
Synergistic Drug Combinations Prevent Resistance in ALK+ Anaplastic Large Cell Lymphoma

 , , , , , , ,
, , , , , , ,  , ,
, ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Chemicals, Antibodies and Cells
2.2. Long-Term Cell Cultures
2.3. Proliferation Assays, Synergy and Statistical Analysis
2.4. Cell Cycle and Apoptosis
2.5. Soft Agar Colony Assay
2.6. Western Blot Analysis
2.7. Quantitative Real-Time PCR
2.8. 3D Matrix-Embedded Cell Cultures
2.9. In Vivo Experiments
3. Results
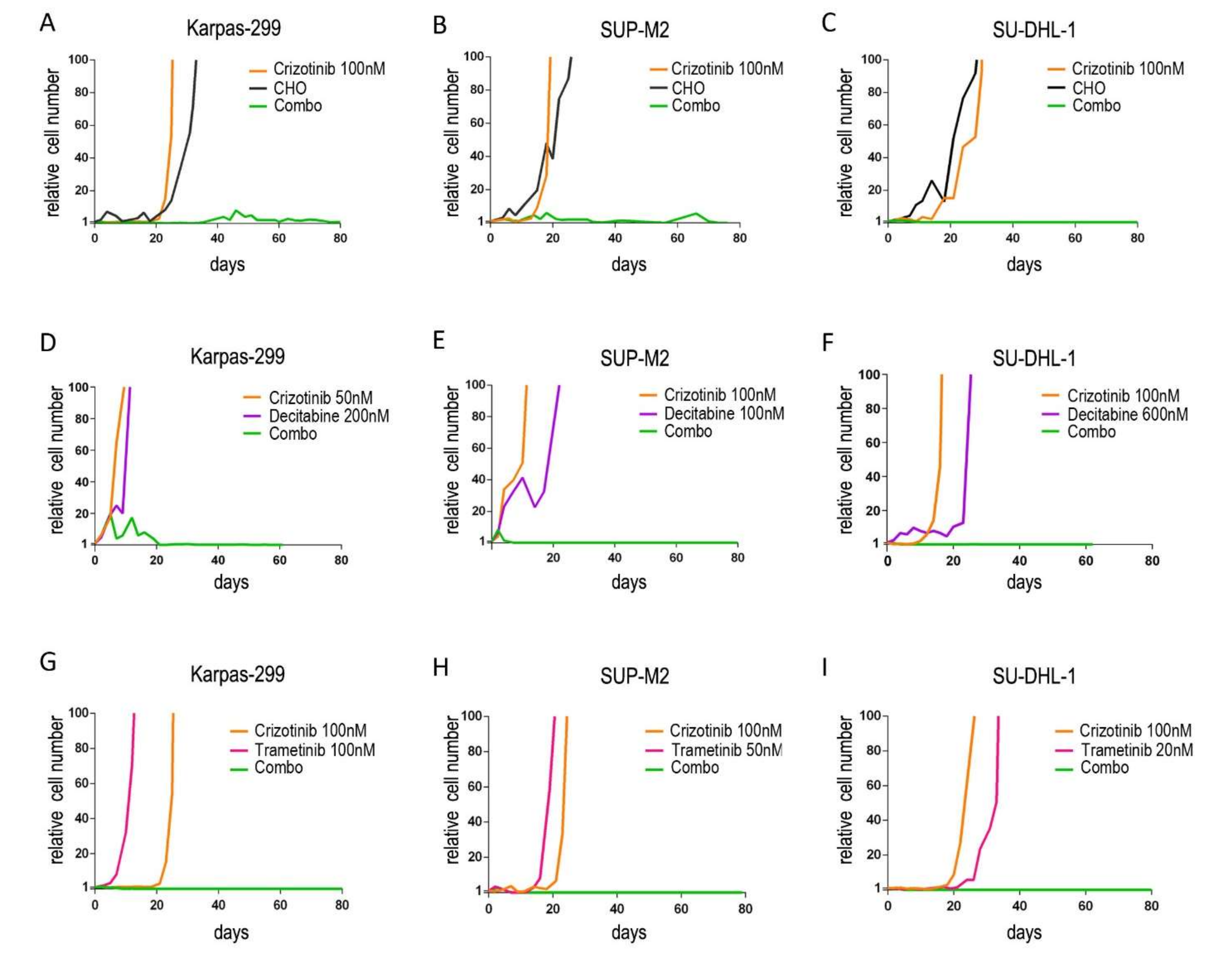
3.1. Upfront Combined Treatments Prevent the Selection of Resistant Clones in ALK+ ALCL Cells
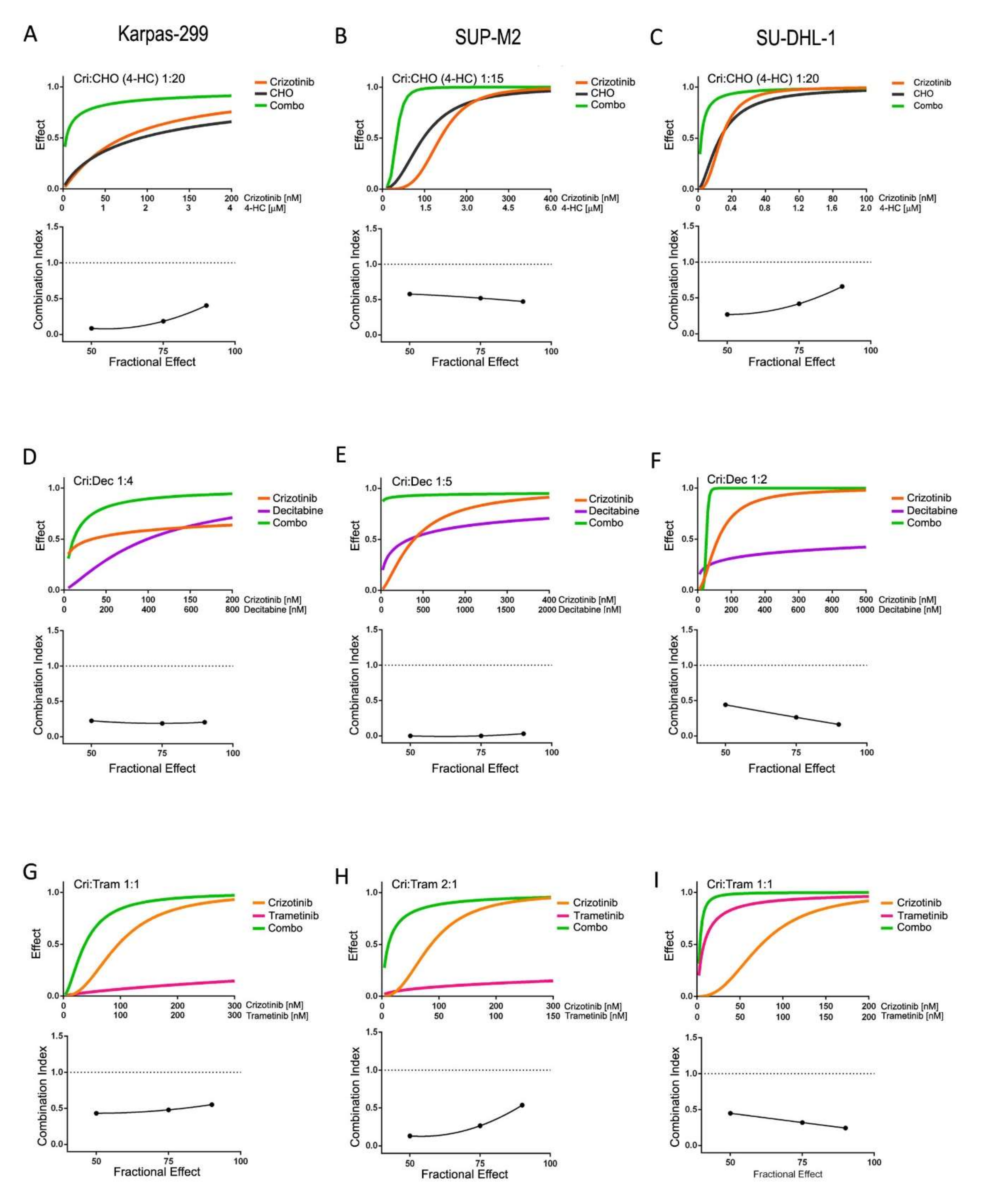
3.2. Synergistic Interaction of Crizotinib with Decitabine, CHO and Trametinib
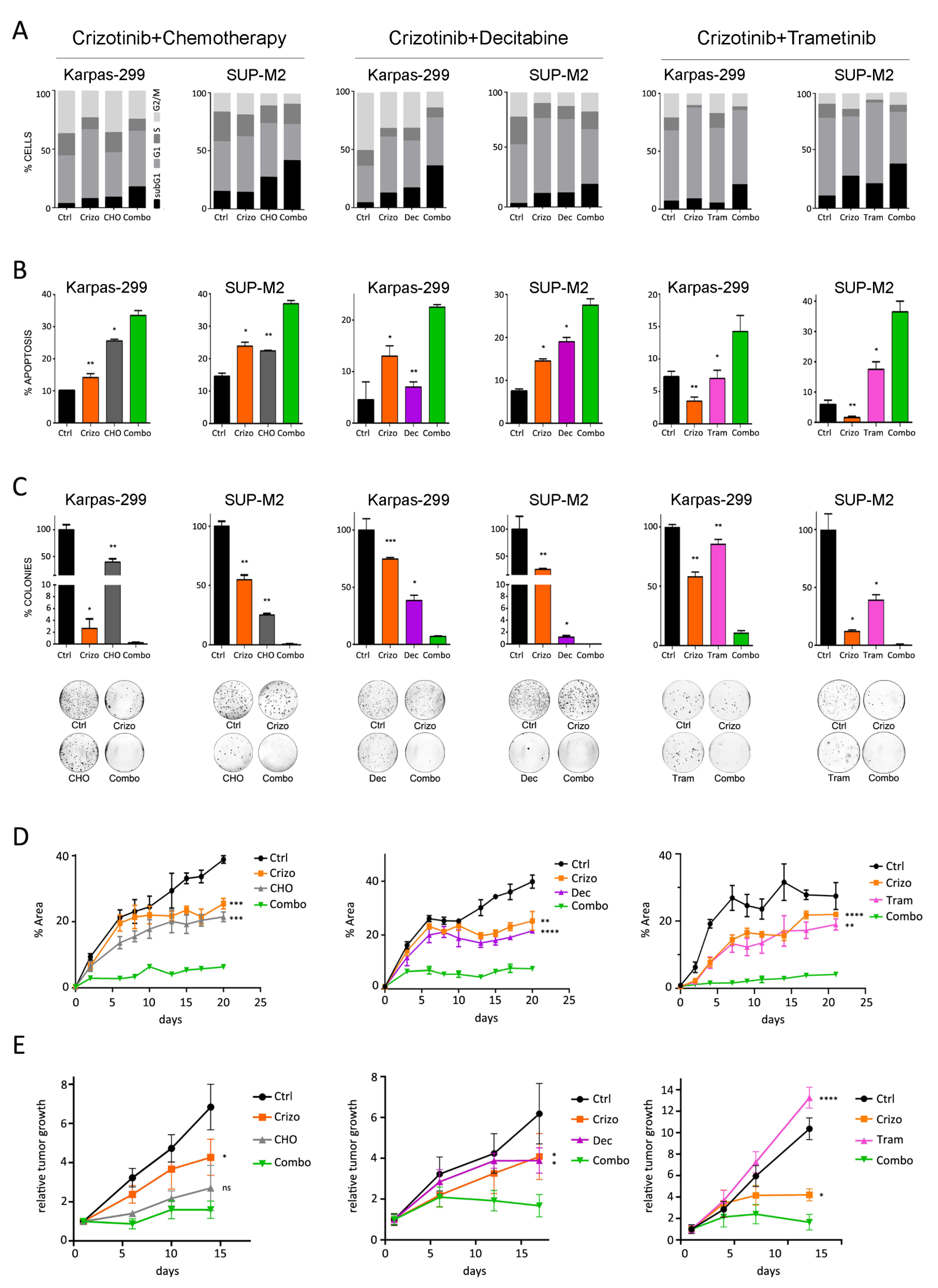
3.3. Combined Treatments Enhance Apoptosis in vitro and Tumor Growth Inhibition in Mice
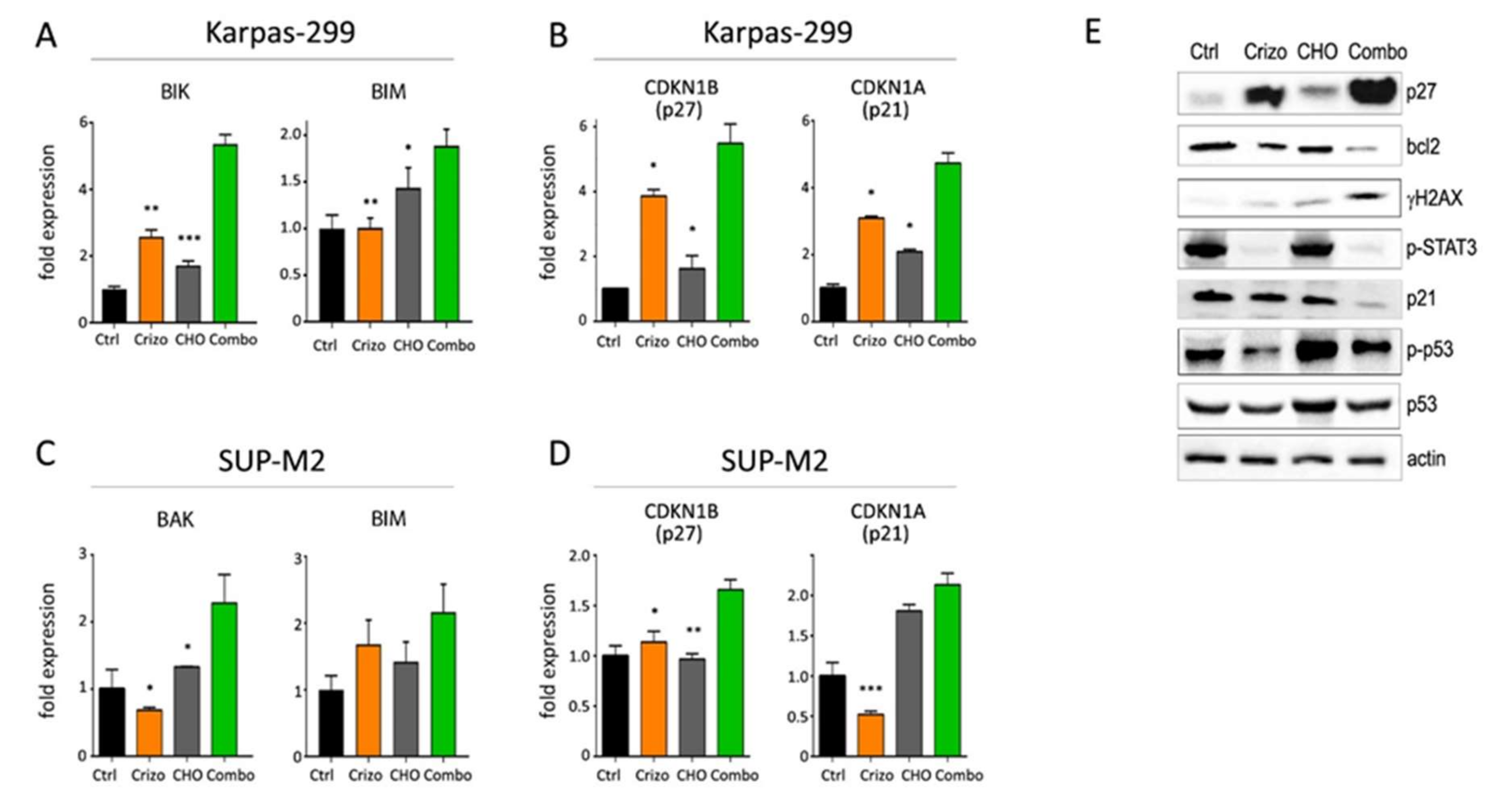
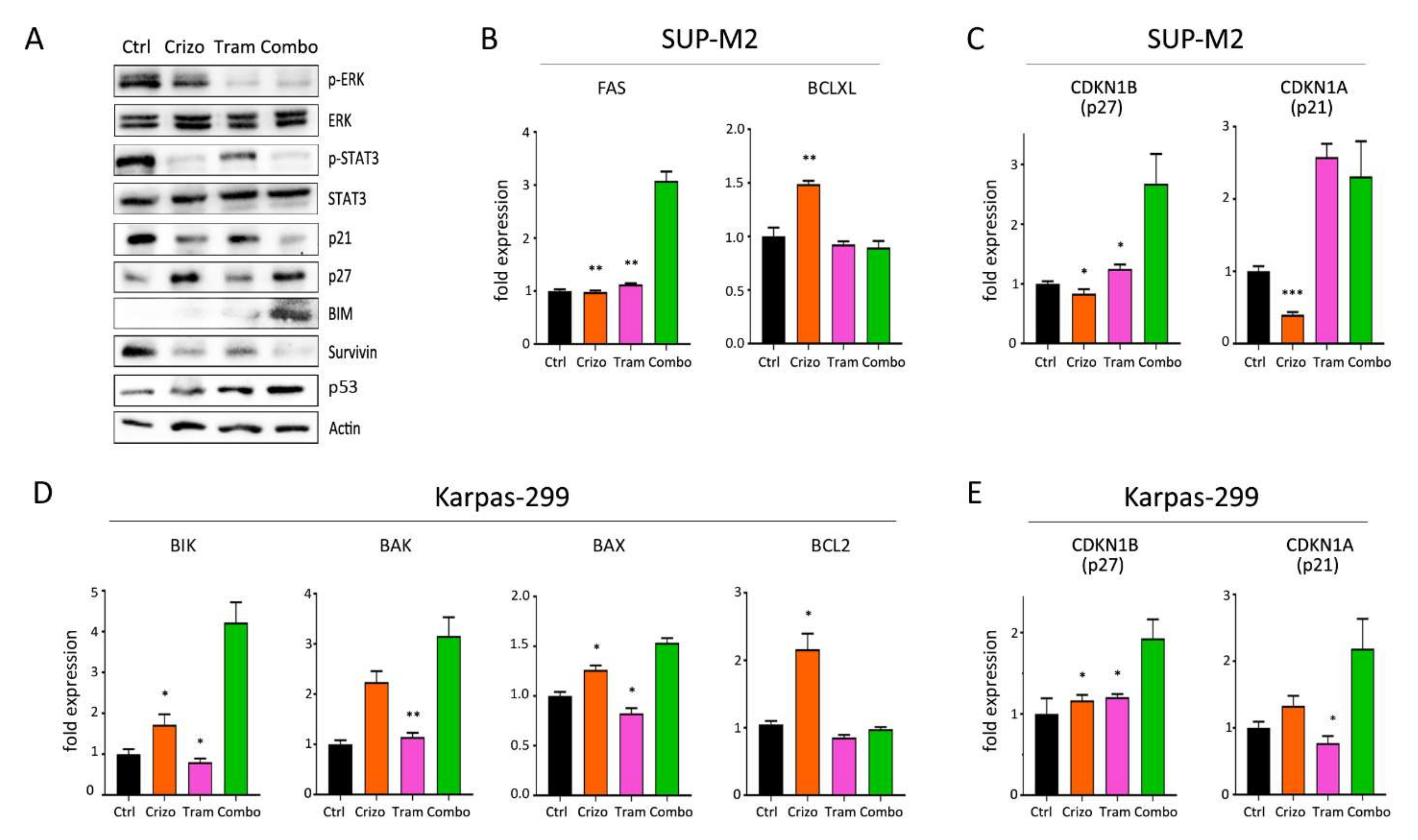
3.4. Molecular Effects of Combined Therapies
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bozic, I.; Reiter, J.G.; Allen, B.; Antal, T.; Chatterjee, K.; Shah, P.; Moon, Y.S.; Yaqubie, A.; Kelly, N.; Le, D.T.; et al. Evolutionary dynamics of cancer in response to targeted combination therapy. eLife 2013, 2, e00747. [Google Scholar] [CrossRef]
- Chiarle, R.; Voena, C.; Ambrogio, C.; Piva, R.; Inghirami, G. The anaplastic lymphoma kinase in the pathogenesis of cancer. Nat. Rev. Cancer 2008, 8, 11–23. [Google Scholar] [CrossRef]
- Sharma, G.G.; Mota, I.; Mologni, L.; Patrucco, E.; Gambacorti-Passerini, C.; Chiarle, R. Tumor resistance against ALK targeted therapy—Where it comes from and where it goes. Cancers 2018, 10, 62. [Google Scholar] [CrossRef] [Green Version]
- Horwitz, S.; O’Connor, O.A.; Pro, B.; Illidge, T.; Fanale, M.; Advani, R.; Bartlett, N.L.; Christensen, J.H.; Morschhauser, F.; Domingo-Domenech, E.; et al. Brentuximab vedotin with chemotherapy for CD30-positive peripheral T-cell lymphoma (ECHELON-2): A global, double-blind, randomised, phase 3 trial. Lancet 2019, 393, 229–240. [Google Scholar] [CrossRef] [Green Version]
- David Sibon, D.; Nguyen, D.-P.; Schmitz, N.; Suzuki, R.; Feldman, A.L.; Gressin, R.; Lamant, L.; Weisenburger, D.D.; Rosenwald, A.; Nakamura, S.; et al. ALK-positive anaplastic large-cell lymphoma in adults: An individual patient data pooled analysis of 263 patients. Haematologica 2019, 104, e562–e565. [Google Scholar] [CrossRef]
