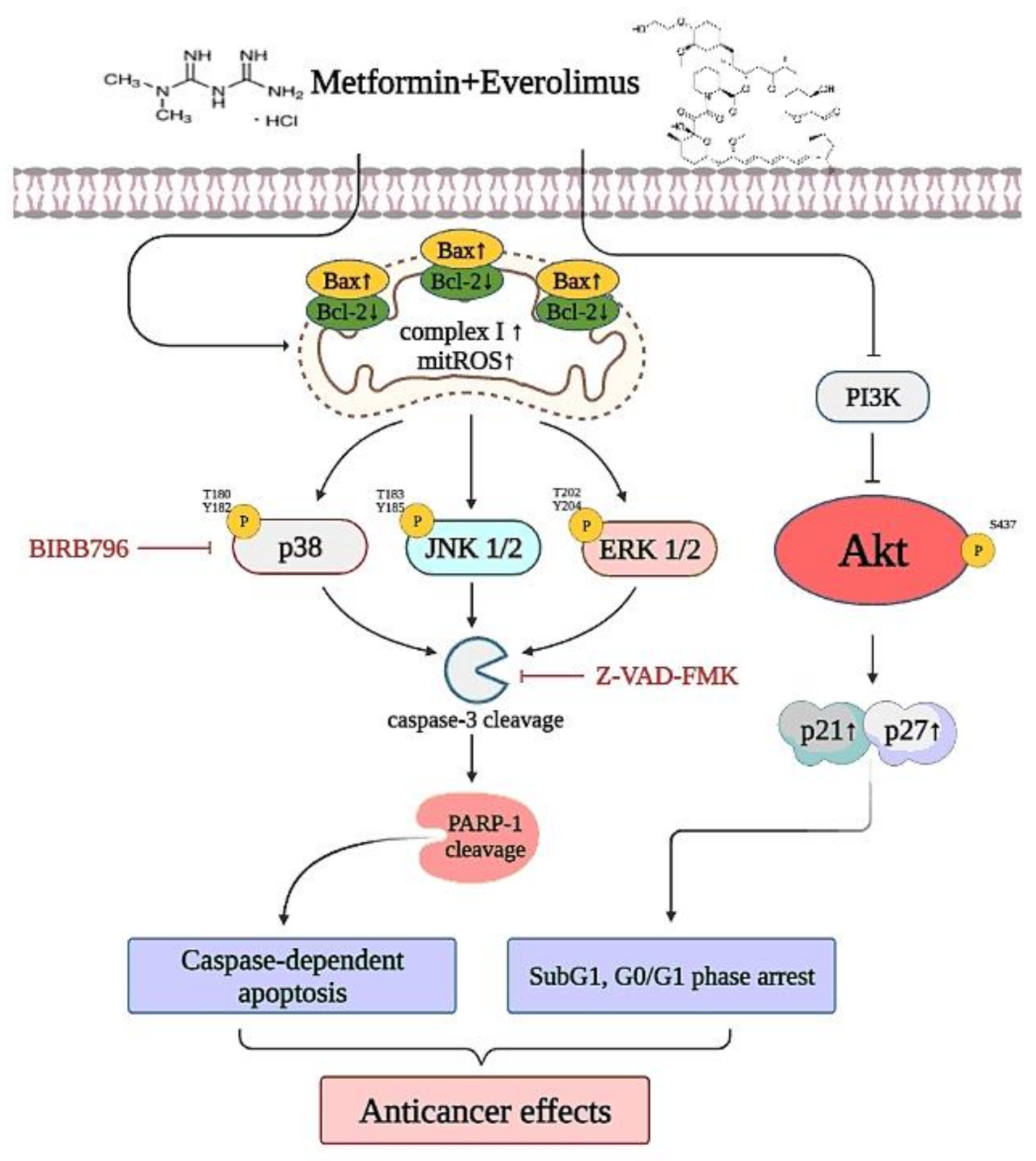
Metformin Potentiates the Anticancer Effect of Everolimus on Cervical Cancer In Vitro and In Vivo


 ,
,
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines and Culture Conditions
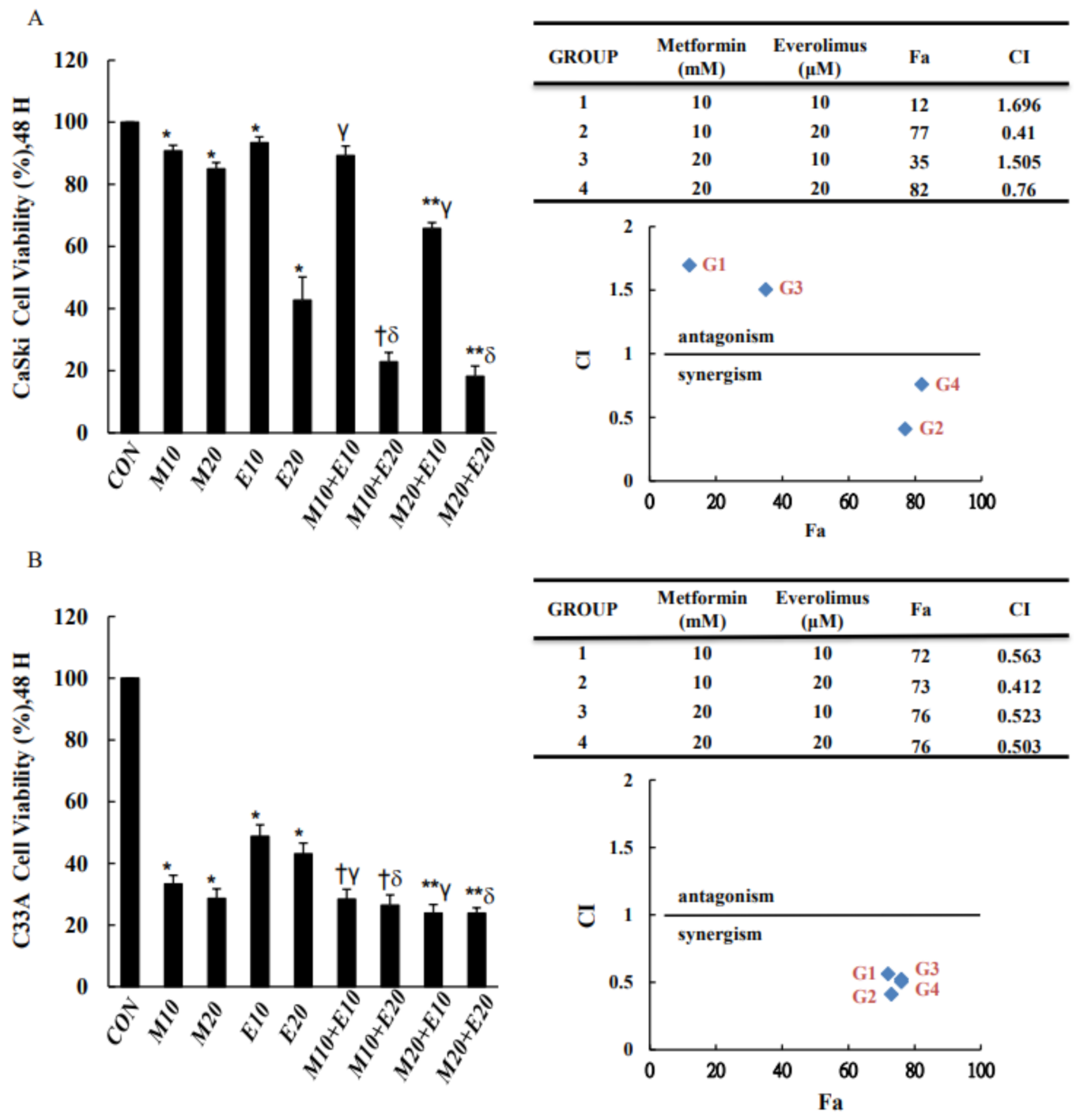
2.2. CCK-8 Assay and Analysis of Drug Synergy
2.3. Cell-Cycle and Apoptosis Analysis
2.4. Mitochondrial ROS Measurement
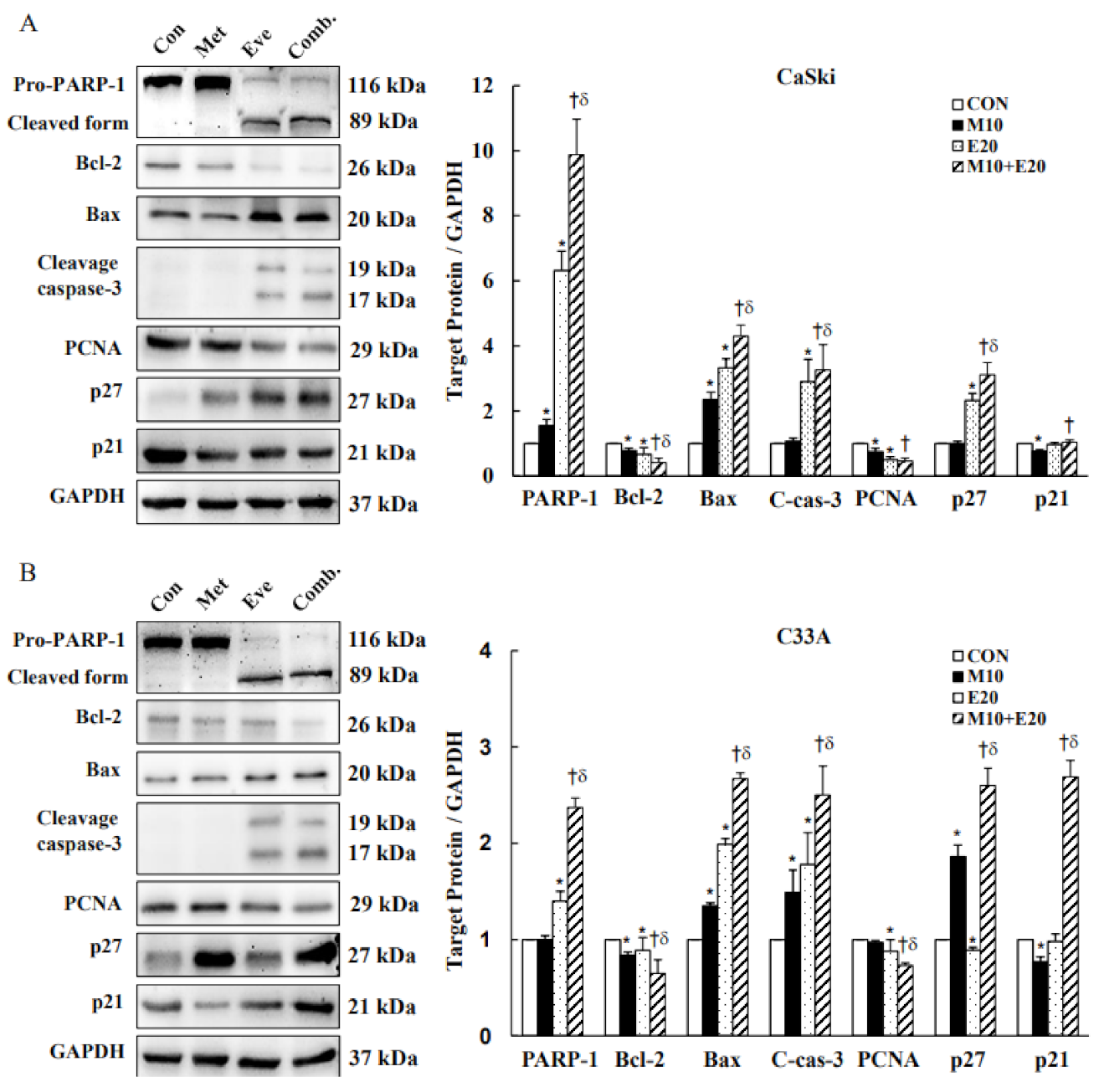
2.5. Western Blot Analysis
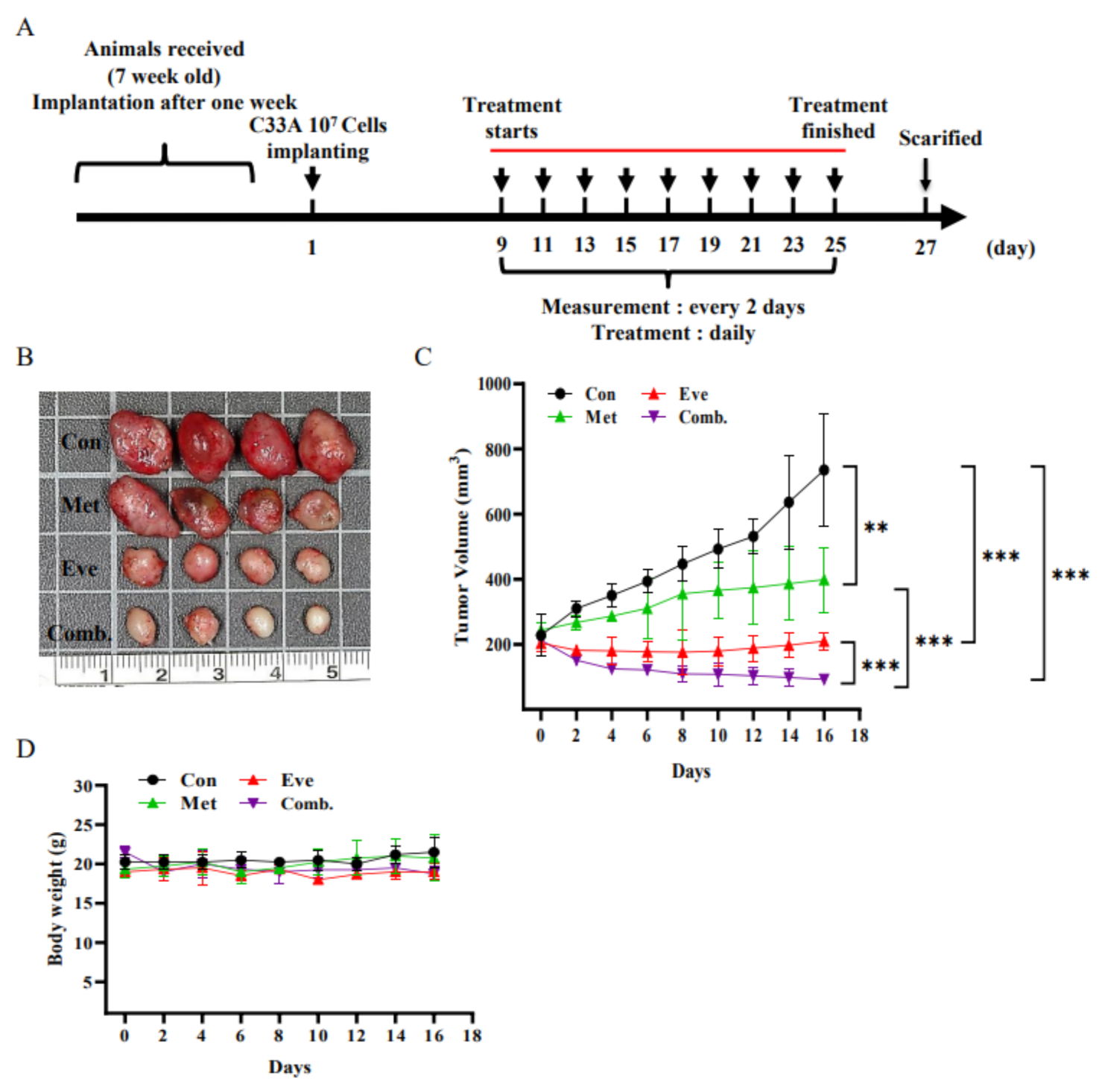
2.6. C33A Tumor Xenograft Model
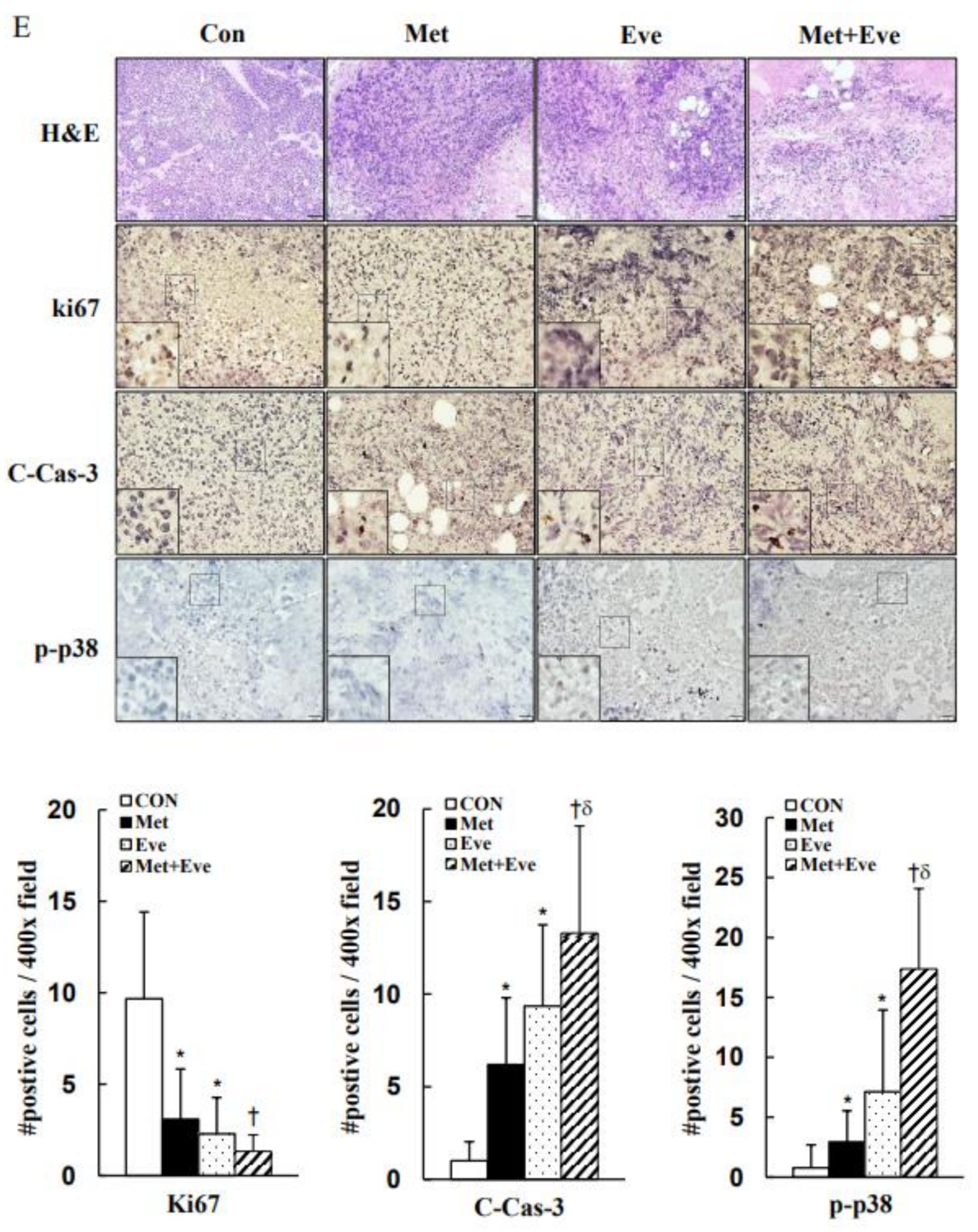
2.7. Histopathology and Immunohistochemistry
2.8. Statistical Analysis
3. Results
3.1. Combined Treatment with Metformin and Everolimus Is More Effective than Either Treatment Alone in Inhibiting Cell Viability
3.2. Combined Treatment with Metformin and Everolimus Is More Effective than Either Treatment Alone in Promoting Cell Apoptosis and Cell-Cycle Arrest
3.3. Combined Treatment with Metformin and Everolimus Synergistically Enhances Mitochondrial ROS and OXPHOS
3.4. Combined Treatment with Metformin and Everolimus Activates Mitochondrial and Caspase-Mediated Apoptosis
3.5. Combined Treatment with Metformin and Everolimus Synergistically Inhibits PI3K(p110α)/AKT Signaling and Activates MAPKs Signaling in Cervical Cancer Cells
3.6. Activations of Caspase-3 and p38 MAPK Are Involved in Metformin- and Everolimus-Induced Apoptosis in Human Cervical Cancer
3.7. Combined Treatment with Metformin and Everolimus Synergistically Suppresses the Growth of C33A Xenograft Tumors
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Arbyn, M.; Weiderpass, E.; Bruni, L.; de Sanjosé, S.; Saraiya, M.; Ferlay, J.; Bray, F. Estimates of incidence and mortality of cervical cancer in 2018: A worldwide analysis. Lancet Glob. Health 2020, 8, e191–e203. [Google Scholar] [CrossRef]
- Bethesda. SEER Cancer Stat Facts: Cervical Cancer. National Cancer Institute. Available online: https://seer.cancer.gov/statfacts/html/cervix.html (accessed on 6 July 2021).
- Global Cancer Observatory. Cancer Tomorrow: A Tool that Predicts the Future Cancer Incidence and Mortality Burden Worldwide from the Current Estimates in 2018 up until 2040. International Agency for Research on Cancer, World Health Organization. 2018. Available online: http://gco.iarc.fr/tomorrow (accessed on 2 October 2020).
- Vizcaino, A.P.; Moreno, V.; Bosch, F.X.; Muñoz, N.; Barros-Dios, X.M.; Parkin, D.M. International trends in the incidence of cervical cancer: I. Adenocarcinoma and adenosquamous cell carcinomas. Int. J. Cancer 1998, 75, 536–545. [Google Scholar] [CrossRef]
- Vizcaino, A.P.; Moreno, V.; Bosch, F.X.; Munoz, N.; Barros-Dios, X.M.; Borras, J.; Parkin, D.M. International trends in incidence of cervical cancer: II. Squamous-cell carcinoma. Int. J. Cancer 2000, 86, 429–435. [Google Scholar] [CrossRef]
- Vinh-Hung, V.; Bourgain, C.; Vlastos, G.; Cserni, G.; De Ridder, M.; Storme, G.; Vlastos, A.-T. Prognostic value of histopathology and trends in cervical cancer: A SEER population study. BMC Cancer 2007, 7, 164. [Google Scholar] [CrossRef]
