
Genomic Determinants of Homologous Recombination Deficiency across Human Cancers


Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Patients
2.2. Genomic Datasets of TCGA Patients
2.3. Association Analyses of Germline and Somatic Mutations Using Multivariate Regression Models
2.4. Association Analyses of Gene-Level CNV Using Multivariate Regression Models
2.5. Independent Contribution of Germline and Somatic Mutations
2.6. Co-Occurrence and Mutual Exclusivity of Germline and Somatic Mutations
2.7. Adjustment for Multiple Comparisons
3. Results
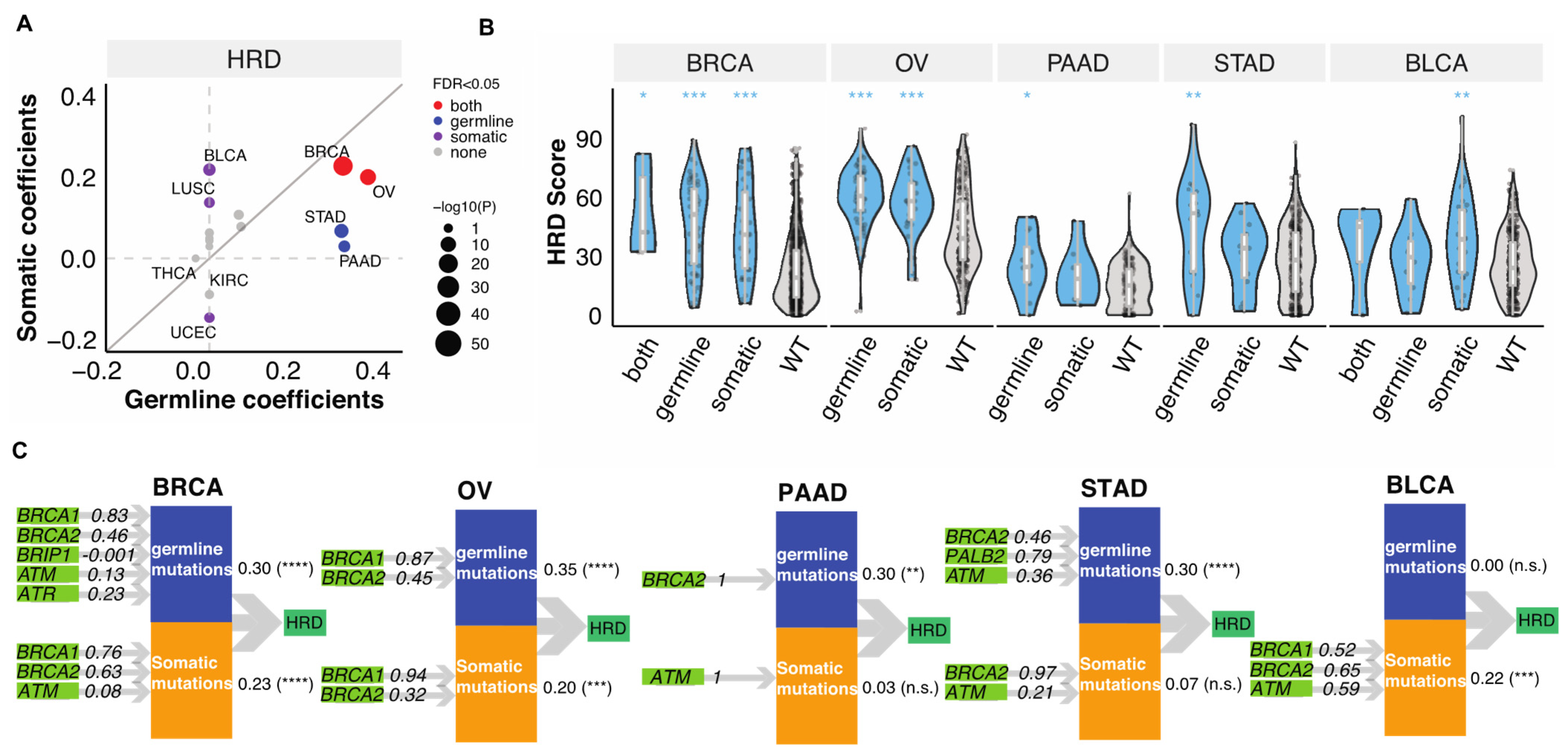
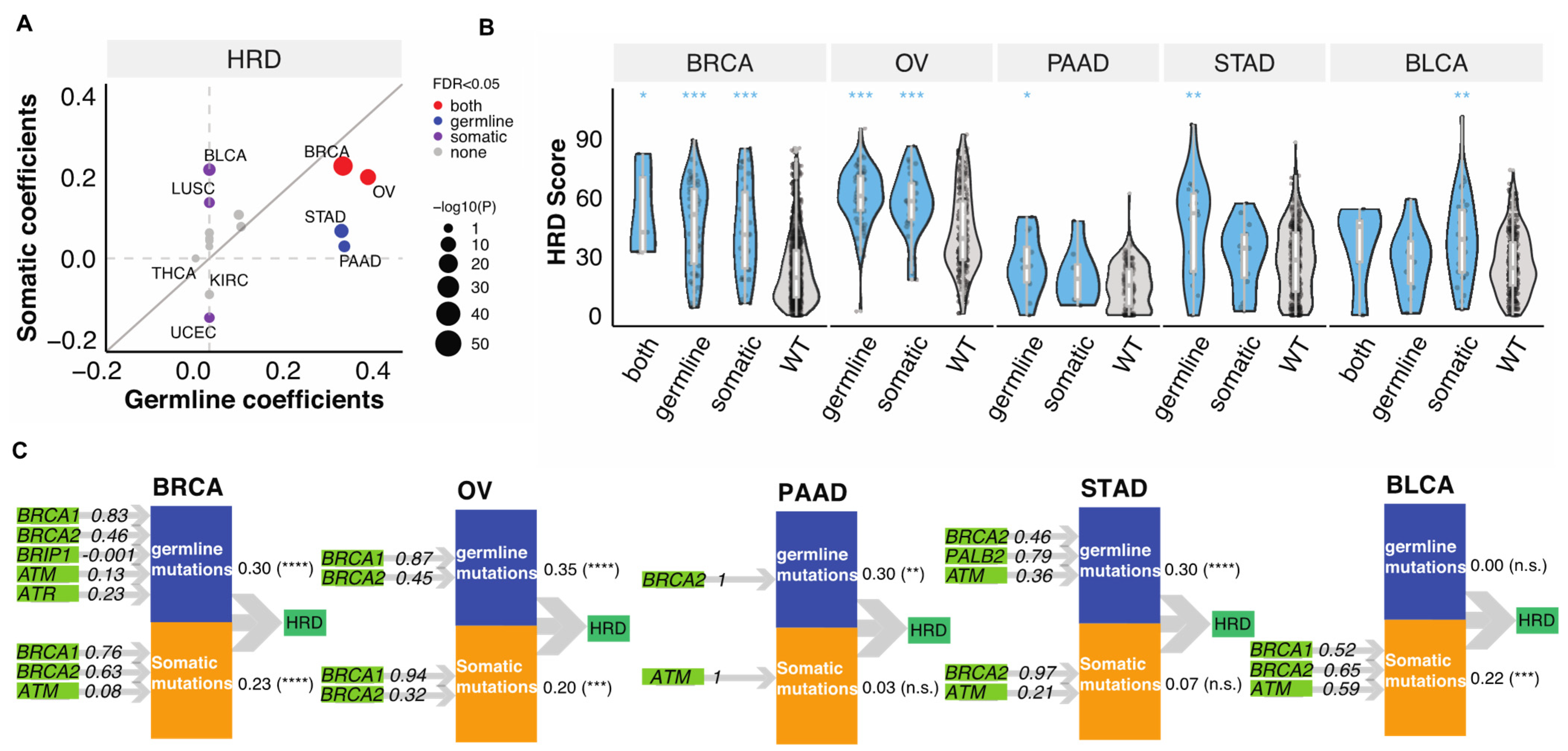
