
NSCLC Biomarkers to Predict Response to Immunotherapy with Checkpoint Inhibitors (ICI): From the Cells to In Vivo Images

 ,
,  , , ,
, , , 
Abstract
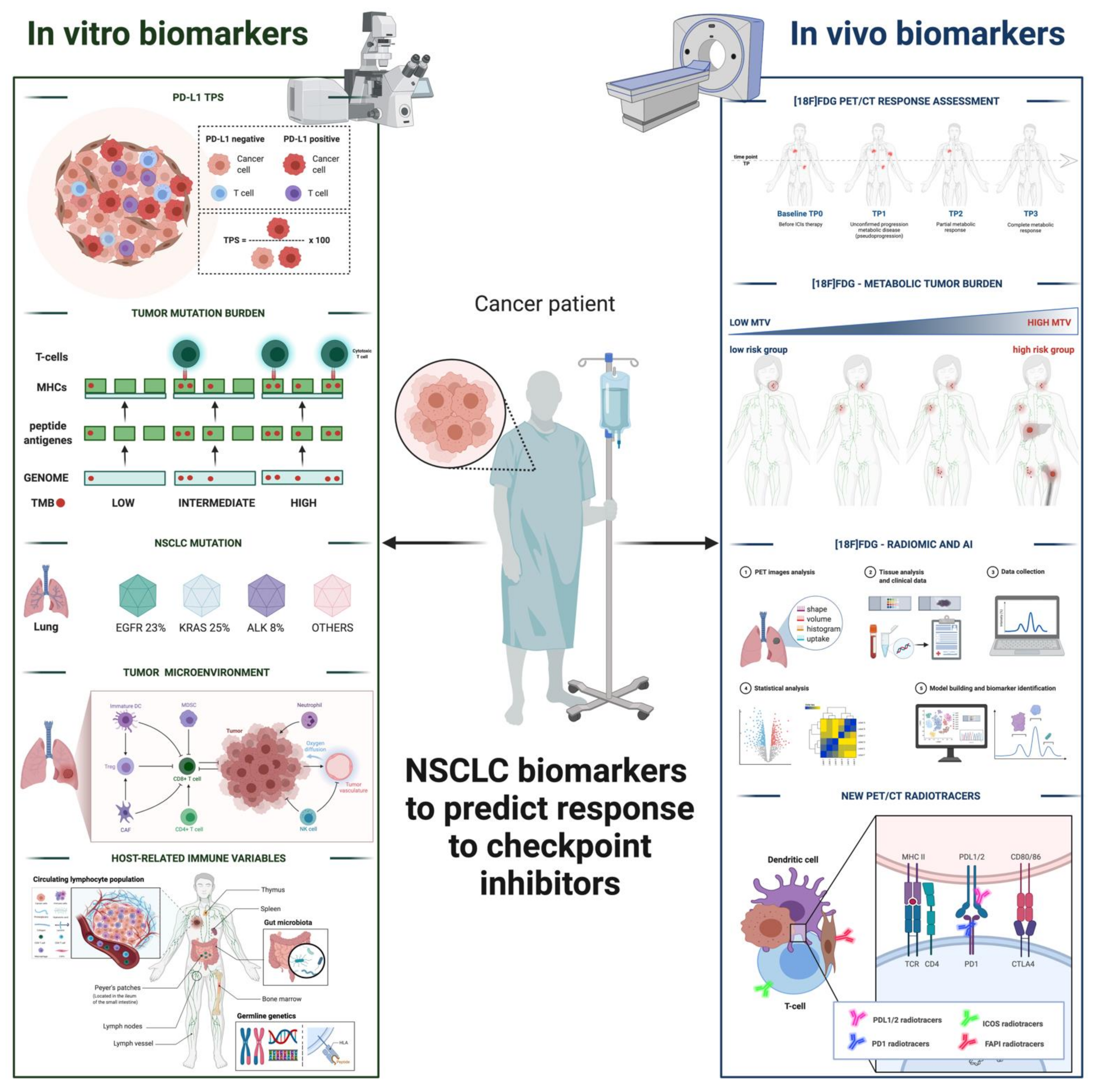
:Simple Summary
Abstract
1. Introduction
2. In Vitro Biomarkers for Immunotherapy
2.1. Tumor-Related Biomarkers
2.1.1. PD-L1 TPS (Tumor Proportion Score)
2.1.2. Tumor Mutational Burden
2.1.3. Tumor Genotype
2.2. Biomarkers Related to Tumor Microenvironment (TME)
2.2.1. T Lymphocyte Infiltration
2.2.2. Tumor Infiltrating CD8+ T Cell: Phenotype and TCR Clonality
2.2.3. Vascularization in Tumor Microenvironment
2.3. Host-Related Biomarkers
2.3.1. Circulating Lymphocyte Population
2.3.2. Innate Immune Populations
2.3.3. Intestinal Microbiota/Microbiome Composition
2.3.4. Germline Genetics
3. In Vivo Biomarkers for Immunotherapy: Molecular Imaging
3.1. 2-Deoxy-2-[18F]fluoro-D-glucose ([18F]FDG) and Immunotherapy in the NSCLC Setting
3.1.1. [18F]FDG and Immunotherapy Response Assessment
3.1.2. [18F]FDG and Immune-Related Adverse Events (irAEs)
3.2. [18F]FDG, Radiomic and AI
3.2.1. Molecular Profiling
3.2.2. Staging and Prognostic Risk Stratification: Prognostic and Predictive Value
3.3. Functional Imaging Probe beyond [18F]FDG
3.3.1. PD-1/PD-L1 Pathways
3.3.2. CD8+ T Lymphocytes
3.3.3. Cancer-Associated Fibroblasts (CAFs)
3.3.4. Other Probes
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| CR | complete response |
| CT | computer tomography |
| CTLA-4 | cytotoxic T-lymphocyte–associated protein 4 |
| EGFR | epidermal growth factor receptor |
| EMA | European Medicines Agency |
| FDA | Food and Drug Administration |
| [18F]FDG | 2-deoxy-2-[18F]fluoro-D-glucose |
| ICIs | immune checkpoint inhibitors |
| IHC | immunohistochemistry |
| irAE | immune-related adverse event |
| mAb | monoclonal antibody |
| MHC-I or -II | major histocompatibility complex-I or -II |
| MR | magnetic resonance |
| OS | overall survival |
| PD | progressive disease |
| PD-1 | programmed cell death protein 1 |
| PD-L1 | programmed cell death ligand 1 |
| PD-L2 | programmed cell death ligand 2 |
| PERCIMT | PET Response Evaluation Criteria for Immunotherapy |
| PERCIST | positron emission tomography response criteria in solid tumors |
| PET | positron emission tomography |
| PFS | progression free survival |
| PR | partial response |
| RECIST | response evaluation criteria in solid tumors |
| SD | stable disease |
| SUV | standardized uptake value |
| TCR | T cell receptor |
| TKI | tyrosine kinase inhibitors |
| TMB | tumor mutational burden |
| TME | tumor microenvironment |
| TPS | tumor proportion score |
References
- WHO (World Health Organization). Oesophageal Source: Globocan 2020 Number of New Cases in 2020, Both Sexes, All Ages; WHO: Geneva, Switzerland, 2020. [Google Scholar]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA. Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- Reck, M.; Heigener, D.F.; Mok, T.; Soria, J.C.; Rabe, K.F. Management of non-small-cell lung cancer: Recent developments. Lancet 2013, 382, 709–719. [Google Scholar] [CrossRef]
- Zhang, N.; Wu, J.; Yu, J.; Zhu, H.; Yang, M.; Li, R. Integrating Imaging, Histologic, and Genetic Features to Predict Tumor Mutation Burden of Non–Small-Cell Lung Cancer. Clin. Lung Cancer 2020, 21, e151–e163. [Google Scholar] [CrossRef] [PubMed]
- Herbst, R.S.; Morgensztern, D.; Boshoff, C. The biology and management of non-small cell lung cancer. Nature 2018, 553, 446–454. [Google Scholar] [CrossRef]
- Planchard, D.; Popat, S.; Kerr, K.; Novello, S.; Smit, E.F.; Faivre-Finn, C.; Mok, T.S.; Reck, M.; Van Schil, P.E.; Hellmann, M.D.; et al. Metastatic non-small cell lung cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2018, 29, iv192–iv237. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, Z. The history and advances in cancer immunotherapy: Understanding the characteristics of tumor-infiltrating immune cells and their therapeutic implications. Cell. Mol. Immunol. 2020, 17, 807–821. [Google Scholar] [CrossRef]
- Waldman, A.D.; Fritz, J.M.; Lenardo, M.J. A guide to cancer immunotherapy: From T cell basic science to clinical practice. Nat. Rev. Immunol. 2020, 20, 651–668. [Google Scholar] [CrossRef]
- Galli, F.; Aguilera, J.V.; Palermo, B.; Markovic, S.N.; Nisticò, P.; Signore, A. Relevance of immune cell and tumor microenvironment imaging in the new era of immunotherapy. J. Exp. Clin. Cancer Res. 2020, 39, 89. [Google Scholar] [CrossRef]
- Gonzalez, H.; Hagerling, C.; Werb, Z. Roles of the immune system in cancer: From tumor initiation to metastatic progression. Genes Dev. 2018, 32, 1267–1284. [Google Scholar] [CrossRef] [Green Version]
- Hashimoto, M.; Kamphorst, A.O.; Im, S.J.; Kissick, H.T.; Pillai, R.N.; Ramalingam, S.S.; Araki, K.; Ahmed, R. CD8 T Cell Exhaustion in Chronic Infection and Cancer: Opportunities for Interventions. Annu. Rev. Med. 2018, 69, 301–318. [Google Scholar] [CrossRef]
- Bouleau, A.; Lebon, V.; Truillet, C. PET imaging of immune checkpoint proteins in oncology. Pharmacol. Ther. 2021, 222, 107786. [Google Scholar] [CrossRef]
- Sun, C.; Mezzadra, R.; Schumacher, T.N. Regulation and Function of the PD-L1 Checkpoint. Immunity 2018, 48, 434–452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barber, D.L.; Wherry, E.J.; Masopust, D.; Zhu, B.; Allison, J.P.; Sharpe, A.H.; Freeman, G.J.; Ahmed, R. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature 2006, 439, 682–687. [Google Scholar] [CrossRef]
- Day, C.L.; Kaufmann, D.E.; Kiepiela, P.; Brown, J.A.; Moodley, E.S.; Reddy, S.; Mackey, E.W.; Miller, J.D.; Leslie, A.J.; DePierres, C.; et al. PD-1 expression on HIV-specific T cells is associated with T-cell exhaustion and disease progression. Nature 2006, 443, 350–354. [Google Scholar] [CrossRef] [PubMed]
- Schildberg, F.A.; Klein, S.R.; Freeman, G.J.; Sharpe, A.H. Coinhibitory Pathways in the B7-CD28 Ligand-Receptor Family. Immunity 2016, 44, 955–972. [Google Scholar] [CrossRef] [Green Version]
- Tumeh, P.C.; Harview, C.L.; Yearley, J.H.; Shintaku, I.P.; Taylor, E.J.M.; Robert, L.; Chmielowski, B.; Spasic, M.; Henry, G.; Ciobanu, V.; et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014, 515, 568–571. [Google Scholar] [CrossRef]
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Carvajal, R.D.; Sosman, J.A.; Atkins, M.B.; et al. Safety, Activity, and Immune Correlates of Anti–PD-1 Antibody in Cancer. N. Engl. J. Med. 2012, 366, 2443–2454. [Google Scholar] [CrossRef]
- Taube, J.M.; Klein, A.; Brahmer, J.R.; Xu, H.; Pan, X.; Kim, J.H.; Chen, L.; Pardoll, D.M.; Topalian, S.L.; Anders, R.A. Association of PD-1, PD-1 ligands, and other features of the tumor immune microenvironment with response to anti-PD-1 therapy. Clin. Cancer Res. 2014, 20, 5064–5074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korman, A.J.; Peggs, K.S.; Allison, J.P. Checkpoint Blockade in Cancer Immunotherapy. Adv. Immunol. 2006, 90, 297–339. [Google Scholar]
- Simpson, T.R.; Li, F.; Montalvo-Ortiz, W.; Sepulveda, M.A.; Bergerhoff, K.; Arce, F.; Roddie, C.; Henry, J.Y.; Yagita, H.; Wolchok, J.D.; et al. Fc-dependent depletion of tumor-infiltrating regulatory t cells co-defines the efficacy of anti-CTLA-4 therapy against melanoma. J. Exp. Med. 2013, 210, 1695–1710. [Google Scholar] [CrossRef] [PubMed]
- Selby, M.J.; Engelhardt, J.J.; Quigley, M.; Henning, K.A.; Chen, T.; Srinivasan, M.; Korman, A.J. Anti-CTLA-4 antibodies of IgG2a isotype enhance antitumor activity through reduction of intratumoral regulatory T cells. Cancer Immunol. Res. 2013, 1, 32–42. [Google Scholar] [CrossRef] [Green Version]
- Hanna, N.H.; Schneider, B.J.; Temin, S.; Baker, S.; Brahmer, J.; Ellis, P.M.; Gaspar, L.E.; Haddad, R.Y.; Hesketh, P.J.; Jain, D.; et al. Therapy for stage IV non–small-cell lung cancer without driver alterations: ASCO and OH (CCO) joint guideline update. J. Clin. Oncol. 2020, 38, 1608–1632. [Google Scholar] [CrossRef]
- Vaddepally, R.K.; Kharel, P.; Pandey, R.; Garje, R.; Chandra, A.B. Review of indications of FDA-approved immune checkpoint inhibitors per NCCN guidelines with the level of evidence. Cancers 2020, 12, 738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- YERVOY (Ipilimumab) Injection, for Intravenous Use Initial U.S. Approval: 2011. Reference ID: 4614238. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/125377s110lbl.pdf (accessed on 6 September 2021).
