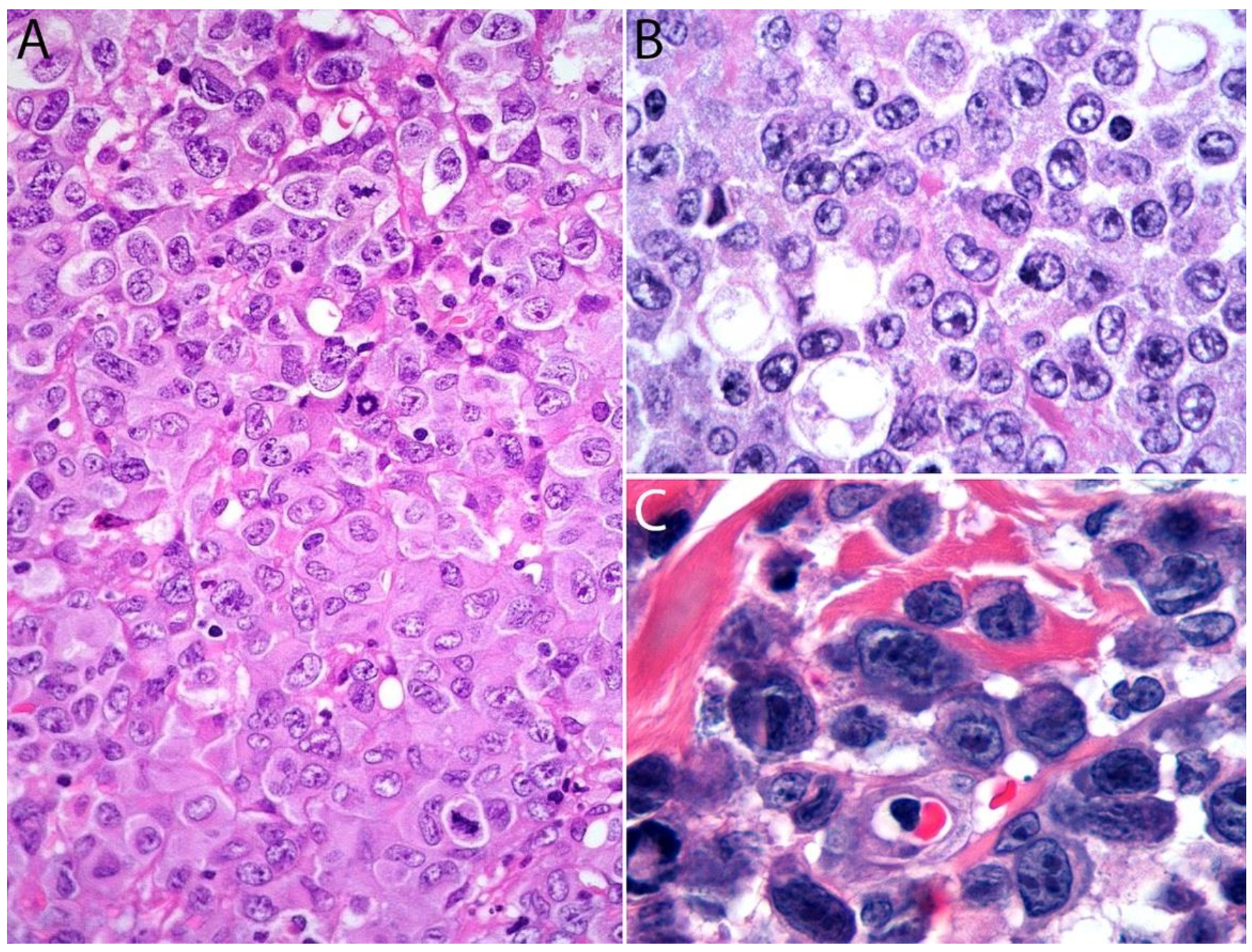
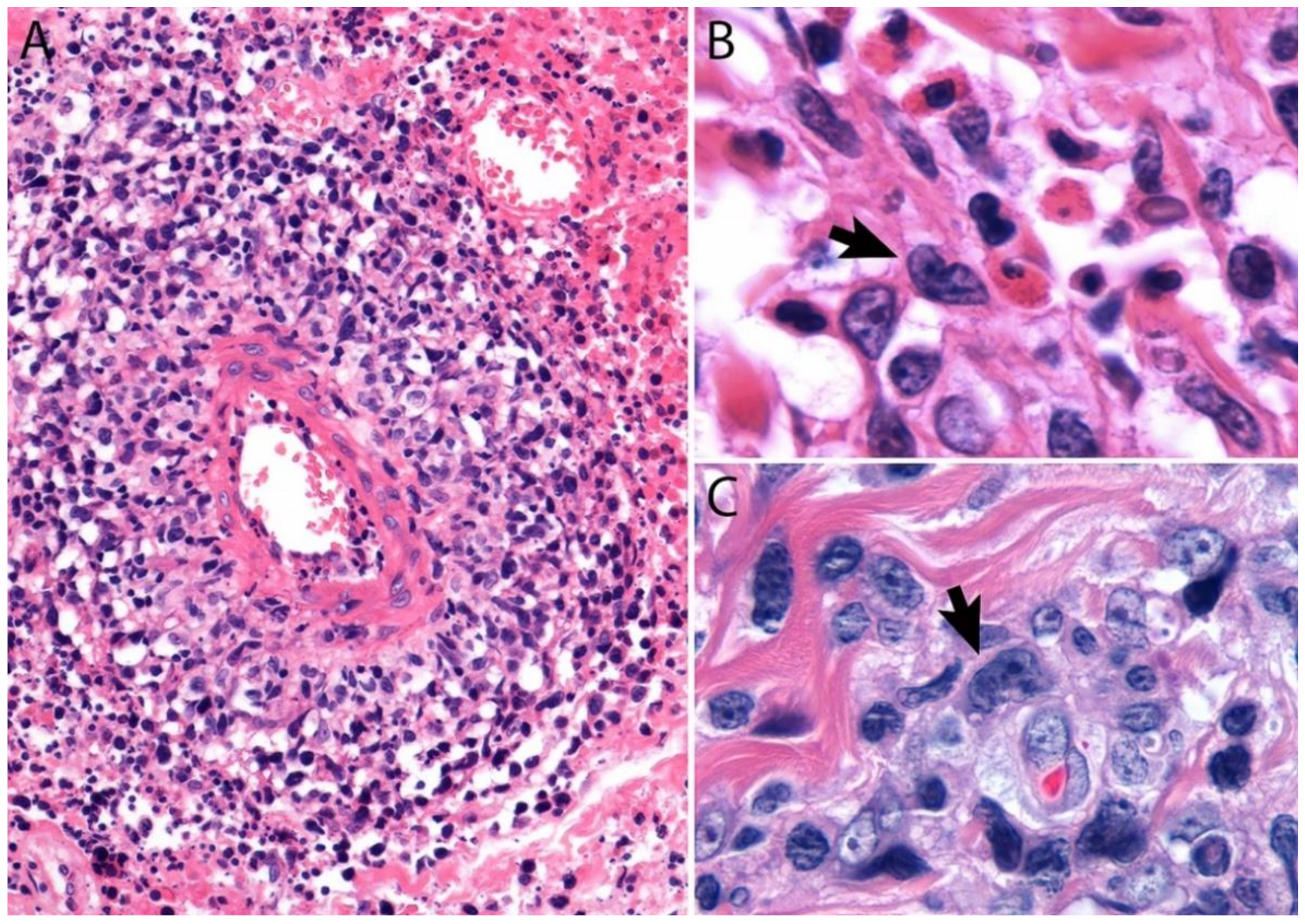
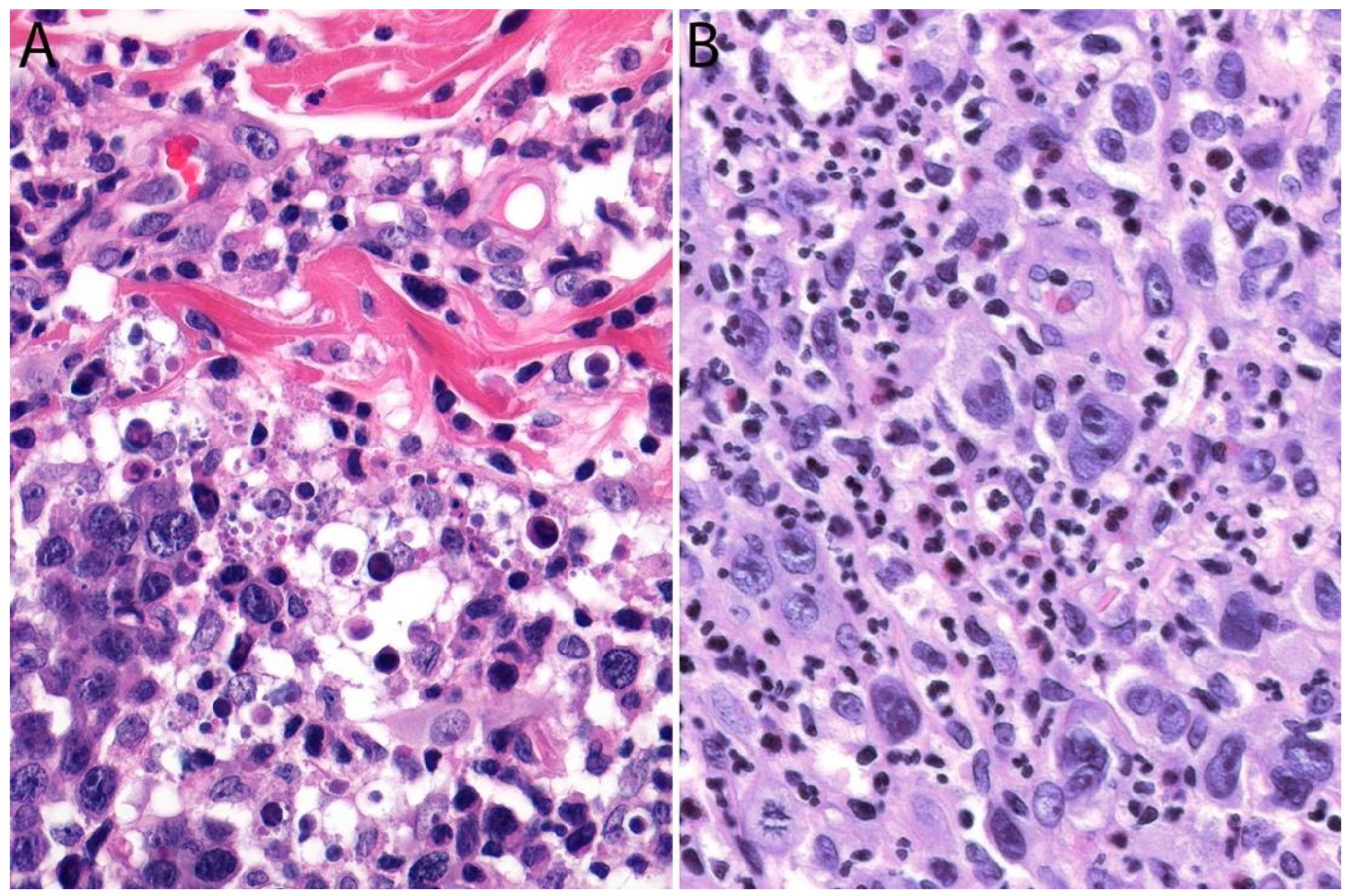
2.4. Pathology
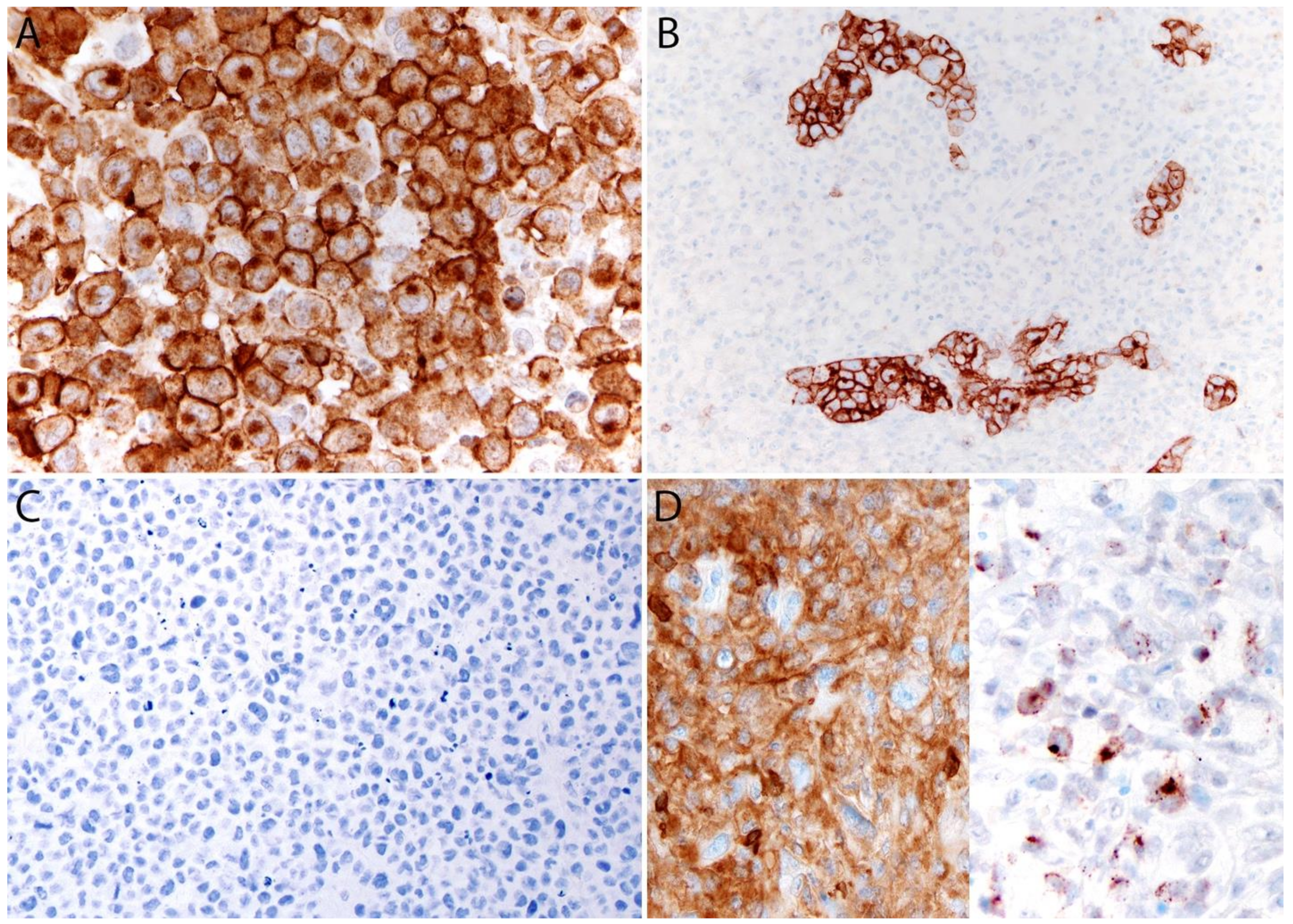
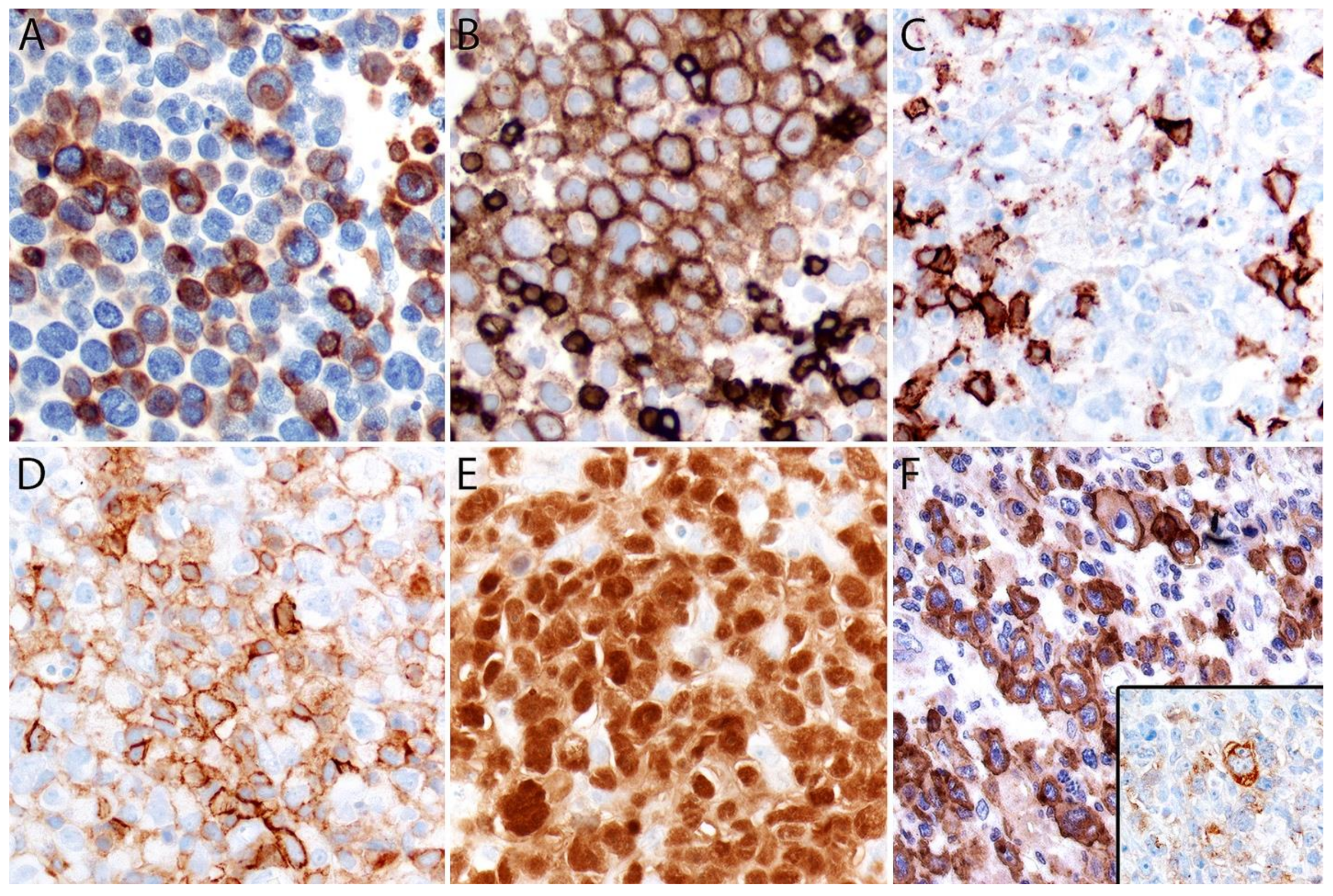
Immunohistochemistry
2.5. Differential Diagnosis
2.5.1. Hematopoietic Tumors
ALK- vs. ALK+ ALCL
ALK- ALCL vs. PTCL, NOS
ALK- ALCL with a “Hodgkin-like Pattern” vs. CHL
ALK- ALCL vs. Large B-Cell Lymphoma, Monocytic Sarcoma and Histiocytic Neoplasms
2.5.2. Non-Hematopoietic Tumors
ALK- ALCL vs. Carcinoma and Embryonal Carcinoma
ALK- ALCL vs. Other Epithelioid Malignancies
Sarcomatoid ALK- ALCL vs. other Spindle Cell Tumors
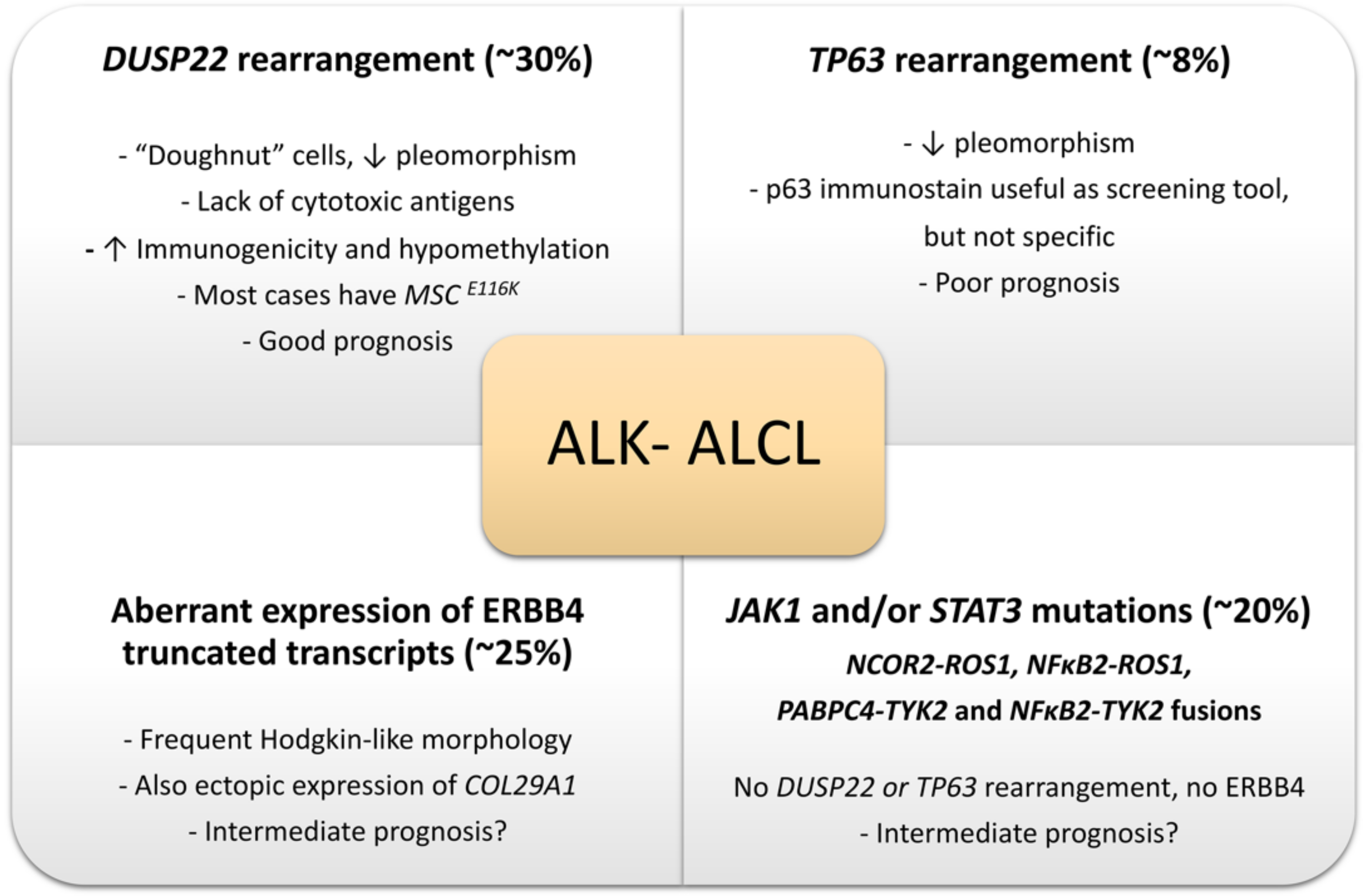
2.6. Genetic Findings and Molecular Alterations
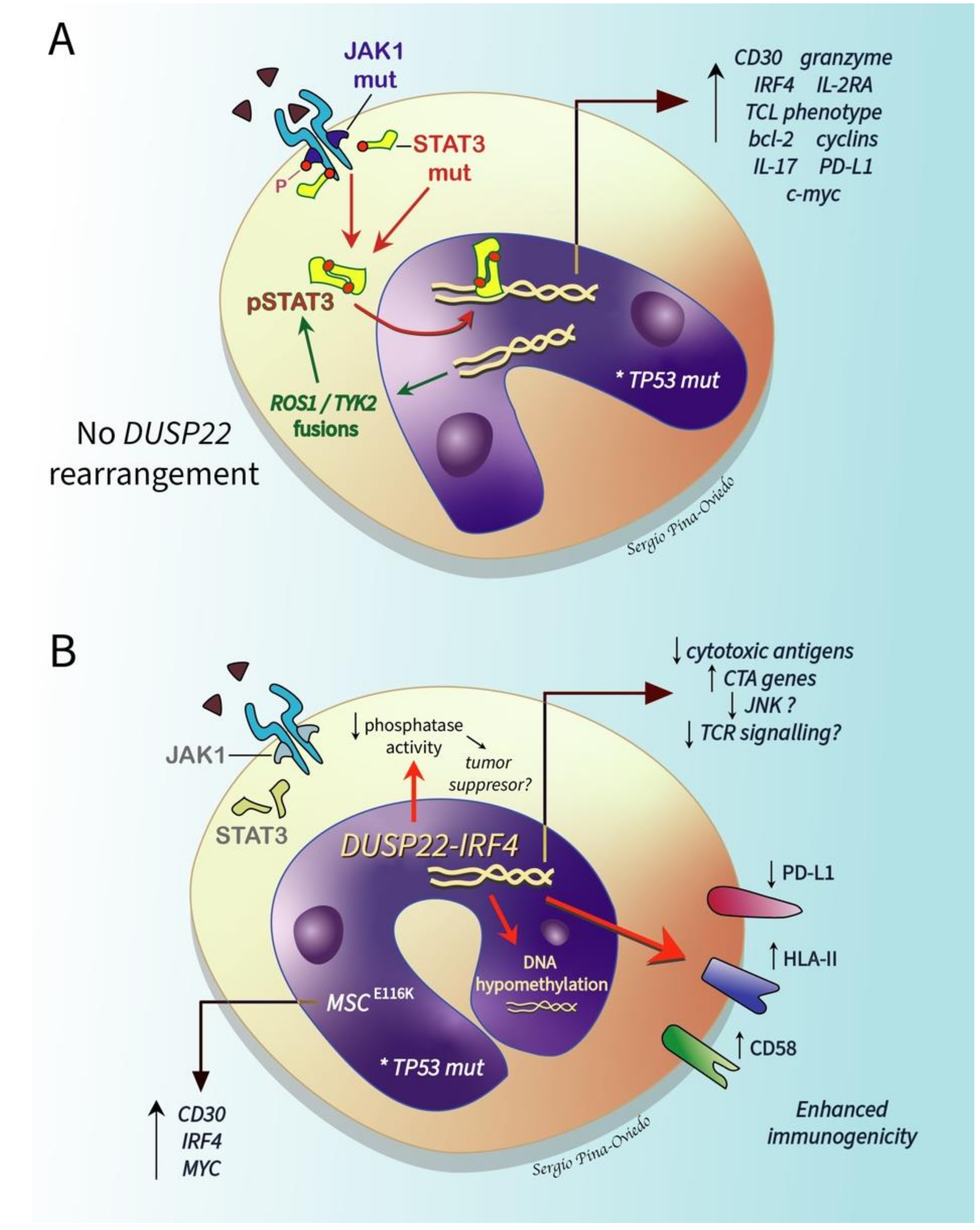
2.6.1. ALK- ALCL, STAT3, and JAK1 Mutations
2.6.2. DUSP22-Rearranged ALK- ALCL
2.6.3. TP63-Rearranged ALK- ALCL
2.6.4. Genetic Abnormalities with a Potential Impact on the Differential Diagnosis of ALK- ALCL vs. CD30+ Large Cell PTCL, NOS
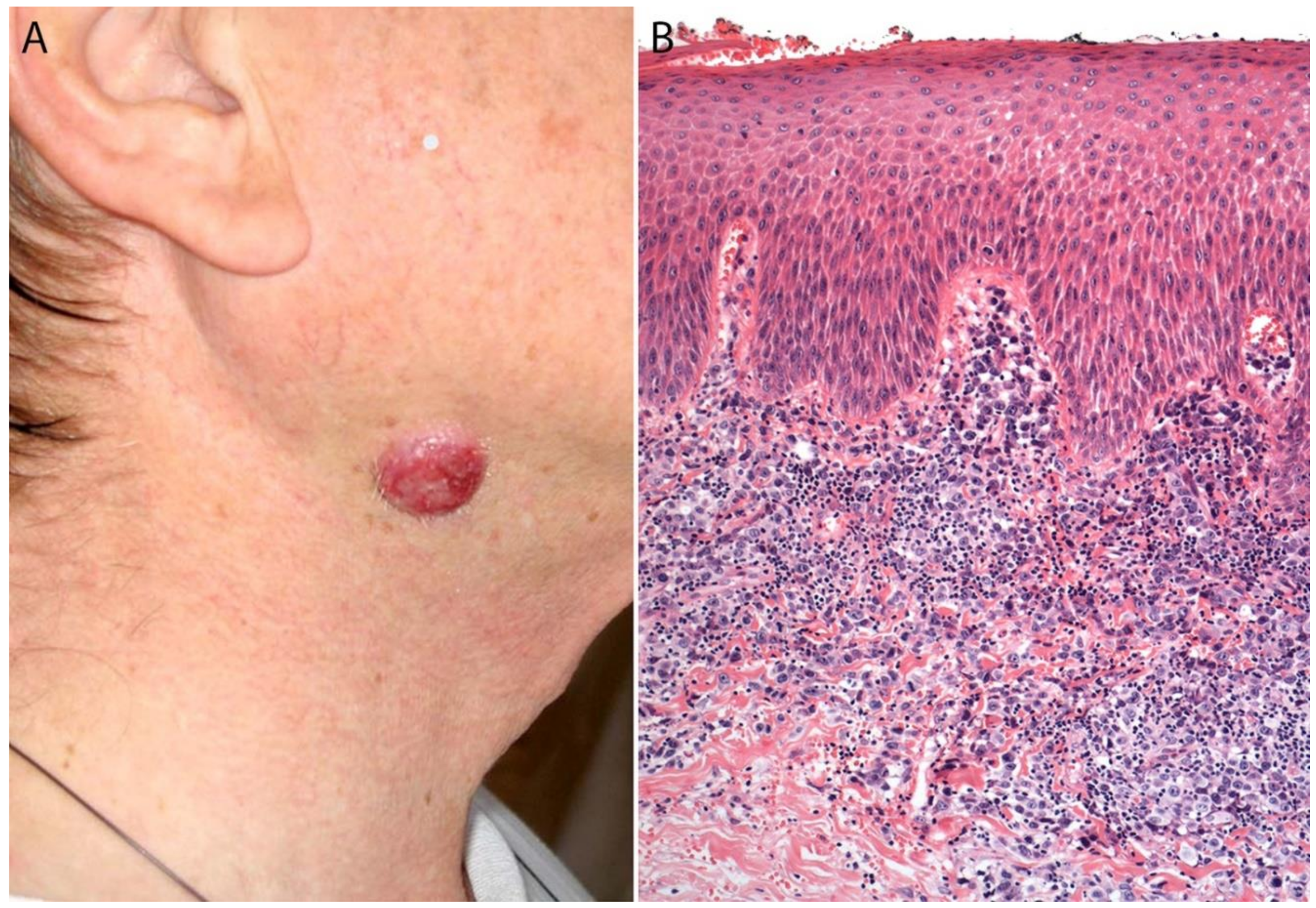
3. Primary Cutaneous ALCL
3.1. Introduction
3.2. Historical Aspects and Definition
3.3. Epidemiology, Risk Factors, and Clinical Features
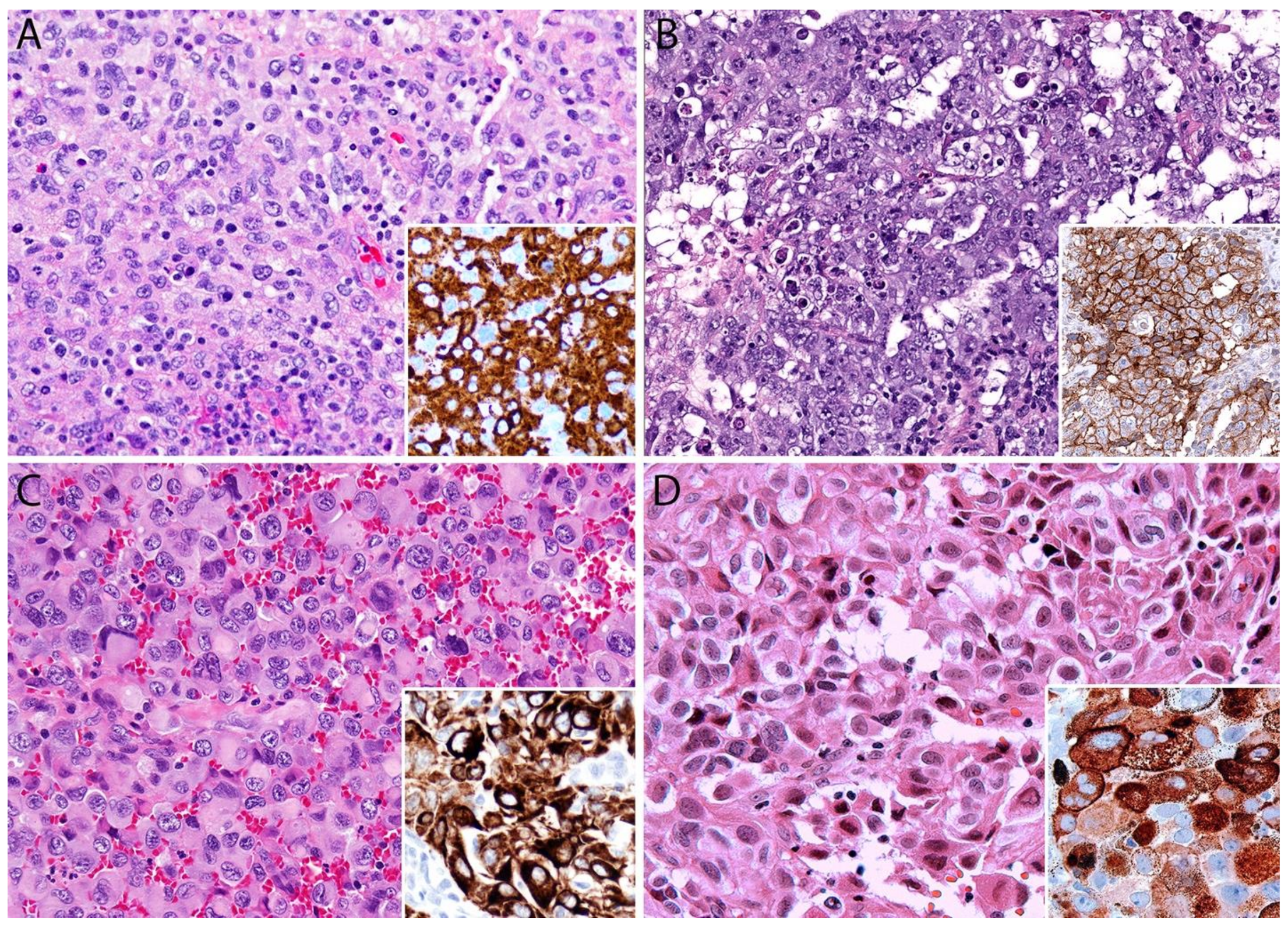
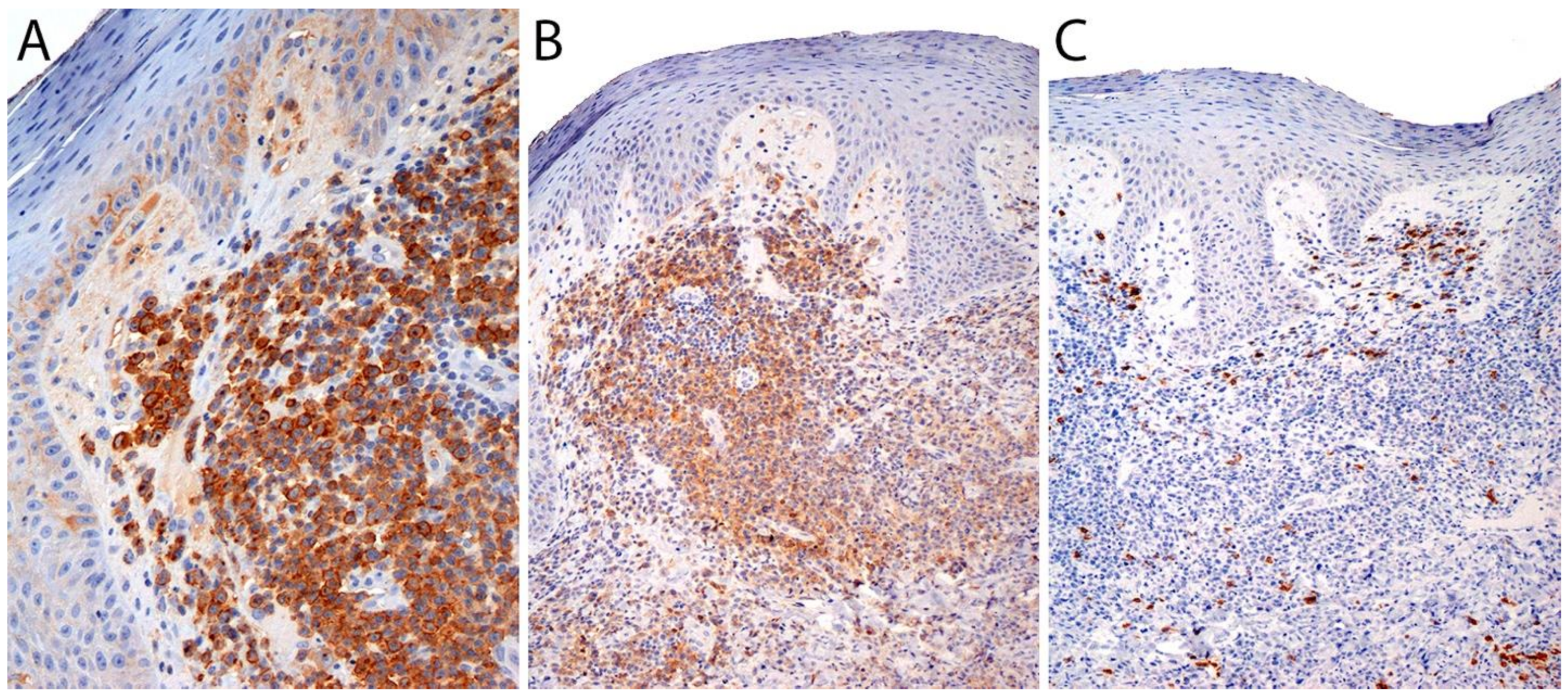
3.4. Pathology
Immunohistochemistry
3.5. Differential Diagnosis
3.6. Genetic Findings and Molecular Alterations
4. Breast Implant-Associated ALCL
4.1. Definition
4.2. Historical Aspects
4.3. Epidemiology, Risk Factors, and Clinical Features
4.4. Pathophysiology
4.5. Handling of Cytology Material and Capsulectomy Specimens of BIA-ALCL
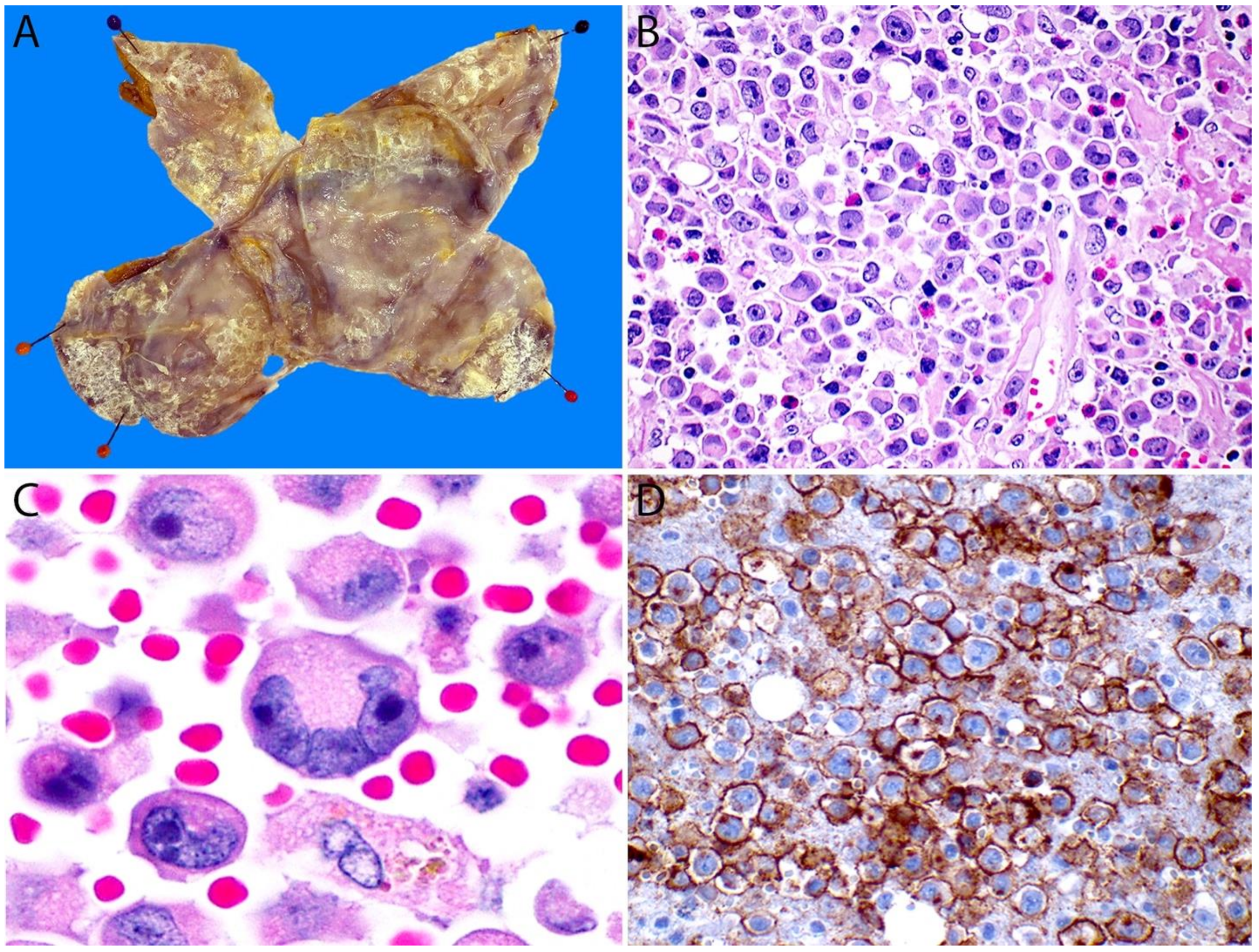
4.6. Pathology
4.6.1. Immunohistochemistry
4.6.2. Staging
4.7. Differential Diagnosis
4.8. Genetic Findings and Molecular Alterations
5. Conclusions
Authors Contributions
Funding
Conflicts of Interest
References
- Feldman, A.L.; Harris, N.L.; Stein, H.; Campo, E.; Kinney, M.C.; Jaffe, E.S.; Falini, B.; Inghirami, G.G.; Pileri, S.A. Anaplastic large cell lymphoma, ALK-negative. In WHO Classification of Tumors of Hematopoietic and Lymphoid Tissues, 4th ed.; Swerdlow, S.H., Campo, E., Harris, N.L., Jaffe, E.S., Pileri, S.A., Stein, H., Thiele, J., Eds.; IARC: Lyon, France, 2017; pp. 418–421. [Google Scholar]
- Pileri, S.A.; Agostinelli, C.; Bacci, F.; Sabattini, E.; Sagramoso, C.; Falini, B.; Piccaluga, P.P. Pathobiology of ALK-negative anaplastic large cell lymphoma. Pediatr. Rep. 2011, 3 (Suppl. S2), e5. [Google Scholar] [CrossRef]
- Savage, K.J.; Harris, N.L.; Vose, J.M.; Ullrich, F.; Jaffe, E.S.; Connors, J.M.; Rimsza, L.; Pileri, S.A.; Chhanabhai, M.; Gascoyne, R.D.; et al. ALK- anaplastic large-cell lymphoma is clinically and immunophenotypically different from both ALK+ ALCL and peripheral T-cell lymphoma, not otherwise specified: Report from the International Peripheral T-Cell Lymphoma Project. Blood 2008, 111, 5496–5504. [Google Scholar] [CrossRef]
- Jaffe, E.S. Anaplastic large cell lymphoma: The shifting sands of diagnostic hematopathology. Mod. Pathol. 2001, 14, 219–228. [Google Scholar] [CrossRef] [PubMed]
- Schwab, U.; Stein, H.; Gerdes, J.; Lemke, H.; Kirchner, H.; Schaadt, M.; Diehl, V. Production of a monoclonal antibody specific for Hodgkin and Sternberg-Reed cells of Hodgkin’s disease and a subset of normal lymphoid cells. Nature 1982, 299, 65–67. [Google Scholar] [CrossRef]
- Stein, H.; Mason, D.Y.; Gerdes, J.; O’Connor, N.; Wainscoat, J.; Pallesen, G.; Gatter, K.; Falini, B.; Delsol, G.; Lemke, H.; et al. The expression of the Hodgkin’s disease associated antigen Ki-1 in reactive and neoplastic lymphoid tissue: Evidence that Reed-Sternberg cells and histiocytic malignancies are derived from activated lymphoid cells. Blood 1985, 66, 848–858. [Google Scholar] [CrossRef]
- Harris, N.L.; Jaffe, E.S.; Stein, H.; Banks, P.M.; Chan, J.K.; Cleary, M.L.; Delsol, G.; De Wolf-Peeters, C.; Falini, B.; Gatter, K.C.; et al. A revised European-American classification of lymphoid neoplasms: A proposal from the International Lymphoma Study Group. Blood 1994, 84, 1361–1392. [Google Scholar] [CrossRef] [PubMed]
- Stansfeld, A.G.; Diebold, J.; Noel, H.; Kapanci, Y.; Rilke, F.; Kelényi, G.; Sundstrom, C.; Lennert, K.; van Unnik, J.A.; Mioduszewska, O.; et al. Updated Kiel classification for lymphomas. Lancet 1988, 1, 292–293. [Google Scholar] [CrossRef]
- Morris, S.W.; Kirstein, M.N.; Valentine, M.B.; Dittmer, K.G.; Shapiro, D.N.; Saltman, D.L.; Look, A.T. Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin’s lymphoma. Science 1994, 263, 1281–1284. [Google Scholar] [CrossRef] [PubMed]
- Mason, D.Y.; Harris, N.L.; Delsol, G.; Stein, H.; Campo, E.; Kinney, M.C.; Jaffe, E.S.; Falini, B. Anaplastic large cell lymphoma, ALK-negative. In WHO Classification of Tumors of Hematopoietic and Lymphoid Tissues; Swerdlow, S.H., Campo, E., Harris, N.L., Jaffe, E.S., Pileri, S.A., Stein, H., Thiele, J., Vardiman, J.W., Eds.; IARC: Lyon, France, 2008; pp. 317–319. [Google Scholar]
- Roman, E.; Smith, A.G. Epidemiology of lymphomas. Histopathology 2011, 58, 4–14. [Google Scholar] [CrossRef] [PubMed]
- Thandra, K.C.; Barsouk, A.; Saginala, K.; Padala, S.A.; Barsouk, A.; Rawla, P. Epidemiology of Non-Hodgkin’s Lymphoma. Med. Sci. 2021, 9, 5. [Google Scholar] [CrossRef]
- Ferreri, A.J.; Govi, S.; Pileri, S.A.; Savage, K.J. Anaplastic large cell lymphoma, ALK-negative. Crit. Rev. Oncol. Hematol. 2013, 85, 206–215. [Google Scholar] [CrossRef]
- Kinney, M.C.; Higgins, R.A.; Medina, E.A. Anaplastic large cell lymphoma: Twenty-five years of discovery. Arch. Pathol. Lab. Med. 2011, 135, 19–43. [Google Scholar] [CrossRef]
- Tsuyama, N.; Sakamoto, K.; Sakata, S.; Dobashi, A.; Takeuchi, K. Anaplastic large cell lymphoma: Pathology, genetics, and clinical aspects. J. Clin. Exp. Hematop. 2017, 57, 120–142. [Google Scholar] [CrossRef]
- Chan, A.C.; Chan, J.K.; Yan, K.W.; Kwong, Y.L. Anaplastic large cell lymphoma presenting as a pleural effusion and mimicking primary effusion lymphoma. A report of 2 cases. Acta Cytol. 2003, 47, 809–816. [Google Scholar] [CrossRef]
- Grandhi, A.; Boros, A.L.; Berardo, N.; Reich, R.F.; Freedman, P.D. Two cases of CD30+, anaplastic lymphoma kinase (ALK)-negative anaplastic large cell lymphoma with oral manifestations. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. 2013, 115, e41–e47. [Google Scholar] [CrossRef] [PubMed]
- Kodama, K.; Hokama, M.; Kawaguchi, K.; Tanaka, Y.; Hongo, K. Primary ALK-1-negative anaplastic large cell lymphoma of the brain: Case report and review of the literature. Neuropathology 2009, 29, 166–171. [Google Scholar] [CrossRef] [PubMed]
- Lagmay, J.; Termuhlen, A.; Fung, B.; Ranalli, M. Primary testicular presentation of ALK-1-negative anaplastic large cell lymphoma in a pediatric patient. J. Pediatr. Hematol. Oncol. 2009, 31, 330–332. [Google Scholar] [CrossRef]