3.1. Germline and Somatic Mutations Associated with HRD in 32 Cancer Types
3.2. Relative Contributions of Germline and Somatic Mutations on HRD
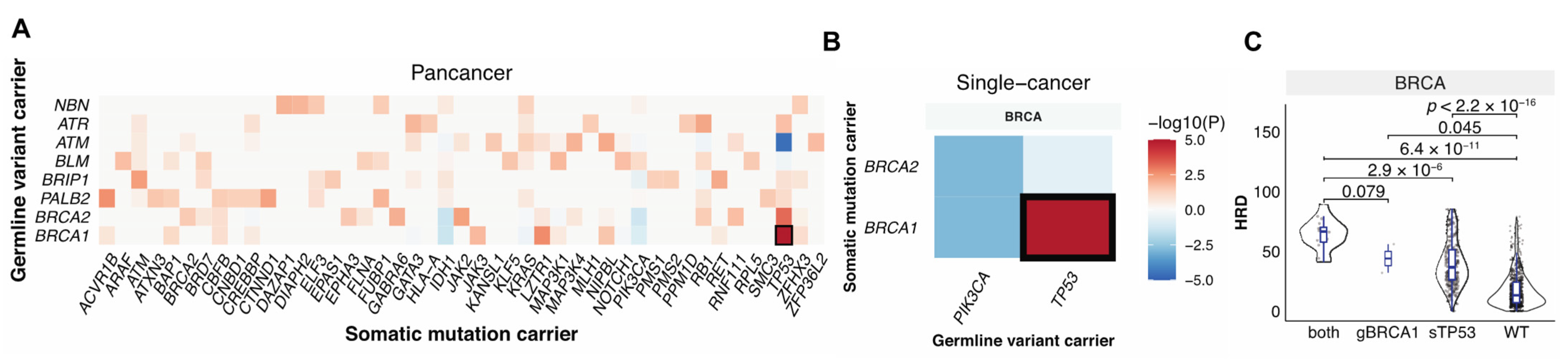
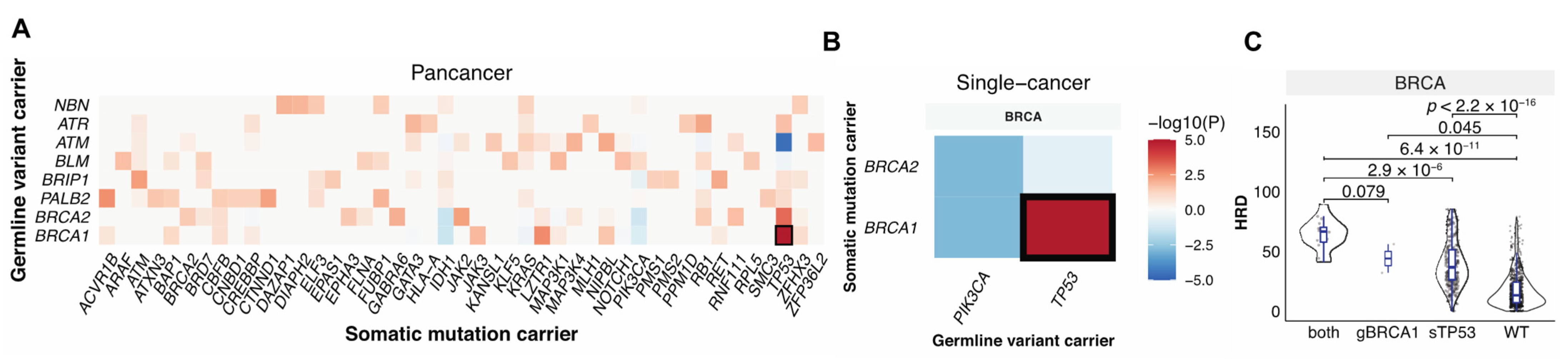
3.3. Germline-Somatic Interactions Shaping HRD
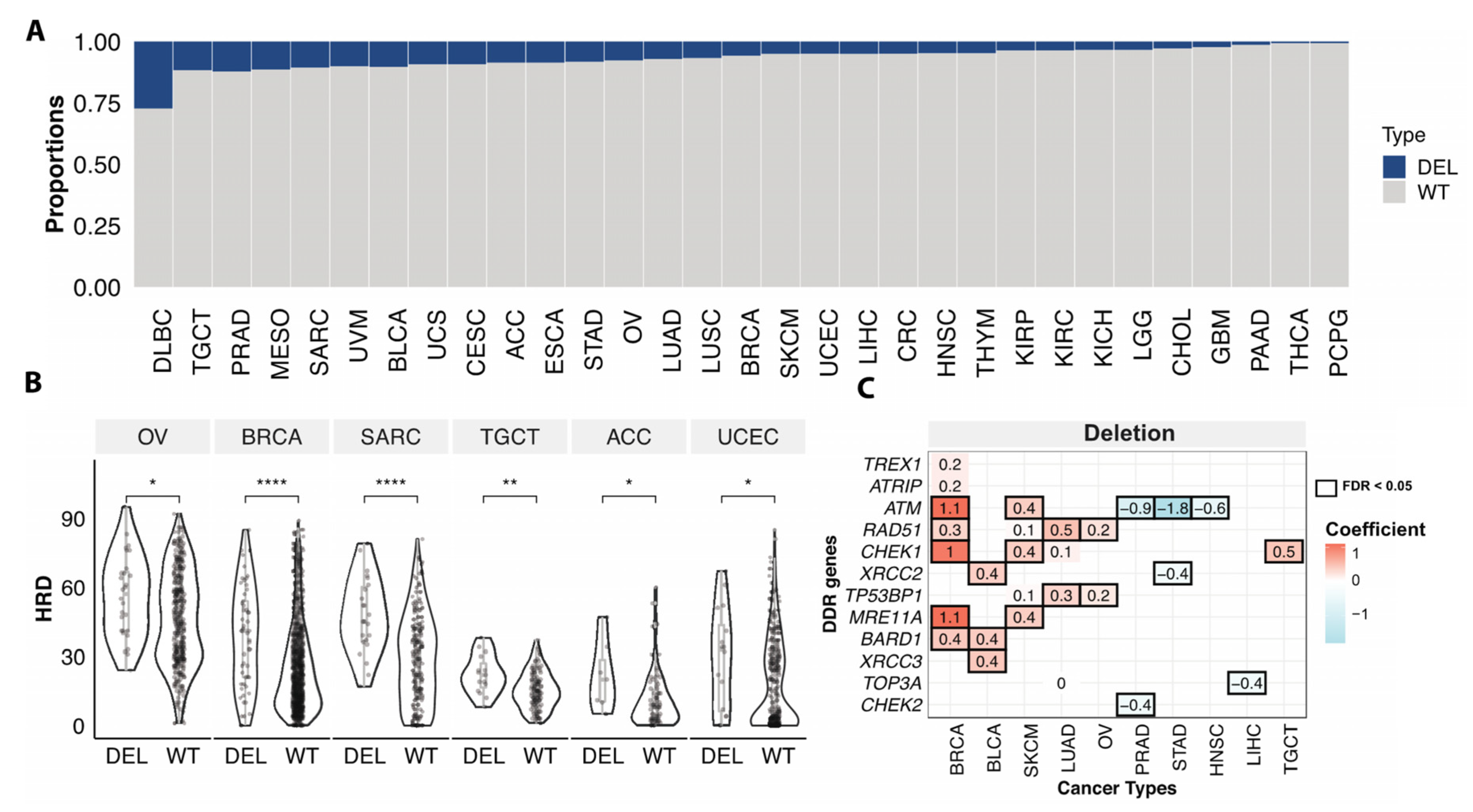
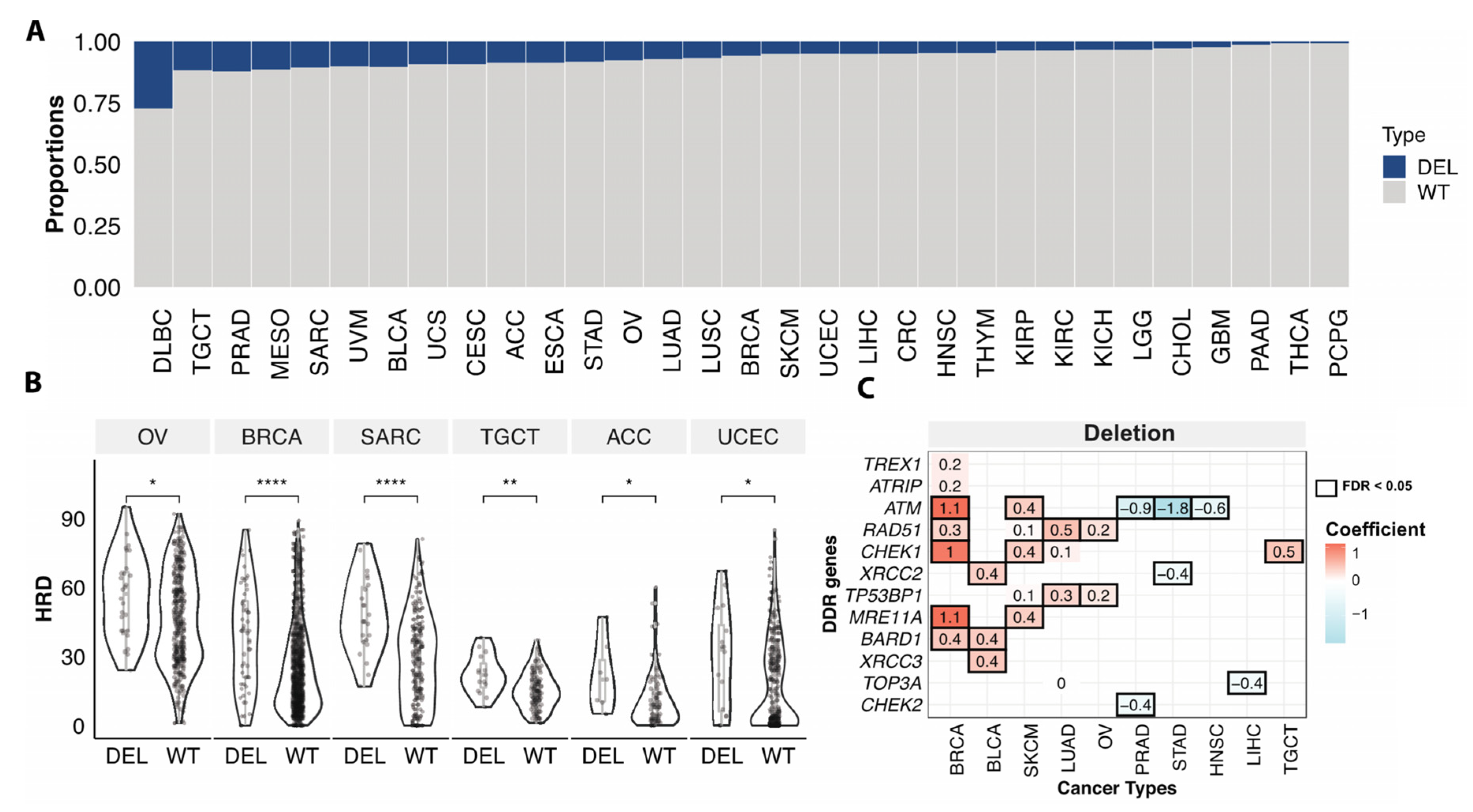
3.4. Somatic Copy Number Variations Associated with HRD
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dottino, J.A.; Moss, H.A.; Lu, K.H.; Secord, A.A.; Havrilesky, L.J. U.S. Food and drug administration-approved poly (Adp-ribose) Polymerase