- Yervoy (Ipilimumab)—EMA/250994/2021. Available online: https://www.ema.europa.eu/en/documents/product-information/yervoy-epar-product-information_en.pdf (accessed on 6 September 2021).
- OPDIVO (Nivolumab) Injection, for Intravenous Use Initial U.S. Approval: 2014. Reference ID: 4734770. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/125554s090lbl.pdf (accessed on 6 September 2021).
- OPDIVO (Nivolumab)—EMA/CHMP/341080/2021. Available online: https://www.ema.europa.eu/en/documents/product-information/opdivo-epar-product-information_en.pdf (accessed on 6 September 2021).
- KEYTRUDA® (Pembrolizumab) Injection, for Intravenous Use Initial U.S. Approval: 2014. Reference ID: 4766009. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/125514s096lbl.pdf (accessed on 6 September 2021).
- KEYTRUDA® (Pembrolizumab)—EMEA/H/C/003820-II/0097. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/keytruda#product-information-section (accessed on 6 September 2021).
- LIBTAYO® (Cemiplimab-Rwlc) Injection, for Intravenous Use Initial U.S. Approval: 2018. Reference ID: 4750303. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/761097s007lbl.pdf (accessed on 6 September 2021).
- LIBTAYO® (Cemiplimab-Rwlc)—EMA/357238/2019-EMEA/H/C/004844. Available online: https://www.ema.europa.eu/en/documents/product-information/libtayo-epar-product-information_en.pdf (accessed on 6 September 2021).
- IMFINZI® (Durvalumab) Injection, for Intravenous Use Initial U.S. Approval: 2017. Reference ID: 4749639. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/761069s029lbl.pdf (accessed on 6 September 2021).
- IMFINZI® (Durvalumab)—EMA/521637/2018 EMEA/H/C/004771. Available online: https://www.ema.europa.eu/en/documents/product-information/imfinzi-epar-product-information_en.pdf (accessed on 6 September 2021).
- TECENTRIQ® (Atezolizumab) Injection, for Intravenous Use Initial U.S. Approval: 2016. Reference ID: 4748227. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/761034s033s034s035s036s037s038lbl.pdf (accessed on 6 September 2021).
- TECENTRIQ® (Atezolizumab)—EMA/234492/2021 EMEA/H/C/004143. Available online: https://www.ema.europa.eu/en/documents/product-information/tecentriq-epar-product-information_en.pdf (accessed on 6 September 2021).
- Park, H.J.; Kim, K.W.; Won, S.E.; Yoon, S.; Chae, Y.K.; Tirumani, S.H.; Ramaiya, N.H. Definition, Incidence, and Challenges for Assessment of Hyperprogressive Disease during Cancer Treatment with Immune Checkpoint Inhibitors: A Systematic Review and Meta-analysis. JAMA Netw. Open 2021, 4. [Google Scholar] [CrossRef]
- Fridman, W.H.; Zitvogel, L.; Sautès-Fridman, C.; Kroemer, G. The immune contexture in cancer prognosis and treatment. Nat. Rev. Clin. Oncol. 2017, 14, 717–734. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.; Hu, X.; Zheng, S.; Yang, A.; Li, X.; Tang, H.; Kong, Y.; Xie, X. Discordance of immunotherapy response predictive biomarkers between primary lesions and paired metastases in tumours: A multidimensional analysis. EBioMedicine 2021, 63. [Google Scholar] [CrossRef]
- Liberati, A.; Altman, D.G.; Tetzlaff, J.; Mulrow, C.; Gøtzsche, P.C.; Ioannidis, J.P.A.; Clarke, M.; Devereaux, P.J.; Kleijnen, J.; Moher, D. The PRISMA statement for reporting systematic reviews and meta-analyses of studies that evaluate health care interventions: Explanation and elaboration. In Proceedings of the Journal of clinical epidemiology. J. Clin. Epidemiol. 2009, 62, e1–e34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borghaei, H.; Paz-Ares, L.; Horn, L.; Spigel, D.R.; Steins, M.; Ready, N.E.; Chow, L.Q.; Vokes, E.E.; Felip, E.; Holgado, E.; et al. Nivolumab versus Docetaxel in Advanced Nonsquamous Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 373, 1627–1639. [Google Scholar] [CrossRef]
- Brahmer, J.; Reckamp, K.L.; Baas, P.; Crinò, L.; Eberhardt, W.E.E.; Poddubskaya, E.; Antonia, S.; Pluzanski, A.; Vokes, E.E.; Holgado, E.; et al. Nivolumab versus Docetaxel in Advanced Squamous-Cell Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 373, 123–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, D.S.; Mellman, I. Elements of cancer immunity and the cancer-immune set point. Nature 2017, 541, 321–330. [Google Scholar] [CrossRef]
- Tsao, M.S.; Kerr, K.M.; Kockx, M.; Beasley, M.B.; Borczuk, A.C.; Botling, J.; Bubendorf, L.; Chirieac, L.; Chen, G.; Chou, T.Y.; et al. PD-L1 Immunohistochemistry Comparability Study in Real-Life Clinical Samples: Results of Blueprint Phase 2 Project. J. Thorac. Oncol. 2018, 13, 1302–1311. [Google Scholar] [CrossRef] [Green Version]
- Herbst, R.S.; Soria, J.C.; Kowanetz, M.; Fine, G.D.; Hamid, O.; Gordon, M.S.; Sosman, J.A.; McDermott, D.F.; Powderly, J.D.; Gettinger, S.N.; et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature 2014, 515, 563–567. [Google Scholar] [CrossRef] [Green Version]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in cancer immunotherapy. Science (80-) 2015, 348, 69–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Havel, J.J.; Chowell, D.; Chan, T.A. The evolving landscape of biomarkers for checkpoint inhibitor immunotherapy. Nat. Rev. Cancer 2019, 19, 133–150. [Google Scholar] [CrossRef]
- Hellmann, M.D.; Nathanson, T.; Rizvi, H.; Creelan, B.C.; Sanchez-Vega, F.; Ahuja, A.; Ni, A.; Novik, J.B.; Mangarin, L.M.B.; Abu-Akeel, M.; et al. Genomic Features of Response to Combination Immunotherapy in Patients with Advanced Non-Small-Cell Lung Cancer. Cancer Cell 2018, 33, 843–852.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.J.R.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Børresen-Dale, A.L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science (80-). 2015, 348, 124–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rizvi, H.; Sanchez-Vega, F.; La, K.; Chatila, W.; Jonsson, P.; Halpenny, D.; Plodkowski, A.; Long, N.; Sauter, J.L.; Rekhtman, N.; et al. Molecular determinants of response to anti-programmed cell death (PD)-1 and anti-programmed death-ligand 1 (PD-L1) blockade in patients with non-small-cell lung cancer profiled with targeted next-generation sequencing. J. Clin. Oncol. 2018, 36, 633–641. [Google Scholar] [CrossRef]
- Gandara, D.R.; Paul, S.M.; Kowanetz, M.; Schleifman, E.; Zou, W.; Li, Y.; Rittmeyer, A.; Fehrenbacher, L.; Otto, G.; Malboeuf, C.; et al. Blood-based tumor mutational burden as a predictor of clinical benefit in non-small-cell lung cancer patients treated with atezolizumab. Nat. Med. 2018, 24. [Google Scholar] [CrossRef]
- Hellmann, M.D.; Ciuleanu, T.-E.; Pluzanski, A.; Lee, J.S.; Otterson, G.A.; Audigier-Valette, C.; Minenza, E.; Linardou, H.; Burgers, S.; Salman, P.; et al. Nivolumab plus Ipilimumab in Lung Cancer with a High Tumor Mutational Burden. N. Engl. J. Med. 2018, 378, 2093–2104. [Google Scholar] [CrossRef]
- Hellmann, M.D.; Paz-Ares, L.; Bernabe Caro, R.; Zurawski, B.; Kim, S.-W.; Carcereny Costa, E.; Park, K.; Alexandru, A.; Lupinacci, L.; de la Mora Jimenez, E.; et al. Nivolumab plus Ipilimumab in Advanced Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2019, 381, 2020–2031. [Google Scholar] [CrossRef]
- Update on the Phase III NEPTUNE Trial of Imfinzi Plus Tremelimumab in Stage IV Non-Small Cell Lung Cancer. Available online: https://www.astrazeneca.com/media-centre/press-releases/2019/update-on-the-phase-iii-neptune-trial-of-imfinzi-plus-tremelimumab-in-stage-iv-non-small-cell-lung-cancer-21082019.html (accessed on 30 March 2020).
