
Prevalence of Germline Pathogenic Variants in Cancer Predisposing Genes in Czech and Belgian Pancreatic Cancer Patients

 , , , , , ,
, , , , , ,  , , , ,
, , , ,  add
Show full author list
add
Show full author list
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Patients and Samples
2.2. NGS and Bioinformatics
2.3. Variant Prioritization
- With low variant allele fraction (VAF < 0.15);
- With a high minor allele frequency (MAF > 0.001) in population databases: Exome Sequencing Project (ESP), 1000 Genomes Project and gnomAD, except variants classified pathogenic/likely pathogenic (P/LP) in ClinVar;
- In UTR, non-splice site intronic, synonymous and non-frameshift insertions/deletions, unless classified as P/LP in ClinVar;
- Classified as benign/likely benign (B/LB) in ClinVar, if marked by at least two stars in ClinVar or if classified as B/LB by an expert panel;
- Low risk variants in CHEK2 (c.470C > T; p.I157T), APC (c.3920T > A; p.I1307K), and heterozygous MUTYH variants.
2.4. Statistical Analysis
3. Results
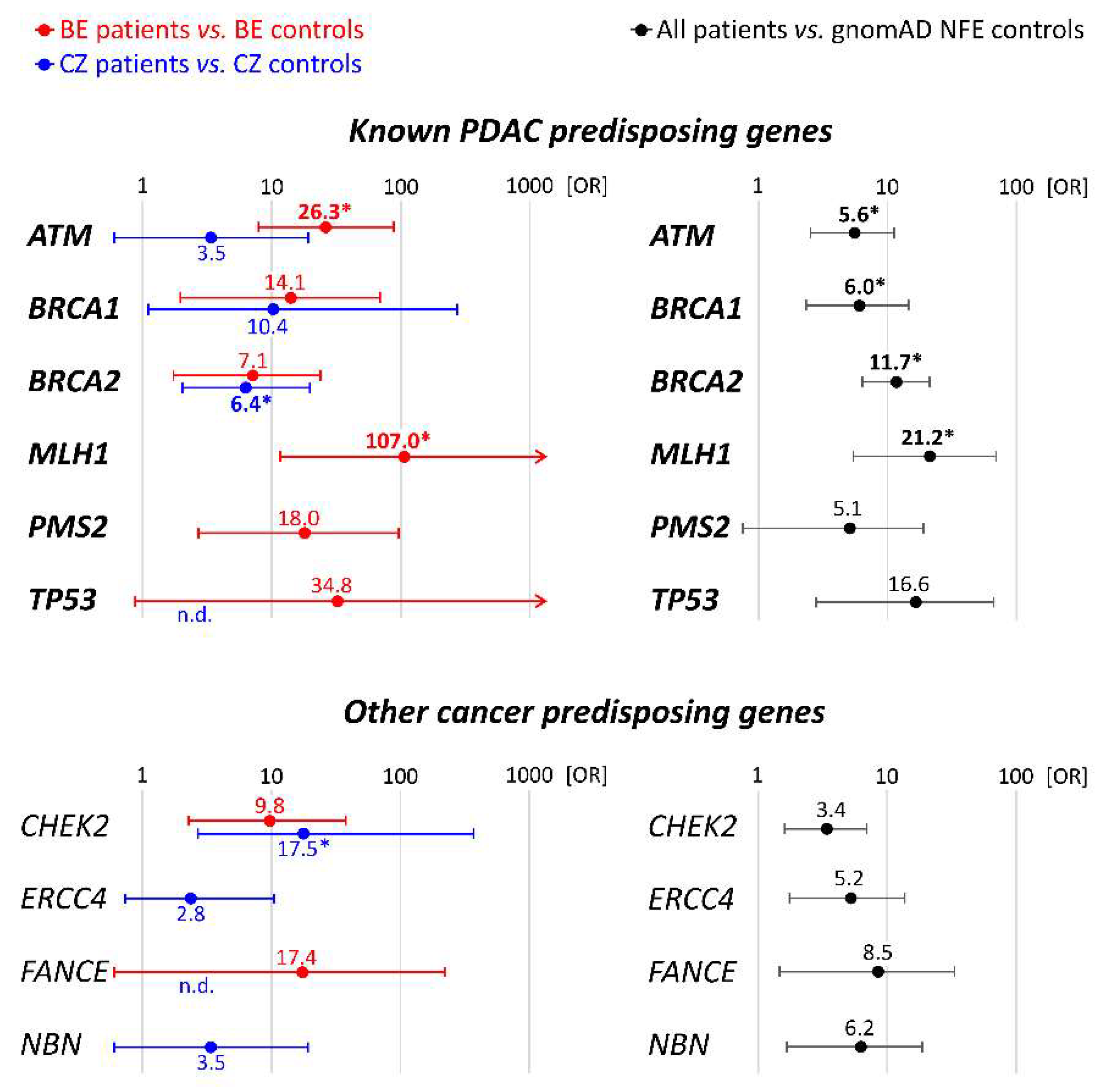
3.1. Spectrum and Frequencies of Germline Alterations in PDAC Patients
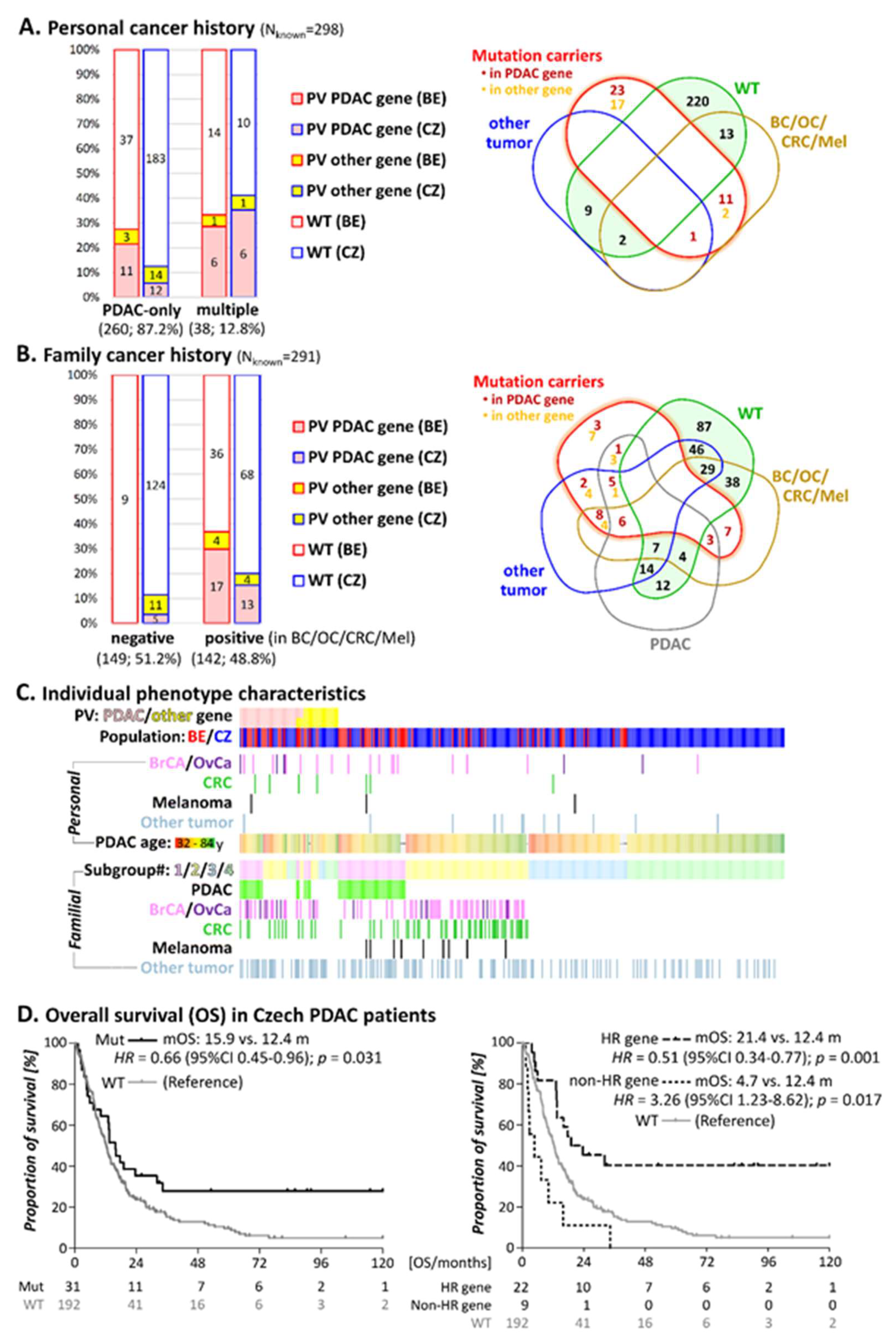
3.2. Personal Cancer History
3.3. Family Cancer History
3.4. Survival in Individuals with PV
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benzel, J.; Fendrich, V. Familial Pancreatic Cancer. Oncol. Res. Treat. 2018, 41, 611–618. [Google Scholar] [CrossRef]
- Llach, J.; Carballal, S.; Moreira, L. Familial Pancreatic Cancer: Current Perspectives. Cancer Manag. Res. 2020, 12, 743–758. [Google Scholar] [CrossRef] [Green Version]
- Goggins, M.; Overbeek, K.A.; Brand, R.; Syngal, S.; Del Chiaro, M.; Bartsch, D.K.; Bassi, C.; Carrato, A.; Farrell, J.; Fishman, E.K.; et al. Management of patients with increased risk for familial pancreatic cancer: Updated recommendations from the International Cancer of the Pancreas Screening (CAPS) Consortium. Gut 2020, 69, 7–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Golan, T.; Kindler, H.L.; Park, J.O.; Reni, M.; Macarulla, T.; Hammel, P.; Van Cutsem, E.; Arnold, D.; Hochhauser, D.; McGuinness, D.; et al. Geographic and Ethnic Heterogeneity of Germline BRCA1 or BRCA2 Mutation Prevalence Among Patients With Metastatic Pancreatic Cancer Screened for Entry Into the POLO Trial. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2020, 38, 1442–1454. [Google Scholar] [CrossRef]