- Gambacorti Passerini, C.; Farina, F.; Stasia, A.; Redaelli, S.; Ceccon, M.; Mologni, L.; Messa, C.; Guerra, L.; Giudici, G.; Sala, E.; et al. Crizotinib in advanced, chemoresistant anaplastic lymphoma kinase-positive lymphoma patients. J. Natl. Cancer Inst. 2014, 106, djt378. [Google Scholar] [CrossRef]
- Gambacorti-Passerini, C.; Orlov, S.; Zhang, L.; Braiteh, F.; Huang, H.; Esaki, T.; Horibe, K.; Ahn, J.-S.; Beck, J.T.; Edenfield, W.J.; et al. Long-term effects of crizotinib in ALK-positive tumors (excluding NSCLC): A phase 1b open-label study. Am. J. Hematol. 2018, 93, 607–614. [Google Scholar] [CrossRef]
- Ceccon, M.; Mologni, L.; Giudici, G.; Piazza, R.; Pirola, A.; Fontana, D.; Gambacorti-Passerini, C. Treatment Efficacy and Resistance Mechanisms Using the Second-Generation ALK Inhibitor AP26113 in Human NPM-ALK-Positive Anaplastic Large Cell Lymphoma. Mol. Cancer Res. 2015, 13, 775–783. [Google Scholar] [CrossRef] [Green Version]
- Redaelli, S.; Ceccon, M.; Zappa, M.; Sharma, G.G.; Mastini, C.; Mauri, M.; Nigoghossian, M.; Massimino, L.; Cordani, N.; Farina, F.; et al. Lorlatinib Treatment Elicits Multiple On- and Off-Target Mechanisms of Resistance in ALK-Driven Cancer. Cancer Res. 2018, 78, 6866–6880. [Google Scholar] [CrossRef] [Green Version]
- Ceccon, M.; Mologni, L.; Bisson, W.; Scapozza, L.; Gambacorti-Passerini, C. Crizotinib-resistant NPM-ALK mutants confer differential sensitivity to unrelated Alk inhibitors. Mol. Cancer Res. 2013, 11, 122–132. [Google Scholar] [CrossRef] [Green Version]
- Mologni, L.; Ceccon, M.; Fontana, D.; Pirola, A.; Piazza, R.; Gambacorti-Passerini, C. Drug-resistant NPM/ALK mutants show different sensitivity to second generation tyrosine kinase inhibitors. In Proceedings of the AACR Annual Meeting, Philadelphia, PA, USA, 18–22 April 2015. [Google Scholar]
- Hrustanovic, G.; Olivas, V.; Pazarentzos, E.; Tulpule, A.; Asthana, S.; Blakely, C.M.; Okimoto, R.A.; Lin, L.; Neel, D.S.; Sabnis, A.; et al. RAS-MAPK dependence underlies a rational polytherapy strategy in EML4-ALK-positive lung cancer. Nat. Med. 2015, 21, 1038–1047. [Google Scholar] [CrossRef]
- Berberich, A.; Schmitt, L.M.; Pusch, S.; Hielscher, T.; Rübmann, P.; Hucke, N.; Latzer, P.; Heßling, B.; Lemke, D.; Kessler, T.; et al. cMyc and ERK activity are associated with resistance to ALK inhibitory treatment in glioblastoma. J. Neurooncol. 2020, 146, 9–23. [Google Scholar] [CrossRef]
- Crystal, A.S.; Shaw, A.T.; Sequist, L.V.; Friboulet, L.; Niederst, M.J.; Lockerman, E.L.; Frias, R.L.; Gainor, J.F.; Amzallag, A.; Greninger, P.; et al. Patient-derived models of acquired resistance can identify effective drug combinations for cancer. Science 2014, 346, 1480–1486. [Google Scholar] [CrossRef] [Green Version]
