Simple Summary
Ovarian cancer (OC) represents the fifth leading cause of cancer-related deaths among women. In the advanced disease setting, OC recurrence after chemotherapy is over 70% in the first 2 years, with few therapeutic options. Immunotherapy with the immune checkpoint inhibitors (ICIs) showed high efficacy and changed the therapeutic scenario of many tumors in the last 10 years. With our review, we aimed to summarize the clinical trials of ICIs in OC. In OC, ICIs clinical trials have reported poor outcomes in terms of patient response and survival, with some studies failing to reach their objectives. Combining immunotherapy with drugs targeting different pathways might enhance efficacy and overcome cancer resistance. The search for biomarkers predicting ICIs response is essential for the identification of patients most likely to benefit from ICI therapy.
Abstract
Background: Ovarian cancer (OC) represents the eighth most common cancer and the fifth leading cause of cancer-related deaths among the female population. In an advanced setting, chemotherapy represents the first-choice treatment, despite a high recurrence rate. In the last ten years, immunotherapy based on immune checkpoint inhibitors (ICIs) has profoundly modified the therapeutic scenario of many solid tumors. We sought to summarize the main findings regarding the clinical use of ICIs in OC. Methods: We searched PubMed, Embase, and Cochrane Databases, and conference abstracts from international congresses (such as ASCO, ESMO, SGO) for clinical trials, focusing on ICIs both as monotherapy and as combinations in the advanced OC. Results: 20 studies were identified, of which 16 were phase I or II and 4 phase III trials. These trials used ICIs targeting PD1 (nivolumab, pembrolizumab), PD-L1 (avelumab, aterolizumab, durvalumab), and CTLA4 (ipilimumab, tremelimumab). There was no reported improvement in survival, and some trials were terminated early due to toxicity or lack of response. Combining ICIs with chemotherapy, anti-VEGF therapy, or PARP inhibitors improved response rates and survival in spite of a worse safety profile. Conclusions: The identification of biomarkers with a predictive role for ICIs’ efficacy is mandatory. Moreover, genomic and immune profiling of OC might lead to better treatment options and facilitate the design of tailored trials.
Keywords:
ovarian cancer; checkpoint inhibitors; ICIs; immunotherapy; PARP; avelumab; pembrolizumab; nivolumab; bevacizumab; platinum 1. Introduction
Ovarian cancer (OC) accounts for about 2% of tumors, representing the eighth most common cancer among the female population. The incidence is around 11 cases/100,000 inhabitants/year, and it is higher among white women [1,2]. The frequency of OC rises with age, being uncommon before 30, and more frequently presenting at 50–70. Globally, ovarian cancer represents the fifth leading cause of female cancer-related deaths, with a 5 y survival rate falling from 90% at stage I to 25% at stage IV [2]. The majority of OCs have an epithelial origin, among whom serous carcinoma has the most aggressive features and is usually diagnosed at advanced stages [3]. Platinum-based chemotherapy regimens represent the mainstay of treatment [4,5,6,7]. The response to these agents and the treatment-free interval (TFI) after platinum define the subsequent treatment, moving from the platinum-refractory (PR) (relapse < 6 months from the platinum end) to the platinum-sensitive (PS) patients (TFI > 12 mos). Despite initial benefits, disease recurrence occurs in over 2/3 of patients within the first two years. Therefore, new drugs were explored, and other agents such as the PARP-inhibitor (PARPi) agents and the anti-vascular endothelial growth factor (VEGF) bevacizumab were approved in the advanced setting [8,9].
Immunotherapy has represented a breakthrough therapy for many solid tumors [10]. Thus far, the best-studied mechanisms for inducing an immune response against tumors rely on inhibiting the immune checkpoint. The immune checkpoint inhibitors (ICIs) consist of monoclonal antibodies targeting Programmed Cell Death Protein 1 (PD-1)/Programmed Death-Ligand 1 (PD-L1) or Cytotoxic T-Lymphocyte Antigen 4 (CTLA-4), expressed by tumor or immune cells. After binding with these ligands, ICIs remove the inhibition signals for the immune system, unlocking the anti-tumor response [11]. However, in OC, ICIs reported modest results, and some phase III trials were prematurely terminated for futility. Combinations with other compounds, such as PARPis or anti-angiogenic drugs, represent promising opportunities to enhance the clinical effectiveness of immunotherapy [12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36].
We hereinafter ought to synthesize the clinical trials involving ICIs that were conducted in advanced OC to discuss the pros and cons and explore future perspectives to maximize the efficacy of immunotherapy for most women with advanced disease.
2. Materials and Methods
We searched the PubMed, EMBASE, and Cochrane databases and abstracts from international conferences (e.g., ASCO, ESMO, SGO). The terms (‘ovarian cancer’ OR ‘ovarian carcinoma’) AND (‘immune checkpoint inhibitor’ OR ICI OR avelumab OR nivolumab OR atezolizumab OR pembrolizumab OR durvalumab OR tremelimumab OR ipilimumab OR ‘anti PD1’ OR ‘anti PD-L1’ OR ‘anti CTLA4’) were used. Papers published in peer-reviewed journals and conference abstracts in the English language up to June 2021 were selected. We included clinical trials, whereas reviews, letters, and personal opinions were excluded. A total of 20 studies were included in our review.
3. Results
Since the FDA approved ipilimumab for advanced melanoma in 2011, the last ten years were characterized by the widespread use of ICIs, revolutionizing the therapeutic algorithm for many solid tumors [10,37]. ICIs are monoclonal antibodies that promote the anti-tumoral response of the host immune system through the inhibition of negative signals for effector T-cells. Among the ICIs tested in clinical trials in OC, pembrolizumab and nivolumab target PD-1, atezolizumab, avelumab, and durvalumab bound PD-L1, ipilimumab and tremelimumab are directed against CTLA-4. Sixteen phase I or II and 4 phase III trials were published (Table 1).

Table 1.
Trials of ICIs in OC.
3.1. Anti PD1 Agents
Nivolumab and Pembrolizumab were tested as single agents and combined with other ICIs, chemotherapy, anti-angiogenic agents, and PARPis.
3.1.1. Pembrolizumab
In the KEYNOTE-100 (NCT02674061) phase II study, pembrolizumab 200 mg q3w was administered to two cohorts of patients with recurrent ovarian cancer (ROC): cohort A enrolled 285 patients after one to three prior therapies with a treatment-free interval (TFI) of 3–12 months; cohort B included 91 progressive patients with up to six previous lines of therapy with a TFI of at least 3 mos. The primary endpoint was overall response rate (ORR) by Response Evaluation Criteria in Solid Tumors (RECIST) criteria and according to PD-L1 expression. Secondary endpoints included: duration of response (DoR), disease-control rate (DCR), progression-free survival (PFS), overall survival (OS), and safety. The combined ORR of the two cohorts was 8.0%, the overall DCR 37%, and around 1/3 of responses lasted more than 6 months. The mDoR was 8.2 mos in cohort A and not reached in cohort B. The mPFS was 2.1 mos [12]. PD-L1 positive patients (defined as a combined positive score-[CPS] ≥ 10) reached better results than PD-L1 negative, in terms of both ORR (17.1%) and mOS (21.9 mos-cohort A, and 24.0 mos-cohort B) [12,13]. The most common adverse events (AEs) were fatigue (33.8%), nausea (15.4%), and decreased appetite (10.6%), with 19.7% of women experiencing >G3 AEs. The most common immune-related AEs (irAEs) were thyroid disorders (17.5%). Two treatment-related deaths were recorded, and 5.1% of patients discontinued the treatment due to toxicity [12].
In the Keynote-028 (NCT02054806) multi-cohort phase Ib trial, only PD-L1 positive patients were included. Twenty-six women were treated with pembrolizumab in the OC cohort. The ORR represented the primary endpoint. After a median follow-up of 15.4 mos, ORR was 11.5%, mPFS 1.9 mos, and mOS was 13.8 mos. A total of 73.1% of patients experienced at least one treatment-related adverse event (TRAE): arthralgia (19.2%), nausea (15.4%), and pruritus (15.4%) were the most common. One G3 hypertransaminasemia was recorded, while no deaths or treatment discontinuation for toxicity occurred [14].
Attempts to combine pembrolizumab were made with chemotherapy, bevacizumab, or PARPis. Pembrolizumab plus liposomal doxorubicin (PLD) resulted in an ORR of 19%, 3 PR, and 1 SD >24 weeks, among 26 platinum-resistant patients, with no G4 or G5 toxicities. G2 pneumonitis occurred in 8% of patients. G3 AEs included rash (19%) and ALT increase (8%) [15]. In the NCT02853318 phase II trial, 40 platinum-progressive OC women were treated with pembrolizumab plus bevacizumab plus oral cyclophosphamide. The study met its primary endpoints, reaching an ORR of 47.5% and an mPFS of 10 mos. The 6-month PFS rate was 100% for the PS-ROC and 59% for the PR-ROC patients (p = 0.024). The most frequent AEs were fatigue (45.0%), diarrhea (32.5%), and hypertension (27.5%), while the most common ≥G3 AEs were hypertension (15.0%) and lymphopenia (7.5%) [16]. The TOPACIO/Keynote-162 (NCT02657889) was a phase I/II study evaluating the combination of pembrolizumab plus the PARPi niraparib, conducted among triple-negative breast cancer and ROC patients. In the PR-ROC cohort, 62 patients were treated. The ORR (primary endpoint of the study) was 25%, the DCR was 68%. In the breast cancer gene (BRCA)-mutant population, ORR and DCR were 45% and 73%, respectively. The most frequent ≥G3 TRAEs were anemia (21%) and thrombocytopenia (9%). No treatment-related deaths were recorded [17].