- Donadon, V.; Balbi, M.; Mas, M.D.; Casarin, P.; Zanette, G. Metformin and reduced risk of hepatocellular carcinoma in diabetic patients with chronic liver disease. Liver Int. 2010, 30, 750–758. [Google Scholar] [CrossRef]
- Kim, H.J.; Kwon, H.; Lee, J.W.; Kim, H.J.; Lee, S.B.; Park, H.S.; Sohn, G.; Lee, Y.; Koh, B.S.; Yu, J.H. Metformin increases survival in hormone receptor-positive, HER2-positive breast cancer patients with diabetes. Breast Cancer Res. 2015, 17, 64. [Google Scholar] [CrossRef]
- Kong, F.; Gao, F.; Liu, H.; Chen, L.; Zheng, R.; Yu, J.; Li, X.; Liu, G.; Jia, Y. Metformin use improves the survival of diabetic combined small-cell lung cancer patients. Tumor Biol. 2015, 36, 8101–8106. [Google Scholar] [CrossRef]
- Hanprasertpong, J.; Jiamset, I.; Geater, A.; Peerawong, T.; Hemman, W.; Kornsilp, S. The effect of metformin on oncological outcomes in patients with cervical cancer with type 2 diabetes mellitus. Int. J. Gynecol. Cancer 2017, 27, 131–137. [Google Scholar] [CrossRef]
- Takiuchi, T.; Machida, H.; Hom, M.S.; Mostofizadeh, S.; Frimer, M.; Brunette, L.L.; Matsuo, K. Association of metformin use and survival outcome in women with cervical cancer. Int. J. Gynecol. Cancer 2017, 27, 1455–1463. [Google Scholar] [CrossRef]
- Vazquez-Martin, A.; Oliveras-Ferraros, C.; Menendez, J.A. The antidiabetic drug metformin suppresses HER2 (erbB-2) oncoprotein overexpression via inhibition of the mTOR effector p70S6K1 in human breast carcinoma cells. Cell Cycle 2009, 8, 88–96. [Google Scholar] [CrossRef]
- Shi, W.; Xiao, D.; Wang, L.; Dong, L.; Yan, Z.; Shen, Z.; Chen, S.; Chen, Y.; Zhao, W. Therapeutic metformin/AMPK activation blocked lymphoma cell growth via inhibition of mTOR pathway and induction of autophagy. Cell Death Dis. 2012, 3, e275. [Google Scholar] [CrossRef]
- Wang, C.-F.; Zhang, G.; Zhao, L.-J.; Qi, W.-J.; Li, X.-P.; Wang, J.-L.; Wei, L.-H. Overexpression of the insulin receptor isoform A promotes endometrial carcinoma cell growth. PLoS ONE 2013, 8, e69001. [Google Scholar] [CrossRef]
- Ling, S.; Xie, H.; Yang, F.; Shan, Q.; Dai, H.; Zhuo, J.; Wei, X.; Song, P.; Zhou, L.; Xu, X. Metformin potentiates the effect of arsenic trioxide suppressing intrahepatic cholangiocarcinoma: Roles of p38 MAPK, ERK3, and mTORC1. J. Hematol. Oncol. 2017, 10, 59. [Google Scholar] [CrossRef]
- He, Y.; Cao, L.; Wang, L.; Liu, L.; Huang, Y.; Gong, X. Metformin inhibits proliferation of human thyroid cancer TPC-1 cells by decreasing LRP2 to suppress the JNK pathway. OncoTargets Ther. 2020, 13, 45. [Google Scholar] [CrossRef]
- Saber, S.; Ghanim, A.M.; El-Ahwany, E.; Abd El-Kader, E.M. Novel complementary antitumour effects of celastrol and metformin by targeting IκBκB, apoptosis and NLRP3 inflammasome activation in diethylnitrosamine-induced murine hepatocarcinogenesis. Cancer Chemother. Pharmacol. 2020, 85, 331–343. [Google Scholar] [CrossRef]