- Research, C. for D.E. and FDA Approves Pembrolizumab for Adults and Children with TMB-H Solid Tumors. Available online: https://www.fda.gov/drugs/drug-approvals-and-databases/fda-approves-pembrolizumab-adults-and-children-tmb-h-solid-tumors (accessed on 6 September 2021).
- Litchfield, K.; Reading, J.L.; Puttick, C.; Thakkar, K.; Abbosh, C.; Bentham, R.; Watkins, T.B.K.; Rosenthal, R.; Biswas, D.; Rowan, A.; et al. Meta-analysis of tumor- and T cell-intrinsic mechanisms of sensitization to checkpoint inhibition. Cell 2021, 184, 596–614.e14. [Google Scholar] [CrossRef]
- Wang, Z.; Duan, J.; Wang, G.; Zhao, J.; Xu, J.; Han, J.; Zhao, Z.; Zhao, J.; Zhu, B.; Zhuo, M.; et al. Allele Frequency–Adjusted Blood-Based Tumor Mutational Burden as a Predictor of Overall Survival for Patients With NSCLC Treated With PD-(L)1 Inhibitors. J. Thorac. Oncol. 2020, 15, 556–567. [Google Scholar] [CrossRef] [PubMed]
- Cai, L.; Bai, H.; Duan, J.; Wang, Z.; Gao, S.; Wang, D.; Wang, S.; Jiang, J.; Han, J.; Tian, Y.; et al. Epigenetic alterations are associated with tumor mutation burden in non-small cell lung cancer. J. Immunother. Cancer 2019, 7. [Google Scholar] [CrossRef] [Green Version]
- Gainor, J.F.; Shaw, A.T.; Sequist, L.V.; Fu, X.; Azzoli, C.G.; Piotrowska, Z.; Huynh, T.G.; Zhao, L.; Fulton, L.; Schultz, K.R.; et al. EGFR mutations and ALK rearrangements are associated with low response rates to PD-1 pathway blockade in non-small cell lung cancer: A retrospective analysis. Clin. Cancer Res. 2016, 22, 4585–4593. [Google Scholar] [CrossRef] [Green Version]
- Bylicki, O.; Paleiron, N.; Margery, J.; Guisier, F.; Vergnenegre, A.; Robinet, G.; Auliac, J.B.; Gervais, R.; Chouaid, C. Targeting the PD-1/PD-L1 Immune Checkpoint in EGFR-Mutated or ALK-Translocated Non-Small-Cell Lung Cancer. Target. Oncol. 2017, 12, 563–569. [Google Scholar] [CrossRef] [PubMed]
- Mazieres, J.; Drilon, A.; Lusque, A.; Mhanna, L.; Cortot, A.B.; Mezquita, L.; Thai, A.A.; Mascaux, C.; Couraud, S.; Veillon, R.; et al. Immune checkpoint inhibitors for patients with advanced lung cancer and oncogenic driver alterations: Results from the IMMUNOTARGET registry. Ann. Oncol. 2019, 30, 1321–1328. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, C.; Thompson, J.C.; Chien, A.L.; Quinn, K.J.; Hwang, W.T.; Black, T.A.; Yee, S.S.; Christensen, T.E.; LaRiviere, M.J.; Silva, B.A.; et al. Baseline Plasma Tumor Mutation Burden Predicts Response to Pembrolizumab-based Therapy in Patients with Metastatic Non–Small Cell Lung Cancer. Clin. Cancer Res. 2020, 26, 2354–2361. [Google Scholar] [CrossRef] [Green Version]
- Lamberti, G.; Spurr, L.F.; Li, Y.; Ricciuti, B.; Recondo, G.; Umeton, R.; Nishino, M.; Sholl, L.M.; Meyerson, M.L.; Cherniack, A.D.; et al. Clinicopathological and genomic correlates of programmed cell death ligand 1 (PD-L1) expression in nonsquamous non-small-cell lung cancer. Ann. Oncol. 2020, 31, 807–814. [Google Scholar] [CrossRef] [PubMed]
- Schoenfeld, A.J.; Rizvi, H.; Bandlamudi, C.; Sauter, J.L.; Travis, W.D.; Rekhtman, N.; Plodkowski, A.J.; Perez-Johnston, R.; Sawan, P.; Beras, A.; et al. Clinical and molecular correlates of PD-L1 expression in patients with lung adenocarcinomas. Ann. Oncol. 2020, 31, 599–608. [Google Scholar] [CrossRef]
- Cho, B.C.; Lopes, G.; Kowalski, D.M.; Kasahara, K.; Wu, Y.-L.; Castro, G.; Turna, H.Z.; Cristescu, R.; Aurora-Garg, D.; Loboda, A.; et al. Abstract CT084: Relationship between STK11 and KEAP1 mutational status and efficacy in KEYNOTE-042: Pembrolizumab monotherapy versus platinum-based chemotherapy as first-line therapy for PD-L1-positive advanced NSCLC. In Proceedings of the Annual Meeting of the American Association for Cancer Research 2020, Philadelphia, PA, USA, 27–28 April 2020; American Association for Cancer Research (AACR): Philadelphia, PA, USA, 2020; Volume 80, p. CT084. [Google Scholar]
- Aviel-Ronen, S.; Blackhall, F.H.; Shepherd, F.A.; Tsao, M.S. K-ras mutations in non-small-cell lung carcinoma: A review. Clin. Lung Cancer 2006, 8, 30–38. [Google Scholar] [CrossRef]
- Pylayeva-Gupta, Y.; Lee, K.E.; Hajdu, C.H.; Miller, G.; Bar-Sagi, D. Oncogenic Kras-Induced GM-CSF Production Promotes the Development of Pancreatic Neoplasia. Cancer Cell 2012, 21, 836–847. [Google Scholar] [CrossRef] [Green Version]
- Dong, Z.Y.; Zhong, W.Z.; Zhang, X.C.; Su, J.; Xie, Z.; Liu, S.Y.; Tu, H.Y.; Chen, H.J.; Sun, Y.L.; Zhou, Q.; et al. Potential predictive value of TP53 and KRAS mutation status for response to PD-1 blockade immunotherapy in lung adenocarcinoma. Clin. Cancer Res. 2017, 23, 3012–3024. [Google Scholar] [CrossRef] [Green Version]
- Toki, M.I.; Mani, N.; Smithy, J.W.; Liu, Y.; Altan, M.; Wasserman, B.; Tuktamyshov, R.; Schalper, K.; Syrigos, K.N.; Rimm, D.L. Immune Marker Profiling and Programmed Death Ligand 1 Expression Across NSCLC Mutations. J. Thorac. Oncol. 2018, 13, 1884–1896. [Google Scholar] [CrossRef] [Green Version]
- Skoulidis, F.; Goldberg, M.E.; Greenawalt, D.M.; Hellmann, M.D.; Awad, M.M.; Gainor, J.F.; Schrock, A.B.; Hartmaier, R.J.; Trabucco, S.E.; Gay, L.; et al. STK11/LKB1 mutations and PD-1 inhibitor resistance in KRAS-mutant lung adenocarcinoma. Cancer Discov. 2018, 8, 822–835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koyama, S.; Akbay, E.A.; Li, Y.Y.; Aref, A.R.; Skoulidis, F.; Herter-Sprie, G.S.; Buczkowski, K.A.; Liu, Y.; Awad, M.M.; Denning, W.L.; et al. STK11/LKB1 deficiency promotes neutrophil recruitment and proinflammatory cytokine production to suppress T-cell activity in the lung tumor microenvironment. Cancer Res. 2016, 76, 999–1008. [Google Scholar] [CrossRef] [Green Version]
- Rizvi, N.; Cho, B.C.; Reinmuth, N.; Lee, K.H.; Luft, A.; Ahn, M.; Papadimitrakopoulou, V.; Heymach, J.; Scheuring, U.; Higgs, B.; et al. OA04.07 Mutations Associated with Sensitivity or Resistance to Immunotherapy in mNSCLC: Analysis from the MYSTIC Trial. J. Thorac. Oncol. 2019, 14, S217. [Google Scholar] [CrossRef]
- Okamura, R.; Kato, S.; Lee, S.; Jimenez, R.E.; Sicklick, J.K.; Kurzrock, R. ARID1A alterations function as a biomarker for longer progression-free survival after anti-PD-1/PD-L1 immunotherapy. J. Immunother. Cancer 2020, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, L.; Ruiz, J.; O’Neill, S.S.; Grant, S.C.; Petty, W.J.; Yang, M.; Chen, K.; Topaloglu, U.; Pasche, B.; Zhang, W. Favorable outcome of patients with lung adenocarcinoma harboring POLE mutations and expressing high PD-L1. Mol. Cancer 2018, 17, 81. [Google Scholar] [CrossRef]
- Chen, H.; Chong, W.; Teng, C.; Yao, Y.; Wang, X.; Li, X. The immune response-related mutational signatures and driver genes in non-small-cell lung cancer. Cancer Sci. 2019, 110, 2348–2356. [Google Scholar] [CrossRef]
- Brambilla, E.; Le Teuff, G.; Marguet, S.; Lantuejoul, S.; Dunant, A.; Graziano, S.; Pirker, R.; Douillard, J.Y.; Le Chevalier, T.; Filipits, M.; et al. Prognostic effect of tumor lymphocytic infiltration in resectable non-small-cell lung cancer. J. Clin. Oncol. 2016, 34, 1223–1230. [Google Scholar] [CrossRef] [PubMed]
- Zeng, D.Q.; Yu, Y.F.; Ou, Q.Y.; Li, X.Y.; Zhong, R.Z.; Xie, C.M.; Hu, Q.G. Prognostic and predictive value of tumor-infiltrating lymphocytes for clinical therapeutic research in patients with non-small cell lung cancer. Oncotarget 2016, 7, 13765–13781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.S.; Ruppin, E. Multiomics Prediction of Response Rates to Therapies to Inhibit Programmed Cell Death 1 and Programmed Cell Death 1 Ligand 1. JAMA Oncol. 2019, 5, 1614–1618. [Google Scholar] [CrossRef] [PubMed]
- Gajewski, T.F.; Schreiber, H.; Fu, Y.X. Innate and adaptive immune cells in the tumor microenvironment. Nat. Immunol. 2013, 14, 1014–1022. [Google Scholar] [CrossRef] [Green Version]
- Hori, S.; Nomura, T.; Sakaguchi, S. Control of regulatory T cell development by the transcription factor Foxp3. J. Immunol. 2017, 198, 981–985. [Google Scholar] [CrossRef] [Green Version]
- Thommen, D.S.; Koelzer, V.H.; Herzig, P.; Roller, A.; Trefny, M.; Dimeloe, S.; Kiialainen, A.; Hanhart, J.; Schill, C.; Hess, C.; et al. A transcriptionally and functionally distinct pd-1 + cd8 + t cell pool with predictive potential in non-small-cell lung cancer treated with pd-1 blockade. Nat. Med. 2018, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riaz, N.; Havel, J.J.; Makarov, V.; Desrichard, A.; Urba, W.J.; Sims, J.S.; Hodi, F.S.; Martín-Algarra, S.; Mandal, R.; Sharfman, W.H.; et al. Tumor and Microenvironment Evolution during Immunotherapy with Nivolumab. Cell 2017, 171, 934–949.e15. [Google Scholar] [CrossRef] [Green Version]