- Lannon, M.; Lu, J.Q.; Chum, M.; Wang, B.H. ALK-negative CNS anaplastic large cell lymphoma: Case report and review of literature. Br. J. Neurosurg. 2020, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Lobo, J.; Henrique, R.; Monteiro, P.; Lobo, C. ALK-negative anaplastic large cell lymphoma with urinary bladder involvement diagnosed in urine cytology: A case report and literature review. Diagn. Cytopathol. 2017, 45, 354–358. [Google Scholar] [CrossRef]
- Saikia, U.N.; Sharma, N.; Duseja, A.; Bhalla, A.; Joshi, K. Anaplastic large cell lymphoma presenting as acute liver failure: A report of two cases with review of literature. Ann. Hepatol 2010, 9, 457–461. [Google Scholar] [CrossRef]
- Sanka, R.K.; Eagle, R.C., Jr.; Wojno, T.H.; Neufeld, K.R.; Grossniklaus, H.E. Spectrum of CD30+ lymphoid proliferations in the eyelid lymphomatoid papulosis, cutaneous anaplastic large cell lymphoma, and anaplastic large cell lymphoma. Ophthalmology 2010, 117, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Karki, N.R.; Badin, K.; Savage, N.; Bryan, L. Leukaemic relapse of anaplastic large cell lymphoma, ALK negative. BMJ Case Rep. 2021, 14, e239213. [Google Scholar] [CrossRef] [PubMed]
- Kasinathan, G. Leukemic phase of ALK-negative anaplastic large cell lymphoma in a patient who is on androgenic steroids: A case report. Ann. Med. Surg. 2020, 49, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Zhao, X.; Wang, E.; Chen, W.; Huang, Q. ALK-negative anaplastic large cell lymphoma with extensive peripheral blood and bone marrow involvements manifested as “leukemic phase”. Leuk. Res. 2010, 34, 475–482. [Google Scholar] [CrossRef] [PubMed]
- Wong, W.S.; Liu, B.W.; Lam, F.S.; Wong, K.F. ALK-negative anaplastic large cell lymphoma in leukemic phase with near-pentaploidy. Leuk. Lymphoma 2010, 51, 1927–1930. [Google Scholar] [CrossRef]
- Wang, S.S.; Flowers, C.R.; Kadin, M.E.; Chang, E.T.; Hughes, A.M.; Ansell, S.M.; Feldman, A.L.; Lightfoot, T.; Boffetta, P.; Melbye, M.; et al. Medical history, lifestyle, family history, and occupational risk factors for peripheral T-cell lymphomas: The InterLymph Non-Hodgkin Lymphoma Subtypes Project. J. Natl. Cancer Inst. Monogr. 2014, 2014, 66–75. [Google Scholar] [CrossRef]
- Brody, G.S.; Deapen, D.; Taylor, C.R.; Pinter-Brown, L.; House-Lightner, S.R.; Andersen, J.S.; Carlson, G.; Lechner, M.G.; Epstein, A.L. Anaplastic large cell lymphoma occurring in women with breast implants: Analysis of 173 cases. Plast. Reconstr. Surg. 2015, 135, 695–705. [Google Scholar] [CrossRef]
- Quesada, A.E.; Medeiros, L.J.; Clemens, M.W.; Ferrufino-Schmidt, M.C.; Pina-Oviedo, S.; Miranda, R.N. Breast implant-associated anaplastic large cell lymphoma: A review. Mod. Pathol. 2019, 32, 166–188. [Google Scholar] [CrossRef]
- Marra, A.; Viale, G.; Pileri, S.A.; Pravettoni, G.; Viale, G.; de Lorenzi, F.; Nolè, F.; Veronesi, P.; Curigliano, G. Breast implant-associated anaplastic large cell lymphoma: A comprehensive review. Cancer Treat. Rev. 2020, 84, 101963. [Google Scholar] [CrossRef]
- Kim, Y.C.; Yang, W.I.; Lee, M.G.; Kim, S.N.; Cho, K.H.; Lee, S.J.; Lee, M.W.; Koh, J.K. Epstein-Barr virus in CD30 anaplastic large cell lymphoma involving the skin and lymphomatoid papulosis in South Korea. Int. J. Dermatol. 2006, 45, 1312–1316. [Google Scholar] [CrossRef]
- Ma, L.; Katz, Y.; Sharan, K.P.; Schwarting, R.; Kim, A.S. Epstein-Barr virus positive anaplastic large cell lymphoma: Myth or reality? Int. J. Clin. Exp. Pathol. 2010, 4, 100–110. [Google Scholar]
- Benharroch, D.; Meguerian-Bedoyan, Z.; Lamant, L.; Amin, C.; Brugières, L.; Terrier-Lacombe, M.J.; Haralambieva, E.; Pulford, K.; Pileri, S.; Morris, S.W.; et al. ALK-positive lymphoma: A single disease with a broad spectrum of morphology. Blood 1998, 91, 2076–2084. [Google Scholar] [CrossRef] [PubMed]
- King, R.L.; Dao, L.N.; McPhail, E.D.; Jaffe, E.S.; Said, J.; Swerdlow, S.H.; Sattler, C.A.; Ketterling, R.P.; Sidhu, J.S.; Hsi, E.D.; et al. Morphologic Features of ALK-negative Anaplastic Large Cell Lymphomas With DUSP22 Rearrangements. Am. J. Surg. Pathol. 2016, 40, 36–43. [Google Scholar] [CrossRef] [PubMed]
- Amador, C.; Feldman, A.L. How I Diagnose Anaplastic Large Cell Lymphoma. Am. J. Clin. Pathol. 2021, 155, 479–497. [Google Scholar] [CrossRef]
- Kadin, M.E. Anaplastic large cell lymphoma and its morphological variants. Cancer Surv. 1997, 30, 77–86. [Google Scholar] [PubMed]
- Chan, J.K.; Buchanan, R.; Fletcher, C.D. Sarcomatoid variant of anaplastic large-cell Ki-1 lymphoma. Am. J. Surg. Pathol. 1990, 14, 983–988. [Google Scholar] [CrossRef]
- Pileri, S.; Bocchia, M.; Baroni, C.D.; Martelli, M.; Falini, B.; Sabattini, E.; Gherlinzoni, F.; Amadori, S.; Poggi, S.; Mazza, P.; et al. Anaplastic large cell lymphoma (CD30 +/Ki-1+): Results of a prospective clinico-pathological study of 69 cases. Br. J. Haematol. 1994, 86, 513–523. [Google Scholar] [CrossRef] [PubMed]
- Parrilla Castellar, E.R.; Jaffe, E.S.; Said, J.W.; Swerdlow, S.H.; Ketterling, R.P.; Knudson, R.A.; Sidhu, J.S.; Hsi, E.D.; Karikehalli, S.; Jiang, L.; et al. ALK-negative anaplastic large cell lymphoma is a genetically heterogeneous disease with widely disparate clinical outcomes. Blood 2014, 124, 1473–1480. [Google Scholar] [CrossRef]
- Foss, H.D.; Anagnostopoulos, I.; Araujo, I.; Assaf, C.; Demel, G.; Kummer, J.A.; Hummel, M.; Stein, H. Anaplastic large-cell lymphomas of T-cell and null-cell phenotype express cytotoxic molecules. Blood 1996, 88, 4005–4011. [Google Scholar] [CrossRef]
- Vega, F.; Amador, C.; Chadburn, A.; Feldman, A.L.; Hsi, E.D.; Wang, W.; Medeiros, L.J. American Registry of Pathology Expert Opinions: Recommendations for the diagnostic workup of mature T cell neoplasms. Ann. Diagn. Pathol. 2020, 49, 151623. [Google Scholar] [CrossRef]
- Feldman, A.L.; Law, M.E.; Inwards, D.J.; Dogan, A.; McClure, R.F.; Macon, W.R. PAX5-positive T-cell anaplastic large cell lymphomas associated with extra copies of the PAX5 gene locus. Mod. Pathol. 2010, 23, 593–602. [Google Scholar] [CrossRef] [PubMed]
- Gustmann, C.; Altmannsberger, M.; Osborn, M.; Griesser, H.; Feller, A.C. Cytokeratin expression and vimentin content in large cell anaplastic lymphomas and other non-Hodgkin’s lymphomas. Am. J. Pathol. 1991, 138, 1413–1422. [Google Scholar] [PubMed]
- Nguyen, T.T.; Kreisel, F.H.; Frater, J.L.; Bartlett, N.L. Anaplastic large-cell lymphoma with aberrant expression of multiple cytokeratins masquerading as metastatic carcinoma of unknown primary. J. Clin. Oncol. 2013, 31, e443–e445. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Ming, J.; Zhang, S.; Li, B.; Han, X.; Qiu, X. Cytokeratin positivity in anaplastic large cell lymphoma: A potential diagnostic pitfall in misdiagnosis of metastatic carcinoma. Int. J. Clin. Exp. Pathol. 2013, 6, 798–801. [Google Scholar]