- Martin-Castillo, B.; Vazquez-Martin, A.; Oliveras-Ferraros, C.; Menendez, J.A. Metformin and cancer: Doses, mechanisms and the dandelion and hormetic phenomena. Cell Cycle 2010, 9, 1057–1064. [Google Scholar] [CrossRef]
- Dhillon, S.S.; Groman, A.; Meagher, A.; Demmy, T.; Warren, G.W.; Yendamuri, S. Metformin and not diabetes influences the survival of resected early stage NSCLC patients. J. Cancer Sci. Ther. 2014, 6, 217. [Google Scholar]
- Sayed, R.; Saad, A.S.; El Wakeel, L.; Elkholy, E.; Badary, O. Metformin addition to chemotherapy in stage IV non-small cell lung cancer: An open label randomized controlled study. Asian Pac. J. Cancer Prev. 2015, 16, 6621–6626. [Google Scholar] [CrossRef]
- Alcusky, M.; Keith, S.W.; Karagiannis, T.; Rabinowitz, C.; Louis, D.Z.; Maio, V. Metformin exposure and survival in head and neck cancer: A large population-based cohort study. J. Clin. Pharm. Ther. 2019, 44, 588–594. [Google Scholar] [CrossRef]
- Tyszka-Czochara, M.; Lasota, M.; Majka, M. Caffeic acid and metformin inhibit invasive phenotype induced by TGF-β1 in C-4I and HTB-35/SiHa human cervical squamous carcinoma cells by acting on different molecular targets. Int. J. Mol. Sci. 2018, 19, 266. [Google Scholar] [CrossRef]
- Xia, C.; Chen, R.; Chen, J.; Qi, Q.; Pan, Y.; Du, L.; Xiao, G.; Jiang, S. Combining metformin and nelfinavir exhibits synergistic effects against the growth of human cervical cancer cells and xenograft in nude mice. Sci. Rep. 2017, 7, 43373. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.Y.; Gulinazi, Y.; Du, Y.; Ning, C.C.; Cheng, Y.L.; Shan, W.W.; Luo, X.Z.; Zhang, H.W.; Zhu, Q.; Ma, F.H. Metformin plus megestrol acetate compared with megestrol acetate alone as fertility-sparing treatment in patients with atypical endometrial hyperplasia and well-differentiated endometrial cancer: A randomised controlled trial. BJOG Int. J. Obstet. Gynaecol. 2020, 127, 848–857. [Google Scholar] [CrossRef] [PubMed]
- Bragagnoli, A.C.; Araujo, R.L.; Ferraz, M.W.; Dos Santos, L.V.; Abdalla, K.C.; Comar, F.; Santos, F.A.; Oliveira, M.A.; Carvalheira, J.B.C.; Cárcano, F.M. Metformin plus lrinotecan in patients with refractory colorectal cancer: A phase 2 clinical trial. Br. J. Cancer 2021, 124, 1072–1078. [Google Scholar] [CrossRef] [PubMed]
- Kordes, S.; Pollak, M.N.; Zwinderman, A.H.; Mathôt, R.A.; Weterman, M.J.; Beeker, A.; Punt, C.J.; Richel, D.J.; Wilmink, J.W. Metformin in patients with advanced pancreatic cancer: A double-blind, randomised, placebo-controlled phase 2 trial. Lancet Oncol. 2015, 16, 839–847. [Google Scholar] [CrossRef]
- Zheng, Y.; Zhu, J.; Zhang, H.; Liu, Y.; Sun, H. Metformin plus first-line chemotherapy versus chemotherapy alone in the treatment of epithelial ovarian cancer: A prospective open-label pilot trial. Cancer Chemother. Pharmacol. 2019, 84, 1349–1357. [Google Scholar] [CrossRef]
- Reni, M.; Dugnani, E.; Cereda, S.; Belli, C.; Balzano, G.; Nicoletti, R.; Liberati, D.; Pasquale, V.; Scavini, M.; Maggiora, P. (Ir) relevance of metformin treatment in patients with metastatic pancreatic cancer: An open-label, randomized phase II trial. Clin. Cancer Res. 2016, 22, 1076–1085. [Google Scholar] [CrossRef]