inhibitor maintenance therapy for recurrent ovarian cancer: A cost-effectiveness analysis. Obstet. Gynecol. 2019, 133, 795–802. [Google Scholar] [CrossRef]
- Hoppe, M.M.; Sundar, R.; Tan, D.S.P.; Jeyasekharan, A.D. Biomarkers for homologous recombination deficiency in cancer. J. Natl. Cancer Inst. 2018, 110, 704–713. [Google Scholar] [CrossRef] [PubMed]
- Robson, M.; Im, S.-A.; Senkus, E.; Xu, B.; Domchek, S.M.; Masuda, N.; Delaloge, S.; Li, W.; Tung, N.; Armstrong, A.; et al. Olaparib for metastatic breast cancer in patients with a germline BRCA mutation. New Engl. J. Med. 2017, 377, 523–533. [Google Scholar] [CrossRef] [PubMed]
- Mirza, M.R.; Monk, B.J.; Herrstedt, J.; Oza, A.; Mahner, S.; Redondo, A.; Fabbro, M.; Ledermann, J.A.; Lorusso, D.; Vergote, I.; et al. Niraparib maintenance therapy in platinum-sensitive, recurrent ovarian cancer. New Engl. J. Med. 2016, 375, 2154–2164. [Google Scholar] [CrossRef] [PubMed]
- FDA U. 2019 FDA Approves Niraparib For HRD-Positive Advanced Ovarian Cancer. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-niraparib-hrd-positive-advanced-ovarian-cancer#:~:text=The%20FDA%20also%20approved%20the,genomic%20instability%20for%20this%20indication (accessed on 23 October 2019).