- Wang, X.; Boddicker, R.L.; Dasari, S.; Sidhu, J.S.; Kadin, M.E.; Macon, W.R.; Ansell, S.M.; Ketterling, R.P.; Rech, K.L.; Feldman, A.L. Expression of p63 protein in anaplastic large cell lymphoma: Implications for genetic subtyping. Hum. Pathol. 2017, 64, 19–27. [Google Scholar] [CrossRef]
- Al-Rohil, R.N.; Torres-Cabala, C.A.; Patel, A.; Tetzlaff, M.T.; Ivan, D.; Nagarajan, P.; Curry, J.L.; Miranda, R.N.; Duvic, M.; Prieto, V.G.; et al. Loss of CD30 expression after treatment with brentuximab vedotin in a patient with anaplastic large cell lymphoma: A novel finding. J. Cutan. Pathol. 2016, 43, 1161–1166. [Google Scholar] [CrossRef] [PubMed]
- Arai, H.; Furuichi, S.; Nakamura, Y.; Nakamura, Y.; Ichikawa, M.; Mitani, K. [ALK-negative anaplastic large cell lymphoma with loss of CD30 expression during treatment with brentuximab vedotin]. Rinsho Ketsueki 2016, 57, 634–637. [Google Scholar] [CrossRef]
- Goyal, A.; Patel, S.; Goyal, K.; Morgan, E.A.; Foreman, R.K. Variable loss of CD30 expression by immunohistochemistry in recurrent cutaneous CD30+ lymphoid neoplasms treated with brentuximab vedotin. J. Cutan. Pathol. 2019, 46, 823–829. [Google Scholar] [CrossRef]
- Nielson, C.; Fischer, R.; Fraga, G.; Aires, D. Loss of CD30 Expression in Anaplastic Large Cell Lymphoma Following Brentuximab Therapy. J. Drugs Dermatol. 2016, 15, 894–895. [Google Scholar]
- Ravindran, A.; Feldman, A.L.; Ketterling, R.P.; Dasari, S.; Rech, K.L.; McPhail, E.D.; Kurtin, P.J.; Shi, M. Striking Association of Lymphoid Enhancing Factor (LEF1) Overexpression and DUSP22 Rearrangements in Anaplastic Large Cell Lymphoma. Am. J. Surg. Pathol. 2021, 45, 550–557. [Google Scholar] [CrossRef]
- Mélard, P.; Idrissi, Y.; Andrique, L.; Poglio, S.; Prochazkova-Carlotti, M.; Berhouet, S.; Boucher, C.; Laharanne, E.; Chevret, E.; Pham-Ledard, A.; et al. Molecular alterations and tumor suppressive function of the DUSP22 (Dual Specificity Phosphatase 22) gene in peripheral T-cell lymphoma subtypes. Oncotarget 2016, 7, 68734–68748. [Google Scholar] [CrossRef]
- Tzankov, A.; Bourgau, C.; Kaiser, A.; Zimpfer, A.; Maurer, R.; Pileri, S.A.; Went, P.; Dirnhofer, S. Rare expression of T-cell markers in classical Hodgkin’s lymphoma. Mod. Pathol. 2005, 18, 1542–1549. [Google Scholar] [CrossRef]
- Pletneva, M.A.; Smith, L.B. Anaplastic large cell lymphoma: Features presenting diagnostic challenges. Arch. Pathol. Lab. Med. 2014, 138, 1290–1294. [Google Scholar] [CrossRef]
- Seitz, V.; Hummel, M.; Marafioti, T.; Anagnostopoulos, I.; Assaf, C.; Stein, H. Detection of clonal T-cell receptor gamma-chain gene rearrangements in Reed-Sternberg cells of classic Hodgkin disease. Blood 2000, 95, 3020–3024. [Google Scholar] [CrossRef]
- Aguilera, N.S.; Chen, J.; Bijwaard, K.E.; Director-Myska, A.E.; Barekman, C.L.; Millward, C.; Lichy, J.; Abbondanzo, S.L. Gene rearrangement and comparative genomic hybridization studies of classic Hodgkin lymphoma expressing T-cell antigens. Arch. Pathol. Lab. Med. 2006, 130, 1772–1779. [Google Scholar] [CrossRef]
- Döring, C.; Hansmann, M.L.; Agostinelli, C.; Piccaluga, P.P.; Facchetti, F.; Pileri, S.; Küppers, R.; Newrzela, S.; Hartmann, S. A novel immunohistochemical classifier to distinguish Hodgkin lymphoma from ALK anaplastic large cell lymphoma. Mod. Pathol. 2014, 27, 1345–1354. [Google Scholar] [CrossRef] [PubMed]
- Han, J.J.; O’Byrne, M.; Stenson, M.J.; Maurer, M.J.; Wellik, L.E.; Feldman, A.L.; McPhail, E.D.; Witzig, T.E.; Gupta, M. Prognostic and therapeutic significance of phosphorylated STAT3 and protein tyrosine phosphatase-6 in peripheral-T cell lymphoma. Blood Cancer J. 2018, 8, 110. [Google Scholar] [CrossRef]
- Leoncini, L.; Del Vecchio, M.T.; Kraft, R.; Megha, T.; Barbini, P.; Cevenini, G.; Poggi, S.; Pileri, S.; Tosi, P.; Cottier, H. Hodgkin’s disease and CD30-positive anaplastic large cell lymphomas--a continuous spectrum of malignant disorders. A quantitative morphometric and immunohistologic study. Am. J. Pathol. 1990, 137, 1047–1057. [Google Scholar] [PubMed]
- Bovio, I.M.; Allan, R.W. The expression of myeloid antigens CD13 and/or CD33 is a marker of ALK+ anaplastic large cell lymphomas. Am. J. Clin. Pathol. 2008, 130, 628–634. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Mariño-Enríquez, A.; Wang, W.L.; Roy, A.; Lopez-Terrada, D.; Lazar, A.J.; Fletcher, C.D.; Coffin, C.M.; Hornick, J.L. Epithelioid inflammatory myofibroblastic sarcoma: An aggressive intra-abdominal variant of inflammatory myofibroblastic tumor with nuclear membrane or perinuclear ALK. Am. J. Surg. Pathol. 2011, 35, 135–144. [Google Scholar] [CrossRef]
- Xing, X.; Feldman, A.L. Anaplastic large cell lymphomas: ALK positive, ALK negative, and primary cutaneous. Adv. Anat. Pathol. 2015, 22, 29–49. [Google Scholar] [CrossRef]
- Salaverria, I.; Beà, S.; Lopez-Guillermo, A.; Lespinet, V.; Pinyol, M.; Burkhardt, B.; Lamant, L.; Zettl, A.; Horsman, D.; Gascoyne, R.; et al. Genomic profiling reveals different genetic aberrations in systemic ALK-positive and ALK-negative anaplastic large cell lymphomas. Br. J. Haematol. 2008, 140, 516–526. [Google Scholar] [CrossRef] [PubMed]
- Hassler, M.R.; Pulverer, W.; Lakshminarasimhan, R.; Redl, E.; Hacker, J.; Garland, G.D.; Merkel, O.; Schiefer, A.I.; Simonitsch-Klupp, I.; Kenner, L.; et al. Insights into the Pathogenesis of Anaplastic Large-Cell Lymphoma through Genome-wide DNA Methylation Profiling. Cell Rep. 2016, 17, 596–608. [Google Scholar] [CrossRef] [PubMed]
- Crescenzo, R.; Abate, F.; Lasorsa, E.; Tabbo, F.; Gaudiano, M.; Chiesa, N.; Di Giacomo, F.; Spaccarotella, E.; Barbarossa, L.; Ercole, E.; et al. Convergent mutations and kinase fusions lead to oncogenic STAT3 activation in anaplastic large cell lymphoma. Cancer Cell 2015, 27, 516–532. [Google Scholar] [CrossRef]
- Lobello, C.; Tichy, B.; Bystry, V.; Radova, L.; Filip, D.; Mraz, M.; Montes-Mojarro, I.A.; Prokoph, N.; Larose, H.; Liang, H.C.; et al. STAT3 and TP53 mutations associate with poor prognosis in anaplastic large cell lymphoma. Leukemia 2021, 35, 1500–1505. [Google Scholar] [CrossRef] [PubMed]
- Richardson, A.I.; Yin, C.C.; Cui, W.; Li, N.; Medeiros, L.J.; Li, L.; Zhang, D. p53 and β-Catenin Expression Predict Poorer Prognosis in Patients With Anaplastic Large-Cell Lymphoma. Clin. Lymphoma Myeloma Leuk. 2019, 19, e385–e392. [Google Scholar] [CrossRef]
- Feldman, A.L.; Dogan, A.; Smith, D.I.; Law, M.E.; Ansell, S.M.; Johnson, S.H.; Porcher, J.C.; Ozsan, N.; Wieben, E.D.; Eckloff, B.W.; et al. Discovery of recurrent t(6;7)(p25.3;q32.3) translocations in ALK-negative anaplastic large cell lymphomas by massively parallel genomic sequencing. Blood 2011, 117, 915–919. [Google Scholar] [CrossRef] [PubMed]