- Alghandour, R.; Ebrahim, M.A.; Elshal, A.M.; Ghobrial, F.; Elzaafarany, M.; ELbaiomy, M.A. Repurposing metformin as anticancer drug: Randomized controlled trial in advanced prostate cancer (MANSMED). In Urologic Oncology: Seminars and Original Investigations, 2021; Elsevier: Amsterdam, The Netherlands, 2021. [Google Scholar]
- Nanni, O.; Amadori, D.; De Censi, A.; Rocca, A.; Freschi, A.; Bologna, A.; Gianni, L.; Rosetti, F.; Amaducci, L.; Cavanna, L. Metformin plus chemotherapy versus chemotherapy alone in the first-line treatment of HER2-negative metastatic breast cancer. The MYME randomized, phase 2 clinical trial. Breast Cancer Res. Treat. 2019, 174, 433–442. [Google Scholar] [CrossRef]
- Hasskarl, J. Everolimus. Small Mol. Oncol. 2018, 211, 101–123. [Google Scholar]
- Karvelas, G.; Roumpi, A.; Komporozos, C.; Syrigos, K. Everolimus as cancer therapy: Cardiotoxic or an unexpected antiatherogenic agent? A narrative review. Hell. J. Cardiol. 2018, 59, 196–200. [Google Scholar] [CrossRef]
- Taylor, S.E.; Chu, T.; Elvin, J.A.; Edwards, R.P.; Zorn, K.K. Phase II study of everolimus and bevacizumab in recurrent ovarian, peritoneal, and fallopian tube cancer. Gynecol. Oncol. 2020, 156, 32–37. [Google Scholar] [CrossRef]
- Pascual, T.; Apellániz-Ruiz, M.; Pernaut, C.; Cueto-Felgueroso, C.; Villalba, P.; Álvarez, C.; Manso, L.; Inglada-Pérez, L.; Robledo, M.; Rodríguez-Antona, C. Polymorphisms associated with everolimus pharmacokinetics, toxicity and survival in metastatic breast cancer. PLoS ONE 2017, 12, e0180192. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wei, J.; Li, L.; Fan, C.; Sun, Y. Combined use of metformin and everolimus is synergistic in the treatment of breast cancer cells. Oncol. Res. 2015, 22, 193. [Google Scholar] [CrossRef]
- Zhang, H.-H.; Guo, X.-L. Combinational strategies of metformin and chemotherapy in cancers. Cancer Chemother. Pharmacol. 2016, 78, 13–26. [Google Scholar] [CrossRef]
- Pusceddu, S.; De Braud, F.; Concas, L.; Bregant, C.; Leuzzi, L.; Formisano, B.; Buzzoni, R. Rationale and protocol of the MetNET-1 trial, a prospective, single center, phase II study to evaluate the activity and safety of everolimus in combination with octreotide LAR and metformin in patients with advanced pancreatic neuroendocrine tumors. Tumori J. 2014, 100, e286–e289. [Google Scholar]
- Chen, Y.-H.; Yang, S.-F.; Yang, C.-K.; Tsai, H.-D.; Chen, T.-H.; Chou, M.-C.; Hsiao, Y.-H. Metformin induces apoptosis and inhibits migration by activating the AMPK/p53 axis and suppressing PI3K/AKT signaling in human cervical cancer cells. Mol. Med. Rep. 2021, 23, 88. [Google Scholar] [CrossRef]
- Chen, Y.H.; Huang, Y.C.; Yang, S.F.; Yen, H.H.; Tsai, H.D.; Hsieh, M.C.; Hsiao, Y.H. Pitavastatin and metformin synergistically activate apoptosis and autophagy in pancreatic cancer cells. Environ. Toxicol. 2021, 36, 1491–1503. [Google Scholar] [CrossRef]