- Hruban, R.H.; Canto, M.I.; Goggins, M.; Schulick, R.; Klein, A.P. Update on familial pancreatic cancer. Adv. Surg. 2010, 44, 293–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salo-Mullen, E.E.; O’Reilly, E.M.; Kelsen, D.P.; Ashraf, A.M.; Lowery, M.A.; Yu, K.H.; Reidy, D.L.; Epstein, A.S.; Lincoln, A.; Saldia, A.; et al. Identification of germline genetic mutations in patients with pancreatic cancer. Cancer 2015, 121, 4382–4388. [Google Scholar] [CrossRef]
- Shindo, K.; Yu, J.; Suenaga, M.; Fesharakizadeh, S.; Cho, C.; Macgregor-Das, A.; Siddiqui, A.; Witmer, P.D.; Tamura, K.; Song, T.J.; et al. Deleterious Germline Mutations in Patients With Apparently Sporadic Pancreatic Adenocarcinoma. J. Clin. Oncol. 2017, 35, 3382–3390. [Google Scholar] [CrossRef]
- Yurgelun, M.B.; Chittenden, A.B.; Morales-Oyarvide, V.; Rubinson, D.A.; Dunne, R.F.; Kozak, M.M.; Qian, Z.R.; Welch, M.W.; Brais, L.K.; Da Silva, A.; et al. Germline cancer susceptibility gene variants, somatic second hits, and survival outcomes in patients with resected pancreatic cancer. Genetic. Med. 2019, 21, 213–223. [Google Scholar] [CrossRef] [Green Version]
- Fountzilas, E.; Eliades, A.; Koliou, G.-A.; Achilleos, A.; Loizides, C.; Tsangaras, K.; Pectasides, D.; Sgouros, J.; Papakostas, P.; Rallis, G.; et al. Clinical Significance of Germline Cancer Predisposing Variants in Unselected Patients with Pancreatic Adenocarcinoma. Cancers 2021, 13, 198. [Google Scholar] [CrossRef]
- Goldstein, J.B.; Zhao, L.; Wang, X.; Ghelman, Y.; Overman, M.J.; Javle, M.M.; Shroff, R.T.; Varadhachary, G.R.; Wolff, R.A.; McAllister, F.; et al. Germline DNA Sequencing Reveals Novel Mutations Predictive of Overall Survival in a Cohort of Patients with Pancreatic Cancer. Clin. Cancer Res. 2020, 26, 1385–1394. [Google Scholar] [CrossRef]
- Pishvaian, M.J.; Blais, E.M.; Brody, J.R.; Lyons, E.; DeArbeloa, P.; Hendifar, A.; Mikhail, S.; Chung, V.; Sahai, V.; Sohal, D.P.S.; et al. Overall survival in patients with pancreatic cancer receiving matched therapies following molecular profiling: A retrospective analysis of the Know Your Tumor registry trial. Lancet Oncol. 2020, 21, 508–518. [Google Scholar] [CrossRef]
- Golan, T.; Hammel, P.; Reni, M.; Van Cutsem, E.; Macarulla, T.; Hall, M.J.; Park, J.-O.; Hochhauser, D.; Arnold, D.; Oh, D.-Y.; et al. Maintenance Olaparib for Germline BRCA-Mutated Metastatic Pancreatic Cancer. N. Engl. J. Med. 2019, 381, 317–327. [Google Scholar] [CrossRef] [PubMed]