- Kaufman, B.; Shapira-Frommer, R.; Schmutzler, R.K.; Audeh, M.W.; Friedlander, M.; Balmana, J.; Mitchell, G.; Fried, G.; Stemmer, S.M.; Hubertet, A.; et al. Olaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutation. J. Clin. Oncol. 2015, 33, 244–250. [Google Scholar] [CrossRef]
- Tung, N.M.; Robson, M.E.; Ventz, S.; Santa-Maria, C.A.; Nanda, R.; Marcom, P.K.; Shah, P.D.; Ballinger, T.J.; Yang, E.S.; Vinayak, S.; et al. TBCRC 048: Phase II study of olaparib for metastatic breast cancer and mutations in homologous recombination-related genes. J. Clin. Oncol. 2020, 38, 4274–4282. [Google Scholar] [CrossRef] [PubMed]
- Bang, Y.-J.; Xu, R.-H.; Chin, K.; Lee, K.-W.; Park, S.H.; Rha, S.Y.; Shen, L.; Qin, S.; Xu, N.; Im, S.-A.; et al. Olaparib in combination with paclitaxel in patients with advanced gastric cancer who have progressed following first-line therapy (GOLD): A double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 2017, 18, 1637–1651. [Google Scholar] [CrossRef]
- Vikas, P.; Borcherding, N.; Chennamadhavuni, A.; Garje, R. Therapeutic potential of combining PARP inhibitor and immunotherapy in solid tumors. Front. Oncol. 2020, 10, 570. [Google Scholar] [CrossRef]
- Sztupinszki, Z.; Diossy, M.; Krzystanek, M.; Borcsok, J.; Pomerantz, M.M.; Tisza, V.; Spisak, S.; Rusz, O.; Csabai, I.; Freedmanet, M.L.; et al. Detection of molecular signatures of homologous recombination deficiency in prostate cancer with or without BRCA1/2 mutations. Clin. Cancer Res. 2020, 26, 2673–2680. [Google Scholar] [CrossRef]
- Rahman, N. Realizing the promise of cancer predisposition genes. Nature 2014, 505, 302–308. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.-L.; Mashl, R.J.; Wu, Y.; Ritter, D.I.; Wang, J.; Oh, C.; Paczkowska, M.; Reynolds, S.; Wyczalkowski, M.; Oak, N.; et al. Pathogenic germline variants in 10,389 adult cancers. Cell 2018, 173, 355–370.e14. [Google Scholar] [CrossRef]
- Knijnenburg, T.A.; Wang, L.; Zimmermann, M.T.; Chambwe, N.; Gao, G.; Cherniack, A.D.; Fan, H.; Shen, H.; Way, G.P.; Greene, C.S.; et al. Genomic and molecular landscape of DNA damage repair deficiency across the cancer genome atlas. Cell Rep. 2018, 23, 239–254.e6. [Google Scholar] [CrossRef] [PubMed]
- Mermel, C.H.; Schumacher, E.S.; Hill, B.; Meyerson, M.L.; Beroukhim, R.; Getz, G. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol. 2011, 12, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Taylor, A.M.; Shih, J.; Ha, G.; Gao, G.F.; Zhang, X.; Berger, A.C.; Schumacher, S.E.; Wang, C.; Hu, H.; Liu, J.; et al. Genomic and Functional Approaches to Understanding Cancer Aneuploidy. Cancer Cell 2018, 33, 676–689 e673. [Google Scholar] [CrossRef] [PubMed]
- Carter, S.L.; Cibulskis, K.; Helman, E.; McKenna, A.; Shen, H.; Zack, T.; Laird, P.W.; Onofrio, R.C.; Winckler, W.; Weir, B.A.; et al. Absolute quantification of somatic DNA alterations in human cancer. Nat. Biotechnol. 2012, 30, 413–421. [Google Scholar] [CrossRef] [PubMed]