- Zakikhani, M.; Dowling, R.; Fantus, I.G.; Sonenberg, N.; Pollak, M. Metformin is an AMP kinase–dependent growth inhibitor for breast cancer cells. Cancer Res. 2006, 66, 10269–10273. [Google Scholar] [CrossRef]
- Zhang, J.W.; Zhao, F.; Sun, Q. Metformin synergizes with rapamycin to inhibit the growth of pancreatic cancer in vitro and in vivo. Oncol. Lett. 2018, 15, 1811–1816. [Google Scholar] [CrossRef]
- Dong, S.; Zhang, X.-C.; Cheng, H.; Zhu, J.-Q.; Chen, Z.-H.; Zhang, Y.-F.; Xie, Z.; Wu, Y.-L. Everolimus synergizes with gefitinib in non-small-cell lung cancer cell lines resistant to epidermal growth factor receptor tyrosine kinase inhibitors. Cancer Chemother. Pharmacol. 2012, 70, 707–716. [Google Scholar] [CrossRef] [PubMed]
- Chou, T.-C.; Talalay, P. Quantitative analysis of dose-effect relationships: The combined effects of multiple drugs or enzyme inhibitors. Adv. Enzym. Regul. 1984, 22, 27–55. [Google Scholar] [CrossRef]
- Esposito, K.; Chiodini, P.; Colao, A.; Lenzi, A.; Giugliano, D. Metabolic syndrome and risk of cancer: A systematic review and meta-analysis. Diabetes Care 2012, 35, 2402–2411. [Google Scholar] [CrossRef] [PubMed]
- Esposito, K.; Chiodini, P.; Capuano, A.; Bellastella, G.; Maiorino, M.I.; Giugliano, D. Metabolic syndrome and endometrial cancer: A meta-analysis. Endocrine 2014, 45, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Tao, M.; Zhao, L.; Zhang, X. The association between diabetes/hyperglycemia and the prognosis of cervical cancer patients: A systematic review and meta-analysis. Medicine 2017, 96, e7981. [Google Scholar] [CrossRef] [PubMed]
- Gillani, S.W.; Zaghloul, H.A.; Ansari, I.A.; Abdul, M.I.M.; Sulaiman, S.A.S.; Baig, M.R.; Rathore, H.A. Multivariate analysis on the effects of diabetes and related clinical parameters on cervical cancer survival probability. Sci. Rep. 2019, 9, 1084. [Google Scholar] [CrossRef]
- Peng, M.; Darko, K.O.; Tao, T.; Huang, Y.; Su, Q.; He, C.; Yin, T.; Liu, Z.; Yang, X. Combination of metformin with chemotherapeutic drugs via different molecular mechanisms. Cancer Treat. Rev. 2017, 54, 24–33. [Google Scholar] [CrossRef]
- Liu, H.; Scholz, C.; Zang, C.; Schefe, J.H.; Habbel, P.; Regierer, A.-C.; Schulz, C.-O.; Possinger, K.; Eucker, J. Metformin and the mTOR inhibitor everolimus (RAD001) sensitize breast cancer cells to the cytotoxic effect of chemotherapeutic drugs in vitro. Anticancer Res. 2012, 32, 1627–1637. [Google Scholar]
- Li, S.-M.H.; Liu, S.-T.; Chang, Y.-L.; Ho, C.-L.; Huang, S.-M. Metformin causes cancer cell death through downregulation of p53-dependent differentiated embryo chondrocyte 1. J. Biomed. Sci. 2018, 25, 81. [Google Scholar]
- Xiao, X.; He, Q.; Lu, C.; Werle, K.D.; Zhao, R.-X.; Chen, J.; Davis, B.C.; Cui, R.; Liang, J.; Xu, Z.-X. Metformin impairs the growth of liver kinase B1-intact cervical cancer cells. Gynecol. Oncol. 2012, 127, 249–255. [Google Scholar] [CrossRef]
- Zhou, X.; Xue, Y.; Zhu, B.; Sha, J. Effects of metformin on proliferation of human colon carcinoma cell line SW-480. J. South. Med. Univ. 2010, 30, 1935–1938. [Google Scholar]