3.1.2. Nivolumab
As a single agent, nivolumab was administered to 20 patients with PR-ROC in the UMIN000005714 phase II trial, evaluating the best overall response (BOR) as a primary endpoint: two complete responses (CR) were recorded, the DCR was 45%, the mPFS 3.5 mos, and the mOS 20.0 mos. Of note, ≥G3 TRAEs occurred in 40% of patients. Two patients (10%) experienced serious TRAEs, and 11% of patients discontinued Nivolumab treatment mainly due to treatment-related thyroid disorders [18].
In the NRG-GY003 phase II study (NCT02498600), nivolumab alone or plus ipilimumab was administered to 100 platinum-progressing ROC patients. The primary endpoint was ORR; secondary endpoints included PFS and OS, stratified by the platinum-free interval (PFI). The combination of nivolumab and ipilimumab vs. nivolumab alone resulted in increased ORR (31.4% vs. 12.2%; (p = 0.034), longer PFS (3.9 vs. 2 mos; HR = 0.53, 95% CI 0.34–0.82) and longer OS (28.1 vs. 21.8 mos; HR = 0.79, 95% CI 0.44–1.42). However, the combination treatment was less tolerated (>G3 AEs 49% vs. 33%). The response was not associated with PD-L1 status [19].
The combination of nivolumab plus bevacizumab was tested in 38 platinum-progressing epithelial ovarian cancer (EOC) patients in the NCT02873962 phase II study. ORR was the primary endpoint, while secondary endpoints were ORR according to platinum sensitivity and PD-L1 expression, PFS, and safety. The combination of nivolumab plus bevacizumab resulted in an ORR of 28.9%, ranging from 16.7% in the platinum-resistant (n = 18) to 40.0% in the platinum-sensitive patients (n = 20). Median PFS was 9.4 mos and 12.1 mos in the overall and platinum-sensitive population, respectively. Of note, better response rates were observed in patients with PD-L1 negative than PD-L1 positive disease. A total of 89.5% of patients developed AEs, of whom the most common were fatigue (47.4%), headache (28.9%), myalgia (28.9%), serum amylases increase (28.9%), aspartate aminotransferase level increase (26.3%), hypertension (26.3%). Four pneumonitis (10.5%) and two colitis (5.3%) cases were reported [20].
A unique phase III trial (NINJA) was conducted in the Japanese population, randomizing patients with PR-ROC to nivolumab (n = 157) versus gemcitabine or PLD (n = 159) at the physician’s choice. However, the trial failed its primary endpoint, as there was no difference between the two groups for OS (HR = 1.03, 95% CI, 0.8–1.32; p = 0.8). Moreover, mPFS was shorter in the nivolumab group (2.04 mos) than in the gem/PLD group (3.84 mos; HR = 1.46, 95% CI, 1.15–1.85; p = 0.002) The incidence of ≥G3 AEs was 22.4% with nivolumab and 68.4% with gem/PLD [21].
3.2. Anti-PD-L1 Agents
3.2.1. Avelumab
In the OC cohort of the JAVELIN (NCT01772004) phase Ib study, avelumab 10 mg/kg q2w determined an objective response in 12/125 patients, including 1 CR and 11 partial responses (PR). The 1 y PFS rate was 10.2% (95% CI, 5.4–16.7%), the mPFS was 2.6 months (95% CI, 1.4–2.8 mos), and the mOS was 11.2 months (95% CI, 8.7–15.4 mos). The responses were recorded independently from PD-L1 expression. The most frequent AEs were fatigue (13.6%), diarrhea (12.0%), and nausea (11.2%). ≥G3 AEs occurred in 7.2% of patients, among which the most frequent was the increase in lipase level (2.4%) [22].
The combination of avelumab plus chemotherapy was tested in two randomized phase III trials. The JAVELIN Ovarian 100 (NCT02718417) evaluated carboplatin-paclitaxel chemotherapy alone versus chemotherapy plus avelumab followed by avelumab maintenance versus chemotherapy plus avelumab in the front line OC setting. Nine hundred and ninety-eight stage III-IV patients were enrolled. The primary endpoint was PFS. However, after a median follow-up of 11 mos, PFS was not improved in both avelumab arms, and the trial was stopped after meeting futility criteria [23]. Based on the absence of benefit from Avelumab in unselected patients, the JAVELIN Ovarian PARP 100 (NCT03642132) phase III study, with three arms consisting of chemotherapy plus avelumab followed by maintenance with avelumab plus talazoparib, chemotherapy followed by talazoparib maintenance, and chemotherapy plus bevacizumab followed by bevacizumab, was also terminated [24].
In the JAVELIN 200 (NCT02580058) phase III study, 566 patients with PR-ROC were randomized 1:1:1 to avelumab alone or avelumab plus PLD or PLD alone. PFS and OS were the co-primary endpoints. The combination group achieved a non significant longer OS (15.7 mos; HR vs. PLD 0.89, 95% CI, 0.74–1.24, p = 0.2) and PFS (3.7 mos, HR 0.78, 95% CI, 0.59–1.24; p = 0.03) with respect to PLD single agent. Avelumab alone compared to PLD did not improve either OS (HR = 1.14, 95% CI, 0.95–1.58; p = 0.82) or PFS (HR = 1.68, 95% CI, 1.32–2.60; p > 0.99). A longer PFS and OS trend was observed in the avelumab + PLD arm vs. PLD arm among the PD-L1+ patients (58%). In the avelumab, avelumab plus PLD, and PLD arms, ≥G3 TRAEs occurred in 49.7%, 68.7%, and 59.3% of patients, respectively [25].
In the phase II ENCORE-603 study, 126 patients with PR-ROC were randomized 2:1 to avelumab plus the class I selective histone deacetylase (HDAC) inhibitor entinostat or avelumab plus placebo (PBO), with PFS as the primary endpoint. No significant differences were detected between the two groups for mPFS (1.64 vs. 1.51 mos; HR = 0.9, 95% CI, 0.58–1.39; p = 0.031), ORR, and OS. AEs were more frequent with the avelumab–Entinostat combination than with avelumab–PBO (93% vs. 78%, ≥G3 AEs 41% vs. 10%), with fatigue, nausea, diarrhea, anemia, and chills being the most frequent [26].
3.2.2. Atezolizumab
In the randomized phase III IMagyn050/GOG 3015/ENGOT-OV39 trial, 1,301 stage III/IV OC patients were treated with the combination of carboplatin, paclitaxel, and bevacizumab plus or minus atezolizumab (1200 mg q3w). The PFS improvement was not significant both in the overall population (19.5 vs. 18.4 mos HR = 0.92; 95% CI, 0.79–1.07; p = 0.28) and among the PD-L1 positive patients (20.8 vs. 18.5 mos HR = 0.8; 95% CI, 0.65–0.99; p = 0.38). Similarly, no benefit emerged at the interim analysis for OS. Anemia, neutropenia, and hypertension were the most common ≥G3 AEs [27].
3.2.3. Durvalumab
Durvalumab was tested in combination with chemotherapy, PARPis, or anti-VEGF agents.
In the NCT02431559 phase I/II trial, 40 PR-ROC women received durvalumab plus PLD, reaching an ORR of 15% and a 6-mos PFS rate of 47.7%. The most frequent TRAEs were palmar–plantar erythrodysesthesia syndrome (PPES), stomatitis, fatigue, abdominal pain, nausea, fever. G3 TRAEs occurred in at least two patients and included lymphopenia, anemia, increased lipase, rash, and stomatitis [28].