- Slavin, T.P.; Niell-Swiller, M.; Solomon, I.; Nehoray, B.; Rybak, C.; Blazer, K.R.; Weitzel, J.N. Clinical Application of Multigene Panels: Challenges of Next-Generation Counseling and Cancer Risk Management. Front. Oncol. 2015, 5, 208. [Google Scholar] [CrossRef] [Green Version]
- Kleiblova, P.; Stolarova, L.; Krizova, K.; Lhota, F.; Hojny, J.; Zemankova, P.; Havranek, O.; Vocka, M.; Cerna, M.; Lhotova, K.; et al. Identification of deleterious germline CHEK2 mutations and their association with breast and ovarian cancer. Int. J. Cancer 2019, 145, 1782–1797. [Google Scholar] [CrossRef] [PubMed]
- Soukupova, J.; Zemankova, P.; Lhotova, K.; Janatova, M.; Borecka, M.; Stolarova, L.; Lhota, F.; Foretova, L.; Machackova, E.; Stranecky, V.; et al. Validation of CZECANCA (CZEch CAncer paNel for Clinical Application) for targeted NGS-based analysis of hereditary cancer syndromes. PLoS ONE 2018, 13, e0195761. [Google Scholar] [CrossRef] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Genome Aggregation Database (gnomAD). Available online: http://gnomad.broadinstitute.org/ (accessed on 30 September 2020).
- Claes, K.; Poppe, B.; Coene, I.; De Paepe, A.; Messiaen, L. BRCA1 and BRCA2 germline mutation spectrum and frequencies in Belgian breast/ovarian cancer families. Br. J. Cancer 2004, 90, 1244–1251. [Google Scholar] [CrossRef] [PubMed]
- Pohlreich, P.; Zikan, M.; Stribrna, J.; Kleibl, Z.; Janatova, M.; Kotlas, J.; Zidovska, J.; Novotny, J.; Petruzelka, L.; Szabo, C.; et al. High proportion of recurrent germline mutations in the BRCA1 gene in breast and ovarian cancer patients from the Prague area. Breast Cancer Res. 2005, 7, R728–R736. [Google Scholar] [CrossRef] [Green Version]
- Hu, C.; Hart, S.N.; Polley, E.C.; Gnanaolivu, R.; Shimelis, H.; Lee, K.Y.; Lilyquist, J.; Na, J.; Moore, R.; Antwi, S.O.; et al. Association Between Inherited Germline Mutations in Cancer Predisposition Genes and Risk of Pancreatic Cancer. JAMA 2018, 319, 2401–2409. [Google Scholar] [CrossRef]
- Armstrong, S.A.; Schultz, C.W.; Azimi-Sadjadi, A.; Brody, J.R.; Pishvaian, M.J. ATM Dysfunction in Pancreatic Adenocarcinoma and Associated Therapeutic Implications. Mol. Cancer Ther. 2019, 18, 1899–1908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cremin, C.; Lee, M.K.