- Luchtel, R.A.; Dasari, S.; Oishi, N.; Pedersen, M.B.; Hu, G.; Rech, K.L.; Ketterling, R.P.; Sidhu, J.; Wang, X.; Katoh, R.; et al. Molecular profiling reveals immunogenic cues in anaplastic large cell lymphomas with DUSP22 rearrangements. Blood 2018, 132, 1386–1398. [Google Scholar] [CrossRef]
- Luchtel, R.A.; Zimmermann, M.T.; Hu, G.; Dasari, S.; Jiang, M.; Oishi, N.; Jacobs, H.K.; Zeng, Y.; Hundal, T.; Rech, K.L.; et al. Recurrent MSC (E116K) mutations in ALK-negative anaplastic large cell lymphoma. Blood 2019, 133, 2776–2789. [Google Scholar] [CrossRef]
- Patterson, K.I.; Brummer, T.; O’Brien, P.M.; Daly, R.J. Dual-specificity phosphatases: Critical regulators with diverse cellular targets. Biochem. J. 2009, 418, 475–489. [Google Scholar] [CrossRef]
- Lang, R.; Raffi, F.A.M. Dual-Specificity Phosphatases in Immunity and Infection: An Update. Int. J. Mol. Sci. 2019, 20, 2710. [Google Scholar] [CrossRef]
- Hamada, N.; Mizuno, M.; Tomita, H.; Iwamoto, I.; Hara, A.; Nagata, K.I. Expression analyses of Dusp22 (Dual-specificity phosphatase 22) in mouse tissues. Med. Mol. Morphol. 2018, 51, 111–117. [Google Scholar] [CrossRef]
- Chen, A.J.; Zhou, G.; Juan, T.; Colicos, S.M.; Cannon, J.P.; Cabriera-Hansen, M.; Meyer, C.F.; Jurecic, R.; Copeland, N.G.; Gilbert, D.J.; et al. The dual specificity JKAP specifically activates the c-Jun N-terminal kinase pathway. J. Biol. Chem. 2002, 277, 36592–36601. [Google Scholar] [CrossRef]
- Li, J.P.; Yang, C.Y.; Chuang, H.C.; Lan, J.L.; Chen, D.Y.; Chen, Y.M.; Wang, X.; Chen, A.J.; Belmont, J.W.; Tan, T.H. The phosphatase JKAP/DUSP22 inhibits T-cell receptor signalling and autoimmunity by inactivating Lck. Nat. Commun. 2014, 5, 3618. [Google Scholar] [CrossRef] [PubMed]
- Sekine, Y.; Tsuji, S.; Ikeda, O.; Sato, N.; Aoki, N.; Aoyama, K.; Sugiyama, K.; Matsuda, T. Regulation of STAT3-mediated signaling by LMW-DSP2. Oncogene 2006, 25, 5801–5806. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Chang, Y.; Liu, J.; Chen, M.; Wang, H.; Huang, M.; Liu, S.; Wang, X.; Zhao, Q. JNK Pathway-Associated Phosphatase/DUSP22 Suppresses CD4(+) T-Cell Activation and Th1/Th17-Cell Differentiation and Negatively Correlates with Clinical Activity in Inflammatory Bowel Disease. Front. Immunol. 2017, 8, 781. [Google Scholar] [CrossRef] [PubMed]
- Chuang, H.C.; Chen, Y.M.; Hung, W.T.; Li, J.P.; Chen, D.Y.; Lan, J.L.; Tan, T.H. Downregulation of the phosphatase JKAP/DUSP22 in T cells as a potential new biomarker of systemic lupus erythematosus nephritis. Oncotarget 2016, 7, 57593–57605. [Google Scholar] [CrossRef]
- Candi, E.; Dinsdale, D.; Rufini, A.; Salomoni, P.; Knight, R.A.; Mueller, M.; Krammer, P.H.; Melino, G. TAp63 and DeltaNp63 in cancer and epidermal development. Cell Cycle 2007, 6, 274–285. [Google Scholar] [CrossRef] [PubMed]
- Melino, G. p63 is a suppressor of tumorigenesis and metastasis interacting with mutant p53. Cell Death Differ. 2011, 18, 1487–1499. [Google Scholar] [CrossRef] [PubMed]
- Fukushima, N.; Satoh, T.; Sueoka, N.; Sato, A.; Ide, M.; Hisatomi, T.; Kuwahara, N.; Tomimasu, R.; Tsuneyoshi, N.; Funai, N.; et al. Clinico-pathological characteristics of p63 expression in B-cell lymphoma. Cancer Sci. 2006, 97, 1050–1055. [Google Scholar] [CrossRef] [PubMed]
- Hallack Neto, A.E.; Siqueira, S.A.; Dulley, F.L.; Ruiz, M.A.; Chamone, D.A.; Pereira, J. p63 protein expression in high risk diffuse large B-cell lymphoma. J. Clin. Pathol. 2009, 62, 77–79. [Google Scholar] [CrossRef] [PubMed]
- Hedvat, C.V.; Teruya-Feldstein, J.; Puig, P.; Capodieci, P.; Dudas, M.; Pica, N.; Qin, J.; Cordon-Cardo, C.; Di Como, C.J. Expression of p63 in diffuse large B-cell lymphoma. Appl. Immunohistochem. Mol. Morphol. 2005, 13, 237–242. [Google Scholar] [CrossRef]
- Hu, W.M.; Jin, J.T.; Wu, C.Y.; Lu, J.B.; Zhang, L.H.; Zeng, J.; Lin, S.X. Expression of P63 and its correlation with prognosis in diffuse large B-cell lymphoma: A single center experience. Diagn. Pathol. 2019, 14, 128. [Google Scholar] [CrossRef]
- Kim, H.J.; Kim, H.K.; Park, G.; Min, S.K.; Cha, H.J.; Lee, H.; Choi, S.J.; Na, H.Y.; Choe, J.Y.; Kim, J.E. Comparative pathologic analysis of mediastinal B-cell lymphomas: Selective expression of p63 but no GATA3 optimally differentiates primary mediastinal large B-cell lymphoma from classic Hodgkin lymphoma. Diagn. Pathol. 2019, 14, 133. [Google Scholar] [CrossRef] [PubMed]
- Robson, A.; Shukur, Z.; Ally, M.; Kluk, J.; Liu, K.; Pincus, L.; Sahni, D.; Sundram, U.; Subtil, A.; Karai, L.; et al. Immunocytochemical p63 expression discriminates between primary cutaneous follicle centre cell and diffuse large B cell lymphoma-leg type, and is of the TAp63 isoform. Histopathology 2016, 69, 11–19. [Google Scholar] [CrossRef]
- Zamò, A.; Malpeli, G.; Scarpa, A.; Doglioni, C.; Chilosi, M.; Menestrina, F. Expression of TP73L is a helpful diagnostic marker of primary mediastinal large B-cell lymphomas. Mod. Pathol. 2005, 18, 1448–1453. [Google Scholar] [CrossRef][Green Version]
- Agnelli, L.; Mereu, E.; Pellegrino, E.; Limongi, T.; Kwee, I.; Bergaggio, E.; Ponzoni, M.; Zamò, A.; Iqbal, J.; Piccaluga, P.P.; et al. Identification of a 3-gene model as a powerful diagnostic tool for the recognition of ALK-negative anaplastic large-cell lymphoma. Blood 2012, 120, 1274–1281. [Google Scholar] [CrossRef]
- Lamant, L.; de Reyniès, A.; Duplantier, M.M.; Rickman, D.S.; Sabourdy, F.; Giuriato, S.; Brugières, L.; Gaulard, P.; Espinos, E.; Delsol, G. Gene-expression profiling of systemic anaplastic large-cell lymphoma reveals differences based on ALK status and two distinct morphologic ALK+ subtypes. Blood 2007, 109, 2156–2164. [Google Scholar] [CrossRef]
- Fitzpatrick, M.J.; Massoth, L.R.; Marcus, C.; Vergilio, J.A.; Severson, E.; Duncan, D.; Ramkissoon, S.H.; Hasserjian, R.P.; Kim, A.S.; Sohani, A.R.; et al. JAK2 Rearrangements Are a Recurrent Alteration in CD30+ Systemic T-Cell Lymphomas With Anaplastic Morphology. Am. J. Surg. Pathol. 2021, 45, 895–904. [Google Scholar] [CrossRef] [PubMed]
- Scarfò, I.; Pellegrino, E.; Mereu, E.; Kwee, I.; Agnelli, L.; Bergaggio, E.; Garaffo, G.; Vitale, N.; Caputo, M.; Machiorlatti, R.; et al. Identification of a new subclass of ALK-negative ALCL expressing aberrant levels of ERBB4 transcripts. Blood 2016, 127, 221–232. [Google Scholar] [CrossRef] [PubMed]
- Martin, J.M.; Wu, H.; Barta, S.K. CD30+ T-cell lymphoproliferative disorders. Chin. Clin. Oncol. 2019, 8, 4. [Google Scholar] [CrossRef]
- Willemze, R.; Paulli, M.; Kadin, M.E. Primary cutaneous CD30-positive T-cell lymphoproliferative disorders. In WHO Classification of Tumors of Hematopoietic and Lymphoid Tissues, 4th ed.; Swerdlow, S.H., Campo, E., Harris, N.L., Jaffe, E.S., Pileri, S.A., Stein, H., Thiele, J., Eds.; IARC: Lyon, France, 2017; pp. 392–396. [Google Scholar]
- Kempf, W. Cutaneous CD30-Positive Lymphoproliferative Disorders. Surg. Pathol. Clin. 2014, 7, 203–228. [Google Scholar] [CrossRef]