- Birkbak, N.J.; Wang, Z.C.; Kim, J.Y.; Eklund, A.C.; Li, Q.; Tian, R.; Bowman-Colin, C.; Li, Y.; Greene-Colozzi, A.; Iglehart, J.D.; et al. Telomeric allelic imbalance indicates defective DNA repair and sensitivity to DNA-damaging agents. Cancer Discov 2012, 2, 366–375. [Google Scholar] [CrossRef] [PubMed]
- Popova, T.; Manie, E.; Rieunier, G.; Caux-Moncoutier, V.; Tirapo, C.; Dubois, T.; Delattre, O.; Sigal-Zafrani, B.; Bollet, M.; Longy, M.; et al. Ploidy and large-scale genomic instability consistently identify basal-like breast carcinomas with BRCA1/2 inactivation. Cancer Res. 2012, 72, 5454–5462. [Google Scholar] [CrossRef] [PubMed]
- Grossman, R.L.; Heath, A.P.; Ferretti, V.; Varmus, H.E.; Lowy, D.R.; Kibbe, W.A.; Staudt, L.M. Toward a Shared Vision for Cancer Genomic Data. N Engl J Med 2016, 375, 1109–1112. [Google Scholar] [CrossRef]
- Ellrott, K.; Bailey, M.H.; Saksena, G.; Covington, K.R.; Kandoth, C.; Stewart, C.; Hess, J.; Ma, S.; Chiotti, K.E.; McLellan, M.; et al. Scalable open science approach for mutation calling of tumor exomes using multiple genomic pipelines. Cell Syst. 2018, 6, 271–281.e7. [Google Scholar] [CrossRef] [PubMed]
- Bailey, M.H.; Tokheim, C.; Porta-Pardo, E.; Sengupta, S.; Bertrand, D.; Weerasinghe, A.; Colaprico, A.; Wendl, M.C.; Kim, J.; Reardon, B.; et al. Comprehensive characterization of cancer driver genes and mutations. Cell 2018, 174, 1034–1035. [Google Scholar] [CrossRef]
- Ding, L.; Bailey, M.H.; Porta-Pardo, E.; Thorsson, V.; Colaprico, A.; Bertrand, D.; Gibbs, D.L.; Weerasinghe, A.; Huang, K.L.; Tokheim, C.; et al. Perspective on Oncogenic Processes at the End of the Beginning of Cancer Genomics. Cell 2018, 173, 305–320.e310. [Google Scholar] [CrossRef] [PubMed]
- Carrot-Zhang, J.; Chambwe, N.; Damrauer, J.S.; Knijnenburg, T.A.; Robertson, A.G.; Yau, C.; Zhou, W.; Berger, A.C.; Huang, K.L.; Newberg, J.Y.; et al. Comprehensive Analysis of Genetic Ancestry and Its Molecular Correlates in Cancer. Cancer Cell 2020, 37, 639–654.e636. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. A Language and Environment for Statistical Computing. R Foundation for Statistical Computing, Vienna, Austria. 2020. Available online: https://www.R-project.org/ (accessed on 11 August 2021).
- Yuan, Y.; Liu, L.; Chen, H.; Wang, Y.; Xu, Y.; Mao, H.; Li, J.; Mills, G.B.; Shu, Y.; Li, L.; et al. Comprehensive characterization of molecular differences in cancer between male and female patients. Cancer Cell 2016, 29, 711–722. [Google Scholar] [CrossRef] [PubMed]
- Wold, H. Causal flows with latent variables: Partings of the ways in the light of NIPALS modelling. Eur. Eco. Rev. 1974, 5, 67–86. [Google Scholar] [CrossRef]
- Gaston Sanchez LTaGR. plspm: Tools for Partial Least Squares Path Modeling (PLS-PM). 2015. Available online: https://github.com/gastonstat/plspm (accessed on 11 August 2021).
- Mateo, J.; Carreira, S.; Sandhu, S.; Miranda, S.; Mossop, H.; Perez-Lopez, R.; Rodrigues, D.N.; Robinson, D.; Omlin, A.; Tunariu, N.; et al. DNA-repair defects and olaparib in metastatic prostate cancer. New Engl. J. Med. 2015, 373, 1697–1708. [Google Scholar] [CrossRef] [PubMed]