-C.; Hong, Q.; Hoeschen, C.; Mackenzie, A.; Dixon, K.; McCullum, M.; Nuk, J.; Kalloger, S.; Karasinska, J.; et al. Burden of hereditary cancer susceptibility in unselected patients with pancreatic ductal adenocarcinoma referred for germline screening. Cancer Med. 2020, 9, 4004–4013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brand, R.; Borazanci, E.; Speare, V.; Dudley, B.; Karloski, E.; Peters, M.L.B.; Stobie, L.; Bahary, N.; Zeh, H.; Zureikat, A.; et al. Prospective study of germline genetic testing in incident cases of pancreatic adenocarcinoma. Cancer 2018, 124, 3520–3527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cybulski, C.; Górski, B.; Huzarski, T.; Masojć, B.; Mierzejewski, M.; Debniak, T.; Teodorczyk, U.; Byrski, T.; Gronwald, J.; Matyjasik, J.; et al. CHEK2 is a multiorgan cancer susceptibility gene. Am. J. Hum. Genet. 2004, 75, 1131–1135. [Google Scholar] [CrossRef] [Green Version]
- Stolarova, L.; Kleiblova, P.; Janatova, M.; Soukupova, J.; Zemankova, P.; Macurek, L.; Kleibl, Z. CHEK2 Germline Variants in Cancer Predisposition: Stalemate Rather than Checkmate. Cells 2020, 9, 2675. [Google Scholar] [CrossRef] [PubMed]
- Lener, M.R.; Kashyap, A.; Kluzniak, W.; Cybulski, C.; Soluch, A.; Pietrzak, S.; Huzarski, T.; Gronwald, J.; Lubinski, J. The Prevalence of Founder Mutations Among Individuals from Families with Familial Pancreatic Cancer Syndrome. Cancer Res. Treat. 2016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhan, W.; Shelton, C.A.; Greer, P.J.; Brand, R.E.; Whitcomb, D.C. Germline Variants and Risk for Pancreatic Cancer: A Systematic Review and Emerging Concepts. Pancreas 2018, 47, 924–936. [Google Scholar] [CrossRef]
- Doi, H.; Koyano, S.; Miyatake, S.; Nakajima, S.; Nakazawa, Y.; Kunii, M.; Tomita-Katsumoto, A.; Oda, K.; Yamaguchi, Y.; Fukai, R.; et al. Cerebellar ataxia-dominant phenotype in patients with ERCC4 mutations. J. Hum. Genet. 2018, 63, 417–423. [Google Scholar] [CrossRef]
- Lener, M.R.; Scott, R.J.; Kluzniak, W.; Baszuk, P.; Cybulski, C.; Wiechowska-Kozlowska, A.; Huzarski, T.; Byrski, T.; Kladny, J.; Pietrzak, S.; et al. Do founder mutations characteristic of some cancer sites also predispose to pancreatic cancer? Int. J. Cancer 2016, 139, 601–606. [Google Scholar] [CrossRef] [Green Version]
- Borecka, M.; Zemankova, P.; Lhota, F.; Soukupova, J.; Kleiblova, P.; Vocka, M.; Soucek, P.; Ticha, I.; Kleibl, Z.; Janatova, M. The c.657del5 variant in the NBN gene predisposes to pancreatic cancer. Gene 2016, 587, 169–172. [Google Scholar] [CrossRef]
- Rump, A.; Benet-Pages, A.; Schubert, S.; Kuhlmann, J.D.; Janavičius, R.; Macháčková, E.; Foretová, L.; Kleibl, Z.; Lhota, F.; Zemankova, P.; et al. Identification and Functional Testing of ERCC2 Mutations in a Multi-national Cohort of Patients with Familial Breast- and Ovarian Cancer. PLoS Genet. 2016, 12, 1–18. [Google Scholar] [CrossRef]