- Ambrogio, C.; Martinengo, C.; Voena, C.; Tondat, F.; Riera, L.; di Celle, P.F.; Inghirami, G.; Chiarle, R. NPM-ALK oncogenic tyrosine kinase controls T-cell identity by transcriptional regulation and epigenetic silencing in lymphoma cells. Cancer Res. 2009, 69, 8611–8619. [Google Scholar] [CrossRef] [Green Version]
- Yun, M.R.; Lim, S.M.; Kim, S.K.; Choi, H.M.; Pyo, K.H.; Kim, S.K.; Lee, J.M.; Lee, Y.W.; Choi, J.W.; Kim, H.R.; et al. Enhancer remodeling and MicroRNA alterations are associated with acquired resistance to ALK inhibitors. Cancer Res. 2018, 78, 3350–3362. [Google Scholar] [CrossRef] [Green Version]
- Hassler, M.R.; Pulverer, W.; Lakshminarasimhan, R.; Redl, E.; Hacker, J.; Garland, G.D.; Merkel, O.; Schiefer, A.-I.; Simonitsch-Klupp, I.; Kenner, L.; et al. Insights into the Pathogenesis of Anaplastic Large-Cell Lymphoma through Genome-wide DNA Methylation Profiling. Cell Rep. 2016, 17, 596–608. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Wang, H.Y.; Liu, X.; Wasik, M.A. STAT5A is epigenetically silenced by the tyrosine kinase NPM1-ALK and acts as a tumor suppressor by reciprocally inhibiting NPM1-ALK expression. Nat. Med. 2007, 13, 1341–1348. [Google Scholar] [CrossRef]
- Piazza, R.; Magistroni, V.; Mogavero, A.; Andreoni, F.; Ambrogio, C.; Chiarle, R.; Mologni, L.; Bachmann, P.S.; Lock, R.B.; Collini, P.; et al. Epigenetic silencing of the proapoptotic gene BIM in anaplastic large cell lymphoma through an MeCP2/SIN3a deacetylating complex. Neoplasia 2013, 15, 511–522. [Google Scholar] [CrossRef] [Green Version]
- Hassler, M.R.; Klisaroska, A.; Kollmann, K.; Steiner, I.; Bilban, M.; Schiefer, A.I.; Sexl, V.; Egger, G. Antineoplastic activity of the DNA methyltransferase inhibitor 5-aza-2′-deoxycytidine in anaplastic large cell lymphoma. Biochimie 2012, 94, 2297–2307. [Google Scholar] [CrossRef] [Green Version]
- Prokoph, N.; Probst, N.A.; Lee, L.C.; Monahan, J.M.; Matthews, J.D.; Liang, H.-C.; Bahnsen, K.; Montes-Mojarro, I.A.; Karaca-Atabay, E.; Sharma, G.G.; et al. IL10RA Modulates Crizotinib Sensitivity in NPM1-ALK-positive Anaplastic Large Cell Lymphoma. Blood 2020, 136, 1657–1669. [Google Scholar] [CrossRef]
- Chou, T.C. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol. Rev. 2006, 58, 621–681. [Google Scholar] [CrossRef] [PubMed]
- Redaelli, S.; Ceccon, M.; Antolini, L.; Rigolio, R.; Pirola, A.; Peronaci, M.; Gambacorti-Passerini, C.; Mologni, L. Synergistic activity of ALK and mTOR inhibitors for the treatment of NPM-ALK positive lymphoma. Oncotarget 2016, 7, 72886–72897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mologni, L.; Brussolo, S.; Ceccon, M.; Gambacorti-Passerini, C. Synergistic Effects of Combined Wnt/KRAS Inhibition in Colorectal Cancer Cells. PLoS ONE 2012, 7, e51449. [Google Scholar] [CrossRef] [Green Version]