The combination of durvalumab and olaparib was tested in three phase II studies. The MEDIOLA study aimed to evaluate 12 w DCR and safety as primary endpoints, plus 28 w DCR, ORR, DOR, PFS, and OS as secondary endpoints. Initially, 32 women with BRCA-mutant PS-ROC were included. The 12 w DCR was 81% [29]. After a median follow-up of 20.4 mos, 28 w DCR was 65.6% with mPFS 11.1 mos, ORR 71.9%, and mOS was not reached [30]. Subsequently, the study included 63 BRCA-wild type patients. Thirty-two patients received durvalumab plus olaparib, 31 patients were treated with olaparib plus durvalumab plus bevacizumab. The doublet cohort reached an ORR of 31.3% (95% CI 16.1–50.0%) vs. 77.4% in the triplet cohort (95% CI 58.9–90.4%). The mPFS was 5.5 mos for the doublet cohort and 14.7 mos for the triplet cohort, respectively. The 24 w DCR was 28.1% in the doublet cohort and 77.4% in the triplet cohort. The most common ≥G3 AEs were anemia, neutropenia, and lipase increased in both cohorts, while in the triplet cohort, hypertension and fatigue were also registered. Six percent and sixteen percent of patients discontinued the treatment in the double and triplet cohorts, respectively [31]. In the dose-escalation phase I/II NCT02484404 trial, among 35 patients with ROC, a DCR of 53% was observed with durvalumab plus olaparib or cediranib (5 PR, 13 stable disease [SD]). ≥G3 AEs included anemia (26%) and lymphopenia (14%) [32]. In a third single-center study (NCT02484404), 35 patients with PR-ROC were included. The (primary endpoint) ORR was 14%. Exploratory analyses showed that an increased gamma-interferon (γ-IFN) production was associated with longer PFS (p = 0.023), whereas increased vascular endothelial growth factor receptor (VEGFR)-3 levels determined shorter PFS (p = 0.017). Haematologic toxicity caused the highest ≥G3 AEs (most frequently anemia, 31%) [33].
3.3. Anti CTLA-4
Few trials have explored the activity of single agents anti-CTLA-4 Ipilimumab or Tremelimumab in the advanced/recurrent OC with unsatisfactory results. In the NCT01611558 phase II trial with ipilimumab at the dose of 10 mg/kg, 38 out of 40 PR-ROC patients did not complete treatment due to PD, severe toxicity, or death [34]. The combination with PARPis is still at an early stage but seems to be tolerated and induces anti-tumor responses. More specifically, 24 PR-ROC patients received tremelimumab alone or combined with olaparib in the NCT02485990 phase II trial, with 1 PR and 9 SD. No G4 AEs were reported, while the most common G3 toxicities were rash (13%), hepatitis, and colitis (both 8%) [35]. The same combination was administered to three BRCA-mutant OC patients in the NCT02571725 phase I trial, with a good safety profile (only G1/2 AEs were reported) and decreased tumor size after three cycles [36].
4. Discussion
Given the impact on morbidity and mortality among the female population, the search for new therapeutic options represents an unmet need for OC. Immunotherapy has revolutionized the treatment landscape of many solid tumors in the last ten years, and it now represents the first therapeutic approach with impressive survival benefits in diseases such as lung cancer, melanoma, renal cell carcinoma [10]. However, limited benefits have emerged in OC, even leading to premature termination due to the futility of some studies. Different components of the OC tumor microenvironment (TME) contribute to this failure, such as myeloid-derived suppressor cells (MDSCs), tumor-associated macrophages (TAMs), T-cells, cytokines, and soluble factors [37,38,39,40]. MDSCs exert immunosuppressive functions, such as the inhibition of T-effector and natural killer (NK)-cells, and are induced under pro-inflammatory cytokines, IFNγ, tumor necrosis factor-alpha (TNFα), interleukin (IL)-6 [41]. In OC, IL-6 plays a negative prognostic role and is associated with high MDSCs, and tumor progression [42,43]. The inflammatory cytokines cooperate to induce cyclooxygenase-2 (COX-2) and lead to prostaglandin E2 (PGE2) synthesis, which limits T-cell recruiting at tumor sites, together with VEGF [44,45]. TAMs are recruited at ovarian tumor sites, and IL-6, IL-10, transforming growth factor (TGF)-β promote their differentiation in M2 macrophages, associated with tumor invasiveness, spread, and angiogenesis [46,47,48]. M2 macrophages increase with the OC stage when contemporary M1 macrophages decrease, playing a negative prognostic role [49,50,51]. Moreover, they promote immunosuppression by producing cytokines (IL-1R, IL-10, C-C Motif Chemokine Ligand [CCL]17, CCL20, CCL22) that inhibit T-effectors proliferation and enhance Tregs function [52,53,54]. Treg cells are associated with advanced stages of OC and have a negative prognostic and immunosuppressive role [54]. They produce IL-10 and TGFβ, contributing to the inhibition of effector T-cells [55]. High levels of immunosuppressive elements within OC TME can also weaken dendritic cells and antigen-presenting cells (APCs) activity [56]. More accurate knowledge of the TME of the primary tumors and the metastatic sites will facilitate the design of more effective treatment combinations (Figure 1).
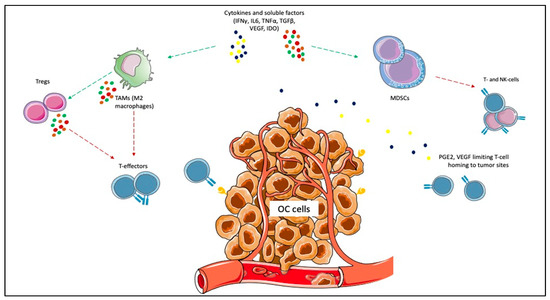
Figure 1.
Immunosuppressive elements of ovarian cancer (OC) microenvironment. Cytokines and other soluble factors, such as interferon-gamma (IFNγ), tumor necrosis factor-alpha (TNFα), interleukin (IL)-6, IL-10, and transforming growth factor-beta (TGFβ) induce the proliferation of myeloid-derived suppressor cells (MDSCs) and the polarization of tumor-associated macrophages (TAMs) towards the M2 subtype. MDSCs exert immunosuppressive functions, such as the inhibition of T-effector and natural killer (NK)-cells. The inflammatory cytokines cooperate to induce cyclooxygenase-2 (COX-2) and lead to prostaglandin E2 (PGE2) synthesis, which limits T-cell recruiting at tumor sites. M2 macrophages promote immunosuppression by producing cytokines (e.g., IL-1R, IL-10, C-C Motif Chemokine Ligand [CCL]17, CCL20, CCL22) that inhibit T-effectors proliferation and enhance Tregs function.
OC encompasses a heterogeneous group of malignancies that in over 95% of cases have an epithelial origin and are more frequently represented by high grade serous ovarian carcinoma (HGSOC) (70% of cases), followed by endometrioid ovarian cancer (EOC) (10%), clear cell OC (ccOC) (10%), low-grade serous OC (LGSOC, less than 5%), and mucinous OC (MOC, around 3%) [3]. Among them, the ccOC seems to be the most immunogenic: it more frequently carries the DNA microsatellite instability (MSI), has higher CD8+ tumor-infiltrating lymphocites (TILs), CD8+/CD4+ ratio, and higher PD-L1 levels [57,58]. Effectively, it is five times more responsive to ICIs than other OC subtypes [19]. Even among HGSOC, at least four different genomic classes were identified in The Cancer Genomic Atlas registry, differing for immunoreactivity. A unique subtype expresses genes related to immune sensitivity such as Toll-like receptor (TLR), TNF and is characterized by higher TILs infiltration [59,60]. Moreover, proteomics studies showed that the four subclasses of HSGOC are characterized by different expressions of proteins involved in DNA replication, ECM and cellular interaction, and cytokine signaling that contributes to immune responsiveness [61]. In our opinion, the different ICIs response observed among OC patients is rooted in the inter-tumor heterogeneity. Therefore, a deeper insight into the genomics characteristics of OC and their relationship with the immunological profile could allow us to better clarify the predictive factors for ICIs response. Ideally, specific immunogenomic scores could be developed for more accurate patients selection.
OC has been indicated as potentially more immune responsive when carrying BRCA mutations or homologous recombination deficiency (HRD). In fact, the impaired DNA repair leads to neo-antigens production, resulting in a higher tumor mutational burden (TMB) (even if <10 mutations per megabase are usually detected) and recruiting TILs at tumor sites. However, HRD or BRCA mutations were not linked to a higher sensitivity to ICIs in the IMagyn050 nor in the Javelin Ovarian 100 trials [23,27]. BRCA-mutant/HRD OC is associated with higher CD3+ and CD8+ TILs, PD1/PD-L1 levels, and genes related to cytotoxicity, such as T-Cell Receptor (TCR), γ-IFN, and TNF-Receptor pathway [62,63,64,65]. As proof of this, in the NCT02484404 trial, durvalumab plus olaparib determined a longer PFS in case of increased γ-IFN production [33]. Another mechanism of immune responsiveness is represented by the mismatch repair (MMR) deficiency, harboring the DNA MSI. MSI tumors produce neo-antigens, with a 10–100-fold higher TMB than MS stable (MSS)-tumors, resulting in high immunogenicity. Some genes triggering MSI were also identified in a percentage ranging from 17% to 59% of OC (more commonly in non-serous subtypes): the oncosuppressor TP53; Dihydropyrimidinase-related protein (DPYSL)-2, involved in microtubules function; Alpha Kinase (ALPK)-2, with a role in apoptosis and DNA repair [66]. In Lynch syndrome, a germline mutation of the MMR genes MutL homolog (MLH)-1, MutS homolog (MSH)-2 and -6, PMS1 homolog (PMS)-2 leads to an increased risk to develop some cancer subtypes, including OC [67]. Therefore, these tumors may be good candidates for ICIs treatment. Other genes could be involved in ICI response, justifying the different results observed among OC patients. The SWItch/Sucrose Non-Fermentable (SWI/SNF) complex consists of around 15 subunits, acting as a chromatin remodeler. In other tumor subtypes, the loss of function of the SWI/SNF complex predicts ICI response, increasing MMR deficiency, TMB, and neo-antigens production [68]. SWI/SNF complex mutations were frequently detected in OC [69]. We can assume that genetic diversity contributes to different ICI responses among OC patients. A more extensive genetic characterization could allow more accurate identification of responders and non-responders.