- Dudley, B.; Karloski, E.; Monzon, F.A.; Singhi, A.D.; Lincoln, S.E.; Bahary, N.; Brand, R.E. Germline mutation prevalence in individuals with pancreatic cancer and a history of previous malignancy. Cancer 2018, 124, 1691–1700. [Google Scholar] [CrossRef] [Green Version]
- Bartsch, D.K.; Matthäi, E.; Mintziras, I.; Bauer, C.; Figiel, J.; Sina-Boemers, M.; Gress, T.M.; Langer, P.; Slater, E.P. The German National Case Collection for Familial Pancreatic Carcinoma (FaPaCa)—Knowledge Gained in 20 Years. Dtsch. Arztebl. Int. 2021, 118, 163–168. [Google Scholar] [CrossRef]
- Chaffee, K.G.; Oberg, A.L.; McWilliams, R.R.; Majithia, N.; Allen, B.A.; Kidd, J.; Singh, N.; Hartman, A.-R.; Wenstrup, R.J.; Petersen, G.M. Prevalence of germ-line mutations in cancer genes among pancreatic cancer patients with a positive family history. Genet. Med. 2018, 20, 119–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takai, E.; Yachida, S.; Shimizu, K.; Furuse, J.; Kubo, E.; Ohmoto, A.; Suzuki, M.; Hruban, R.H.; Okusaka, T.; Morizane, C.; et al. Germline mutations in Japanese familial pancreatic cancer patients. Oncotarget 2016, 7, 74227–74235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rustgi, A.K. Familial pancreatic cancer: Genetic advances. Genes Dev. 2014, 28, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grant, R.C.; Selander, I.; Connor, A.A.; Selvarajah, S.; Borgida, A.; Briollais, L.; Petersen, G.M.; Lerner-Ellis, J.; Holter, S.; Gallinger, S. Prevalence of germline mutations in cancer predisposition genes in patients with pancreatic cancer. Gastroenterology 2015, 148, 556–564. [Google Scholar] [CrossRef] [Green Version]
- Smith, A.L.; Alirezaie, N.; Connor, A.; Chan-Seng-Yue, M.; Grant, R.; Selander, I.; Bascuñana, C.; Borgida, A.; Hall, A.; Whelan, T.; et al. Candidate DNA repair susceptibility genes identified by exome sequencing in high-risk pancreatic cancer. Cancer Lett. 2016, 370, 302–312. [Google Scholar] [CrossRef] [Green Version]
- Yadav, S.; Kasi, P.M.; Bamlet, W.R.; Ho, T.P.; Polley, E.C.; Hu, C.; Hart, S.N.; Rabe, K.G.; Boddicker, N.J.; Gnanaolivu, R.D.; et al. Effect of Germline Mutations in Homologous Recombination Repair Genes on Overall Survival of Patients with Pancreatic Adenocarcinoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2020, 26, 6505–6512. [Google Scholar] [CrossRef]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