- McCabe, N.; Turner, N.C.; Lord, C.; Kluzek, K.; Białkowska, A.; Swift, S.; Giavara, S.; O’Connor, M.J.; Tutt, A.N.; Zdzienicka, M.Z.; et al. Deficiency in the repair of DNA damage by homologous recombination and sensitivity to poly(ADP-ribose) Polymerase inhibition. Cancer Res. 2006, 66, 8109–8115. [Google Scholar] [CrossRef]
- Moudry, P.; Watanabe, K.; Wolanin, K.M.; Bartkova, J.; Wassing, I.E.; Watanabe, S.; Strauss, R.; Pedersen, R.T.; Oestergaard, V.; Lisby, M.; et al. TOPBP1 regulates RAD51 phosphorylation and chromatin loading and determines PARP inhibitor sensitivity. J. Cell Biol. 2016, 212, 281–288. [Google Scholar] [CrossRef]
- Qing, T.; Mohsen, H.; Marczyk, M.; Ye, Y.; O’Meara, T.; Zhao, H.; Townsend, J.P.; Gerstein, M.; Hatzis, C.; Kluger, Y.; et al. Germline variant burden in cancer genes correlates with age at diagnosis and somatic mutation burden. Nat. Commun. 2020, 11, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Carter, H.; Marty, R.; Hofree, M.; Gross, A.M.; Jensen, J.; Fisch, K.; Wu, X.; DeBoever, C.; Van Nostrand, E.; Song, Y.; et al. Interaction landscape of inherited polymorphisms with somatic events in cancer. Cancer Discov. 2017, 7, 410–423. [Google Scholar] [CrossRef] [PubMed]
- Hoy, S.M. Talazoparib: First global approval. Drugs 2018, 78, 1939–1946. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.; Ison, G.; McKee, A.E.; Zhang, H.; Tang, S.; Gwise, T.; Sridhara, R.; Lee, E.; Tzou, A.; Philip, R.; et al. FDA approval summary: Olaparib monotherapy in patients with deleterious germline brca-mutated advanced ovarian cancer treated with three or more lines of chemotherapy. Clin. Cancer Res. 2015, 21, 4257–4261. [Google Scholar] [CrossRef] [PubMed]
- Fewings, E.; Larionov, A.; Redman, J.; Goldgraben, M.; Scarth, J.; Richardson, S.; Brewer, C.; Davidson, R.; Ellis, I.; Evans, D.G.; et al. Germline pathogenic variants in PALB2 and other cancer-predisposing genes in families with hereditary diffuse gastric cancer without CDH1 mutation: A whole-exome sequencing study. Lancet Gastroenterol. Hepatol. 2018, 3, 489–498. [Google Scholar] [CrossRef]
- Watkins, J.A.; Irshad, S.; Grigoriadis, A.; Tutt, A.N.J. Genomic scars as biomarkers of homologous recombination deficiency and drug response in breast and ovarian cancers. Breast Cancer Res. 2014, 16, 211. [Google Scholar] [CrossRef] [PubMed]
- Den Brok, W.D.; Schrader, K.A.; Sun, S.; Tinker, A.V.; Zhao, E.Y.; Aparicio, S.; Gelmon, K.A. Homologous recombination deficiency in breast cancer: A clinical review. JCO Precision Oncol. 2017, 1, 1–13. [Google Scholar] [CrossRef]
- Rafiei, S.; Fitzpatrick, K.; Liu, D.; Cai, M.-Y.; Elmarakeby, H.A.; Park, J.; Ricker, C.; Kochupurakkal, B.S.; Choudhury, A.D.; Hahn, W.C.; et al. ATM Loss Confers Greater Sensitivity to ATR Inhibition Than PARP Inhibition in Prostate Cancer. Cancer Res. 2020, 80, 2094–2100. [Google Scholar] [CrossRef] [PubMed]


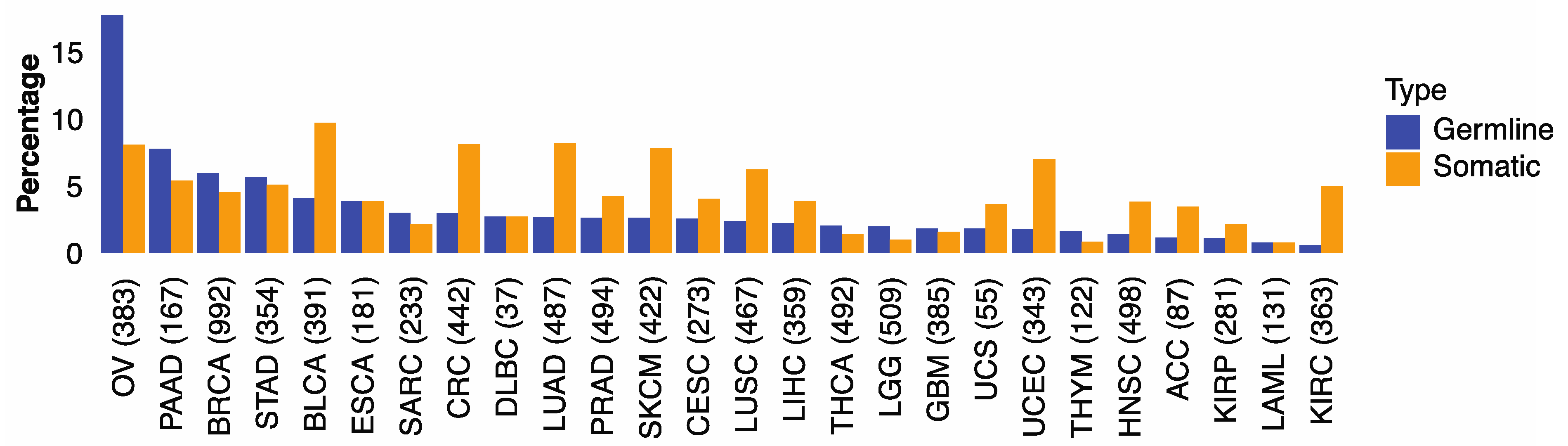{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Index | Cancer | Gene | Mutation Type | Number of Cases Affected (%) | Correlation Coefficients | FDR |
|---|---|---|---|---|---|---|