- Berti, E.; Gianotti, R.; Alessi, E. Primary anaplastic large cell lymphoma of the skin. Dermatologica 1989, 178, 225–227. [Google Scholar] [CrossRef]
- King, R.L.; Musiek, A.C.; Feldman, A.L. Primary cutaneous CD30+ T-cell lymphoproliferative disorders. In Hematopathology of the Skin. Clinical & Pathological Approach; Gru, A.A., Schaffer, A., Eds.; Wolters Kluwer Health: Philadelphia, PA, USA, 2017; pp. 141–158. [Google Scholar]
- Brown, R.A.; Fernandez-Pol, S.; Kim, J. Primary cutaneous anaplastic large cell lymphoma. J. Cutan. Pathol. 2017, 44, 570–577. [Google Scholar] [CrossRef]
- Sevilla-Lizcano, D.B.; Ortiz-Hidalgo, C. Linfoma anaplasico de celulas grandes: Estudio clinico-patologico e inmunohistoquimica de 20 casos clasificados de acuerdo a la revision de la Organizacion Mundial de la Salud de neoplasias linfoides de 2016. Gac. Mex. Oncol. 2017, 16, 215–225. [Google Scholar] [CrossRef]
- Sarfraz, H.; Gentille, C.; Ensor, J.; Wang, L.; Wong, S.; Ketcham, M.S.; Joshi, J.; Pingali, S.R.K. Primary cutaneous anaplastic large-cell lymphoma: A review of the SEER database from 2005 to 2016. Clin. Exp. Dermatol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Pittaluga, S.; Raffeld, M.; Guerrera, M.; Seibel, N.L.; Jaffe, E.S. Primary cutaneous CD30-positive anaplastic large cell lymphoma in childhood: Report of 4 cases and review of the literature. Pediatr. Dev. Pathol. 2005, 8, 52–60. [Google Scholar] [CrossRef] [PubMed]
- Seo, A.N.; Lee, S.J.; Choi, Y.H.; Chung, H.Y.; Huh, J.; Yoon, G.S. Congenital primary cutaneous anaplastic large-cell lymphoma: A case report. Am. J. Dermatopathol. 2015, 37, 398–400. [Google Scholar] [CrossRef]
- Prieto-Torres, L.; Rodriguez-Pinilla, S.M.; Onaindia, A.; Ara, M.; Requena, L.; Piris, M. CD30-positive primary cutaneous lymphoproliferative disorders: Molecular alterations and targeted therapies. Haematologica 2019, 104, 226–235. [Google Scholar] [CrossRef] [PubMed]
- Philippe, E.; Creech, K.T.; Cook, N.; Segura, J. Recurrent Primary Cutaneous Anaplastic Large Cell Lymphoma With Systemic Involvement: A Case Report and Literature Review. Cureus 2021, 13, e14284. [Google Scholar] [CrossRef]
- Hruska, C.J.; Bertoli, R.J.; Young, Y.D.; Burkhart, P.H.; Googe, P.B. Primary cutaneous anaplastic large cell lymphoma in a patient receiving adalimumab. JAAD Case Rep. 2015, 1, 56–59. [Google Scholar] [CrossRef] [PubMed]
- Papathemeli, D.; Gräfe, R.; Hildebrandt, U.; Zettl, U.K.; Ulrich, J. Development of a primary cutaneous CD30(+) anaplastic large-cell T-cell lymphoma during treatment of multiple sclerosis with fingolimod. Mult. Scler. 2016, 22, 1888–1890. [Google Scholar] [CrossRef] [PubMed]
- Querfeld, C.; Khan, I.; Mahon, B.; Nelson, B.P.; Rosen, S.T.; Evens, A.M. Primary cutaneous and systemic anaplastic large cell lymphoma: Clinicopathologic aspects and therapeutic options. Oncology 2010, 24, 574–587. [Google Scholar] [PubMed]
- Di Raimondo, C.; Parekh, V.; Song, J.Y.; Rosen, S.T.; Querfeld, C.; Zain, J.; Martinez, X.U.; Abdulla, F.R. Primary Cutaneous CD30+ Lymphoproliferative Disorders: A Comprehensive Review. Curr. Hematol. Malig. Rep. 2020, 15, 333–342. [Google Scholar] [CrossRef] [PubMed]
- Pandiar, D.; Smitha, T. The “hallmark” cells. J. Oral Maxillofac. Pathol. 2019, 23, 176–177. [Google Scholar] [CrossRef]
- Irshaid, L.; Xu, M.L. ALCL by any other name: The many facets of anaplastic large cell lymphoma. Pathology 2020, 52, 100–110. [Google Scholar] [CrossRef] [PubMed]
- Magro, C.M.; Momtahen, S.; Kiuru, M. Primary Cutaneous Small Cell Variant of Anaplastic Large Cell Lymphoma: A Case Series and Review of the Literature. Am. J. Dermatopathol. 2017, 39, 877–889. [Google Scholar] [CrossRef]
- Kong, Y.Y.; Dai, B.; Kong, J.C.; Lu, H.F.; Shi, D.R. Neutrophil/eosinophil-rich type of primary cutaneous anaplastic large cell lymphoma: A clinicopathological, immunophenotypic and molecular study of nine cases. Histopathology 2009, 55, 189–196. [Google Scholar] [CrossRef]
- Burg, G.; Kempf, W.; Kazakov, D.V.; Dummer, R.; Frosch, P.J.; Lange-Ionescu, S.; Nishikawa, T.; Kadin, M.E. Pyogenic lymphoma of the skin: A peculiar variant of primary cutaneous neutrophil-rich CD30+ anaplastic large-cell lymphoma. Clinicopathological study of four cases and review of the literature. Br. J. Dermatol. 2003, 148, 580–586. [Google Scholar] [CrossRef]
- Guitart, J.; Martinez-Escala, M.E.; Deonizio, J.M.; Gerami, P.; Kadin, M.E. CD30(+) cutaneous lymphoproliferative disorders with pseudocarcinomatous hyperplasia are associated with a T-helper-17 cytokine profile and infiltrating granulocytes. J. Am. Acad. Dermatol. 2015, 72, 508–515. [Google Scholar] [CrossRef]
- Kempf, W.; Kazakov, D.V.; Paredes, B.E.; Laeng, H.R.; Palmedo, G.; Kutzner, H. Primary cutaneous anaplastic large cell lymphoma with angioinvasive features and cytotoxic phenotype: A rare lymphoma variant within the spectrum of CD30+ lymphoproliferative disorders. Dermatology 2013, 227, 346–352. [Google Scholar] [CrossRef] [PubMed]
- Onaindia, A.; de Villambrosía, S.G.; Prieto-Torres, L.; Rodríguez-Pinilla, S.M.; Montes-Moreno, S.; González-Vela, C.; Piris, M.A. DUSP22-rearranged anaplastic lymphomas are characterized by specific morphological features and a lack of cytotoxic and JAK/STAT surrogate markers. Haematologica 2019, 104, e158–e162. [Google Scholar] [CrossRef]
- Xue, Y.N.; Wang, Z.; Sun, J.F.; Chen, H. Primary cutaneous anaplastic large-cell lymphoma with 6p25.3 rearrangement exhibits a biphasic histopathologic pattern: Two case reports and literature review. J. Cutan. Pathol. 2021. [Google Scholar] [CrossRef]
- Ferrara, G.; Ena, L.; Cota, C.; Cerroni, L. Intralymphatic Spread Is a Common Finding in Cutaneous CD30+ Lymphoproliferative Disorders. Am. J. Surg. Pathol. 2015, 39, 1511–1517. [Google Scholar] [CrossRef] [PubMed]
- Metcalf, R.A.; Bashey, S.; Wysong, A.; Kim, J.; Kim, Y.H.; Gratzinger, D. Intravascular ALK-negative anaplastic large cell lymphoma with localized cutaneous involvement and an indolent clinical course: Toward recognition of a distinct clinicopathologic entity. Am. J. Surg. Pathol. 2013, 37, 617–623. [Google Scholar] [CrossRef]
- Resnik, K.S.; Kutzner, H. Of lymphocytes and cutaneous epithelium: Keratoacanthomatous hyperplasia in CD30+ lymphoproliferative disorders and CD30+ cells associated with keratoacanthoma. Am. J. Dermatopathol. 2010, 32, 314–315. [Google Scholar] [CrossRef]
- Misra, M.; Raghuvanshi, S.; Goel, M.M.; Verma, S.P. ALK-negative primary cutaneous T-cell anaplastic large cell lymphoma, myxoid variant; masquerading as sarcoma: Unveiling the diagnostic dilemma. BMJ Case Rep. 2021, 14, e239350. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Sun, N.C.; Nozawa, Y.; Arber, D.A.; Chu, P.; Chang, K.L.; Weiss, L.M. Histological and immunohistochemical characterization of extranodal diffuse large-cell lymphomas with prominent spindle cell features. Histopathology 2001, 39, 476–481. [Google Scholar] [CrossRef]