| Characteristics | All Patients (n = 298) | Belgian Patients (n = 72) | Czech Patients (n = 226) | p-Value * |
|---|---|---|---|---|
| Gender | ||||
| Female (%) | 150 (50.3) | 41 (56.9) | 109 (48.2) | 0.22 a |
| Male (%) | 148 (49.7) | 31 (43.1) | 117 (51.8) | |
| Age at PDAC diagnosis; Mean age (SE) | 61.9 (0.6) | 58.0 (1.4) | 63.11 (0.6) | 0.002 b |
| <50 (%) | 40 (13.4) | 19 (26.4) | 21 (9.3) | |
| 50–59 (%) | 77 (25.8) | 17 (23.6) | 60 (26.6) | |
| 60–69 (%) | 120 (40.3) | 26 (36.1) | 94 (41.6) | |
| ≥70 (%) | 61 (20.5) | 10 (13.9) | 51 (22.6) | |
| Multiple primary tumors in personal history | ||||
| Absent (%) | 260 (87.2) | 51 (70.8) | 209 (92.5) | <0.0001 a |
| Present (%) | 38 (12.8) | 21 (29.2) | 17 (7.5) | |
| Multiple primary tumors in personal history | ||||
| Breast (%) | 19 (6.4) | 11 (15.3) | 8 (3.5) | 0.001 a |
| Ovarian/endometrial (%) | 6 (1.7) | 2 (2.8) | 4 (1.8) | 0.63 a |
| Colon (%) | 7 (2.3) | 7 (9.7) | 0 | <0.0001 a |
| Melanoma (%) | 3 (1.0) | 3 (4.2) | 0 | 0.01 a |
| Other (%) | 12 (4.0) | 4 (5.6) | 8 (3.5) | 0.73 a |
| Family cancer history ‡ (first/second-degree relatives) | ||||
| Negative (%) | 149 (51.2) | 9 (13.6) | 140 (62.2) | <0.0001 a |
| Positive (%) | 142 (48.8) | 57 (86.4) | 85 (37.8) | |
| Unknown | 7 | 6 | 1 | |
| Syndromic ‡ tumors in family cancer history | ||||
| Pancreatic (%) | 56 (19.2) | 34 (51.5) | 22 (9.8) | <0.0001 a |
| Breast (%) | 59 (20.3) | 27 (40.9) | 32 (14.2) | <0.0001 a |
| Ovarian/endometrial (%) | 13 (4.5) | 4 (6.1) | 9 (4.0) | 0.50 a |
| Colon (%) | 52 (17.9) | 16 (24.2) | 36 (15.9) | 0.14 a |
| Melanoma (%) | 7 (2.4) | 3 (4.5) | 4 (1.8) | 0.19 a |
| PDAC Patients | Population-Matched Controls | ||||
|---|---|---|---|---|---|
| Germline PV | All; n = 298 | Belgian; n = 72 | Czech; n = 226 | Belgian; n = 2485 | Czech; n = 777 |
| Known PDAC-Predisposition Genes | |||||
| ATM * | 8 (2.68%) | 5 (6.94%) | 3 (1.32%) | 7 (0.28%) | 3 (0.39%) |
| BRCA1 | 5 (1.67%) | 2 (2.78%) | 3 (1.32%) | 5 (0.20%) | 1 (0.13%) |
| BRCA2 * | 12 (4.01%) | 3 (4.17%) | 9 (3.98%) | 15 (0.60%) | 5 (0.64%) |
| CDKN2A | 1 (0.33%) | 1 (1.39%) | 0 | 0 | 0 |
| MLH1 * | 3 (1.00%) | 3 (4.17%) | 0 | 1 (0.04%) | 0 |
| MSH2 | 0 | 0 | 0 | 2 (0.08%) | 3 (0.39%) |
| MSH6 * | 1 (0.33%) | 1 (1.39%) | 0 | 2 (0.08%) | 0 |
| PALB2 | 2 (0.67%) | 0 | 2 (0.88%) | 4 (0.16%) | 2 (0.26%) |
| PMS2 | 2 (0.67%) | 2 (2.78%) | 0 | 4 (0.16%) | 0 |