The possible relationship between platinum- and immunotherapy-sensitivity/resistance is also a field that merits further investigation [70]. A series of genetic and epigenetic elements were identified to drive platinum response: alterations of p53, specific microRNAs, elements driving the epithelial-to-mesenchymal transition (EMT), HRD, and BRCA mutations [71]. Since BRCA mutation and HRD were proposed to correlate with platinum sensitivity in contemporary deficient nucleotide excision repair, the co-administration of PARPis and ICIs in PS-ROC could result in higher ORR and survival rates [29,30,31]. PARPis enhance ICIs activity because they induce the release of neoantigens, increasing the TMB, promote PD-L1 expression, and directly activate the IFN genes; however, this was determined in OC [17,29,30,31,72,73]. Many ongoing trials are addressing this combination strategy in the advanced setting (Table 2).
Table 2.
Ongoing trials of ICIs combinations in the metastatic OC.
As ICIs monotherapies showed only minimal results in terms of response rate and survival in OC, the combination with agents with different mechanisms of action appears a promising strategy to increase efficacy. Although chemotherapy represents a cornerstone in the treatment of advanced OC, it was historically perceived to play an immunosuppressive role. On the contrary, more recently, it has emerged that platinum derivatives promote APCs and their function, activating the immune response [74,75,76,77]. Doxorubicin plays an immunomodulatory effect, reducing the immunosuppressive state and improving tumor sensitivity to NK and CD8+ T-cells [78]. Low-dose cyclophosphamide also holds immunomodulatory properties, such as Tregs reduction and CD8+ cells induction [79,80]. However, the studies conducted so far did not lead to survival improvements. Besides the immunological potential, timing and schedule should be more deeply investigated and optimized for improving efficacy. The combination of ICIs and anti-VEGF agents seems attractive because the anti-angiogenic drugs directly influence OC TME [20,31,32,81,82,83]. Other combinations with multikinase inhibitors targeting VEGF/VEGFRpathway, such as cabozantinib or lenvatinib, are now under evaluation. The association with other agents with immunotherapeutics role, such as the anti-Lymphocyte-activation gene 3 (LAG-3) Relatlimab, as well as monoclonal antibodies such as the anti-Cluster of differentiation (CD)27 Varlilumab, the anti-CD47 Magrolimab, is under investigation (Table 2). Actually, overcoming the immunosuppressive pathways in the TME could represent a complementary way to potentiate ICIs effect on the immune system. Therapeutic vaccines were administered in OC, inducing cellular and humoral responses but rarely survival improvement as monotherapies [84]. Hence, several tumor-associated antigens were found in OC, such as p53, folate receptor (FR), New York Esophageal Squamous Cell Carcinoma-1 (NY-ESO-1), and Ca125 [85,86,87,88]. Therefore, combinations of ICIs and vaccines need to be explored. New approaches such as autologous TILs, cancer cell therapy, and adoptive cell therapy (ACT) also represent future possibilities for improving ICIs efficacy (Table 2).
Currently, a uniformly accepted predictive role of PD-L1 for ICIs response was not yet identified in solid tumors, including OC. PD-L1 expression varies between primary tumors and metastases, implying heterogeneity [89]. However, even if PD-L1 positivity was retrieved in around 1/3 OCs, the clinical impact was not elucidated, with conflicting results regarding the association with higher tumor stage/grade or shorter survival [90,91,92,93,94]. Indeed, some of the published trials reported better results for PD-L1 positive than PD-L1 negative patients [12,13,27]. In other studies, PD-L1 positivity was not predictive of ICIs response [19,20]. Recent research has focused on the post-transcriptional modifications of PD1, and even more PD-L1, which N-glycosylation of specific sites functionally modulates. PD-L1 and PD1 N-glycosylation ensure stability, prevents clearance, and influences mutual interactions [95,96]. The N-glycosylation of the PD1/PD-L1 receptors and its aberrations should be better investigated as possible immune resistance mechanisms in OC since specific glycoproteomic signatures were found in HGSOC: the immunoreactive subtype was richer in mannose than the mesenchymal, which was mainly fucosylated [97]. Moreover, it was evidenced that the antibodies used in the immunohistochemical analysis for PD-L1 accessed the highly glycosylated PD-L1 with difficulty, resulting in a certain percentage of PD-L1 false-negative results partially explaining ICIs efficacy also in PD-L1 negative patients [98]. More profound knowledge of the post-transcriptional status of PD1/PD-L1 and the search for biomarkers with a predictive role for ICIs’ efficacy is warranted to ensure the best patient selection.
5. Conclusions
Thus far, OC remains one of the few malignancies in which ICIs have not changed the standard of care, and neither monotherapies nor combinations have been approved. Effectively, significant heterogeneity was identified across OC patients at the genomic, proteomic, glycoproteomic, and immunologic levels, that in our opinion, should be further investigated to improve ICIs efficacy. We also believe that the combinations of ICIs with agents with different mechanisms of action will strengthen ICIs efficacy in OC. The combinations of ICIs with anti-VEGF agents or PARP-inhibitors represent potentially very effective associations, and several studies examine this strategy. However, schedule and timing should be optimized in order to preserve tolerability. Combinations with other agents, such as multikinase inhibitors, immunotherapies targeting the immunosuppressive network in the TME, or vaccines, should be further explored to maximize the efficacy with minimal toxicity.
Besides PD-L1, biomarkers with a predictive role to ICIs should be investigated. Integrating such biomarkers with genomic and immunologic profiling will provide a comprehensive understanding of OC, guiding clinical trials towards rational therapy combinations and sequencing.
Author Contributions
Conceptualization, methodology: B.A.M.; software, formal analysis, investigation, data curation, writing—original draft preparation: B.A.M. and M.F.P.M.; writing—review and editing: D.L. and E.M.; visualization, validation: B.A.M., M.F.P.M., D.L., and E.M.; supervision: E.M. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Acknowledgments
Servier Medical Art.
Conflicts of Interest
The authors declare no conflict of interest with regard to this work.
References
- Globocan 2020. Ovary. Available online: https://gco.iarc.fr/today/data/factsheets/cancers/25-Ovary-fact-sheet.pdf (accessed on 15 June 2021).