- Mohammad, R.M.; Wall, N.R.; Dutcher, J.A.; Al-Katib, A.M. The addition of bryostatin 1 to cyclophosphamide, doxorubicin, vincristine, and prednisone (CHOP) chemotherapy improves response in a CHOP-resistant human diffuse large cell lymphoma xenograft model. Clin. Cancer Res. 2000, 6, 4950–4956. [Google Scholar]
- Gainor, J.F.; Dardaei, L.; Yoda, S.; Friboulet, L.; Leshchiner, I.; Katayama, R.; Dagogo-Jack, I.; Gadgeel, S.; Schultz, K.; Singh, M.; et al. Molecular Mechanisms of Resistance to First- and Second-Generation ALK Inhibitors in ALK-Rearranged Lung Cancer. Cancer Discov. 2016, 6, 1118–1133. [Google Scholar] [CrossRef] [Green Version]
- Redaelli, S.; Boschelli, F.; Perini, P.; Pirola, A.; Viltadi, M.; Gambacorti-Passerini, C. Synergistic activity of the Src/Abl inhibitor bosutinib in combination with imatinib. Leukemia 2010, 24, 1223–1227. [Google Scholar] [CrossRef] [Green Version]
- Khan, S.; Jena, G. Sodium valproate, a histone deacetylase inhibitor ameliorates cyclophosphamide-induced genotoxicity and cytotoxicity in the colon of mice. J. Basic Clin. Physiol. Pharmacol. 2014, 25, 329–339. [Google Scholar] [CrossRef]
- Zhang, Q.; Wang, H.Y.; Liu, X.; Bhutani, G.; Kantekure, K.; Wasik, M. IL-2R common gamma-chain is epigenetically silenced by nucleophosphin-anaplastic lymphoma kinase (NPM-ALK) and acts as a tumor suppressor by targeting NPM-ALK. Proc. Natl. Acad. Sci. USA 2011, 108, 11977–11982. [Google Scholar] [CrossRef] [Green Version]
- Vijayachandra, K.; Higgins, W.; Lee, J.; Glick, A. Induction of p16ink4a and p19ARF by TGFbeta1 contributes to growth arrest and senescence response in mouse keratinocytes. Mol. Carcinog. 2009, 48, 181–186. [Google Scholar] [CrossRef]
- Zhang, Y.E. Non-Smad pathways in TGF-beta signaling. Cell Res. 2009, 19, 128–139. [Google Scholar] [CrossRef] [Green Version]
- Garbin, A.; Lovisa, F.; Holmes, A.B.; Damanti, C.C.; Gallingani, I.; Carraro, E.; Accordi, B.; Veltri, G.; Pizzi, M.; d’Amore, E.S.G.; et al. miR-939 acts as tumor suppressor by modulating JUNB transcriptional activity in pediatric anaplastic large cell lymphoma. Haematologica 2021, 106, 610–613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoareau-Aveilla, C.; Quelen, C.; Congras, A.; Caillet, N.; Labourdette, D.; Dozier, C.; Brousset, P.; Lamant, L.; Meggetto, F. miR-497 suppresses cycle progression through an axis involving CDK6 in ALK-positive cells. Haematologica 2019, 104, 347–359. [Google Scholar] [CrossRef]