| STK11 | 0 | 0 | 0 | 1 (0.04%) | 0 |
| TP53 | 2 (0.67%) | 1 (1.39%) | 1 (0.34%) | 1 (0.04%) | 0 |
| PDAC gene PV | 36 | 18 | 18 | 42 | 14 |
| Number of individuals with PDAC PV * | 35 * (11.74%) | 17 * (23.61%) | 18 (7.96%) | 42 (1.69%) | 14 (1.80%) |
| Other cancer predisposition genes for which the association with PDAC is not firmly established | |||||
| BARD1 | 0 | 0 | 0 | 1 (0.04%) | 0 |
| BLM | 0 | 0 | 0 | 3 (0.12%) | 3 (0.39%) |
| BRIP1 | 2 (0.67%) | 0 | 2 (0.88%) | 1 (0.04%) | 0 |
| CDK4 | 0 | 0 | 0 | 1 (0.04%) | 0 |
| CHEK1 | 0 | 0 | 0 | 1 (0.04%) | 0 |
| CHEK2 * | 8 (2.68%) | 3 (4.17%) | 5 (2.21%) | 11 (0.40%) | 1 (0.13%) |
| ERCC4 * | 4 (1.34%) | 0 | 4 (1.76%) | 6 (0.24%) | 5 (0.64%) |
| FANCA | 0 | 0 | 0 | 6 (0.24%) | 3 (0.39%) |
| FANCC | 0 | 0 | 0 | 1 (0.04%) | 0 |
| FANCD2 | 0 | 0 | 0 | 3 (0.12%) | 1 (0.13%) |
| FANCE * | 2 (0.67%) | 1 (1.39%) | 1 (0.44%) | 2 (0.08%) | 0 |
| FANCG * | 1 (0.33%) | 0 | 1 (0.44%) | 3 (0.12%) | 0 |
| FANCI | 0 | 0 | 0 | 3 (0.12%) | 1 (0.13%) |
| FANCL | 0 | 0 | 0 | 1 (0.04%) | 0 |
| FANCM | 1 (0.33%) | 1 (1.39%) | 0 | 10 (0.40%) | 4 (0.51%) |
| HOXB13 | 1 (0.33%) | 0 | 1 (0.44%) | 7 (0.28%) | 0 |
| MRE11 | 0 | 0 | 0 | 5 (0.20%) | 2 (0.26%) |
| NBN | 3 (1.00%) | 0 | 3 (1.32%) | 7 (0.28%) | 3 (0.39%) |
| POLD1 | 1 (0.33%) | 0 | 1 (0.44%) | 1 (0.04%) | 0 |
| POLE | 1 (0.33%) | 1 (1.39%) | 0 | 1 (0.04%) | 0 |
| PTEN | 0 | 0 | 0 | 1 (0.04%) | 0 |
| RAD50 | 0 | 0 | 0 | 3 (0.12%) | 1 (0.13%) |
| RAD51C | 0 | 0 | 0 | 1 (0.04%) | 0 |
| RAD51D | 0 | 0 | 0 | 2 (0.08%) | 0 |
| RAD54L | 0 | 0 | 0 | 7 (0.28%) | 2 (0.26%) |
| RECQL | 0 | 0 | 0 | 3 (0.12%) | 3 (0.39%) |
| SLX4 * | 1 (0.33%) | 1 (1.39%) | 0 | 6 (0.24%) | 1 (0.13%) |
| XRCC2 | 0 | 0 | 0 | 1 (0.04%) | 0 |
| Other gene PV | 25 | 7 | 16 | 98 | 30 |
| Number of individuals with PV in other genes * | 23* (7.72%) | 7 (9.72%) | 16 (7.08%) | 98 (3.94%) | 30 (3.86%) |
| All PV | 61 | 25 | 36 | 148 | 45 |
| All PV carriers * | 54 * (18.12%) | 21 * (29.17%) | 33 * (14.60%) | 140 (5.63%) | 45 (5.79%) |
| PDAC Patients Group | All (n = 291); n | Patients with PV | Patients without PV n (%) | p-Value 2 | |
|---|---|---|---|---|---|
| in PDAC Gene 1; n (%) | in Other Gene Only; n (%) | ||||
| Familial cancer patients | 142 | 30 (21.1) | 8 (5.6%) | 104 (73.2) | |
| Belgian | 57 | 17 (29.8) | 4 (7.0) | 36 (63.2) | 0.07 |
| Czech | 85 | 13 (15.3) | 4 (4.7) | 68 (80.0) | |
| Subgroup#1: ≥1 PDAC in first/second degree relative | 56 | 15 (26.8) | 4 (7.1) | 37 (66.1) | |