- Torre, L.A.; Trabert, B.; DeSantis, C.E.; Miller, K.D.; Samimi, G.; Runowicz, C.D.; Gaudet, M.M.; Jemal, A.; Siegel, R.L. Ovarian Cancer Statistics, 2018. CA A Cancer J. Clin. 2018, 68, 284–296. [Google Scholar] [CrossRef] [PubMed]
- Prat, J.; D’Angelo, E.; Espinosa, I. Ovarian Carcinomas: At Least Five Different Diseases with Distinct Histological Features and Molecular Genetics. Hum. Pathol. 2018, 80, 11–27. [Google Scholar] [CrossRef] [PubMed]
- Piccart, M.J. Randomized Intergroup Trial of Cisplatin-Paclitaxel vs. Cisplatin-Cyclophosphamide in Women with Advanced Epithelial Ovarian Cancer: Three-Year Results. J. Natl. Cancer Inst. 2000, 92, 699–708. [Google Scholar] [CrossRef] [Green Version]
- Ozols, R.F.; Bundy, B.N.; Greer, B.E.; Fowler, J.M.; Clarke-Pearson, D.; Burger, R.A.; Mannel, R.S.; DeGeest, K.; Hartenbach, E.M.; Baergen, R. Phase III Trial of Carboplatin and Paclitaxel Compared with Cisplatin and Paclitaxel in Patients with Optimally Resected Stage III Ovarian Cancer: A Gynecologic Oncology Group Study. JCO 2003, 21, 3194–3200. [Google Scholar] [CrossRef] [PubMed]
- Neijt, J.P.; Engelholm, S.A.; Tuxen, M.K.; Sørensen, P.G.; Hansen, M.; Sessa, C.; de Swart, C.A.M.; Hirsch, F.R.; Lund, B.; van Houwelingen, H.C. Exploratory Phase III Study of Paclitaxel and Cisplatin vs. Paclitaxel and Carboplatin in Advanced Ovarian Cancer. JCO 2000, 18, 3084–3092. [Google Scholar] [CrossRef] [PubMed]
- McGuire, W.P.; Hoskins, W.J.; Brady, M.F.; Kucera, P.R.; Partridge, E.E.; Look, K.Y.; Clarke-Pearson, D.L.; Davidson, M. Cyclophosphamide and Cisplatin Compared with Paclitaxel and Cisplatin in Patients with Stage III and Stage IV Ovarian Cancer. N. Engl. J. Med. 1996, 334, 1–6. [Google Scholar] [CrossRef]
- Perren, T.J.; Swart, A.M.; Pfisterer, J.; Ledermann, J.A.; Pujade-Lauraine, E.; Kristensen, G.; Carey, M.S.; Beale, P.; Cervantes, A.; Kurzeder, C.; et al. A Phase 3 Trial of Bevacizumab in Ovarian Cancer. N. Engl. J. Med. 2011, 365, 2484–2496. [Google Scholar] [CrossRef] [Green Version]
- Moore, K.; Colombo, N.; Scambia, G.; Kim, B.-G.; Oaknin, A.; Friedlander, M.; Lisyanskaya, A.; Floquet, A.; Leary, A.; Sonke, G.S.; et al. Maintenance Olaparib in Patients with Newly Diagnosed Advanced Ovarian Cancer. N. Engl. J. Med. 2018, 379, 2495–2505. [Google Scholar] [CrossRef]
- Robert, C. A Decade of Immune-Checkpoint Inhibitors in Cancer Therapy. Nat. Commun. 2020, 11, 3801. [Google Scholar] [CrossRef]
- Postow, M.A.; Callahan, M.K.; Wolchok, J.D. Immune Checkpoint Blockade in Cancer Therapy. JCO 2015, 33, 1974–1982. [Google Scholar] [CrossRef] [Green Version]
- Matulonis, U.A.; Shapira-Frommer, R.; Santin, A.D.; Lisyanskaya, A.S.; Pignata, S.; Vergote, I.; Raspagliesi, F.; Sonke, G.S.; Birrer, M.; Provencher, D.M.; et al. Antitumor Activity and Safety of Pembrolizumab in Patients with Advanced Recurrent Ovarian Cancer: Results from the Phase II KEYNOTE-100 Study. Ann. Oncol. 2019, 30, 1080–1087. [Google Scholar] [CrossRef]
- Matulonis, U.A.; Shapira, R.; Santin, A.; Lisyanskaya, A.S.; Pignata, S.; Vergote, S.; Raspagliesi, F.; Sonke, G.S.; Birrer, M.; Sehouli, J.; et al. Final Results from the KEYNOTE-100 Trial of Pembrolizumab in Patients with Advanced Recurrent Ovarian Cancer. JCO 2020, 38, 6005. [Google Scholar] [CrossRef]
- Varga, A.; Piha-Paul, S.; Ott, P.A.; Mehnert, J.M.; Berton-Rigaud, D.; Morosky, A.; Yang, P.; Ruman, J.; Matei, D. Pembrolizumab in Patients with Programmed Death Ligand 1–Positive Advanced Ovarian Cancer: Analysis of KEYNOTE-028. Gynecol. Oncol. 2019, 152, 243–250. [Google Scholar] [CrossRef]
- Matulonis, U.A.; Barry, W.; Penson, R.T.; Campos, S.M.; Krasner, C.; Liu, J.F. Phase II Study of Pembrolizumab (Pembro) Combined with Pegylated Liposomal Doxorubicin (PLD) for Recurrent Platinum-Resistant Ovarian, Fallopian Tube or Peritoneal Cancer. Gynecol. Oncol. 2018, 149, 24. [Google Scholar] [CrossRef]
- Zsiros, E.; Lynam, S.; Attwood, K.M.; Wang, C.; Chilakapati, S.; Gomez, E.C.; Liu, S.; Akers, S.; Lele, S.; Frederick, P.J.; et al. Efficacy and Safety of Pembrolizumab in Combination with Bevacizumab and Oral Metronomic Cyclophosphamide in the Treatment of Recurrent Ovarian Cancer: A Phase 2 Nonrandomized Clinical Trial. JAMA Oncol. 2021, 7, 78. [Google Scholar] [CrossRef]
- Konstantinopoulos, P.A.; Waggoner, S.; Vidal, G.A.; Mita, M.; Moroney, J.W.; Holloway, R.; Van Le, L.; Sachdev, J.C.; Chapman-Davis, E.; Colon-Otero, G.; et al. Single-Arm Phases 1 and 2 Trial of Niraparib in Combination with Pembrolizumab in Patients with Recurrent Platinum-Resistant Ovarian Carcinoma. JAMA Oncol. 2019, 5, 1141. [Google Scholar] [CrossRef] [Green Version]
- Hamanishi, J.; Mandai, M.; Ikeda, T.; Minami, M.; Kawaguchi, A.; Murayama, T.; Kanai, M.; Mori, Y.; Matsumoto, S.; Chikuma, S.; et al. Safety and Antitumor Activity of Anti–PD-1 Antibody, Nivolumab, in Patients with Platinum-Resistant Ovarian Cancer. JCO 2015, 33, 4015–4022. [Google Scholar] [CrossRef] [PubMed]
- Zamarin, D.; Burger, R.A.; Sill, M.W.; Powell, D.J.; Lankes, H.A.; Feldman, M.D.; Zivanovic, O.; Gunderson, C.; Ko, E.; Mathews, C.; et al. Randomized Phase II Trial of Nivolumab vs. Nivolumab and Ipilimumab for Recurrent or Persistent Ovarian Cancer: An NRG Oncology Study. JCO 2020, 38, 1814–1823. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.F.; Herold, C.; Gray, K.P.; Penson, R.T.; Horowitz, N.; Konstantinopoulos, P.A.; Castro, C.M.; Hill, S.J.; Curtis, J.; Luo, W.; et al. Assessment of Combined Nivolumab and Bevacizumab in Relapsed Ovarian Cancer: A Phase 2 Clinical Trial. JAMA Oncol. 2019, 5, 1731. [Google Scholar] [CrossRef]
- Omatsu, K.; Hamanishi, J.; Katsumata, N.; Nishio, S.; Sawada, K.; Takeuchi, S.; Aoki, D.; Fujiwara, K.; Sugiyama, T.; Konishi, I. 807O Nivolumab vs. Gemcitabine or Pegylated Liposomal Doxorubicin for Patients with Platinum-Resistant (Advanced or Recurrent) Ovarian Cancer: Open-Label, Randomized Trial in Japan (NINJA Trial). Ann. Oncol. 2020, 31, 611. [Google Scholar] [CrossRef]
- Disis, M.L.; Taylor, M.H.; Kelly, K.; Beck, J.T.; Gordon, M.; Moore, K.M.; Patel, M.R.; Chaves, J.; Park, H.; Mita, A.C.; et al. Efficacy and Safety of Avelumab for Patients with Recurrent or Refractory Ovarian Cancer: Phase 1b Results from the JAVELIN Solid Tumor Trial. JAMA Oncol. 2019, 5, 393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ledermann, J.A.; Colombo, N.; Oza, A.M.; Fujiwara, K.; Birrer, M.J.; Randall, L.M.; Poddubskaya, E.V.; Scambia, G.; Shparyk, Y.V.; Lim, M.C.; et al. Avelumab in Combination with and/or Following Chemotherapy vs Chemotherapy Alone in Patients with Previously Untreated Epithelial Ovarian Cancer: Results from the Phase 3 Javelin Ovarian 100 Trial. Gynecol. Oncol. 2020, 159, 13–14. [Google Scholar] [CrossRef]
- NCT03642132: Avelumab and Talazoparib in Untreated Advanced Ovarian Cancer (JAVELIN OVARIAN PARP 100). Available online: https://clinicaltrials.gov/ct2/show/NCT03642132 (accessed on 15 June 2021).