- Forde, P.M.; Chaft, J.E.; Smith, K.N.; Anagnostou, V.; Cottrell, T.R.; Hellmann, M.D.; Zahurak, M.; Yang, S.C.; Jones, D.R.; Broderick, S.; et al. Neoadjuvant PD-1 Blockade in Resectable Lung Cancer. N. Engl. J. Med. 2018, 378, 1976–1986. [Google Scholar] [CrossRef] [PubMed]
- Roh, W.; Chen, P.L.; Reuben, A.; Spencer, C.N.; Prieto, P.A.; Miller, J.P.; Gopalakrishnan, V.; Wang, F.; Cooper, Z.A.; Reddy, S.M.; et al. Integrated molecular analysis of tumor biopsies on sequential CTLA-4 and PD-1 blockade reveals markers of response and resistance. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef] [Green Version]
- Snyder, A.; Nathanson, T.; Funt, S.A.; Ahuja, A.; Buros Novik, J.; Hellmann, M.D.; Chang, E.; Aksoy, B.A.; Al-Ahmadie, H.; Yusko, E.; et al. Contribution of systemic and somatic factors to clinical response and resistance to PD-L1 blockade in urothelial cancer: An exploratory multi-omic analysis. PLoS Med. 2017, 14, e1002309. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, A.C.; Yarchoan, M.; Durham, J.N.; Yusko, E.C.; Rytlewski, J.A.; Robins, H.S.; Laheru, D.A.; Le, D.T.; Lutz, E.R.; Jaffee, E.M. T cell receptor repertoire features associated with survival in immunotherapy-treated pancreatic ductal adenocarcinoma. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [Green Version]
- Bellone, M.; Calcinotto, A. Ways to enhance lymphocyte trafficking into tumors and fitness of tumor infiltrating lymphocytes. Front. Oncol. 2013, 3, 231. [Google Scholar] [CrossRef] [Green Version]
- Beatty, G.L.; Moon, E.K. Chimeric antigen receptor T cells are vulnerable to immunosuppressive mechanisms present within the tumor microenvironment. Oncoimmunology 2014, 3, e970027-1–e970027-3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campesato, L.F.; Merghoub, T. Antiangiogenic therapy and immune checkpoint blockade go hand in hand. Ann. Transl. Med. 2017, 5, 497. [Google Scholar] [CrossRef] [PubMed]
- Fukumura, D.; Kloepper, J.; Amoozgar, Z.; Duda, D.G.; Jain, R.K. Enhancing cancer immunotherapy using antiangiogenics: Opportunities and challenges. Nat. Rev. Clin. Oncol. 2018, 15, 325–340. [Google Scholar] [CrossRef]
- Socinski, M.A.; Jotte, R.M.; Cappuzzo, F.; Orlandi, F.; Stroyakovskiy, D.; Nogami, N.; Rodríguez-Abreu, D.; Moro-Sibilot, D.; Thomas, C.A.; Barlesi, F.; et al. Atezolizumab for First-Line Treatment of Metastatic Nonsquamous NSCLC. N. Engl. J. Med. 2018, 378, 2288–2301. [Google Scholar] [CrossRef]
- Rahma, O.E.; Hodi, F.S. The intersection between tumor angiogenesis and immune suppression. Clin. Cancer Res. 2019, 25, 5449–5457. [Google Scholar] [CrossRef] [Green Version]
- Voron, T.; Colussi, O.; Marcheteau, E.; Pernot, S.; Nizard, M.; Pointet, A.L.; Latreche, S.; Bergaya, S.; Benhamouda, N.; Tanchot, C.; et al. VEGF-A modulates expression of inhibitory checkpoints on CD8+ T cells in tumors. J. Exp. Med. 2015, 212, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Gabrilovich, D.; Ishida, T.; Oyama, T.; Ran, S.; Kravtsov, V.; Nadaf, S.; Carbone, D.P. Vascular endothelial growth factor inhibits the development of dendritic cells and dramatically affects the differentiation of multiple hematopoietic lineages in vivo. Blood 1998, 92, 4150–4166. [Google Scholar] [CrossRef]
- Huang, Y.; Yuan, J.; Righi, E.; Kamoun, W.S.; Ancukiewicz, M.; Nezivar, J.; Santosuosso, M.; Martin, J.D.; Martin, M.R.; Vianello, F.; et al. Vascular normalizing doses of antiangiogenic treatment reprogram the immunosuppressive tumor microenvironment and enhance immunotherapy. Proc. Natl. Acad. Sci. USA 2012, 109, 17561–17566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallin, J.J.; Bendell, J.C.; Funke, R.; Sznol, M.; Korski, K.; Jones, S.; Hernandez, G.; Mier, J.; He, X.; Hodi, F.S.; et al. Atezolizumab in combination with bevacizumab enhances antigen-specific T-cell migration in metastatic renal cell carcinoma. Nat. Commun. 2016, 7. [Google Scholar] [CrossRef]
- Lanitis, E.; Dangaj, D.; Irving, M.; Coukos, G. Mechanisms regulating T-cell infiltration and activity in solid tumors. Ann. Oncol. 2017, 28, xii18–xii32. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Giobbie-Hurder, A.; Liao, X.; Connelly, C.; Connolly, E.M.; Li, J.; Manos, M.P.; Lawrence, D.; McDermott, D.; Severgnini, M.; et al. Angiopoietin-2 as a biomarker and target for immune checkpoint therapy. Cancer Immunol. Res. 2017, 5, 17–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamphorst, A.O.; Pillai, R.N.; Yang, S.; Nasti, T.H.; Akondy, R.S.; Wieland, A.; Sica, G.L.; Yu, K.; Koenig, L.; Patel, N.T.; et al. Proliferation of PD-1+ CD8 T cells in peripheral blood after PD-1-targeted therapy in lung cancer patients. Proc. Natl. Acad. Sci. USA 2017, 114, 4993–4998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, K.H.; Cho, J.; Ku, B.M.; Koh, J.; Sun, J.M.; Lee, S.H.; Ahn, J.S.; Cheon, J.; Min, Y.J.; Park, S.H.; et al. The first-week proliferative response of peripheral blood PD-1þCD8þ T cells predicts the response to Anti-PD-1 therapy in solid tumors. Clin. Cancer Res. 2019, 25, 2144–2154. [Google Scholar] [CrossRef]
- Ferrara, R.; Naigeon, M.; Auclin, E.; Duchemann, B.; Cassard, L.; Jouniaux, J.M.; Boselli, L.; Grivel, J.; Desnoyer, A.; Mezquita, L.; et al. Circulating T-cell immunosenescence in patients with advanced non⇓small cell lung cancer treated with single-agent PD-1/PD-L1 inhibitors or platinum-based chemotherapy. Clin. Cancer Res. 2021, 27, 492–503. [Google Scholar] [CrossRef]
- Carnio, S.; Mariniello, A.; Pizzutilo, P.; Numico, G.; Borra, G.; Lunghi, A.; Soto Parra, H.; Buosi, R.; Vavalà, T.; Stura, I.; et al. ROC Analysis Identifies Baseline and Dynamic NLR and dNLR Cut-Offs to Predict ICI Outcome in 402 Advanced NSCLC Patients. J. Mol. Pathol. 2020, 1, 4. [Google Scholar] [CrossRef]
- Mezquita, L.; Auclin, E.; Ferrara, R.; Charrier, M.; Remon, J.; Planchard, D.; Ponce, S.; Ares, L.P.; Leroy, L.; Audigier-Valette, C.; et al. Association of the lung immune prognostic index with immune checkpoint inhibitor outcomes in patients with advanced non-small cell lung cancer. JAMA Oncol. 2018, 4, 351–357. [Google Scholar] [CrossRef]
- Möller, M.; Turzer, S.; Schütte, W.; Seliger, B.; Riemann, D. Blood Immune Cell Biomarkers in Patient with Lung Cancer Undergoing Treatment with Checkpoint Blockade. J. Immunother. 2020, 43, 57–66. [Google Scholar] [CrossRef]
- Sanmamed, M.F.; Perez-Gracia, J.L.; Schalper, K.A.; Fusco, J.P.; Gonzalez, A.; Rodriguez-Ruiz, M.E.; Oñate, C.; Perez, G.; Alfaro, C.; Martín-Algarra, S.; et al. Changes in serum interleukin-8 (IL-8) levels reflect and predict response to anti-PD-1 treatment in melanoma and non-small-cell lung cancer patients. Ann. Oncol. 2017, 28, 1988–1995. [Google Scholar] [CrossRef]
- Schalper, K.A.; Carleton, M.; Zhou, M.; Chen, T.; Feng, Y.; Huang, S.P.; Walsh, A.M.; Baxi, V.; Pandya, D.; Baradet, T.; et al. Elevated serum interleukin-8 is associated with enhanced intratumor neutrophils and reduced clinical benefit of immune-checkpoint inhibitors. Nat. Med. 2020, 26, 688–692. [Google Scholar] [CrossRef]
- Krieg, C.; Nowicka, M.; Guglietta, S.; Schindler, S.; Hartmann, F.J.; Weber, L.M.; Dummer, R.; Robinson, M.D.; Levesque, M.P.; Becher, B. High-dimensional single-cell analysis predicts response to anti-PD-1 immunotherapy. Nat. Med. 2018, 24, 144–153. [Google Scholar] [CrossRef]
- Kumar, V.; Patel, S.; Tcyganov, E.; Gabrilovich, D.I. The Nature of Myeloid-Derived Suppressor Cells in the Tumor Microenvironment. Trends Immunol. 2016, 37, 208–220. [Google Scholar] [CrossRef] [Green Version]
- Weber, R.; Fleming, V.; Hu, X.; Nagibin, V.; Groth, C.; Altevogt, P.; Utikal, J.; Umansky, V. Myeloid-derived suppressor cells hinder the anti-cancer activity of immune checkpoint inhibitors. Front. Immunol. 2018, 9, 1310. [Google Scholar] [CrossRef] [Green Version]
- Weide, B.; Martens, A.; Zelba, H.; Stutz, C.; Derhovanessian, E.; Di Giacomo, A.M.; Maio, M.; Sucker, A.; Schilling, B.; Schadendorf, D.; et al. Myeloid-derived suppressor cells predict survival of patients with advanced melanoma: Comparison with regulatory T cells and NY-ESO-1- or melan-A-specific T cells. Clin. Cancer Res. 2014, 20, 1601–1609. [Google Scholar] [CrossRef] [Green Version]