| 1 | BRCA | BRCA1 | germline | 18 (1.8) | 0.91 | <1.0 × 10−50 |
| 2 | BRCA | BRCA2 | germline | 16 (1.6) | 0.63 | <1.0 × 10−50 |
| 3 | OV | BRCA1 | germline | 35 (9.1) | 0.32 | 1.3 × 10−42 |
| 4 | STAD | PALB2 | germline | 4 (1.1) | 0.84 | 1.2 × 10−41 |
| 5 | PAAD | BRCA2 | germline | 5 (3) | 0.70 | 1.3 × 10−17 |
| 6 | STAD | BRCA2 | germline | 4 (1.1) | 0.61 | 6.7 × 10−17 |
| 7 | OV | BRCA2 | germline | 26 (6.8) | 0.20 | 5.0 × 10−12 |
| 8 | BRCA | ATR | germline | 5 (0.5) | 0.47 | 7.7 × 10−12 |
| 9 | BRCA | ATM | germline | 9 (0.9) | 0.32 | 8.5 × 10−8 |
| 10 | PRAD | ATM | germline | 6 (1.2) | 0.45 | 9.0 × 10−6 |
| 11 | LUAD | ATM | germline | 6 (1.2) | 0.30 | 5.8 × 10−5 |
| 12 | STAD | ATM | germline | 6 (1.7) | 0.25 | 0.007 |
| 13 | BRCA | BRCA1 | somatic | 15 (1.5) | 0.79 | <1.0 × 10−50 |
| 14 | BRCA | BRCA2 | somatic | 11 (1.1) | 0.81 | <1.0 × 10−50 |
| 15 | BLCA | BRCA2 | somatic | 7 (1.8) | 0.56 | 1.6 × 10−21 |
| 16 | OV | BRCA1 | somatic | 17 (4.4) | 0.27 | 5.1 × 10−17 |
| 17 | UCEC | ATM | somatic | 15 (4.4) | −0.70 | 2.9 × 10−13 |
| 18 | LUSC | BRCA2 | somatic | 7 (1.5) | 0.35 | 6.4 × 10−10 |
| 19 | BLCA | BRCA1 | somatic | 6 (1.5) | 0.37 | 1.5 × 10−8 |
| 20 | STAD | BRCA2 | somatic | 4 (1.1) | 0.44 | 1.5 × 10−8 |
| 21 | UCEC | BRCA2 | somatic | 7 (2) | −0.80 | 1.5 × 10−8 |
| 22 | BLCA | ATM | somatic | 23 (5.9) | 0.20 | 1.7 × 10−7 |
| 23 | UCEC | CHEK2 | somatic | 4 (1.2) | 0.51 | 3.3 × 10−7 |
| 24 | LUAD | ATM | somatic | 24 (4.9) | 0.18 | 1.3 × 10−5 |
| 25 | OV | BRCA2 | somatic | 7 (1.8) | 0.23 | 2.2 × 10−5 |
| 26 | PRAD | BRCA2 | somatic | 5 (1) | 0.42 | 7.8 × 10−5 |
| 27 | CRC | ATM | somatic | 23 (5.2) | 0.20 | 0.0003 |
| 28 | PRAD | ATM | somatic | 15 (3) | 0.24 | 0.0007 |
| 29 | LUSC | BRCA1 | somatic | 6 (1.3) | 0.21 | 0.002 |
| 30 | LUAD | BRCA2 | somatic | 4 (0.8) | −0.46 | 0.004 |
| 31 | HNSC | ATR | somatic | 4 (0.8) | 0.27 | 0.005 |
| 32 | BRCA | ATM | somatic | 14 (1.4) | 0.14 | 0.007 |
| 33 | KIRC | ATR | somatic | 4 (1.1) | 0.47 | 0.008 |
| 34 | CRC | BRCA2 | somatic | 6 (1.4) | 0.27 | 0.01 |
| 35 | SKCM | BRCA2 | somatic | 4 (0.9) | −0.39 | 0.02 |
| 36 | LUSC | ATR | somatic | 4 (0.9) | −0.22 | 0.03 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qing, T.; Wang, X.; Jun, T.; Ding, L.; Pusztai, L.; Huang, K.-L. Genomic Determinants of Homologous Recombination Deficiency across Human Cancers. Cancers 2021, 13, 4572. https://doi.org/10.3390/cancers13184572
Qing T, Wang X, Jun T, Ding L, Pusztai L, Huang K-L. Genomic Determinants of Homologous Recombination Deficiency across Human Cancers. Cancers. 2021; 13(18):4572. https://doi.org/10.3390/cancers13184572
Chicago/Turabian StyleQing, Tao, Xinfeng Wang, Tomi Jun, Li Ding, Lajos Pusztai, and Kuan-Lin Huang. 2021. "Genomic Determinants of Homologous Recombination Deficiency across Human Cancers" Cancers 13, no. 18: 4572. https://doi.org/10.3390/cancers13184572
APA StyleQing, T., Wang, X., Jun, T., Ding, L., Pusztai, L., & Huang, K.-L. (2021). Genomic Determinants of Homologous Recombination Deficiency across Human Cancers. Cancers, 13(18), 4572. https://doi.org/10.3390/cancers13184572