- Pujade-Lauraine, E.; Fujiwara, K.; Ledermann, J.A.; Oza, A.M.; Kristeleit, R.S.; Ray-Coquard, I.L.; Richardson, G.E.; Sessa, C.; Yonemori, K.; Banerjee, S.; et al. Avelumab Alone or in Combination with Pegylated Liposomal Doxorubicin vs. Pegylated Liposomal Doxorubicin Alone in Platinum-Resistant or Refractory Epithelial Ovarian Cancer: Primary and Biomarker Analysis of the Phase III JAVELIN Ovarian 200 Trial. Gynecol. Oncol. 2019, 154, 21–22. [Google Scholar] [CrossRef]
- Cadoo, K.A.; Meyers, M.L.; Burger, R.A.; Armstrong, D.K.; Penson, R.T.; Gordon, M.S.; Fleming, G.F.; Moroney, J.W.; Hamilton, E.P.; Duska, L.R.; et al. A Phase II Randomized Study of Avelumab plus Entinostat vs. Avelumab plus Placebo in Patients (Pts) with Advanced Epithelial Ovarian Cancer (EOC). JCO 2019, 37, 5511. [Google Scholar] [CrossRef]
- Moore, K.N.; Bookman, M.; Sehouli, J.; Miller, A.; Anderson, C.; Scambia, G.; Myers, T.; Taskiran, C.; Robison, K.; Mäenpää, J.; et al. Atezolizumab, Bevacizumab, and Chemotherapy for Newly Diagnosed Stage III or IV Ovarian Cancer: Placebo-Controlled Randomized Phase III Trial (IMagyn050/GOG 3015/ENGOT-OV39). JCO 2021, 39, 1842–1855. [Google Scholar] [CrossRef] [PubMed]
- O’Cearbhaill, R.E.; Wolfer, A.; Disilvestro, P.; O’Malley, D.M.; Sabbatini, P.; Shohara, L.; Schwarzenberger, P.O.; Ricciardi, T.; Macri, M.; Ryan, A.; et al. A Phase I/II Study of Chemo-Immunotherapy with Durvalumab (Durva) and Pegylated Liposomal Doxorubicin (PLD) in Platinum-Resistant Recurrent Ovarian Cancer (PROC). Ann. Oncol. 2018, 29, 337. [Google Scholar] [CrossRef]
- Drew, Y.; de Jonge, M.; Hong, S.H.; Park, Y.H.; Wolfer, A.; Brown, J.; Ferguson, M.; Gore, M.E.; Alvarez, R.H.; Gresty, C.; et al. An Open-Label, Phase II Basket Study of Olaparib and Durvalumab (MEDIOLA): Results in Germline BRCA -Mutated ( GBRCA m) Platinum-Sensitive Relapsed (PSR) Ovarian Cancer (OC). Gynecol. Oncol. 2018, 149, 246–247. [Google Scholar] [CrossRef]
- Drew, Y.; Kaufman, B.; Banerjee, S.; Lortholary, A.; Hong, S.H.; Park, Y.H.; Zimmermann, S.; Roxburgh, P.; Ferguson, M.; Alvarez, R.H.; et al. Phase II study of olaparib 1 durvalumab (MEDIOLA): Updated Results in Germline BRCA-Mutated Platinum-Sensitive Relapsed (PSR) Ovarian Cancer (OC). Ann. Oncol. 2019, 30, 485–486. [Google Scholar] [CrossRef]
- Drew, Y.; Penson, R.T.; O’Malley, D.M.; Kim, J.-W.; Zimmermann, S.; Roxburgh, P.; Sohn, J.; Stemmer, S.M.; Bastian, S.; Ferguson, M.; et al. 814MO Phase II Study of Olaparib (O) plus Durvalumab (D) and Bevacizumab (B) (MEDIOLA): Initial Results in Patients (Pts) with Non-Germline BRCA-Mutated (Non-GBRCAm) Platinum Sensitive Relapsed (PSR) Ovarian Cancer (OC). Ann. Oncol. 2020, 31, 615–616. [Google Scholar] [CrossRef]
- Lee, J.M.; Cimino-Mathews, A.; Peer, C.J.; Zimmer, A.; Lipkowitz, S.; Annunziata, C.M.; Cao, L.; Harrell, M.I.; Swisher, E.M.; Houston, N.; et al. Safety and Clinical Activity of the Programmed Death-Ligand 1 Inhibitor Durvalumab in Combination with Poly (ADP-Ribose) Polymerase Inhibitor Olaparib or Vascular Endothelial Growth Factor Receptor 1–3 Inhibitor Cediranib in Women’s Cancers: A Dose-Escalation, Phase I Study. JCO 2017, 35, 2193–2202. [Google Scholar] [CrossRef]
- Lampert, E.J.; Zimmer, A.; Padget, M.; Cimino-Mathews, A.; Nair, J.R.; Liu, Y.; Swisher, E.M.; Hodge, J.W.; Nixon, A.B.; Nichols, E.; et al. Combination of PARP Inhibitor Olaparib, and PD-L1 Inhibitor Durvalumab, in Recurrent Ovarian Cancer: A Proof-of-Concept Phase II Study. Clin. Cancer Res. 2020, 26, 4268–4279. [Google Scholar] [CrossRef] [PubMed]
- NCT01611558: Phase II Study of Ipilimumab Monotherapy in Recurrent Platinum-Sensitive Ovarian Cancer-Study Results. Available online: https://clinicaltrials.gov/ct2/show/results/NCT01611558 (accessed on 15 June 2021).
- Gaillard, S.; Berg, M.; Harrison, J.; Huang, P.; Leatherman, J.M.; Doucet, M.; Sen, R.; Suru, A.; Cai, H.; Durham, J.N.; et al. A Clinical Study of Tremelimumab Alone or in Combination with Olaparib in Patients with Advanced Epithelial Ovarian Cancer. JCO 2020, 38, 6045. [Google Scholar] [CrossRef]
- Adams, S.F.; Rixe, O.; Lee, J.-H.; McCance, D.J.; Westgate, S.; Eberhardt, S.C.; Rutledge, T.; Muller, C. Phase I Study Combining Olaparib and Tremelimumab for the Treatment of Women with BRCA-Deficient Recurrent Ovarian Cancer. JCO 2017, 35, 17052. [Google Scholar] [CrossRef]
- Hartnett, E.G.; Knight, J.; Radolec, M.; Buckanovich, R.J.; Edwards, R.P.; Vlad, A.M. Immunotherapy Advances for Epithelial Ovarian Cancer. Cancers 2020, 12, 3733. [Google Scholar] [CrossRef]
- Ning, F.; Cole, C.B.; Annunziata, C.M. Driving Immune Responses in the Ovarian Tumor Microenvironment. Front. Oncol. 2021, 10, 604084. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Yang, Y.; Yang, J.; Zhao, X.; Wei, X. Tumor Microenvironment in Ovarian Cancer: Function and Therapeutic Strategy. Front. Cell Dev. Biol. 2020, 8, 758. [Google Scholar] [CrossRef]
- Konstantinopoulos, P.A.; Cannistra, S.A. Immune Checkpoint Inhibitors in Ovarian Cancer: Can We Bridge the Gap Between IMagynation and Reality? JCO 2021, 10, 1833–1838. [Google Scholar] [CrossRef]
- Kapka-Skrzypczak, L.; Wolinska, E.; Szparecki, G.; Czajka, M.; Skrzypczak, M. The Immunohistochemical Analysis of Membrane-Bound CD55, CD59 and Fluid-Phase FH and FH-like Complement Inhibitors in Cancers of Ovary and Corpus Uteri Origin. Cent. Eur. J. Immunol. 2015, 3, 349–353. [Google Scholar] [CrossRef] [Green Version]
- Wouters, M.; Dijkgraaf, E.; Kuijjer, M.; Jordanova, E.; Hollema, H.; Welters, M.; van der Hoeven, J.; Daemen, T.; Kroep, J.; Nijman, H.; et al. Interleukin-6 Receptor and Its Ligand Interleukin-6 Are Opposite Markers for Survival and Infiltration with Mature Myeloid Cells in Ovarian Cancer. OncoImmunology 2014, 3, e962397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolomeyevskaya, N.; Eng, K.H.; Khan, A.N.H.; Grzankowski, K.S.; Singel, K.L.; Moysich, K.; Segal, B.H. Cytokine Profiling of Ascites at Primary Surgery Identifies an Interaction of Tumor Necrosis Factor-α and Interleukin-6 in Predicting Reduced Progression-Free Survival in Epithelial Ovarian Cancer. Gynecol. Oncol. 2015, 138, 352–357. [Google Scholar] [CrossRef] [Green Version]
- Wong, J.L.; Obermajer, N.; Odunsi, K.; Edwards, R.P.; Kalinski, P. Synergistic COX2 Induction by IFNγ and TNFα Self-Limits Type-1 Immunity in the Human Tumor Microenvironment. Cancer Immunol. Res. 2016, 4, 303–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Motz, G.T.; Santoro, S.P.; Wang, L.-P.; Garrabrant, T.; Lastra, R.R.; Hagemann, I.S.; Lal, P.; Feldman, M.D.; Benencia, F.; Coukos, G. Tumor Endothelium FasL Establishes a Selective Immune Barrier Promoting Tolerance in Tumors. Nat. Med. 2014, 20, 607–615. [Google Scholar] [CrossRef]