- Meyer, C.; Cagnon, L.; Costa-Nunes, C.M.; Baumgaertner, P.; Montandon, N.; Leyvraz, L.; Michielin, O.; Romano, E.; Speiser, D.E. Frequencies of circulating MDSC correlate with clinical outcome of melanoma patients treated with ipilimumab. Cancer Immunol. Immunother. 2014, 63, 247–257. [Google Scholar] [CrossRef] [Green Version]
- Youn, J.I.; Park, S.M.; Park, S.; Kim, G.; Lee, H.J.; Son, J.; Hong, M.H.; Ghaderpour, A.; Baik, B.; Islam, J.; et al. Peripheral natural killer cells and myeloid-derived suppressor cells correlate with anti-PD-1 responses in non-small cell lung cancer. Sci. Rep. 2020, 10. [Google Scholar] [CrossRef]
- Sivan, A.; Corrales, L.; Hubert, N.; Williams, J.B.; Aquino-Michaels, K.; Earley, Z.M.; Benyamin, F.W.; Lei, Y.M.; Jabri, B.; Alegre, M.L.; et al. Commensal Bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science (80-) 2015, 350, 1084–1089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iida, N.; Dzutsev, A.; Stewart, C.A.; Smith, L.; Bouladoux, N.; Weingarten, R.A.; Molina, D.A.; Salcedo, R.; Back, T.; Cramer, S.; et al. Commensal bacteria control cancer response to therapy by modulating the tumor microenvironment. Science (80-) 2013, 342, 967–970. [Google Scholar] [CrossRef] [PubMed]
- Chaput, N.; Lepage, P.; Coutzac, C.; Soularue, E.; Le Roux, K.; Monot, C.; Boselli, L.; Routier, E.; Cassard, L.; Collins, M.; et al. Corrigendum: Baseline gut microbiota predicts clinical response and colitis in metastatic melanoma patients treated with ipilimumab (Annals of Oncology (2017) 28:6 (1368–1379) DOI: 10.1093/annonc/mdx108). Ann. Oncol. 2019, 30, 2012. [Google Scholar] [CrossRef] [PubMed]
- Matson, V.; Fessler, J.; Bao, R.; Chongsuwat, T.; Zha, Y.; Alegre, M.L.; Luke, J.J.; Gajewski, T.F. The commensal microbiome is associated with anti-PD-1 efficacy in metastatic melanoma patients. Science (80-) 2018, 359, 104–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Routy, B.; Le Chatelier, E.; Derosa, L.; Duong, C.P.M.; Alou, M.T.; Daillère, R.; Fluckiger, A.; Messaoudene, M.; Rauber, C.; Roberti, M.P.; et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science (80-) 2018, 359, 91–97. [Google Scholar] [CrossRef] [Green Version]
- Parham, P.; Ohta, T. Population biology of antigen presentation by MHC class I molecules. Science (80-) 1996, 272, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Chowell, D.; Morris, L.G.T.; Grigg, C.M.; Weber, J.K.; Samstein, R.M.; Makarov, V.; Kuo, F.; Kendall, S.M.; Requena, D.; Riaz, N.; et al. Patient HLA class I genotype influences cancer response to checkpoint blockade immunotherapy. Science (80-) 2018, 359, 582–587. [Google Scholar] [CrossRef] [Green Version]
- Vargas, F.A.; Furness, A.J.S.; Litchfield, K.; Joshi, K.; Rosenthal, R.; Ghorani, E.; Solomon, I.; Lesko, M.H.; Ruef, N.; Roddie, C.; et al. Fc Effector Function Contributes to the Activity of Human Anti-CTLA-4 Antibodies. Cancer Cell 2018, 33, 649–663.e4. [Google Scholar] [CrossRef] [Green Version]
- Aide, N.; Lasnon, C.; Kesner, A.; Levin, C.S.; Buvat, I.; Iagaru, A.; Hermann, K.; Badawi, R.D.; Cherry, S.R.; Bradley, K.M.; et al. New PET technologies–embracing progress and pushing the limits. Eur. J. Nucl. Med. Mol. Imaging 2021. [Google Scholar] [CrossRef] [PubMed]
- Luke, J.J.; Flaherty, K.T.; Ribas, A.; Long, G.V. Targeted agents and immunotherapies: Optimizing outcomes in melanoma. Nat. Rev. Clin. Oncol. 2017, 14, 463–482. [Google Scholar] [CrossRef] [Green Version]
- Grootjans, W.; De Geus-Oei, L.F.; Troost, E.G.C.; Visser, E.P.; Oyen, W.J.G.; Bussink, J. PET in the management of locally advanced and metastatic NSCLC. Nat. Rev. Clin. Oncol. 2015, 12, 395–407. [Google Scholar] [CrossRef]
- Salaün, P.Y.; Abgral, R.; Malard, O.; Querellou-Lefranc, S.; Quere, G.; Wartski, M.; Coriat, R.; Hindie, E.; Taieb, D.; Tabarin, A.; et al. Good clinical practice recommendations for the use of PET/CT in oncology. Eur. J. Nucl. Med. Mol. Imaging 2020, 47, 28–50. [Google Scholar] [CrossRef]
- Fuchs, S.; Grössmann, N.; Ferch, M.; Busse, R.; Wild, C. Evidence-based indications for the planning of PET or PET/CT capacities are needed. Clin. Transl. Imaging 2019, 7, 65–81. [Google Scholar] [CrossRef] [Green Version]
- Costa, L.B.; Queiroz, M.A.; Barbosa, F.G.; Nunes, R.F.; Zaniboni, E.C.; Ruiz, M.M.; Jardim, D.; Marin, J.F.G.; Cerri, G.G.; Buchpiguel, C.A. Reassessing patterns of response to immunotherapy with pet: From morphology to metabolism. Radiographics 2021, 41, 120–143. [Google Scholar] [CrossRef]
- Aide, N.; De Pontdeville, M.; Lopci, E. Evaluating response to immunotherapy with 18F-FDG PET/CT: Where do we stand? Eur. J. Nucl. Med. Mol. Imaging 2020, 47, 1019–1021. [Google Scholar] [CrossRef] [Green Version]
- Rossi, G.; Bauckneht, M.; Genova, C.; Rijavec, E.; Biello, F.; Mennella, S.; Dal Bello, M.G.; Cittadini, G.; Bruzzi, P.; Piva, R.; et al. Comparison Between 18F-FDG PET-Based and CT-Based Criteria in Non-Small Cell Lung Cancer Patients Treated with Nivolumab. J. Nucl. Med. 2020, 61, 990–998. [Google Scholar] [CrossRef] [PubMed]
- Liberini, V.; Laudicella, R.; Capozza, M.; Huellner, M.W.; Burger, I.A.; Baldari, S.; Terreno, E.; Deandreis, D. The future of cancer diagnosis, treatment and surveillance: A systemic review on immunotherapy and immuno-pet radiotracers. Molecules 2021, 26, 2201. [Google Scholar] [CrossRef] [PubMed]
- Kandathil, A.; Kay, F.U.; Butt, Y.M.; Wachsmann, J.W.; Subramaniam, R.M. Role of FDG PET/CT in the eighth edition of TNM staging of non– Small cell lung cancer. Radiographics 2018, 38, 2134–2149. [Google Scholar] [CrossRef] [PubMed]
- Ettinger, D.S.; Wood, D.E.; Chair, V.; Aisner, D.L.; Akerley, W.; Bauman, J.R.; Bharat, A.; Lurie, R.H.; Bruno, D.S.; Chang, J.Y.; et al. Continue NCCN Guidelines Panel Disclosures NCCN Guidelines Version 4. Non-Small Cell Lung Cancer 2021, 19, 254–266. [Google Scholar]
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef] [Green Version]
- Kaira, K.; Kuji, I.; Kagamu, H. Value of 18F-FDG-PET to predict PD-L1 expression and outcomes of PD-1 inhibition therapy in human cancers. Cancer Imaging 2021, 21, 11. [Google Scholar] [CrossRef]
- Cho, S.Y.; Huff, D.T.; Jeraj, R.; Albertini, M.R. FDG PET/CT for Assessment of Immune Therapy: Opportunities and Understanding Pitfalls. Semin. Nucl. Med. 2020, 50, 518–531. [Google Scholar] [CrossRef]
- Iravani, A.; Hicks, R.J. Imaging the cancer immune environment and its response to pharmacologic intervention, Part 1: The role of 18F-FDG PET/CT. J. Nucl. Med. 2020, 61, 943–950. [Google Scholar] [CrossRef]
- Beer, L.; Hochmair, M.; Haug, A.R.; Schwabel, B.; Kifjak, D.; Wadsak, W.; Fuereder, T.; Fabikan, H.; Fazekas, A.; Schwab, S.; et al. Comparison of RECIST, iRECIST, and PERCIST for the evaluation of response to PD-1/PD-L1 blockade therapy in patients with non-small cell lung cancer. Clin. Nucl. Med. 2019, 44, 535–543. [Google Scholar] [CrossRef] [PubMed]
- Mulkey, F.; Theoret, M.R.; Keegan, P.; Pazdur, R.; Sridhara, R. Comparison of iRECIST versus RECIST V.1.1 in patients treated with an anti-PD-1 or PD-L1 antibody: Pooled FDA analysis. J. Immunother. Cancer 2020, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tazdait, M.; Mezquita, L.; Lahmar, J.; Ferrara, R.; Bidault, F.; Ammari, S.; Balleyguier, C.; Planchard, D.; Gazzah, A.; Soria, J.C.; et al. Patterns of responses in metastatic NSCLC during PD-1 or PDL-1 inhibitor therapy: Comparison of RECIST 1.1, irRECIST and iRECIST criteria. Eur. J. Cancer 2018, 88, 38–47. [Google Scholar] [CrossRef]
- Boellaard, R.; Delgado-Bolton, R.; Oyen, W.J.G.; Giammarile, F.; Tatsch, K.; Eschner, W.; Verzijlbergen, F.J.; Barrington, S.F.; Pike, L.C.; Weber, W.A.; et al. FDG PET/CT: EANM procedure guidelines for tumour imaging: Version 2.0. Eur. J. Nucl. Med. Mol. Imaging 2015, 42, 328–354. [Google Scholar] [CrossRef] [PubMed]
- Stephen Hodi, F.; Ballinger, M.; Lyons, B.; Soria, J.C.; Nishino, M.; Tabernero, J.; Powles, T.; Smith, D.; Hoos, A.; McKenna, C.; et al. Immune-modified response evaluation criteria in solid tumors (imrecist): Refining guidelines to assess the clinical benefit of cancer immunotherapy. J. Clin. Oncol. 2018, 36, 850–858. [Google Scholar] [CrossRef]
- Ito, K.; Teng, R.; Schöder, H.; Humm, J.L.; Ni, A.; Michaud, L.; Nakajima, R.; Yamashita, R.; Wolchok, J.D.; Weber, W.A. 18 F-FDG PET/CT for monitoring of ipilimumab therapy in patients with metastatic melanoma. J. Nucl. Med. 2019, 60, 335–341. [Google Scholar] [CrossRef] [Green Version]