| Belgian | 34 | 9 (26.5) | 3 (8.8) | 22 (64.7) | 0.83 |
| Czech | 22 | 6 (27.3) | 1 (4.5) | 15 (68.2) | |
| Subgroup#2: ≥1 Tumor associated with increased PDAC risk in First/second degree relative | 86 | 15 (17.4) | 4 (4.7) | 67 (77.9) | |
| Belgian | 23 | 8 (34.8) | 1 (4.3) | 14 (60.9) | 0.04 |
| Czech | 63 | 7 (11.1) | 3 (4.8) | 53 (84.1) | |
| Sporadic PDAC patients 3 | 149 | 5 (3.4) | 11 (7.4) | 133 (89.3) | |
| Belgian | 9 | 0 | 0 | 9 (100) | n.d. |
| Czech | 140 | 5 (3.6) | 11 (7.9) | 124 (88.5) | |
| Subgroup#3: Sporadic PDAC, early onset (≤60 years) | 53 | 1 (1.9) | 4 (7.5) | 48 (90.6) | |
| Belgian | 9 | 0 | 0 | 9 (100.0) | n.d. |
| Czech | 44 | 1 (2.3) | 4 (9.1) | 39 (88.6) | |
| Subgroup#4: Sporadic PDAC, later onset (>60years) | 96 | 4 (4.2) | 7 (7.3) | 85 (89.6) | |
| Belgian | 0 | 0 | 0 | 0 | n.d. |
| Czech | 96 | 4 (4.2) | 7 (7.3) | 85 (89.6) | |
| Sum | 291 | 35 | 19 | 237 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wieme, G.; Kral, J.; Rosseel, T.; Zemankova, P.; Parton, B.; Vocka, M.; Van Heetvelde, M.; Kleiblova, P.; Blaumeiser, B.; Soukupova, J.; et al. Prevalence of Germline Pathogenic Variants in Cancer Predisposing Genes in Czech and Belgian Pancreatic Cancer Patients. Cancers 2021, 13, 4430. https://doi.org/10.3390/cancers13174430
Wieme G, Kral J, Rosseel T, Zemankova P, Parton B, Vocka M, Van Heetvelde M, Kleiblova P, Blaumeiser B, Soukupova J, et al. Prevalence of Germline Pathogenic Variants in Cancer Predisposing Genes in Czech and Belgian Pancreatic Cancer Patients. Cancers. 2021; 13(17):4430. https://doi.org/10.3390/cancers13174430
Chicago/Turabian StyleWieme, Greet, Jan Kral, Toon Rosseel, Petra Zemankova, Bram Parton, Michal Vocka, Mattias Van Heetvelde, Petra Kleiblova, Bettina Blaumeiser, Jana Soukupova, and et al. 2021. "Prevalence of Germline Pathogenic Variants in Cancer Predisposing Genes in Czech and Belgian Pancreatic Cancer Patients" Cancers 13, no. 17: 4430. https://doi.org/10.3390/cancers13174430
APA StyleWieme, G., Kral, J., Rosseel, T., Zemankova, P., Parton, B., Vocka, M., Van Heetvelde, M., Kleiblova, P., Blaumeiser, B., Soukupova, J., van den Ende, J., Nehasil, P., Tejpar, S., Borecka, M., Gómez García, E. B., Blok, M. J., Safarikova, M., Kalousova, M., Geboes, K., ... Claes, K. B. M. (2021). Prevalence of Germline Pathogenic Variants in Cancer Predisposing Genes in Czech and Belgian Pancreatic Cancer Patients. Cancers, 13(17), 4430. https://doi.org/10.3390/cancers13174430