- Kempf, W. A new era for cutaneous CD30-positive T-cell lymphoproliferative disorders. Semin. Diagn. Pathol. 2017, 34, 22–35. [Google Scholar] [CrossRef] [PubMed]
- van der Weyden, C.A.; Pileri, S.A.; Feldman, A.L.; Whisstock, J.; Prince, H.M. Understanding CD30 biology and therapeutic targeting: A historical perspective providing insight into future directions. Blood Cancer J. 2017, 7, e603. [Google Scholar] [CrossRef]
- Collins, K.; Gu, J.; Aung, P.P.; Nagarajan, P.; Curry, J.L.; Huen, A.; Ivan, D.; Prieto, V.G.; Tetzlaff, M.T.; Duvic, M.; et al. Is immunohistochemical expression of GATA3 helpful in the differential diagnosis of transformed mycosis fungoides and primary cutaneous CD30-positive T cell lymphoproliferative disorders? Virchows Arch. 2021, 479, 377–383. [Google Scholar] [CrossRef]
- Mitteldorf, C.; Robson, A.; Tronnier, M.; Pfaltz, M.C.; Kempf, W. Galectin-3 Expression in Primary Cutaneous CD30-Positive Lymphoproliferative Disorders and Transformed Mycosis Fungoides. Dermatology 2015, 231, 164–170. [Google Scholar] [CrossRef]
- Olsen, S.H.; Ma, L.; Schnitzer, B.; Fullen, D.R. Clusterin expression in cutaneous CD30-positive lymphoproliferative disorders and their histologic simulants. J. Cutan. Pathol. 2009, 36, 302–307. [Google Scholar] [CrossRef]
- Geller, S.; Canavan, T.N.; Pulitzer, M.; Moskowitz, A.J.; Myskowski, P.L. ALK-positive primary cutaneous anaplastic large cell lymphoma: A case report and review of the literature. Int. J. Dermatol. 2018, 57, 515–520. [Google Scholar] [CrossRef] [PubMed]
- Oschlies, I.; Lisfeld, J.; Lamant, L.; Nakazawa, A.; d’Amore, E.S.; Hansson, U.; Hebeda, K.; Simonitsch-Klupp, I.; Maldyk, J.; Müllauer, L.; et al. ALK-positive anaplastic large cell lymphoma limited to the skin: Clinical, histopathological and molecular analysis of 6 pediatric cases. A report from the ALCL99 study. Haematologica 2013, 98, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Pulitzer, M.; Ogunrinade, O.; Lin, O.; Steinherz, P. ALK-positive (2p23 rearranged) anaplastic large cell lymphoma with localization to the skin in a pediatric patient. J. Cutan. Pathol. 2015, 42, 182–187. [Google Scholar] [CrossRef] [PubMed]
- Magro, C.M.; Dyrsen, M.E. Cutaneous lymphocyte antigen expression in benign and neoplastic cutaneous B- and T-cell lymphoid infiltrates. J. Cutan. Pathol. 2008, 35, 1040–1049. [Google Scholar] [CrossRef] [PubMed]
- Hagiwara, M.; Tomita, A.; Takata, K.; Shimoyama, Y.; Yoshino, T.; Tomita, Y.; Nakamura, S. Primary cutaneous CD30 positive T-cell lymphoproliferative disorders with aberrant expression of PAX5: Report of three cases. Pathol. Int. 2012, 62, 264–270. [Google Scholar] [CrossRef] [PubMed]
- Yu, B.H.; Zhang, Y.; Xue, T.; Shui, R.H.; Lu, H.F.; Zhou, X.Y.; Zhu, X.Z.; Li, X.Q. The clinicopathological relevance of uniform CD56 expression in anaplastic large cell lymphoma: A retrospective analysis of 18 cases. Diagn. Pathol. 2021, 16, 1. [Google Scholar] [CrossRef]
- Macgrogan, G.; Vergier, B.; Dubus, P.; Beylot-Barry, M.; Belleannee, G.; Delaunay, M.M.; Eghbali, H.; Beylot, C.; Rivel, J.; Trojani, M.; et al. CD30-positive cutaneous large cell lymphomas. A comparative study of clinicopathologic and molecular features of 16 cases. Am. J. Clin. Pathol. 1996, 105, 440–450. [Google Scholar] [CrossRef]
- Mao, X.; Orchard, G.; Lillington, D.M.; Russell-Jones, R.; Young, B.D.; Whittaker, S. Genetic alterations in primary cutaneous CD30+ anaplastic large cell lymphoma. Genes Chromosomes Cancer 2003, 37, 176–185. [Google Scholar] [CrossRef]
- Szuhai, K.; van Doorn, R.; Tensen, C.P.; Van, K. Array-CGH analysis of cutaneous anaplastic large cell lymphoma. Methods Mol. Biol. 2013, 973, 197–212. [Google Scholar] [CrossRef]
- Karai, L.J.; Kadin, M.E.; Hsi, E.D.; Sluzevich, J.C.; Ketterling, R.P.; Knudson, R.A.; Feldman, A.L. Chromosomal rearrangements of 6p25.3 define a new subtype of lymphomatoid papulosis. Am. J. Surg. Pathol. 2013, 37, 1173–1181. [Google Scholar] [CrossRef]
- Wood, G.S. Analysis of the t(2;5) (p23;q35) translocation in CD30+ primary cutaneous lymphoproliferative disorders and Hodgkin’s disease. Leuk. Lymphoma 1998, 29, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Feldman, A.L.; Harris, N.L.; Stein, H.; Campo, E.; Kinney, M.C.; Jaffe, E.S.; Falini, B.; Inghirami, G.G.; Pileri, S.A. Breast implant-associated anaplastic large cell lymphoma. In WHO Classification of Tumors of Hematopoietic and Lymphoid Tissues, 4th ed.; Swerdlow, S.H., Campo, E., Harris, N.L., Jaffe, E.S., Pileri, S.A., Stein, H., Thiele, J., Eds.; IARC: Lyon, France, 2017; pp. 421–422. [Google Scholar]
- Miranda, R.N.; Feldman, A.L.; Soares, F.A. Breast implant-associated anaplastic large cell lymphoma. In Breast Tumors, 5th ed.; WHO Classification ot Tumors Editorial Board, Ed.; IARC: Lyon, France, 2019; pp. 245–248. [Google Scholar]
- Keech, J.A., Jr.; Creech, B.J. Anaplastic T-cell lymphoma in proximity to a saline-filled breast implant. Plast. Reconstr. Surg. 1997, 100, 554–555. [Google Scholar] [CrossRef] [PubMed]
- American Society of Plastic Surgeons. BIA-ALCL Physician Resources. Available online: https://www.plasticsurgery.org/for-medical-professionals/health-policy/bia-alcl-physician-resources (accessed on 12 August 2021).
- Roden, A.C.; Macon, W.R.; Keeney, G.L.; Myers, J.L.; Feldman, A.L.; Dogan, A. Seroma-associated primary anaplastic large-cell lymphoma adjacent to breast implants: An indolent T-cell lymphoproliferative disorder. Mod. Pathol. 2008, 21, 455–463. [Google Scholar] [CrossRef] [PubMed]
- Miranda, R.N.; Aladily, T.N.; Prince, H.M.; Kanagal-Shamanna, R.; de Jong, D.; Fayad, L.E.; Amin, M.B.; Haideri, N.; Bhagat, G.; Brooks, G.S.; et al. Breast implant-associated anaplastic large-cell lymphoma: Long-term follow-up of 60 patients. J. Clin. Oncol. 2014, 32, 114–120. [Google Scholar] [CrossRef]
- Talwalkar, S.S.; Miranda, R.N.; Valbuena, J.R.; Routbort, M.J.; Martin, A.W.; Medeiros, L.J. Lymphomas involving the breast: A study of 106 cases comparing localized and disseminated neoplasms. Am. J. Surg. Pathol. 2008, 32, 1299–1309. [Google Scholar] [CrossRef]
- Miranda, R.N.; Lin, L.; Talwalkar, S.S.; Manning, J.T.; Medeiros, L.J. Anaplastic large cell lymphoma involving the breast: A clinicopathologic study of 6 cases and review of the literature. Arch. Pathol. Lab. Med. 2009, 133, 1383–1390. [Google Scholar] [CrossRef]
- U. S. Food & Drug Administration. Medical Device Reports of Breast Implant-Associated Anaplastic Large Cell Lymphoma. Available online: https://www.fda.gov/medical-devices/breast-implants/medical-device-reports-breast-implant-associated-anaplastic-large-cell-lymphoma (accessed on 12 August 2021).