- Anwar, H.; Sachpekidis, C.; Winkler, J.; Kopp-Schneider, A.; Haberkorn, U.; Hassel, J.C.; Dimitrakopoulou-Strauss, A. Absolute number of new lesions on 18F-FDG PET/CT is more predictive of clinical response than SUV changes in metastatic melanoma patients receiving ipilimumab. Eur. J. Nucl. Med. Mol. Imaging 2018, 45, 376–383. [Google Scholar] [CrossRef]
- Wolchok, J.D.; Hoos, A.; O’Day, S.; Weber, J.S.; Hamid, O.; Lebbé, C.; Maio, M.; Binder, M.; Bohnsack, O.; Nichol, G.; et al. Guidelines for the evaluation of immune therapy activity in solid tumors: Immune-related response criteria. Clin. Cancer Res. 2009, 15, 7412–7420. [Google Scholar] [CrossRef] [Green Version]
- Therasse, P.; Arbuck, S.G.; Eisenhauer, E.A.; Wanders, J.; Kaplan, R.S.; Rubinstein, L.; Verweij, J.; Van Glabbeke, M.; Van Oosterom, A.T.; Christian, M.C.; et al. New guidelines to evaluate the response to treatment in solid tumors. J. Natl. Cancer Inst. 2000, 92, 205–216. [Google Scholar] [CrossRef] [Green Version]
- Wahl, R.L.; Jacene, H.; Kasamon, Y.; Lodge, M.A. From RECIST to PERCIST: Evolving considerations for PET response criteria in solid tumors. J. Nucl. Med. 2009, 50, 122S. [Google Scholar] [CrossRef] [Green Version]
- Park, H.J.; Kim, K.W.; Pyo, J.; Suh, C.H.; Yoon, S.; Hatabu, H.; Nishino, M. Incidence of pseudoprogression during immune checkpoint inhibitor therapy for solid tumors: A systematic review and meta-Analysis. Radiology 2020, 297, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Humbert, O.; Cadour, N.; Paquet, M.; Schiappa, R.; Poudenx, M.; Chardin, D.; Borchiellini, D.; Benisvy, D.; Ouvrier, M.J.; Zwarthoed, C.; et al. 18FDG PET/CT in the early assessment of non-small cell lung cancer response to immunotherapy: Frequency and clinical significance of atypical evolutive patterns. Eur. J. Nucl. Med. Mol. Imaging 2020, 47, 1158–1167. [Google Scholar] [CrossRef] [PubMed]
- Champiat, S.; Dercle, L.; Ammari, S.; Massard, C.; Hollebecque, A.; Postel-Vinay, S.; Chaput, N.; Eggermont, A.; Marabelle, A.; Soria, J.C.; et al. Hyperprogressive disease is a new pattern of progression in cancer patients treated by anti-PD-1/PD-L1. Clin. Cancer Res. 2017, 23, 1920–1928. [Google Scholar] [CrossRef] [Green Version]
- Castello, A.; Rossi, S.; Mazziotti, E.; Toschi, L.; Lopci, E. Hyperprogressive Disease in Patients with Non-Small Cell Lung Cancer Treated with Checkpoint Inhibitors: The Role of 18F-FDG PET/CT. J. Nucl. Med. 2020, 61, 821–826. [Google Scholar] [CrossRef]
- Donegani, M.I.; Ferrarazzo, G.; Marra, S.; Miceli, A.; Raffa, S.; Bauckneht, M.; Morbelli, S. Positron emission tomography-based response to target and immunotherapies in oncology. Medicina 2020, 56, 373. [Google Scholar] [CrossRef]
- Aide, N.; Hicks, R.J.; Le Tourneau, C.; Lheureux, S.; Fanti, S.; Lopci, E. FDG PET/CT for assessing tumour response to immunotherapy: Report on the EANM symposium on immune modulation and recent review of the literature. Eur. J. Nucl. Med. Mol. Imaging 2019, 46, 238–250. [Google Scholar] [CrossRef] [Green Version]
- Eze, C.; Schmidt-Hegemann, N.S.; Sawicki, L.M.; Kirchner, J.; Roengvoraphoj, O.; Käsmann, L.; Mittlmeier, L.M.; Kunz, W.G.; Tufman, A.; Dinkel, J.; et al. PET/CT imaging for evaluation of multimodal treatment efficacy and toxicity in advanced NSCLC—current state and future directions. Eur. J. Nucl. Med. Mol. Imaging 2021. [Google Scholar] [CrossRef]
- Eshghi, N.; Garland, L.L.; Nia, E.; Betancourt, R.; Krupinski, E.; Kuo, P.H. 18F-FDG PET/CT can predict development of thyroiditis due to immunotherapy for lung cancer. J. Nucl. Med. Technol. 2018, 46, 260–264. [Google Scholar] [CrossRef] [Green Version]
- Rizwan, S.; Alhamad, K.; Abel, S.; Bakalov, V.; Rodriguez, R.R.; Sethi, A.; Khan, T.; Attah, A.A.; Yasir, M.; Shankar, K.; et al. Impact of immune-related adverse events on survival in patients with advanced non-small cell lung cancer treated with immune checkpoint inhibitors: A real-world perspective. J. Clin. Oncol. 2021, 39, e21213. [Google Scholar] [CrossRef]
- Prigent, K.; Aide, N. 18F-Fludeoxyglucose PET/Computed Tomography for Assessing Tumor Response to Immunotherapy and Detecting Immune-Related Side Effects: A Checklist for the PET Reader. PET Clin. 2020, 15, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Nobashi, T.; Baratto, L.; Reddy, S.A.; Srinivas, S.; Toriihara, A.; Hatami, N.; Yohannan, T.K.; Mittra, E. Predicting Response to Immunotherapy by Evaluating Tumors, Lymphoid Cell-Rich Organs, and Immune-Related Adverse Events Using FDG-PET/CT. Clin. Nucl. Med. 2019, 44, e272–e279. [Google Scholar] [CrossRef] [PubMed]
- Lambin, P.; Leijenaar, R.T.H.; Deist, T.M.; Peerlings, J.; De Jong, E.E.C.; Van Timmeren, J.; Sanduleanu, S.; Larue, R.T.H.M.; Even, A.J.G.; Jochems, A.; et al. Radiomics: The Bridge between Medical Imaging and Personalized Medicine. Available online: https://www.ncbi.nlm.nih.gov/pubmed/?term=Lambin++Radiomics%3A+the+bridge+between+medical+imaging+and+personalized+medicine (accessed on 7 February 2020).
- Zwanenburg, A.; Leger, S.; Vallières, M.; Löck, S. Image Biomarker Standardisation Initiative. arXiv 2016, arXiv:1612.07003. [Google Scholar]
- Sanduleanu, S.; Woodruff, H.C.; de Jong, E.E.C.; van Timmeren, J.E.; Jochems, A.; Dubois, L.; Lambin, P. Tracking tumor biology with radiomics: A systematic review utilizing a radiomics quality score. Radiother. Oncol. 2018, 127, 349–360. [Google Scholar] [CrossRef]
- Mayerhoefer, M.E.; Materka, A.; Langs, G.; Häggström, I.; Szczypiński, P.; Gibbs, P.; Cook, G. Introduction to Radiomics. J. Nucl. Med. 2020, 61, 488–495. [Google Scholar] [CrossRef] [PubMed]
- Gatta, R.; Depeursinge, A.; Ratib, O.; Michielin, O.; Leimgruber, A. Integrating radiomics into holomics for personalised oncology: From algorithms to bedside. Eur. Radiol. Exp. 2020, 4, 11. [Google Scholar] [CrossRef]
- Froelich, J.W.; Salavati, A. Artificial intelligence in PET/CT is about to make whole-body tumor burden measurements a clinical reality. Radiology 2020, 294, 453–454. [Google Scholar] [CrossRef]
- Laudicella, R.; Comelli, A.; Stefano, A.; Szostek, M.; Crocè, L.; Vento, A.; Spataro, A.; Comis, A.D.; La Torre, F.; Gaeta, M.; et al. Artificial Neural Networks in Cardiovascular Diseases and its Potential for Clinical Application in Molecular Imaging. Curr. Radiopharm. 2020, 13. [Google Scholar] [CrossRef]
- Piñeiro-Fiel, M.; Moscoso, A.; Pubul, V.; Ruibal, Á.; Silva-Rodríguez, J.; Aguiar, P. A Systematic Review of PET Textural Analysis and Radiomics in Cancer. Diagnostics 2021, 11, 380. [Google Scholar] [CrossRef] [PubMed]
- Yoon, H.J.; Sohn, I.; Cho, J.H.; Lee, H.Y.; Kim, J.H.; Choi, Y.L.; Kim, H.; Lee, G.; Lee, K.S.; Kim, J. Decoding tumor phenotypes for ALK, ROS1, and RET fusions in lung adenocarcinoma using a radiomics approach. Medicine 2015, 94. [Google Scholar] [CrossRef]
- Yip, S.S.F.; Kim, J.; Coroller, T.P.; Parmar, C.; Velazquez, E.R.; Huynh, E.; Mak, R.H.; Aerts, H.J.W.L. Associations between somatic mutations and metabolic imaging phenotypes in non-small cell lung cancer. J. Nucl. Med. 2017, 58, 569–576. [Google Scholar] [CrossRef]
- Li, X.; Yin, G.; Zhang, Y.; Dai, D.; Liu, J.; Chen, P.; Zhu, L.; Ma, W.; Xu, W. Predictive Power of a Radiomic Signature Based on 18F-FDG PET/CT Images for EGFR Mutational Status in NSCLC. Front. Oncol. 2019, 9. [Google Scholar] [CrossRef] [Green Version]
- Jiang, M.; Zhang, Y.; Xu, J.; Ji, M.; Guo, Y.Y.; Guo, Y.Y.; Xiao, J.; Yao, X.; Shi, H.; Zeng, M. Assessing EGFR gene mutation status in non-small cell lung cancer with imaging features from PET/CT. Nucl. Med. Commun. 2019, 40, 842–849. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.J.; Zhao, X.; Zhao, Y.; Zhang, J.J.; Zhang, Z.; Wang, J.; Wang, Y.; Dai, M.; Han, J. Value of pre-therapy 18F-FDG PET/CT radiomics in predicting EGFR mutation status in patients with non-small cell lung cancer. Eur. J. Nucl. Med. Mol. Imaging 2020, 47, 1137–1146. [Google Scholar] [CrossRef]
- Koyasu, S.; Nishio, M.; Isoda, H.; Nakamoto, Y.; Togashi, K. Usefulness of gradient tree boosting for predicting histological subtype and EGFR mutation status of non-small cell lung cancer on 18F FDG-PET/CT. Ann. Nucl. Med. 2020, 34, 49–57. [Google Scholar] [CrossRef]