- Zhou, X.; Kuang, Y.; Liang, S.; Wang, L. Metformin inhibits cell proliferation in SKM-1 cells via AMPK-mediated cell cycle arrest. J. Pharmacol. Sci. 2019, 141, 146–152. [Google Scholar] [CrossRef]
- Zhao, B.; Luo, J.; Wang, Y.; Zhou, L.; Che, J.; Wang, F.; Peng, S.; Zhang, G.; Shang, P. Metformin suppresses self-renewal ability and tumorigenicity of osteosarcoma stem cells via reactive oxygen species-mediated apoptosis and autophagy. Oxidative Med. Cell. Longev. 2019. [Google Scholar] [CrossRef]
- Kamarudin, M.N.A.; Sarker, M.M.R.; Zhou, J.-R.; Parhar, I. Metformin in colorectal cancer: Molecular mechanism, preclinical and clinical aspects. J. Exp. Clin. Cancer Res. 2019, 38, 491. [Google Scholar] [CrossRef]
- Du, L.; Li, X.; Zhen, L.; Chen, W.; Mu, L.; Zhang, Y.; Song, A. Everolimus inhibits breast cancer cell growth through PI3K/AKT/mTOR signaling pathway. Mol. Med. Rep. 2018, 17, 7163–7169. [Google Scholar] [CrossRef]
- Alves, R.; Gonçalves, A.C.; Jorge, J.; Alves, J.; da Silva, A.A.; Freitas-Tavares, P.; Costa, J.M.N.; Almeida, A.M.; Sarmento-Ribeiro, A.B. Everolimus in combination with Imatinib overcomes resistance in Chronic myeloid leukaemia. Med. Oncol. 2019, 36, 30. [Google Scholar] [CrossRef]
- Marinelli, O.; Romagnoli, E.; Maggi, F.; Nabissi, M.; Amantini, C.; Morelli, M.B.; Santoni, M.; Battelli, N.; Santoni, G. Exploring treatment with Ribociclib alone or in sequence/combination with Everolimus in ER+ HER2− Rb wild-type and knock-down in breast cancer cell lines. BMC Cancer 2020, 20, 1119. [Google Scholar] [CrossRef]
- Peng, T.; Dou, Q.P. Everolimus Inhibits Growth of Gemcitabine-Resistant Pancreatic Cancer Cells via Induction of Caspase-Dependent Apoptosis and G2/M Arrest. J. Cell. Biochem. 2017, 118, 2722–2730. [Google Scholar] [CrossRef]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Payen, V.L.; Zampieri, L.X.; Porporato, P.E.; Sonveaux, P. Pro-and antitumor effects of mitochondrial reactive oxygen species. Cancer Metastasis Rev. 2019, 38, 189–203. [Google Scholar] [CrossRef]
- Li, B.; Zhou, P.; Xu, K.; Chen, T.; Jiao, J.; Wei, H.; Yang, X.; Xu, W.; Wan, W.; Xiao, J. Metformin induces cell cycle arrest, apoptosis and autophagy through ROS/JNK signaling pathway in human osteosarcoma. Int. J. Biol. Sci. 2020, 16, 74. [Google Scholar] [CrossRef]
- Imhoff, B.R.; Hansen, J.M. Extracellular redox status regulates Nrf2 activation through mitochondrial reactive oxygen species. Biochem. J. 2009, 424, 491–500. [Google Scholar] [CrossRef]
- Cahova, M.; Palenickova, E.; Dankova, H.; Sticova, E.; Burian, M.; Drahota, Z.; Cervinkova, Z.; Kucera, O.; Gladkova, C.; Stopka, P. Metformin prevents ischemia reperfusion-induced oxidative stress in the fatty liver by attenuation of reactive oxygen species formation. Am. J. Physiol. -Gastrointest. Liver Physiol. 2015, 309, G100–G111. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Lu, Q.-J.; Zhao, J.; Wu, G.-Y. Metformin inhibits growth of hepatocellular carcinoma cells by inducing apoptosis via mitochondrion-mediated pathway. Asian Pac. J. Cancer Prev. 2012, 13, 3275–3279. [Google Scholar] [CrossRef] [PubMed]