- Pollard, J.W. Tumour-Educated Macrophages Promote Tumour Progression and Metastasis. Nat. Rev. Cancer 2004, 4, 71–78. [Google Scholar] [CrossRef]
- Reinartz, S.; Schumann, T.; Finkernagel, F.; Wortmann, A.; Jansen, J.M.; Meissner, W.; Krause, M.; Schwörer, A.; Wagner, U.; Müller-Brüsselbach, S.; et al. Mixed-polarization Phenotype of Ascites-associated Macrophages in Human Ovarian Carcinoma: Correlation of CD163 Expression, Cytokine Levels and Early Relapse. Int. J. Cancer 2014, 134, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Yin, M.; Li, X.; Tan, S.; Zhou, H.J.; Ji, W.; Bellone, S.; Xu, X.; Zhang, H.; Santin, A.D.; Lou, G.; et al. Tumor-Associated Macrophages Drive Spheroid Formation during Early Transcoelomic Metastasis of Ovarian Cancer. J. Clin. Investig. 2016, 126, 4157–4173. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; He, Y.; Sun, X.; Li, Q.; Wang, W.; Zhao, A.; Di, W. A High M1/M2 Ratio of Tumor-Associated Macrophages Is Associated with Extended Survival in Ovarian Cancer Patients. J. Ovarian Res. 2014, 7, 19. [Google Scholar] [CrossRef] [Green Version]
- Lan, C.; Huang, X.; Lin, S.; Huang, H.; Cai, Q.; Wan, T.; Lu, J.; Liu, J. Expression of M2-Polarized Macrophages Is Associated with Poor Prognosis for Advanced Epithelial Ovarian Cancer. Technol. Cancer Res. Treat. 2013, 12, 259–267. [Google Scholar] [CrossRef] [Green Version]
- Yuan, X.; Zhang, J.; Li, D.; Mao, Y.; Mo, F.; Du, W.; Ma, X. Prognostic Significance of Tumor-Associated Macrophages in Ovarian Cancer: A Meta-Analysis. Gynecol. Oncol. 2017, 147, 181–187. [Google Scholar] [CrossRef] [PubMed]
- Gordon, S. Alternative Activation of Macrophages. Nat. Rev. Immunol. 2003, 3, 23–35. [Google Scholar] [CrossRef]
- Li, H.; Fan, X.; Houghton, J. Tumor Microenvironment: The Role of the Tumor Stroma in Cancer. J. Cell. Biochem. 2007, 101, 805–815. [Google Scholar] [CrossRef]
- Curiel, T.J.; Coukos, G.; Zou, L.; Alvarez, X.; Cheng, P.; Mottram, P.; Evdemon-Hogan, M.; Conejo-Garcia, J.R.; Zhang, L.; Burow, M.; et al. Specific Recruitment of Regulatory T Cells in Ovarian Carcinoma Fosters Immune Privilege and Predicts Reduced Survival. Nat. Med. 2004, 10, 942–949. [Google Scholar] [CrossRef]
- Schmidt, A.; Oberle, N.; Krammer, P.H. Molecular Mechanisms of Treg-Mediated T Cell Suppression. Front. Immun. 2012, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Obermajer, N.; Muthuswamy, R.; Lesnock, J.; Edwards, R.P.; Kalinski, P. Positive Feedback between PGE2 and COX2 Redirects the Differentiation of Human Dendritic Cells toward Stable Myeloid-Derived Suppressor Cells. Blood 2011, 118, 5498–5505. [Google Scholar] [CrossRef] [PubMed]
- Howitt, B.E.; Strickland, K.C.; Sholl, L.M.; Rodig, S.; Ritterhouse, L.L.; Chowdhury, D.; D’Andrea, A.D.; Matulonis, U.A.; Konstantinopoulos, P.A. Clear Cell Ovarian Cancers with Microsatellite Instability: A Unique Subset of Ovarian Cancers with Increased Tumor-Infiltrating Lymphocytes and PD-1/PD-L1 Expression. OncoImmunology 2017, 6, e1277308. [Google Scholar] [CrossRef] [Green Version]
- Oda, K.; Hamanishi, J.; Matsuo, K.; Hasegawa, K. Genomics to Immunotherapy of Ovarian Clear Cell Carcinoma: Unique Opportunities for Management. Gynecol. Oncol. 2018, 151, 381–389. [Google Scholar] [CrossRef] [PubMed]
- Verhaak, R.G.W.; Tamayo, P.; Yang, J.-Y.; Hubbard, D.; Zhang, H.; Creighton, C.J.; Fereday, S.; Lawrence, M.; Carter, S.L.; Mermel, C.H.; et al. Prognostically Relevant Gene Signatures of High-Grade Serous Ovarian Carcinoma. J. Clin. Invest. 2012. [Google Scholar] [CrossRef]
- Przybytkowski, E.; Davis, T.; Hosny, A.; Eismann, J.; Matulonis, U.A.; Wulf, G.M.; Nabavi, S. An Immune-Centric Exploration of BRCA1 and BRCA2 Germline Mutation Related Breast and Ovarian Cancers. BMC Cancer 2020, 20, 197. [Google Scholar] [CrossRef]
- Zhang, H.; Liu, T.; Zhang, Z.; Payne, S.H.; Zhang, B.; McDermott, J.E.; Zhou, J.Y.; Petyuk, V.A.; Chen, L.; Ray, D.; et al. CPTAC Investigators. Integrated Proteogenomic Characterization of Human High-Grade Serous Ovarian Cancer. Cell 2016, 166, 755–765. [Google Scholar] [CrossRef] [Green Version]
- Strickland, K.C.; Howitt, B.E.; Shukla, S.A.; Rodig, S.; Ritterhouse, L.L.; Liu, J.F.; Garber, J.E.; Chowdhury, D.; Wu, C.J.; D’Andrea, A.D.; et al. Association and Prognostic Significance of BRCA1/2-Mutation Status with Neoantigen Load, Number of Tumor-Infiltrating Lymphocytes and Expression of PD-1/PD-L1 in High Grade Serous Ovarian Cancer. Oncotarget 2016, 7, 13587–13598. [Google Scholar] [CrossRef] [Green Version]
- Morse, C.B.; Toukatly, M.N.; Kilgore, M.R.; Agnew, K.J.; Bernards, S.S.; Norquist, B.M.; Pennington, K.P.; Garcia, R.L.; Liao, J.B.; Swisher, E.M. Tumor Infiltrating Lymphocytes and Homologous Recombination Deficiency Are Independently Associated with Improved Survival in Ovarian Carcinoma. Gynecol. Oncol. 2019, 153, 217–222. [Google Scholar] [CrossRef]
- McAlpine, J.N.; Porter, H.; Köbel, M.; Nelson, B.H.; Prentice, L.M.; Kalloger, S.E.; Senz, J.; Milne, K.; Ding, J.; Shah, S.P.; et al. BRCA1 and BRCA2 Mutations Correlate with TP53 Abnormalities and Presence of Immune Cell Infiltrates in Ovarian High-Grade Serous Carcinoma. Mod. Pathol. 2012, 25, 740–750. [Google Scholar] [CrossRef] [Green Version]
- Gadducci, A.; Guerrieri, M.E. PARP Inhibitors Alone and in Combination with Other Biological Agents in Homologous Recombination Deficient Epithelial Ovarian Cancer: From the Basic Research to the Clinic. Crit. Rev. Oncol. Hematol. 2017, 114, 153–165. [Google Scholar] [CrossRef] [PubMed]
- Deshpande, M.; Romanski, P.A.; Rosenwaks, Z.; Gerhardt, J. Gynecological Cancers Caused by Deficient Mismatch Repair and Microsatellite Instability. Cancers 2020, 12, 3319. [Google Scholar] [CrossRef]
- Helder-Woolderink, J.M.; Blok, E.A.; Vasen, H.F.A.; Hollema, H.; Mourits, M.J.; De Bock, G.H. Ovarian Cancer in Lynch Syndrome; a Systematic Review. Eur. J. Cancer 2016, 55, 65–73. [Google Scholar] [CrossRef]
- Shen, J.; Ju, Z.; Zhao, W.; Wang, L.; Peng, Y.; Ge, Z.; Nagel, Z.D.; Zou, J.; Wang, C.; Kapoor, P.; et al. ARID1A Deficiency Promotes Mutability and Potentiates Therapeutic Antitumor Immunity Unleashed by Immune Checkpoint Blockade. Nat. Med. 2018, 24, 556–562. [Google Scholar] [CrossRef]
- Wang, Y.; Hoang, L.; Ji, J.X.; Huntsman, D.G. SWI/SNF Complex Mutations in Gynecologic Cancers: Molecular Mechanisms and Models. Annu. Rev. Pathol. 2020, 15, 467–492. [Google Scholar] [CrossRef] [Green Version]
- Van Zyl, B.; Tang, D.; Bowden, N.A. Biomarkers of Platinum Resistance in Ovarian Cancer: What Can We Use to Improve Treatment. Endocr. Relat. Cancer 2018, 25, 303–318. [Google Scholar] [CrossRef]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, J.; Zhao, W.; Ju, Z.; Wang, L.; Peng, Y.; Labrie, M.; Yap, T.A.; Mills, G.B.; Peng, G. PARPi Triggers the STING-Dependent Immune Response and Enhances the Therapeutic Efficacy of Immune Checkpoint Blockade Independent of BRCAness. Cancer Res. 2019, 79, 311–319. [Google Scholar] [CrossRef] [Green Version]