- Liu, Q.; Sun, D.; Li, N.; Kim, J.; Feng, D.; Huang, G.; Wang, L.; Song, S. Predicting EGFR mutation subtypes in lung adenocarcinoma using 18F-FDG PET/CT radiomic features. Transl. Lung Cancer Res. 2020, 9, 549–562. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Ji, H.S.; Zhou, C.S.; Dong, H.; Ma, L.; Ge, Y.Q.; Zhu, C.H.; Tian, J.H.; Zhang, L.J.; Zhu, H.; et al. 18F-fluorodeoxyglucose positron emission tomography/computed tomography-based radiomic features for prediction of epidermal growth factor receptor mutation status and prognosis in patients with lung adenocarcinoma. Transl. Lung Cancer Res. 2020, 9, 563–574. [Google Scholar] [CrossRef]
- Jiang, M.; Sun, D.; Guo, Y.; Guo, Y.; Xiao, J.; Wang, L.; Yao, X. Assessing PD-L1 Expression Level by Radiomic Features From PET/CT in Nonsmall Cell Lung Cancer Patients: An Initial Result. Acad. Radiol. 2020, 27, 171–179. [Google Scholar] [CrossRef]
- Lopci, E.; Toschi, L.; Grizzi, F.; Rahal, D.; Olivari, L.; Castino, G.F.; Marchetti, S.; Cortese, N.; Qehajaj, D.; Pistillo, D.; et al. Correlation of metabolic information on FDG-PET with tissue expression of immune markers in patients with non-small cell lung cancer (NSCLC) who are candidates for upfront surgery. Eur. J. Nucl. Med. Mol. Imaging 2016, 43, 1954–1961. [Google Scholar] [CrossRef] [PubMed]
- Takada, K.; Toyokawa, G.; Yoneshima, Y.; Tanaka, K.; Okamoto, I.; Shimokawa, M.; Wakasu, S.; Haro, A.; Osoegawa, A.; Tagawa, T.; et al. 18F-FDG uptake in PET/CT is a potential predictive biomarker of response to anti-PD-1 antibody therapy in non-small cell lung cancer. Sci. Rep. 2019, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grizzi, F.; Castello, A.; Lopci, E. Is it time to change our vision of tumor metabolism prior to immunotherapy? Eur. J. Nucl. Med. Mol. Imaging 2018, 45, 1072–1075. [Google Scholar] [CrossRef]
- Monaco, L.; Gemelli, M.; Gotuzzo, I.; Bauckneht, M.; Crivellaro, C.; Genova, C.; Cortinovis, D.; Zullo, L.; Ammoni, L.C.; Bernasconi, D.P.; et al. Metabolic parameters as biomarkers of response to immunotherapy and prognosis in non-small cell lung cancer (Nsclc): A real world experience. Cancers 2021, 13, 1634. [Google Scholar] [CrossRef] [PubMed]
- Quartuccio, N.; Evangelista, L.; Alongi, P.; Caobelli, F.; Altini, C.; Cistaro, A.; Lambertini, A.; Schiorlin, I.; Popescu, C.E.; Linguanti, F.; et al. Prognostic and diagnostic value of [18F]FDG-PET/CT in restaging patients with small cell lung carcinoma: An Italian multicenter study. Nucl. Med. Commun. 2019, 40, 808–814. [Google Scholar] [CrossRef] [PubMed]
- Lee, P.; Weerasuriya, D.K.; Lavori, P.W.; Quon, A.; Hara, W.; Maxim, P.G.; Le, Q.T.; Wakelee, H.A.; Donington, J.S.; Graves, E.E.; et al. {A figure is presented}Metabolic Tumor Burden Predicts for Disease Progression and Death in Lung Cancer. Int. J. Radiat. Oncol. Biol. Phys. 2007, 69, 328–333. [Google Scholar] [CrossRef] [PubMed]
- Lee, P.; Bazan, J.G.; Lavori, P.W.; Weerasuriya, D.K.; Quon, A.; Le, Q.T.; Wakelee, H.A.; Graves, E.E.; Loo, B.W. Metabolic tumor volume is an independent prognostic factor in patients treated definitively for nonsmall-cell lung cancer. Clin. Lung Cancer 2012, 13, 52–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polverari, G.; Ceci, F.; Bertaglia, V.; Reale, M.L.; Rampado, O.; Gallio, E.; Passera, R.; Liberini, V.; Scapoli, P.; Arena, V.; et al. 18F-FDG pet parameters and radiomics features analysis in advanced nsclc treated with immunotherapy as predictors of therapy response and survival. Cancers 2020, 12, 1163. [Google Scholar] [CrossRef]
- Kim, B.S.; Kang, J.; Jun, S.; Kim, H.; Pak, K.; Kim, G.H.; Heo, B.J.; Kim, Y.H. Association between immunotherapy biomarkers and glucose metabolism from F-18 FDG PET. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 8288–8295. [Google Scholar] [CrossRef]
- Valentinuzzi, D.; Vrankar, M.; Boc, N.; Ahac, V.; Zupancic, Z.; Unk, M.; Skalic, K.; Zagar, I.; Studen, A.; Simoncic, U.; et al. FDG PET immunotherapy radiomics signature (iRADIOMICS) predicts response of non-small-cell lung cancer patients treated with pembrolizumab. Radiol. Oncol. 2020, 54, 285–294. [Google Scholar] [CrossRef]
- Seban, R.D.; Mezquita, L.; Berenbaum, A.; Dercle, L.; Botticella, A.; Le Pechoux, C.; Caramella, C.; Deutsch, E.; Grimaldi, S.; Adam, J.; et al. Baseline metabolic tumor burden on FDG PET/CT scans predicts outcome in advanced NSCLC patients treated with immune checkpoint inhibitors. Eur. J. Nucl. Med. Mol. Imaging 2020, 47, 1147–1157. [Google Scholar] [CrossRef]
- Bauckneht, M.; Genova, C.; Rossi, G.; Rijavec, E.; Bello, M.G.D.; Ferrarazzo, G.; Tagliamento, M.; Donegani, M.I.; Biello, F.; Chiola, S.; et al. The role of the immune metabolic prognostic index in patients with non-small cell lung cancer (Nsclc) in radiological progression during treatment with nivolumab. Cancers 2021, 13, 3117. [Google Scholar] [CrossRef]
- Ziv, E.; Durack, J.C.; Solomon, S.B. The Importance of Biopsy in the Era of Molecular Medicine. Cancer J. 2016, 22, 418–422. [Google Scholar] [CrossRef] [Green Version]
- McQuerry, J.A.; Chang, J.T.; Bowtell, D.D.L.; Cohen, A.; Bild, A.H. Mechanisms and clinical implications of tumor heterogeneity and convergence on recurrent phenotypes. J. Mol. Med. 2017, 95, 1167–1178. [Google Scholar] [CrossRef]
- Kather, J.N.; Suarez-Carmona, M.; Charoentong, P.; Weis, C.A.; Hirsch, D.; Bankhead, P.; Horning, M.; Ferber, D.; Kel, I.; Herpel, E.; et al. Topography of cancer-associated immune cells in human solid tumors. eLife 2018, 7, e36967. [Google Scholar] [CrossRef]
- Pietrobon, V.; Cesano, A.; Marincola, F.; Kather, J.N. Next Generation Imaging Techniques to Define Immune Topographies in Solid Tumors. Front. Immunol. 2021, 11, 3519. [Google Scholar] [CrossRef]
- Pandit-Taskar, N.; Postow, M.A.; Hellmann, M.D.; Harding, J.J.; Barker, C.A.; O’Donoghue, J.A.; Ziolkowska, M.; Ruan, S.; Lyashchenko, S.K.; Tsai, F.; et al. First-in-Humans Imaging with 89Zr-Df-IAB22M2C Anti-CD8 Minibody in Patients with Solid Malignancies: Preliminary Pharmacokinetics, Biodistribution, and Lesion Targeting. J. Nucl. Med. 2020, 61, 512–519. [Google Scholar] [CrossRef] [PubMed]
- Barrett, R.L.; Pure, E. Cancer-associated fibroblasts and their influence on tumor immunity and immunotherapy. eLife 2020, 9, e57243. [Google Scholar] [CrossRef]
- Natarajan, A.; Mayer, A.T.; Xu, L.; Reeves, R.E.; Gano, J.; Gambhir, S.S. Novel Radiotracer for ImmunoPET Imaging of PD-1 Checkpoint Expression on Tumor Infiltrating Lymphocytes. Bioconjug. Chem. 2015, 26, 2062–2069. [Google Scholar] [CrossRef] [PubMed]
- Bensch, F.; van der Veen, E.L.; Lub-de Hooge, M.N.; Jorritsma-Smit, A.; Boellaard, R.; Kok, I.C.; Oosting, S.F.; Schröder, C.P.; Hiltermann, T.J.N.; van der Wekken, A.J.; et al. 89Zr-atezolizumab imaging as a non-invasive approach to assess clinical response to PD-L1 blockade in cancer. Nat. Med. 2018, 24, 1852–1858. [Google Scholar] [CrossRef] [PubMed]
- Niemeijer, A.N.; Leung, D.; Huisman, M.C.; Bahce, I.; Hoekstra, O.S.; van Dongen, G.A.M.S.; Boellaard, R.; Du, S.; Hayes, W.; Smith, R.; et al. Whole body PD-1 and PD-L1 positron emission tomography in patients with non-small-cell lung cancer. Nat. Commun. 2018, 9, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Sliker, B.H.; Campbell, P.M. Fibroblasts influence the efficacy, resistance, and future use of vaccines and immunotherapy in cancer treatment. Vaccines 2021, 9, 634. [Google Scholar] [CrossRef]
- Kraman, M.; Bambrough, P.J.; Arnold, J.N.; Roberts, E.W.; Magiera, L.; Jones, J.O.; Gopinathan, A.; Tuveson, D.A.; Fearon, D.T. Suppression of antitumor immunity by stromal cells expressing fibroblast activation protein-α. Science (80-) 2010, 330, 827–830. [Google Scholar] [CrossRef] [Green Version]
- Loktev, A.; Lindner, T.; Mier, W.; Debus, J.; Altmann, A.; Jäger, D.; Giesel, F.; Kratochwil, C.; Barthe, P.; Roumestand, C.; et al. A tumor-imaging method targeting cancer-associated fibroblasts. J. Nucl. Med. 2018, 59, 1423–1429. [Google Scholar] [CrossRef]
- Kratochwil, C.; Flechsig, P.; Lindner, T.; Abderrahim, L.; Altmann, A.; Mier, W.; Adeberg, S.; Rathke, H.; Röhrich, M.; Winter, H.; et al. 68Ga-FAPI PET/CT: Tracer uptake in 28 different kinds of cancer. J. Nucl. Med. 2019, 60, 801–805. [Google Scholar] [CrossRef] [Green Version]
- Flechsig, P.; Lindner, T.; Abderrahim, L.; Altmann, A.; Mier, W.; Adeberg, S.; Rathke, H.; Winter, H.; Plinkert, P.; Marme, F.; et al. SNMMI Image of the Year: Novel Radiotracer Detects 28 Cancer Types, Paving the Way for Development of New Therapies-SNMMI. Available online: https://www.snmmi.org/NewsPublications/NewsDetail.aspx?ItemNumber=32020 (accessed on 15 August 2021).