- El-Mir, M.-Y.; Nogueira, V.; Fontaine, E.; Avéret, N.; Rigoulet, M.; Leverve, X. Dimethylbiguanide inhibits cell respiration via an indirect effect targeted on the respiratory chain complex I. J. Biol. Chem. 2000, 275, 223–228. [Google Scholar] [CrossRef] [PubMed]
- Owen, M.R.; Doran, E.; Halestrap, A.P. Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem. J. 2000, 348, 607–614. [Google Scholar] [CrossRef]
- Bridges, H.R.; Jones, A.J.; Pollak, M.N.; Hirst, J. Effects of metformin and other biguanides on oxidative phosphorylation in mitochondria. Biochem. J. 2014, 462, 475–487. [Google Scholar] [CrossRef]
- Pecinova, A.; Drahota, Z.; Kovalcikova, J.; Kovarova, N.; Pecina, P.; Alan, L.; Zima, M.; Houstek, J.; Mracek, T. Pleiotropic effects of biguanides on mitochondrial reactive oxygen species production. Oxidative Med. Cell. Longev. 2017. [Google Scholar] [CrossRef]
- Pollak, M. Investigating metformin for cancer prevention and treatment: The end of the beginning. Cancer Discov. 2012, 2, 778–790. [Google Scholar] [CrossRef]
- Cadeddu, G.; Hervás-Morón, A.; Martín-Martín, M.; Pelari-Mici, L.; de Kirsch, K.Y.-C.; Hernández-Corrales, A.; Vallejo-Ocaña, C.; Sastre-Gallego, S.; Carrasco-Esteban, E.; Sancho-García, S. Metformin and statins: A possible role in high-risk prostate cancer. Rep. Pract. Oncol. Radiother. 2020, 25, 163–167. [Google Scholar] [CrossRef]
- Liu, Y.; Zhang, Y.; Jia, K.; Dong, Y.; Ma, W. Metformin inhibits the proliferation of A431 cells by modulating the PI3K/Akt signaling pathway. Exp. Ther. Med. 2015, 9, 1401–1406. [Google Scholar] [CrossRef][Green Version]
- Heckman-Stoddard, B.M.; Gandini, S.; Puntoni, M.; Dunn, B.K.; DeCensi, A.; Szabo, E. Repurposing old drugs to chemoprevention: The case of metformin. In Seminars in Oncology; Elsevier: Amsterdam, The Netherlands, 2016; pp. 123–133. [Google Scholar]
- Alshaker, H.; Wang, Q.; Böhler, T.; Mills, R.; Winkler, M.; Arafat, T.; Kawano, Y.; Pchejetski, D. Combination of RAD001 (everolimus) and docetaxel reduces prostate and breast cancer cell VEGF production and tumour vascularisation independently of sphingosine-kinase-1. Sci. Rep. 2017, 7, 3493. [Google Scholar] [CrossRef]
- Wu, N.; Gu, C.; Gu, H.; Hu, H.; Han, Y.; Li, Q. Metformin induces apoptosis of lung cancer cells through activating JNK/p38 MAPK pathway and GADD153. Neoplasma 2011, 58, 482–490. [Google Scholar] [CrossRef]
- Carracedo, A.; Ma, L.; Teruya-Feldstein, J.; Rojo, F.; Salmena, L.; Alimonti, A.; Egia, A.; Sasaki, A.T.; Thomas, G.; Kozma, S.C. Inhibition of mTORC1 leads to MAPK pathway activation through a PI3K-dependent feedback loop in human cancer. J. Clin. Investig. 2008, 118, 3065–3074. [Google Scholar] [CrossRef]
- Liu, H.; Yao, Y.; Zhang, J.; Li, J. MEK inhibition overcomes everolimus resistance in gastric cancer. Cancer Chemother. Pharmacol. 2020, 85, 1079–1087. [Google Scholar] [CrossRef]