- Ramos-Gallardo, G.; Vélez-Benítez, E.; Cuenca-Pardo, J.; Cárdenas-Camarena, L.; Ramírez-Montañana, A.; Carballo-Zarate, A.; Contreras-Bulnes, L.; Bucio-Duarte, J.; Morales-Olivera, M.; Iribarren-Moreno, R. What is the Process for Breast Implant Manufacturing? Inside Eight Breast Implant Factories. Aesthetic. Plast. Surg. 2020, 44, 2063–2074. [Google Scholar] [CrossRef]
- de Boer, M.; van Leeuwen, F.E.; Hauptmann, M.; Overbeek, L.I.H.; de Boer, J.P.; Hijmering, N.J.; Sernee, A.; Klazen, C.A.H.; Lobbes, M.B.I.; van der Hulst, R.; et al. Breast Implants and the Risk of Anaplastic Large-Cell Lymphoma in the Breast. JAMA Oncol. 2018, 4, 335–341. [Google Scholar] [CrossRef]
- Nelson, J.A.; Dabic, S.; Mehrara, B.J.; Cordeiro, P.G.; Disa, J.J.; Pusic, A.L.; Matros, E.; Dayan, J.H.; Allen, R.J., Jr.; Coriddi, M.; et al. Breast Implant-associated Anaplastic Large Cell Lymphoma Incidence: Determining an Accurate Risk. Ann. Surg. 2020, 272, 403–409. [Google Scholar] [CrossRef]
- de Boer, M.; van der Sluis, W.B.; de Boer, J.P.; Overbeek, L.I.H.; van Leeuwen, F.E.; Rakhorst, H.A.; van der Hulst, R.; Hijmering, N.J.; Bouman, M.B.; de Jong, D. Breast Implant-Associated Anaplastic Large-Cell Lymphoma in a Transgender Woman. Aesthet. Surg. J. 2017, 37, Np83–Np87. [Google Scholar] [CrossRef][Green Version]
- Alcalá, R.; Llombart, B.; Lavernia, J.; Traves, V.; Guillén, C.; Sanmartín, O. Skin involvement as the first manifestation of breast implant-associated anaplastic large cell lymphoma. J. Cutan. Pathol. 2016, 43, 602–608. [Google Scholar] [CrossRef] [PubMed]
- Di Napoli, A.; Pepe, G.; Giarnieri, E.; Cippitelli, C.; Bonifacino, A.; Mattei, M.; Martelli, M.; Falasca, C.; Cox, M.C.; Santino, I.; et al. Cytological diagnostic features of late breast implant seromas: From reactive to anaplastic large cell lymphoma. PLoS ONE 2017, 12, e0181097. [Google Scholar] [CrossRef]
- Adrada, B.E.; Miranda, R.N.; Rauch, G.M.; Arribas, E.; Kanagal-Shamanna, R.; Clemens, M.W.; Fanale, M.; Haideri, N.; Mustafa, E.; Larrinaga, J.; et al. Breast implant-associated anaplastic large cell lymphoma: Sensitivity, specificity, and findings of imaging studies in 44 patients. Breast Cancer Res. Treat. 2014, 147, 1–14. [Google Scholar] [CrossRef]
- Sharma, B.; Jurgensen-Rauch, A.; Pace, E.; Attygalle, A.D.; Sharma, R.; Bommier, C.; Wotherspoon, A.C.; Sharma, S.; Iyengar, S.; El-Sharkawi, D. Breast Implant-associated Anaplastic Large Cell Lymphoma: Review and Multiparametric Imaging Paradigms. Radiographics 2020, 40, 609–628. [Google Scholar] [CrossRef]
- Ferrufino-Schmidt, M.C.; Medeiros, L.J.; Liu, H.; Clemens, M.W.; Hunt, K.K.; Laurent, C.; Lofts, J.; Amin, M.B.; Ming Chai, S.; Morine, A.; et al. Clinicopathologic Features and Prognostic Impact of Lymph Node Involvement in Patients With Breast Implant-associated Anaplastic Large Cell Lymphoma. Am. J. Surg. Pathol. 2018, 42, 293–305. [Google Scholar] [CrossRef] [PubMed]
- Laurent, C.; Delas, A.; Gaulard, P.; Haioun, C.; Moreau, A.; Xerri, L.; Traverse-Glehen, A.; Rousset, T.; Quintin-Roue, I.; Petrella, T.; et al. Breast implant-associated anaplastic large cell lymphoma: Two distinct clinicopathological variants with different outcomes. Ann. Oncol. 2016, 27, 306–314. [Google Scholar] [CrossRef] [PubMed]
- Bizjak, M.; Selmi, C.; Praprotnik, S.; Bruck, O.; Perricone, C.; Ehrenfeld, M.; Shoenfeld, Y. Silicone implants and lymphoma: The role of inflammation. J. Autoimmun. 2015, 65, 64–73. [Google Scholar] [CrossRef] [PubMed]
- Cohen Tervaert, J.W.; Kappel, R.M. Silicone implant incompatibility syndrome (SIIS): A frequent cause of ASIA (Shoenfeld’s syndrome). Immunol. Res. 2013, 56, 293–298. [Google Scholar] [CrossRef] [PubMed]
- Joseph, J.; Variathu, K.T.; Mohanty, M. Mediatory role of interleukin-6 in α smooth muscle actin induction and myofibroblast formation around silicone tissue expander. J. Biomed. Mater. Res. A 2013, 101, 2967–2973. [Google Scholar] [CrossRef] [PubMed]
- Wolfram, D.; Rabensteiner, E.; Grundtman, C.; Böck, G.; Mayerl, C.; Parson, W.; Almanzar, G.; Hasenöhrl, C.; Piza-Katzer, H.; Wick, G. T regulatory cells and TH17 cells in peri-silicone implant capsular fibrosis. Plast. Reconstr. Surg. 2012, 129, 327e–337e. [Google Scholar] [CrossRef] [PubMed]
- Kadin, M.E.; Morgan, J.; Xu, H.; Epstein, A.L.; Sieber, D.; Hubbard, B.A.; Adams, W.P., Jr.; Bacchi, C.E.; Goes, J.C.S.; Clemens, M.W.; et al. IL-13 is produced by tumor cells in breast implant-associated anaplastic large cell lymphoma: Implications for pathogenesis. Hum. Pathol. 2018, 78, 54–62. [Google Scholar] [CrossRef]
- Ramos-Gallardo, G.; Carballo-Zarate, A.A.; Cuenca-Pardo, J.; Cárdenas-Camarena, L.; Solano-Genesta, M.; Beltrán, J.A.C.; Gallagher-Hernandez, S.; Contreras-Bulnes, L.; Vélez-Benitez, E.; Bucio-Duarte, J.J.; et al. What is the Evidence of Lymphoma in Patients with Prostheses Other Than Breast Implants? Aesthetic. Plast. Surg. 2020, 44, 286–294. [Google Scholar] [CrossRef]
- Barr, S.; Hill, E.W.; Bayat, A. Functional biocompatibility testing of silicone breast implants and a novel classification system based on surface roughness. J. Mech. Behav. Biomed. Mater. 2017, 75, 75–81. [Google Scholar] [CrossRef]
- Hu, H.; Johani, K.; Almatroudi, A.; Vickery, K.; Van Natta, B.; Kadin, M.E.; Brody, G.; Clemens, M.; Cheah, C.Y.; Lade, S.; et al. Bacterial Biofilm Infection Detected in Breast Implant-Associated Anaplastic Large-Cell Lymphoma. Plast. Reconstr. Surg. 2016, 137, 1659–1669. [Google Scholar] [CrossRef]
- Loch-Wilkinson, A.; Beath, K.J.; Knight, R.J.W.; Wessels, W.L.F.; Magnusson, M.; Papadopoulos, T.; Connell, T.; Lofts, J.; Locke, M.; Hopper, I.; et al. Breast Implant-Associated Anaplastic Large Cell Lymphoma in Australia and New Zealand: High-Surface-Area Textured Implants Are Associated with Increased Risk. Plast. Reconstr. Surg. 2017, 140, 645–654. [Google Scholar] [CrossRef]
- Deva, A.K.; Turner, S.D.; Kadin, M.E.; Magnusson, M.R.; Prince, H.M.; Miranda, R.N.; Inghirami, G.G.; Adams, W.P., Jr. Etiology of Breast Implant-Associated Anaplastic Large Cell Lymphoma (BIA-ALCL): Current Directions in Research. Cancers 2020, 12, 3861. [Google Scholar] [CrossRef]
- Jaffe, E.S.; Ashar, B.S.; Clemens, M.W.; Feldman, A.L.; Gaulard, P.; Miranda, R.N.; Sohani, A.R.; Stenzel, T.; Yoon, S.W. Best Practices Guideline for the Pathologic Diagnosis of Breast Implant-Associated Anaplastic Large-Cell Lymphoma. J. Clin. Oncol. 2020, 38, 1102–1111. [Google Scholar] [CrossRef]
- Lyapichev, K.A.; Piña-Oviedo, S.; Medeiros, L.J.; Evans, M.G.; Liu, H.; Miranda, A.R.; Hunt, K.K.; Clemens, M.W.; Stewart, J.M.; Amin, M.B.; et al. A proposal for pathologic processing of breast implant capsules in patients with suspected breast implant anaplastic large cell lymphoma. Mod. Pathol. 2020, 33, 367–379. [Google Scholar] [CrossRef]
- Clemens, M.W.; Jacobsen, E.D.; Horwitz, S.M. 2019 NCCN Consensus Guidelines on the Diagnosis and Treatment of Breast Implant-Associated Anaplastic Large Cell Lymphoma (BIA-ALCL). Aesthet. Surg. J. 2019, 39, S3–S13. [Google Scholar] [CrossRef]
- Barbé, E.; de Boer, M.; de Jong, D. A practical cytological approach to the diagnosis of breast-implant associated anaplastic large cell lymphoma. Cytopathology 2019, 30, 363–369. [Google Scholar] [CrossRef]
- Clemens, M.W.; Medeiros, L.J.; Butler, C.E.; Hunt, K.K.; Fanale, M.A.; Horwitz, S.; Weisenburger, D.D.; Liu, J.; Morgan, E.A.; Kanagal-Shamanna, R.; et al. Complete Surgical Excision Is Essential for the Management of Patients With Breast Implant-Associated Anaplastic Large-Cell Lymphoma. J. Clin. Oncol. 2016, 34, 160–168. [Google Scholar] [CrossRef]
- Shepard, E.; Kamenko, S.; Snir, O.L.; Hansen, J. Silicone granuloma mimicking Breast Implant Associated Large Cell Lymphoma (BIA-ALCL): A case report. Case Rep. Plast. Surg. Hand Surg. 2020, 7, 63–67. [Google Scholar] [CrossRef]
- Kadin, M.E.; Morgan, J.; Xu, H.; Glicksman, C.A. CD30+ T Cells in Late Seroma May Not Be Diagnostic of Breast Implant-Associated Anaplastic Large Cell Lymphoma. Aesthet. Surg. J. 2017, 37, 771–775. [Google Scholar] [CrossRef]
- Ryan, C.; Ged, Y.; Quinn, F.; Walker, J.; Kennedy, J.; Gillham, C.; Pittaluga, S.; McDermott, R.; Vandenberghe, E.; Grant, C.; et al. Classical Hodgkin Lymphoma Arising Adjacent to a Breast Implant. Int. J. Surg. Pathol. 2016, 24, 448–455. [Google Scholar] [CrossRef]
- Evans, M.G.; Miranda, R.N.; Young, P.A.; Pai, L.; Wang, H.Y.; Konoplev, S.N.; Medeiros, L.J.; Pinter-Brown, L.C. B-cell lymphomas associated with breast implants: Report of three cases and review of the literature. Ann. Diagn. Pathol. 2020, 46, 151512. [Google Scholar] [CrossRef] [PubMed]
- Medeiros, L.J.; Marques-Piubelli, M.L.; Sangiorgio, V.F.I.; Ruiz-Cordero, R.; Vega, F.; Feldman, A.L.; Chapman, J.R.; Clemens, M.W.; Hunt, K.K.; Evans, M.G.; et al. Epstein-Barr-virus-positive large B-cell lymphoma associated with breast implants: An analysis of eight patients suggesting a possible pathogenetic relationship. Mod. Pathol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Mescam, L.; Camus, V.; Schiano, J.M.; Adélaïde, J.; Picquenot, J.M.; Guille, A.; Bannier, M.; Ruminy, P.; Viailly, P.J.; Jardin, F.; et al. EBV+ diffuse large B-cell lymphoma associated with chronic inflammation expands the spectrum of breast implant-related lymphomas. Blood 2020, 135, 2004–2009. [Google Scholar] [CrossRef] [PubMed]
- Lechner, M.G.; Lade, S.; Liebertz, D.J.; Prince, H.M.; Brody, G.S.; Webster, H.R.; Epstein, A.L. Breast implant-associated, ALK-negative, T-cell, anaplastic, large-cell lymphoma: Establishment and characterization of a model cell line (TLBR-1) for this newly emerging clinical entity. Cancer 2011, 117, 1478–1489. [Google Scholar] [CrossRef]
- Los-de Vries, G.T.; de Boer, M.; van Dijk, E.; Stathi, P.; Hijmering, N.J.; Roemer, M.G.M.; Mendeville, M.; Miedema, D.M.; de Boer, J.P.; Rakhorst, H.A.; et al. Chromosome 20 loss is characteristic of breast implant-associated anaplastic large cell lymphoma. Blood 2020, 136, 2927–2932. [Google Scholar] [CrossRef]
- Blombery, P.; Thompson, E.; Ryland, G.L.; Joyce, R.; Byrne, D.J.; Khoo, C.; Lade, S.; Hertzberg, M.; Hapgood, G.; Marlton, P.; et al. Frequent activating STAT3 mutations and novel recurrent genomic abnormalities detected in breast implant-associated anaplastic large cell lymphoma. Oncotarget 2018, 9, 36126–36136. [Google Scholar] [CrossRef]