- Calais, J.; Mona, C.E. Will FAPI PET/CT replace FDG PET/CT in the next decade? Point—An important diagnostic, phenotypic, and biomarker role. Am. J. Roentgenol. 2021, 216, 305–306. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Pang, Y.; Wu, J.; Zhao, L.; Hao, B.; Wu, J.; Wei, J.; Wu, S.; Zhao, L.; Luo, Z.; et al. Comparison of [68Ga]Ga-DOTA-FAPI-04 and [18F] FDG PET/CT for the diagnosis of primary and metastatic lesions in patients with various types of cancer. Eur. J. Nucl. Med. Mol. Imaging 2020, 47, 1820–1832. [Google Scholar] [CrossRef] [PubMed]
- Watabe, T.; Liu, Y.; Kaneda-Nakashima, K.; Shirakami, Y.; Lindner, T.; Ooe, K.; Toyoshima, A.; Nagata, K.; Shimosegawa, E.; Haberkorn, U.; et al. Theranostics targeting fibroblast activation protein in the tumor stroma: 64Cu- And 225Ac-labeled FAPI-04 in pancreatic cancer xenograft mouse models. J. Nucl. Med. 2020, 61, 563–569. [Google Scholar] [CrossRef] [PubMed]
- Kuyumcu, S.; Kovan, B.; Sanli, Y.; Buyukkaya, F.; Has Simsek, D.; Özkan, Z.G.; Isik, E.G.; Ekenel, M.; Turkmen, C. Safety of Fibroblast Activation Protein-Targeted Radionuclide Therapy by a Low-Dose Dosimetric Approach Using 177Lu-FAPI04. Clin. Nucl. Med. 2021, 46, 641–646. [Google Scholar] [CrossRef]
- Eryilmaz, K.; Kilbas, B. Fully-automated synthesis of 177Lu labelled FAPI derivatives on the module modular lab-Eazy. EJNMMI Radiopharm. Chem. 2021, 6. [Google Scholar] [CrossRef]
- Moradi, F.; Iagaru, A. Will FAPI PET/CT replace FDG PET/CT in the next decade? Counterpoint—No, not so fast! Am. J. Roentgenol. 2021, 216, 307–308. [Google Scholar] [CrossRef]
- Sollini, M.; Kirienko, M.; Gelardi, F.; Fiz, F.; Gozzi, N.; Chiti, A. State-of-the-art of FAPI-PET imaging: A systematic review and meta-analysis. Eur. J. Nucl. Med. Mol. Imaging 2021. [Google Scholar] [CrossRef]
- Finke, D.; Heckmann, M.B.; Herpel, E.; Katus, H.A.; Haberkorn, U.; Leuschner, F.; Lehmann, L.H. Early Detection of Checkpoint Inhibitor-Associated Myocarditis Using 68Ga-FAPI PET/CT. Front. Cardiovasc. Med. 2021, 8, 614997. [Google Scholar] [CrossRef]
- Hamson, E.J.; Keane, F.M.; Tholen, S.; Schilling, O.; Gorrell, M.D. Understanding fibroblast activation protein (FAP): Substrates, activities, expression and targeting for cancer therapy. Proteom.-Clin. Appl. 2014, 8, 454–463. [Google Scholar] [CrossRef]
- Theodoropoulos, A.S.; Gkiozos, I.; Kontopyrgias, G.; Charpidou, A.; Kotteas, E.; Kyrgias, G.; Tolia, M. Modern radiopharmaceuticals for lung cancer imaging with positron emission tomography/computed tomography scan: A systematic review. SAGE Open Med. 2020, 8. [Google Scholar] [CrossRef] [PubMed]
- Padhani, A. PET imaging of tumour hypoxia. Cancer Imaging 2006, 6, S117. [Google Scholar] [CrossRef] [PubMed]
- Pietrobon, V.; Marincola, F.M. Hypoxia and the phenomenon of immune exclusion. J. Transl. Med. 2021, 19, 9. [Google Scholar] [CrossRef]
- Rühle, A.; Grosu, A.L.; Wiedenmann, N.; Mix, M.; Stoian, R.; Niedermann, G.; Baltas, D.; Werner, M.; Weber, W.A.; Kayser, G.; et al. Hypoxia dynamics on FMISO-PET in combination with PD-1/PD-L1 expression has an impact on the clinical outcome of patients with Head-and-neck Squamous Cell Carcinoma undergoing Chemoradiation. Theranostics 2020, 10, 9395–9406. [Google Scholar] [CrossRef] [PubMed]
- Masłowska, K.; Halik, P.K.; Tymecka, D.; Misicka, A.; Gniazdowska, E. The role of vegf receptors as molecular target in nuclear medicine for cancer diagnosis and combination therapy. Cancers 2021, 13, 1072. [Google Scholar] [CrossRef]
- Bahce, I.; Yaqub, M.; Smit, E.F.; Lammertsma, A.A.; van Dongen, G.A.M.S.; Hendrikse, N.H. Personalizing NSCLC therapy by characterizing tumors using TKI-PET and immuno-PET. Lung Cancer 2017, 107, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bahce, I.; Huisman, M.C.; Verwer, E.E.; Ooijevaar, R.; Boutkourt, F.; Vugts, D.J.; van Dongen, G.A.M.S.; Boellaard, R.; Smit, E.F. Pilot study of 89Zr-bevacizumab positron emission tomography in patients with advanced non-small cell lung cancer. EJNMMI Res. 2014, 4, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
| ICI Class | DRUG | Stage | Line | FDA Indication | EMA Indication |
|---|---|---|---|---|---|
| CTLA-4 inhibitor | Ipilimumab | IV | 1st |
|
|
| PD-1 inhibitors | Nivolumab | IV | 1st |
|
|
| 2nd-N |
|
| |||
| Pembrolizumab | III *-IV | 1st |
As a single agent if tumor PD-L1 ≥1% (no EGFR or ALK mutations) [29] |
| |
| 2nd-N |
|
| |||
| Cemiplimab | III *-IV | 1st | |||
| PD-L1 inhibitors | Durvalumab | IIIA **-B | Consolidation after CH-RT |
|
|
| Atezolizumab | IV | 1st |
|
| |
| 2nd-N |
|
|
| Radiological Response Criteria | RECIST 1.1 [145] | irRC [144] | imRECIST [141] |
| Complete response (CR) | Disappearance of all target and non-target lesions without any new lesions. Any pathological lymph nodes must have reduction in short axis to <10 mm. Determined by two observations not less than 4 weeks apart. | Disappearance of all target lesions. Determined by two observations not less than 4 weeks apart. | Disappearance of all target and non-target lesions without any new lesions. Any pathological lymph nodes must have reduction in short axis to <10 mm. Determined by two observations not less than 4 weeks apart. |
| Partial response (PR) | At least a 30% decrease of the sum of maximum diameters of target lesions; no new lesions; no progression of disease. | Sum of product of all lesions decreased by >50% for at least 4 weeks; no new lesions; no progression of any lesions. | At least a 30% decrease of the sum of maximum diameters of target lesions; no new lesions; no progression of disease. |
| Stable disease (SD) | Does not meet the criteria for CR, PR, or PD, taking the smallest sum of the maximum diameters of target lesions as reference. | Sum of product of all lesions decreased by <50% or increased by <25% in the size of one or more lesions. | Does not meet the criteria for CR, PR, or PD, taking the smallest sum of maximum diameters of target lesions as reference. |
| Progressive disease (PD) | Sum of the maximum diameter of lesions increased by >20% over the smallest achieved sum of maximum diameter. The appearance of one or more new lesions is always considered progression. | A single lesion increased by >25% (over the smallest measurement achieved for the single lesion) or the appearance of new lesions that has to be confirmed in two consecutive observations at least 4 weeks apart. | Sum of the maximum diameter of lesions increased by >20% over the smallest achieved sum of maximum diameter. The appearance of new lesions and/or progression of non-target lesions are considered iUPD and must be confirmed 4–8 weeks later as iCPD. Progression is not confirmed in case of the shrinkage of these lesions at 4–8 weeks, and evaluation must be reset. |
| Functional Response Criteria | PERCIST [146] | imPERCIST [142] | PERCIMT [143] |
| Complete metabolic response (CMR) | Complete resolution of [18F]FDG uptake within all lesions to a level of less than or equal to that of the mean liver activity and that is indistinguishable from the background (blood pool uptake). | Complete resolution of [18F]FDG uptake within all lesions to a level of less than or equal to that of the mean liver activity and that is indistinguishable from the background (blood pool uptake). | Complete resolution of [18F]FDG uptake within all lesions to a level of less than or equal to that of the mean liver activity and that is indistinguishable from the background (blood pool uptake). |
| Partial metabolic response (PMR) | Reduction of at least 30% in the sum of SULpeak of all target lesions detected at baseline and an absolute drop of 0.8 SULpeak units. | Reduction of at least 30% in the sum of SULpeak of all target lesions detected at baseline and an absolute drop of 0.8 SULpeak units. | Reduction of at least 30% in the sum of SULpeak of all target lesions detected at baseline and an absolute drop of 0.8 SULpeak units. |
| Stable metabolic disease (SMD) | Does not meet the criteria for CR, PR, or PD. | Does not meet the criteria for CR, PR, or PD. | Does not meet the criteria for CR, PR, or PD. |
| Progressive metabolic disease (PMD) | Increase of at least 30% in the sum of SULpeak of all target lesions detected at baseline and an absolute increase of 0.8 SULpeak units. Or 75% increase in total lesions glycolysis (TLG) with no decrease in SUL. Or The appearance of one or more new FDG-avid lesions that are typical of cancer and that are not related to inflammation or infection is always considered progression. | Increase of at least 30% in the sum of SULpeak of all target lesions detected at baseline, or new FDG-avid lesions are considered UPMD and must be confirmed 4–8 weeks later as CPMD. Progression is not confirmed in case of PMR or SMD at 4–8 weeks, and evaluation must be reset. | Progressive disease if: 4 new lesions (<1 cm in functional diameter); 3 new lesions (>1 cm in functional diameter); 2 new lesions (>1.5 cm in functional diameter). |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liberini, V.; Mariniello, A.; Righi, L.; Capozza, M.; Delcuratolo, M.D.; Terreno, E.; Farsad, M.; Volante, M.; Novello, S.; Deandreis, D. NSCLC Biomarkers to Predict Response to Immunotherapy with Checkpoint Inhibitors (ICI): From the Cells to In Vivo Images. Cancers 2021, 13, 4543. https://doi.org/10.3390/cancers13184543
Liberini V, Mariniello A, Righi L, Capozza M, Delcuratolo MD, Terreno E, Farsad M, Volante M, Novello S, Deandreis D. NSCLC Biomarkers to Predict Response to Immunotherapy with Checkpoint Inhibitors (ICI): From the Cells to In Vivo Images. Cancers. 2021; 13(18):4543. https://doi.org/10.3390/cancers13184543
Chicago/Turabian StyleLiberini, Virginia, Annapaola Mariniello, Luisella Righi, Martina Capozza, Marco Donatello Delcuratolo, Enzo Terreno, Mohsen Farsad, Marco Volante, Silvia Novello, and Désirée Deandreis. 2021. "NSCLC Biomarkers to Predict Response to Immunotherapy with Checkpoint Inhibitors (ICI): From the Cells to In Vivo Images" Cancers 13, no. 18: 4543. https://doi.org/10.3390/cancers13184543
APA StyleLiberini, V., Mariniello, A., Righi, L., Capozza, M., Delcuratolo, M. D., Terreno, E., Farsad, M., Volante, M., Novello, S., & Deandreis, D. (2021). NSCLC Biomarkers to Predict Response to Immunotherapy with Checkpoint Inhibitors (ICI): From the Cells to In Vivo Images. Cancers, 13(18), 4543. https://doi.org/10.3390/cancers13184543