- Schlette, E.J.; Medeiros, L.J.; Goy, A.; Lai, R.; Rassidakis, G.Z. Survivin expression predicts poorer prognosis in anaplastic large-cell lymphoma. J. Clin. Oncol. 2004, 22, 1682–1688. [Google Scholar] [CrossRef] [PubMed]
- Torossian, A.; Broin, N.; Frentzel, J.; Daugrois, C.; Gandarillas, S.; Al Saati, T.; Lamant, L.; Brousset, P.; Giuriato, S.; Espinos, E. Blockade of crizotinib-induced BCL2 elevation in ALK-positive anaplastic large cell lymphoma triggers autophagy associated with cell death. Haematologica 2019, 104, 1428–1439. [Google Scholar] [CrossRef]
- Moore, N.F.; Azarova, A.M.; Bhatnagar, N.; Ross, K.N.; Drake, L.E.; Frumm, S.; Liu, Q.S.; Christie, A.L.; Sanda, T.; Chesler, L.; et al. Molecular rationale for the use of PI3K/AKT/mTOR pathway inhibitors in combination with crizotinib in ALK-mutated neuroblastoma. Oncotarget 2014, 5, 8737–8749. [Google Scholar] [CrossRef] [Green Version]
- Shrestha, N.; Nimick, M.; Dass, P.; Rosengren, R.J.; Ashton, J.C. Mechanisms of suppression of cell growth by dual inhibition of ALK and MEK in ALK-positive non-small cell lung cancer. Sci. Rep. 2019, 9, 18842. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.J.; Noh, K.H.; Lee, Y.-H.; Hong, S.-O.; Song, K.-H.; Lee, H.-J.; Kim, S.; Kim, T.M.; Jeon, J.-H.; Seo, J.H.; et al. Targeting stemness is an effective strategy to control EML4-ALK+ non-small cell lung cancer cells. Oncotarget 2015, 6, 40255–40267. [Google Scholar] [CrossRef] [Green Version]
- Pomari, E.; Basso, G.; Bresolin, S.; Pillon, M.; Carraro, E.; d’Amore, E.S.; Viola, G.; Frasson, C.; Basso, K.; Bonvini, P.; et al. NPM-ALK expression levels identify two distinct subtypes of paediatric anaplastic large cell lymphoma. Leukemia 2017, 31, 498–501. [Google Scholar] [CrossRef] [PubMed]
- Bendjennat, M.; Boulaire, J.; Jascur, T.; Brickner, H.; Barbier, V.; Sarasin, A.; Fotedar, A.; Fotedar, R. UV irradiation triggers ubiquitin-dependent degradation of p21(WAF1) to promote DNA repair. Cell 2003, 114, 599–610. [Google Scholar] [CrossRef]
- Mattiussi, S.; Turrini, P.; Testolin, L.; Martelli, F.; Zaccagnini, G.; Mangoni, A.; Barlucchi, L.M.; Antonini, A.; Illi, B.; Cirielli, C.; et al. p21(Waf1/Cip1/Sdi1) mediates shear stress-dependent antiapoptotic function. Cardiovasc. Res. 2004, 61, 693–704. [Google Scholar] [CrossRef] [Green Version]