- Vikas, P.; Borcherding, N.; Chennamadhavuni, A.; Garje, R. Therapeutic Potential of Combining PARP Inhibitor and Immunotherapy in Solid Tumors. Front. Oncol. 2020, 10, 570. [Google Scholar] [CrossRef] [PubMed]
- Rébé, C.; Demontoux, L.; Pilot, T.; Ghiringhelli, F. Platinum Derivatives Effects on Anticancer Immune Response. Biomolecules 2019, 10, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grabosch, S.; Bulatovic, M.; Zeng, F.; Ma, T.; Zhang, L.; Ross, M.; Brozick, J.; Fang, Y.; Tseng, G.; Kim, E.; et al. Cisplatin-Induced Immune Modulation in Ovarian Cancer Mouse Models with Distinct Inflammation Profiles. Oncogene 2019, 38, 2380–2393. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Feng, Q.-M.; Wang, Y.; Shi, J.; Ge, H.-L.; Di, W. The Immunologic Aspects in Advanced Ovarian Cancer Patients Treated with Paclitaxel and Carboplatin Chemotherapy. Cancer Immunol. Immunother. 2010, 59, 279–291. [Google Scholar] [CrossRef]
- De Biasi, A.R.; Villena-Vargas, J.; Adusumilli, P.S. Cisplatin-Induced Antitumor Immunomodulation: A Review of Preclinical and Clinical Evidence. Clin. Cancer Res. 2014, 20, 5384–5391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Yu, X.; Wang, Z.; Wu, P.; Huang, J. Anthracyclines Potentiate Anti-Tumor Immunity: A New Opportunity for Chemoimmunotherapy. Cancer Letters 2015, 369, 331–335. [Google Scholar] [CrossRef]
- Madondo, M.T.; Quinn, M.; Plebanski, M. Low Dose Cyclophosphamide: Mechanisms of T-Cell Modulation. Cancer Treat. Rev. 2016, 42, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Mkrtichyan, M.; Najjar, Y.G.; Raulfs, E.C.; Abdalla, M.Y.; Samara, R.; Rotem-Yehudar, R.; Cook, L.; Khleif, S.N. Anti-PD-1 Synergizes with Cyclophosphamide to Induce Potent Anti-Tumor Vaccine Effects through Novel Mechanisms. Eur. J. Immunol. 2011, 41, 2977–2986. [Google Scholar] [CrossRef]
- Ziogas, A.C.; Gavalas, N.G.; Tsiatas, M.; Tsitsilonis, O.; Politi, E.; Terpos, E.; Rodolakis, A.; Vlahos, G.; Thomakos, N.; Haidopoulos, D.; et al. VEGF Directly Suppresses Activation of T-Cells from Ovarian Cancer Patients and Healthy Individuals via VEGF Receptor Type 2. Int. J. Cancer 2012, 130, 857–864. [Google Scholar] [CrossRef]
- Shrimali, R.K.; Yu, Z.; Theoret, M.R.; Chinnasamy, D.; Restifo, N.P.; Rosenberg, S.A. Antiangiogenic Agents Can Increase Lymphocyte Infiltration into Tumor and Enhance the Effectiveness of Adoptive Immunotherapy of Cancer. Cancer Res. 2010, 70, 6171–6180. [Google Scholar] [CrossRef] [Green Version]
- Fukumura, D.; Kloepper, J.; Amoozgar, Z.; Duda, D.G.; Jain, R.K. Enhancing Cancer Immunotherapy Using Antiangiogenics: Opportunities and Challenges. Nat. Rev. Clin. Oncol. 2018, 15, 325–340. [Google Scholar] [CrossRef]
- Chow, S.; Berek, J.S.; Dorigo, O. Development of Therapeutic Vaccines for Ovarian Cancer. Vaccines 2020, 8, 657. [Google Scholar] [CrossRef]
- Shih, I.; Kurman, R.J. Ovarian Tumorigenesis: A Proposed Model Based on Morphological and Molecular Genetic Analysis. Am. J. Pathol. 2004, 164, 1511–1518. [Google Scholar] [CrossRef]
- Peoples, G.E.; Anderson, B.W.; Fisk, B.; Kudelka, A.P.; Wharton, J.T.; Ioannides, C.G. Ovarian Cancer-Associated Lymphocyte Recognition of Folate Binding Protein Peptides. Ann. Surg. Oncol. 1998, 5, 743–750. [Google Scholar] [CrossRef] [PubMed]
- Odunsi, K.; Jungbluth, A.A.; Stockert, E.; Qian, F.; Gnjatic, S.; Tammela, J.; Intengan, M.; Beck, A.; Keitz, B.; Santiago, D.; et al. NY-ESO-1 and LAGE-1 Cancer-Testis Antigens Are Potential Targets for Immunotherapy in Epithelial Ovarian Cancer. Cancer Res. 2003, 63, 6076–6083. [Google Scholar] [PubMed]
- Felder, M.; Kapur, A.; Gonzalez-Bosquet, J.; Horibata, S.; Heintz, J.; Albrecht, R.; Fass, L.; Kaur, J.; Hu, K.; Shojaei, H.; et al. MUC16 (CA125): Tumor Biomarker to Cancer Therapy, a Work in Progress. Mol. Cancer. 2014, 13, 129. [Google Scholar] [CrossRef] [Green Version]
- Parvathareddy, S.K.; Siraj, A.K.; Al-Badawi, I.A.; Tulbah, A.; Al-Dayel, F.; Al-Kuraya, K.S. Differential Expression of PD-L1 between Primary and Metastatic Epithelial Ovarian Cancer and Its Clinico-Pathological Correlation. Sci. Rep. 2021, 11, 3750. [Google Scholar] [CrossRef]
- Wang, Q.; Lou, W.; Di, W.; Wu, X. Prognostic Value of Tumor PD-L1 Expression Combined with CD8 + Tumor Infiltrating Lymphocytes in High Grade Serous Ovarian Cancer. Int. Immunopharmacol. 2017, 52, 7–14. [Google Scholar] [CrossRef]
- Zhu, J.; Wen, H.; Bi, R.; Wu, Y.; Wu, X. Prognostic Value of Programmed Death-Ligand 1 (PD-L1) Expression in Ovarian Clear Cell Carcinoma. J. Gynecol. Oncol. 2017, 28, e77. [Google Scholar] [CrossRef] [Green Version]
- Webb, J.R.; Milne, K.; Kroeger, D.R.; Nelson, B.H. PD-L1 Expression Is Associated with Tumor-Infiltrating T Cells and Favorable Prognosis in High-Grade Serous Ovarian Cancer. Gynecol. Oncol. 2016, 141, 293–302. [Google Scholar] [CrossRef] [Green Version]
- Mesnage, S.J.L.; Auguste, A.; Genestie, C.; Dunant, A.; Pain, E.; Drusch, F.; Gouy, S.; Morice, P.; Bentivegna, E.; Lhomme, C.; et al. Neoadjuvant Chemotherapy (NACT) Increases Immune Infiltration and Programmed Death-Ligand 1 (PD-L1) Expression in Epithelial Ovarian Cancer (EOC). Ann. Oncol. 2017, 28, 651–657. [Google Scholar] [CrossRef]
- Kim, H.-S.; Kim, J.-Y.; Lee, Y.J.; Kim, S.H.; Lee, J.-Y.; Nam, E.J.; Kim, S.; Kim, S.W.; Kim, Y.T. Expression of Programmed Cell Death Ligand 1 and Immune Checkpoint Markers in Residual Tumors after Neoadjuvant Chemotherapy for Advanced High-Grade Serous Ovarian Cancer. Gynecol. Oncol. 2018, 151, 414–421. [Google Scholar] [CrossRef] [PubMed]
- Li, C.W.; Lim, S.O.; Xia, W.; Lee, H.H.; Chan, L.C.; Kuo, C.W.; Khoo, K.H.; Chang, S.S.; Cha, J.H.; Kim, T.; et al. Glycosylation and Stabilization of Programmed Death Ligand-1 Suppresses T-Cell Activity. Nat. Commun. 2016, 30, 12632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.N.; Lee, H.H.; Hsu, J.L.; Yu, D.; Hung, M. The Impact of PD-L1 N-Linked Glycosylation on Cancer Therapy and Clinical Diagnosis. J. Biomed. Sci. 2020, 27, 77. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.; Hu, Y.; Sun, S.; Chen, L.; Schnaubelt, M.; Clark, D.; Ao, M.; Zhang, Z.; Chan, D.; Qian, J.; et al. Glycoproteomics-Based Signatures for Tumor Subtyping and Clinical Outcome Prediction of High-Grade Serous Ovarian Cancer. Nat. Commun. 2020, 11, 6139. [Google Scholar] [CrossRef]
- Lee, H.H.; Wang, Y.N.; Xia, W.; Chen, C.H.; Rau, K.M.; Ye, L.; Wei, Y.; Chou, C.K.; Wang, S.C.; Yan, M.; et al. Removal of N-Linked Glycosylation Enhances PD-L1 Detection and Predicts Anti-PD-1/PD-L1 Therapeutic Efficacy. Cancer Cell 2019, 36, 168–178. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).