- Oishi, N.; Brody, G.S.; Ketterling, R.P.; Viswanatha, D.S.; He, R.; Dasari, S.; Mai, M.; Benson, H.K.; Sattler, C.A.; Boddicker, R.L.; et al. Genetic subtyping of breast implant-associated anaplastic large cell lymphoma. Blood 2018, 132, 544–547. [Google Scholar] [CrossRef] [PubMed]
- Di Napoli, A.; Jain, P.; Duranti, E.; Margolskee, E.; Arancio, W.; Facchetti, F.; Alobeid, B.; Santanelli di Pompeo, F.; Mansukhani, M.; Bhagat, G. Targeted next generation sequencing of breast implant-associated anaplastic large cell lymphoma reveals mutations in JAK/STAT signalling pathway genes, TP53 and DNMT3A. Br. J. Haematol. 2018, 180, 741–744. [Google Scholar] [CrossRef] [PubMed]
- Oishi, N.; Miranda, R.N.; Feldman, A.L. Genetics of Breast Implant-Associated Anaplastic Large Cell Lymphoma (BIA-ALCL). Aesthet. Surg. J. 2019, 39, S14–S20. [Google Scholar] [CrossRef] [PubMed]
- Quesada, A.E.; Zhang, Y.; Ptashkin, R.; Ho, C.; Horwitz, S.; Benayed, R.; Dogan, A.; Arcila, M.E. Next generation sequencing of breast implant-associated anaplastic large cell lymphomas reveals a novel STAT3-JAK2 fusion among other activating genetic alterations within the JAK-STAT pathway. Breast. J. 2021, 27, 314–321. [Google Scholar] [CrossRef]
- Laurent, C.; Nicolae, A.; Laurent, C.; Le Bras, F.; Haioun, C.; Fataccioli, V.; Amara, N.; Adélaïde, J.; Guille, A.; Schiano, J.M.; et al. Gene alterations in epigenetic modifiers and JAK-STAT signaling are frequent in breast implant-associated ALCL. Blood 2020, 135, 360–370. [Google Scholar] [CrossRef]
- Murray, P.J. The JAK-STAT signaling pathway: Input and output integration. J. Immunol. 2007, 178, 2623–2629. [Google Scholar] [CrossRef]
- Adlard, J.; Burton, C.; Turton, P. Increasing Evidence for the Association of Breast Implant-Associated Anaplastic Large Cell Lymphoma and Li Fraumeni Syndrome. Case Rep. Genet. 2019, 2019, 5647940. [Google Scholar] [CrossRef]
- Weilemann, A.; Grau, M.; Erdmann, T.; Merkel, O.; Sobhiafshar, U.; Anagnostopoulos, I.; Hummel, M.; Siegert, A.; Hayford, C.; Madle, H.; et al. Essential role of IRF4 and MYC signaling for survival of anaplastic large cell lymphoma. Blood 2015, 125, 124–132. [Google Scholar] [CrossRef] [PubMed]
- de Boer, M.; Hauptmann, M.; Hijmering, N.J.; van Noesel, C.J.M.; Rakhorst, H.A.; Meijers-Heijboer, H.E.J.; de Boer, J.P.; van der Hulst, R.; de Jong, D.; van Leeuwen, F.E. Increased prevalence of BRCA1/2 mutations in women with macrotextured breast implants and anaplastic large cell lymphoma of the breast. Blood 2020, 136, 1368–1372. [Google Scholar] [CrossRef] [PubMed]



















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
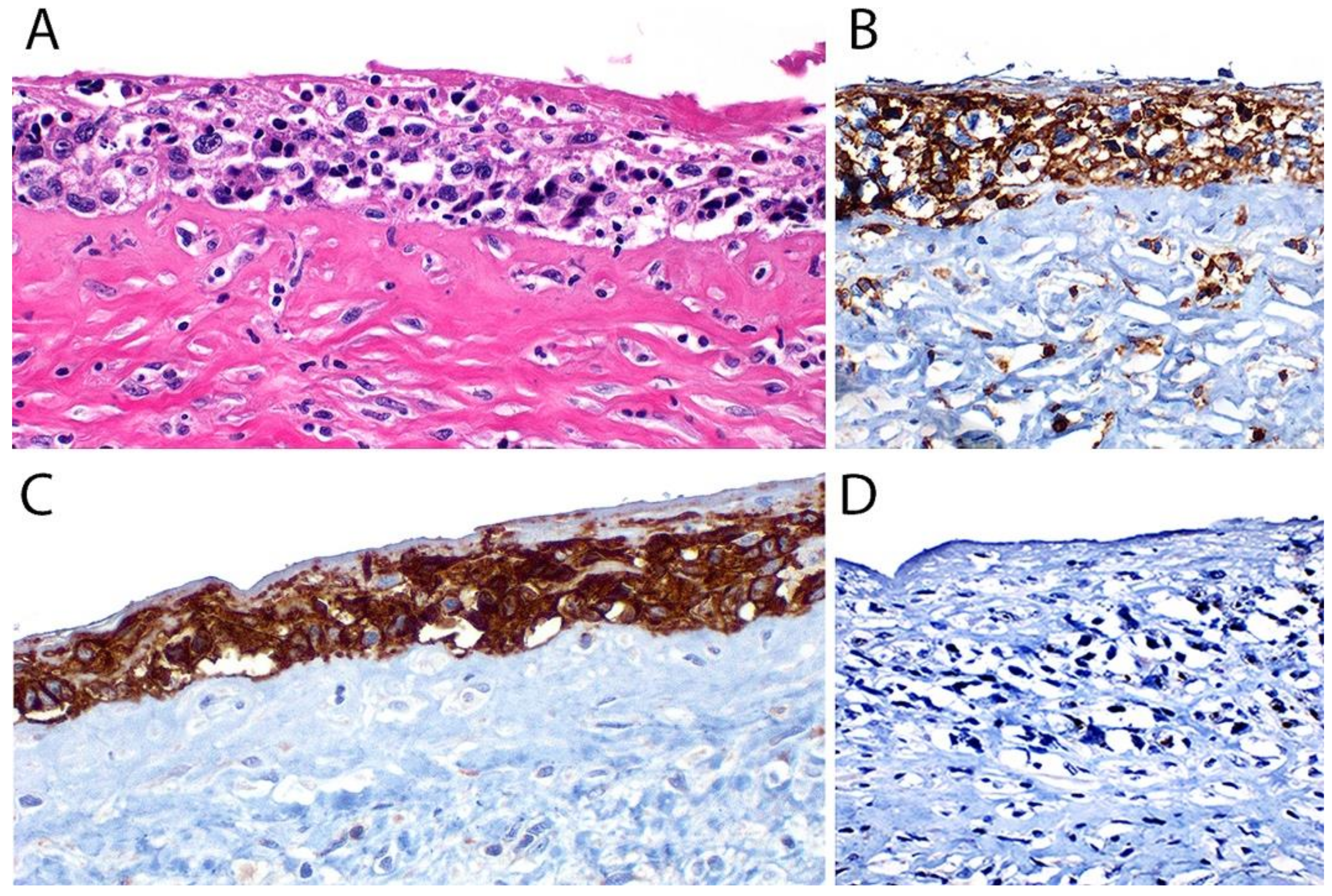{kind=link}
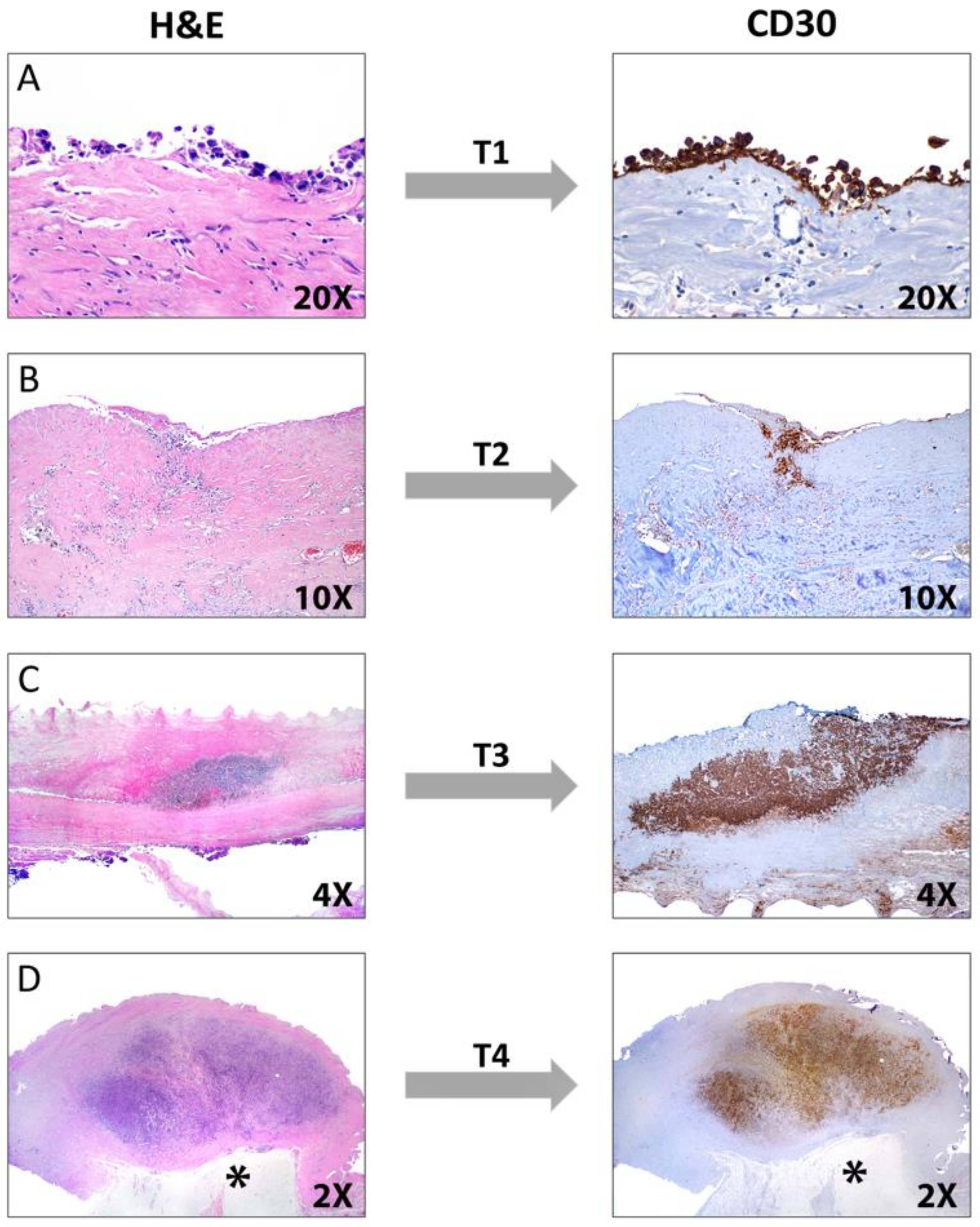{kind=link}
| Entity | Morphologic Features | CD30 | CD15 | CD4 | CD8 | Other T-Cell Markers: CD2, CD3, CD5, CD7 | Cytotoxic Markers:Granzyme B, TIA-1, Perforin | MUM1 | ALK | PAX5 | EBER ISH | EMA, Other |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ALK- ALCL | Hallmark cells Pleomorphism Sinusoidal pattern May show starry-sky pattern DUSP22-rearranged: >Doughnut cells <Pleomorphism | + (>75% of cells) | Usually -rarely + | Usually + | Usually - | Variable for all | Usually + except in DUSP22-rearranged cases | + | - | Usually -Very rarely + (PAX5 extracopies) | - | Variable + EMA (40%) |
| ALK+ ALCL | Hallmark cells Pleomorphism Doughnut cells Sinusoidal pattern | + (>75% of cells) | - Very rarely + | Usually + | Usually - | Variable for all | Usually + | + | + variable patterns | Usually -Very rarely + (PAX5 extracopies) | - | Usually + EMA (>80%) |
| Large cell PTCL, NOS | Usually no hallmark cells Variable pleomorphism | Variable <75% of cells | Usually -rarely + | Variable | Variable | Variable | Usually - unless CD8+ | Variable | - | - | - Rarely + | - EMA If CD30+ difficult to distinguish from ALK- ALCL |
| CHL | Sclerosis and cellular nodules Polymorphic infiltrate Usually no hallmark cells Usually no sinusoidal patternSolid growth in syncytial variant | + | Usually + (60–70%) | Usually – Few + | Usually – Few + | Usually – Few variable | Variable | + | - | Dim + | Variable NS-CHL (20%) MC-CHL (80%) | - EMA |
| Anaplastic LBCL | Similar to ALCL, usually no hallmark cells May show sinusoidal pattern | + (30% of cases) | - | - | - | - | - | Variable | - unless ALK+ LBCL (cytoplasmic granular) | + Other B-cell markers + | - unless EBV+ LBCL | - EMA unless ALK+ LBCL B-cell markers variable or–in ALK+ LBCL |
| Myeloid/monocytic sarcoma | Usually no hallmark cells Variable pleomorphism May show starry-sky pattern | Usually - | + | + if monocytic | - | - | Variable | - | - | - + if t(8;21) | - | - EMA + CD13, CD14, CD64, MPO, lysozyme |
| Histiocytic sarcoma | Usually no hallmark cells Variable pleomorphism Usually no sinusoidal pattern | - | Variable | + | - | - | Variable | - | - | - | - | - EMA + monocyte/ macrophagemarkers CD68, CD163 |
| Entity | Morphologic Features | CD43/ CD45 | CD30 | T-Cell Markers | ALK | CKs | p63 | EMA | Melanoma Markers: S100, MART1, HBM-45, SOX10 | OCT3/4, SALL4 | INI1/SMARCB1 | Desmin | Vascular Markers, Other |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ALK- ALCL | Hallmark cells PleomorphismSinusoidal pattern May show starry-sky pattern DUSP22-rearranged: >Doughnut cells <Pleomorphism | Usually + Few - | + | Variable, usually CD4+ All may be –(null phenotype) | - | - Very rarely + | Variable, + in TP63-rearranged cases | Variable + EMA (40%) | - | - | Retained | - | - |
| Carcinoma | May show hallmark-like cells Variable pleomorphism Sinusoidal pattern | - | - | - | - | + | + if squamous cell carcinoma | + | - | - | Retained | - | - Other markers may be + depending on subtype: TTF1, PAX8 |
| Embryonal carcinoma | May show hallmark-like cells Variable pleomorphism Pseudoglandular, alveolar, solid patterns | - | + | - | - | + | - | + | - | + | Retained | - | - |
| Melanoma | May show hallmark-like cells Variable pleomorphism Sinusoidal pattern | - | - Very rarely + | - | - | - | - | - | + | - | Retained | - | - |
| Epithelioid sarcoma, other rhabdoid tumors | May show hallmark-like cells Variable pleomorphism Nodular and solid growth, central necrosis Subcutaneous, soft tissue mass | - | - | - | - | + | - | + | - | - | Loss | - | + ERG |
| Alveolar soft part sarcoma | May show hallmark-like cells Variable pleomorphism Nodular and alveolar growthSoft tissue mass | - | - | - | - | - | - | - | - | - | Retained | - | - + TFE3 and cathepsin K |
| Epithelioid myofibroblastic sarcoma | Variable pleomorphism Intraabdominal mass Very rare | - | + | - | + nuclear membrane or perinucleolar | - | - | - | - | - | N/A | + | - - caldesmon variable + SMA |
| Entity | Main Clinical and Histopathologic Features |
|---|---|
| Systemic ALK+ ALCL with cutaneous involvement | Cutaneous involvement occurs in up to 60% of cases Peripheral lymph nodes and extranodal sites also affected CD30+, ALK+, EMA+ (strong) ALK gene rearrangement |
| Systemic ALK- ALCL with cutaneous involvement | Peripheral lymph nodes and/or extranodal sites affected Morphologically and immunophenotypically indistinguishable from pc-ALCL |
| MF with large cell transformation | Clinical history of MF with patches, plaques, and tumors Histological characteristics of MF may be present pan T-cell antigens +, may be CD7- and/or CD5- CD30 may be variable +, GATA3+ |
| Classic Hodgkin lymphoma with cutaneous involvement | Peripheral lymphadenopathy, very rarely presents with skin involvement PAX5+ (weak), often CD15+, lack of CD45 Often Epstein-Barr virus + (EBER ISH, LMP-1) T-cell markers usually - |
| Lymphomatoid papulosis (LyP) type C | Multiple papular and papulonecrotic eruptions < 10 mm History of spontaneous regression favors LyP Morphologically and immunophenotypically indistinguishable from pc-ALCL |
| PTCL, NOS, with cutaneous involvement | B symptoms and evidence of disseminated lymphadenopathy Very rarely presents with skin involvement CD30- or weakly + |
| Subcutaneous panniculitis-like TCL | Median age: 35 years, F > M Infiltrate confined to subcutaneous tissue with fat necrosis; dermis and epidermis typically uninvolved CD30-, CD8+, cytotoxic proteins + |
| Primary cutaneous gamma-delta TCL | Common hemophagocytic syndrome Three histologic patterns: epidermotropic, dermal, and subcutaneous Apoptosis, necrosis, and angioinvasion present TCRγ/δ+, CD2+, CD5-, CD7+/-, CD56+, cytotoxic proteins +, CD30 variable |
| Primary cutaneous CD8+ aggressive epidermotropic cytotoxic TCL | Extensive ulcerated cutaneous nodules with an aggressive clinical course Epidermotropic proliferation with pagetoid pattern Ulceration, necrosis, and angioinvasion CD3+, CD8+, cytotoxic proteins +, TCRαβ+, CD2-, CD30-, high Ki-67 |
| Primary cutaneous CD4+ small/medium T-cell lymphoproliferative disorder | Solitary plaque or nodule in face, neck, or upper trunk <30% of large cells CD3+, CD4+, cytotoxic proteins -, CD30- TFH phenotype: PDL-1 (CD279)+, bcl6+, CXCL13+ |
| Genetic Alteration | Frequency |
|---|---|
| Clonal T-cell receptor gamma genes | ~90% |
| ALK and t(2;5) translocation | Absent in 80–90% |
| Gains of 7q31, 17q, and 21 Losses of 3p, 6q16–6q21, 6q27, 8p, and 13q34 | ~50% |
| Rearrangement of the DUSP22-IRF4 locus on 6p25.3 | ~25% Detection of 6p25.3 rearrangement by fluorescence in situ hybridization is highly specific for pc-ALCL (specificity of 99%) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pina-Oviedo, S.; Ortiz-Hidalgo, C.; Carballo-Zarate, A.A.; Zarate-Osorno, A. ALK-Negative Anaplastic Large Cell Lymphoma: Current Concepts and Molecular Pathogenesis of a Heterogeneous Group of Large T-Cell Lymphomas. Cancers 2021, 13, 4667. https://doi.org/10.3390/cancers13184667
Pina-Oviedo S, Ortiz-Hidalgo C, Carballo-Zarate AA, Zarate-Osorno A. ALK-Negative Anaplastic Large Cell Lymphoma: Current Concepts and Molecular Pathogenesis of a Heterogeneous Group of Large T-Cell Lymphomas. Cancers. 2021; 13(18):4667. https://doi.org/10.3390/cancers13184667
Chicago/Turabian StylePina-Oviedo, Sergio, Carlos Ortiz-Hidalgo, Adrian Alejandro Carballo-Zarate, and Alejandra Zarate-Osorno. 2021. "ALK-Negative Anaplastic Large Cell Lymphoma: Current Concepts and Molecular Pathogenesis of a Heterogeneous Group of Large T-Cell Lymphomas" Cancers 13, no. 18: 4667. https://doi.org/10.3390/cancers13184667
APA StylePina-Oviedo, S., Ortiz-Hidalgo, C., Carballo-Zarate, A. A., & Zarate-Osorno, A. (2021). ALK-Negative Anaplastic Large Cell Lymphoma: Current Concepts and Molecular Pathogenesis of a Heterogeneous Group of Large T-Cell Lymphomas. Cancers, 13(18), 4667. https://doi.org/10.3390/cancers13184667