- Sala, E.; Mologni, L.; Truffa, S.; Gaetano, C.; Bollag, G.E.; Gambacorti-Passerini, C. BRAF silencing by short hairpin RNA or chemical blockade by PLX4032 leads to different responses in melanoma and thyroid carcinoma cells. Mol. Cancer Res. 2008, 6, 751–759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ravandi, F.; O’Brien, S.M.; Cortes, J.E.; Thomas, D.M.; Garris, R.; Faderl, S.; Burger, J.A.; Rytting, M.E.; Ferrajoli, A.; Wierda, W.G.; et al. Long-term follow-up of a phase 2 study of chemotherapy plus dasatinib for the initial treatment of patients with Philadelphia chromosome-positive acute lymphoblastic leukemia. Cancer 2015, 121, 4158–4164. [Google Scholar] [CrossRef]
- Zhang, Z.; Zeng, K.; Zhao, S.; Zhao, Y.; Hou, X.; Luo, F.; Lu, F.; Zhang, Y.; Zhou, T.; Ma, Y.; et al. Pemetrexed/carboplatin plus gefitinib as a first-line treatment for EGFR-mutant advanced nonsmall cell lung cancer: A Bayesian network meta-analysis. Ther. Adv. Med. Oncol. 2019, 11, 1758835919891652. [Google Scholar] [CrossRef]
- Leary, M.; Heerboth, S.; Lapinska, K.; Sarkar, S. Sensitization of Drug Resistant Cancer Cells: A Matter of Combination Therapy. Cancers 2018, 10, 483. [Google Scholar] [CrossRef] [Green Version]
- Abaza, Y.; Kantarjian, H.; Alwash, Y.; Borthakur, G.; Champlin, R.; Kadia, T.; Daver, N.; Ravandi, F.; Verstovsek, S.; Burger, J.; et al. Phase I/II study of dasatinib in combination with decitabine in patients with accelerated or blast phase chronic myeloid leukemia. Am. J. Hematol. 2020, 95, 1288–1295. [Google Scholar] [CrossRef]
- Biondi, A.; Gandemer, V.; De Lorenzo, P.; Cario, G.; Campbell, M.; Castor, A.; Pieters, R.; Baruchel, A.; Vora, A.; Leoni, V.; et al. Imatinib treatment of paediatric Philadelphia chromosome-positive acute lymphoblastic leukaemia (EsPhALL2010): A prospective, intergroup, open-label, single-arm clinical trial. Lancet Haematol. 2018, 5, e641–e652. [Google Scholar] [CrossRef] [Green Version]
- Jabbour, E.; Short, N.J.; Ravandi, F.; Huang, X.; Daver, N.; DiNardo, C.D.; Konopleva, M.; Pemmaraju, N.; Wierda, W.; Garcia-Manero, G.; et al. Combination of hyper-CVAD with ponatinib as first-line therapy for patients with Philadelphia chromosome-positive acute lymphoblastic leukaemia: Long-term follow-up of a single-centre, phase 2 study. Lancet Haematol. 2018, 5, e618–e627. [Google Scholar] [CrossRef]
- Menotti, M.; Ambrogio, C.; Cheong, T.-C.; Pighi, C.; Mota, I.; Cassel, S.H.; Compagno, M.; Wang, Q.; Dall’Olio, R.; Minero, V.G.; et al. Wiskott–Aldrich syndrome protein (WASP) is a tumor suppressor in T cell lymphoma. Nat. Med. 2019, 25, 130–140. [Google Scholar] [CrossRef]