- Tseng, S.-C.; Huang, Y.-C.; Chen, H.-J.; Chiu, H.-C.; Huang, Y.-J.; Wo, T.-Y.; Weng, S.-H.; Lin, Y.-W. Metformin-mediated downregulation of p38 mitogen-activated protein kinase-dependent excision repair cross-complementing 1 decreases DNA repair capacity and sensitizes human lung cancer cells to paclitaxel. Biochem. Pharmacol. 2013, 85, 583–594. [Google Scholar] [CrossRef]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disease | Combined Drug(s) | No. of Patients | Finding | Ref. |
|---|---|---|---|---|
| Endometrial cancer | Megestrol acetate compared with megestrol acetate alone | 150 | Significant in 102 atypical endometrial hyperplasia (AEH) patients (39.6 versus 20.4%, OR 2.56, p = 0.04). | [24] |
| Refractory colorectal cancer | lrinotecan | 41 | In the multivariate analysis, disease control at 12 weeks impacted overall survival HR 0.21, p = 0.001. | [25] |
| Pancreatic cancer | Gemcitabine, erlotinib | 121 | Overall survival at 6 months was 56.7% (95% CI 44.1–69.2) in the metformin group and 63.9% (51.9–75.9) in the placebo group (p = 0.41). | [26] |
| Epithelial ovarian cancer | Chemotherapy versus chemotherapy alone | 44 | The disease-free survival (DFS) and progression-free survival (PFS) of patients with metformin use versus without metformin use was 29 versus 26 months (p = 0.61) and 23 versus 21 months (p = 0.68), respectively. | [27] |
| Metastatic pancreatic cancer | (Ir)relevance of metformin treatment | 60 | Six-months progression-free survival (PFS-6) was 52% (95% CI 33–69) in the control group and 42% (24–59) in the metformin group (p = 0.61). | [28] |
| Prostate cancer | metformin plus standard of care or standard of care alone | 124 | Castration-resistant prostate cancer-free survival was improved with metformin (29 months (95% CI 25–33) versus 20 months (16–24); p = 0.01). | [29] |
| Breast cancer | Chemotherapy versus chemotherapy alone | 122 | Median Progression-free survival (PFS) was 9.4 months (95% CI 7.8–10.4) in with metformin intake group and 9.9 (7.4–11.5) in without metformin intake group (p = 0.651). | [30] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, Y.-H.; Wu, J.-X.; Yang, S.-F.; Chen, M.-L.; Chen, T.-H.; Hsiao, Y.-H. Metformin Potentiates the Anticancer Effect of Everolimus on Cervical Cancer In Vitro and In Vivo. Cancers 2021, 13, 4612. https://doi.org/10.3390/cancers13184612
Chen Y-H, Wu J-X, Yang S-F, Chen M-L, Chen T-H, Hsiao Y-H. Metformin Potentiates the Anticancer Effect of Everolimus on Cervical Cancer In Vitro and In Vivo. Cancers. 2021; 13(18):4612. https://doi.org/10.3390/cancers13184612
Chicago/Turabian StyleChen, Ya-Hui, Jyun-Xue Wu, Shun-Fa Yang, Mei-Ling Chen, Tze-Ho Chen, and Yi-Hsuan Hsiao. 2021. "Metformin Potentiates the Anticancer Effect of Everolimus on Cervical Cancer In Vitro and In Vivo" Cancers 13, no. 18: 4612. https://doi.org/10.3390/cancers13184612
APA StyleChen, Y.-H., Wu, J.-X., Yang, S.-F., Chen, M.-L., Chen, T.-H., & Hsiao, Y.-H. (2021). Metformin Potentiates the Anticancer Effect of Everolimus on Cervical Cancer In Vitro and In Vivo. Cancers, 13(18), 4612. https://doi.org/10.3390/cancers13184612