- Russo, A.; Cardona, A.F.; Caglevic, C.; Manca, P.; Ruiz-Patiño, A.; Arrieta, O.; Rolfo, C. Overcoming TKI resistance in fusion-driven NSCLC: New generation inhibitors and rationale for combination strategies. Transl. Lung Cancer Res. 2020, 9, 2581–2598. [Google Scholar] [CrossRef]
- Lowe, E.J.; Reilly, A.F.; Lim, M.S.; Gross, T.G.; Saguilig, L.; Barkauskas, D.A.; Wu, R.; Alexander, S.; Bollard, C.M. Brentuximab vedotin in combination with chemotherapy for pediatric patients with ALK+ALCL: Results of COG trial ANHL12P1. Blood 2021, 137, 3595–3603. [Google Scholar] [CrossRef] [PubMed]
- Hudson, S.; Wang, D.; Middleton, F.; Nevaldine, B.H.; Naous, R.; Hutchison, R.E. Crizotinib induces apoptosis and gene expression changes in ALK+ anaplastic large cell lymphoma cell lines; brentuximab synergizes and doxorubicin antagonizes. Pediatr. Blood Cancer 2018, 65, e27094. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Combination | Cell Line | Ratio | EC50 | EC75 | EC90 | Mean | Level of Synergism | |
|---|---|---|---|---|---|---|---|---|
| CRI:DEC | Karpas 299 | 1:1 | 0.000 | 0.006 | 0.007 | 0.004 | Very Strong Synergism | |
| Karpas 299 | 1:4 | 0.226 | 0.189 | 0.206 | 0.207 | Strong Synergism | ||
| Karpas 299 | 1:20 | 0.509 | 0.512 | 0.560 | 0.527 | Synergism | ||
| Karpas 299 | 1:100 | 0.142 | 0.336 | 0.811 | 0.429 | Synergism | ||
| SU-DHL-1 | 5:1 | 0.448 | 0.301 | 0.203 | 0.317 | Synergism | ||
| SU-DHL-1 | 1:1 | 0.644 | 0.632 | 0.631 | 0.636 | Synergism | ||
| SU-DHL-1 | 1:2 | 0.442 | 0.265 | 0.165 | 0.291 | Strong Synergism | ||
| SU-DHL-1 | 1:5 | 0.779 | 0.675 | 0.641 | 0.698 | Synergism | ||
| SUP-M2 | 5:1 | 1.080 | 0.713 | 0.481 | 0.758 | Moderate Synergism | ||
| SUP-M2 | 1:1 | 0.711 | 0.839 | 1.110 | 0.887 | Slight Synergism | ||
| SUP-M2 | 1:2 | 0.536 | 0.391 | 0.351 | 0.426 | Synergism | ||
| SUP-M2 | 1:5 | 0.000 | 0.001 | 0.038 | 0.013 | Very Strong Synergism | ||
| U937 | 1:1 | >10 | >10 | >10 | >10 | Very Strong Antagonism | ||
| U937 | 1:2 | >10 | >10 | >10 | >10 | Very Strong Antagonism | ||
| CRI:CHO | Karpas 299 | 1:22 | 0.087 | 0.185 | 0.405 | 0.226 | Strong Synergism | |
| Karpas 299 | 1:50 | 0.426 | 0.545 | 0.698 | 0.556 | Synergism | ||
| Karpas 299 | 1:66 | 0.432 | 0.346 | 0.284 | 0.354 | Synergism | ||
| Karpas 299 | 1:150 | 0.500 | 0.625 | 0.781 | 0.635 | Synergism | ||
| SU-DHL-1 | 1:40 | 0.236 | 0.359 | 0.555 | 0.383 | Synergism | ||
| SU-DHL-1 | 1:20 | 0.268 | 0.418 | 0.662 | 0.449 | Synergism | ||
| SU-DHL-1 | 1:10 | 1.298 | 1.420 | 1.571 | 1.430 | Moderate Antagonism | ||
| SUP-M2 | 1:16 | 0.579 | 0.521 | 0.473 | 0.524 | Synergism | ||
| SUP-M2 | 1:32 | 0.845 | 0.715 | 0.611 | 0.724 | Moderate Synergism | ||
| SUP-M2 | 1:64 | 0.869 | 0.759 | 0.667 | 0.765 | Moderate Synergism | ||
| U937 | 1:40 | 1.015 | 0.915 | 0.830 | 0.920 | Nearly Additive | ||
| U937 | 1:20 | 1.096 | 1.044 | 1.008 | 1.050 | Nearly Additive | ||
| U937 | 1:10 | 1.207 | 1.287 | 1.407 | 1.300 | Moderate Antagonism | ||
| CRI:TRAM | Karpas 299 | 1:1 | 0.616 | 0.589 | 0.563 | 0.589 | Synergism | |
| Karpas 299 | 1:2 | 0.510 | 0.477 | 0.448 | 0.478 | Synergism | ||
| Karpas 299 | 1:4 | 0.228 | 0.336 | 0.538 | 0.367 | Synergism | ||
| Karpas 299 | 1:3 | 0.433 | 0.480 | 0.553 | 0.489 | Synergism | ||
| SU-DHL-1 | 1:9 | 0.967 | 0.665 | 0.461 | 0.698 | Synergism | ||
| SU-DHL-1 | 1:3 | 0.758 | 0.572 | 0.438 | 0.589 | Synergism | ||
| SU-DHL-1 | 1:1 | 0.450 | 0.321 | 0.244 | 0.338 | Synergism | ||
| SU-DHL-1 | 3:1 | 0.046 | 0.053 | 0.068 | 0.056 | Very Strong Synergism | ||
| SUP-M2 | 1:2 | 0.483 | 0.542 | 0.615 | 0.546 | Synergism | ||
| SUP-M2 | 1:1 | 0.579 | 0.599 | 0.624 | 0.600 | Synergism | ||
| SUP-M2 | 2:1 | 0.132 | 0.266 | 0.538 | 0.312 | Synergism | ||
| U937 | 1:1 | >10 | >10 | >10 | >10 | Very Strong Antagonism | ||
| U937 | 1:3 | >10 | >10 | >10 | >10 | Very Strong Antagonism | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arosio, G.; Sharma, G.G.; Villa, M.; Mauri, M.; Crespiatico, I.; Fontana, D.; Manfroni, C.; Mastini, C.; Zappa, M.; Magistroni, V.; et al. Synergistic Drug Combinations Prevent Resistance in ALK+ Anaplastic Large Cell Lymphoma. Cancers 2021, 13, 4422. https://doi.org/10.3390/cancers13174422
Arosio G, Sharma GG, Villa M, Mauri M, Crespiatico I, Fontana D, Manfroni C, Mastini C, Zappa M, Magistroni V, et al. Synergistic Drug Combinations Prevent Resistance in ALK+ Anaplastic Large Cell Lymphoma. Cancers. 2021; 13(17):4422. https://doi.org/10.3390/cancers13174422
Chicago/Turabian StyleArosio, Giulia, Geeta G. Sharma, Matteo Villa, Mario Mauri, Ilaria Crespiatico, Diletta Fontana, Chiara Manfroni, Cristina Mastini, Marina Zappa, Vera Magistroni, and et al. 2021. "Synergistic Drug Combinations Prevent Resistance in ALK+ Anaplastic Large Cell Lymphoma" Cancers 13, no. 17: 4422. https://doi.org/10.3390/cancers13174422
APA StyleArosio, G., Sharma, G. G., Villa, M., Mauri, M., Crespiatico, I., Fontana, D., Manfroni, C., Mastini, C., Zappa, M., Magistroni, V., Ceccon, M., Redaelli, S., Massimino, L., Garbin, A., Lovisa, F., Mussolin, L., Piazza, R., Gambacorti-Passerini, C., & Mologni, L. (2021). Synergistic Drug Combinations Prevent Resistance in ALK+ Anaplastic Large Cell Lymphoma. Cancers, 13(17), 4422. https://doi.org/10.3390/cancers13174422