
NK Cells in Myeloproliferative Neoplasms (MPN)


{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
1.1. Introduction to MPNs
1.1.1. Disease Characteristics
1.1.2. Ph+ MPNs
1.1.3. Classical MPNs
2. Immunological Changes in MPNs
3. Natural Killer Cells
4. NK Cells in MPN
4.1. CML
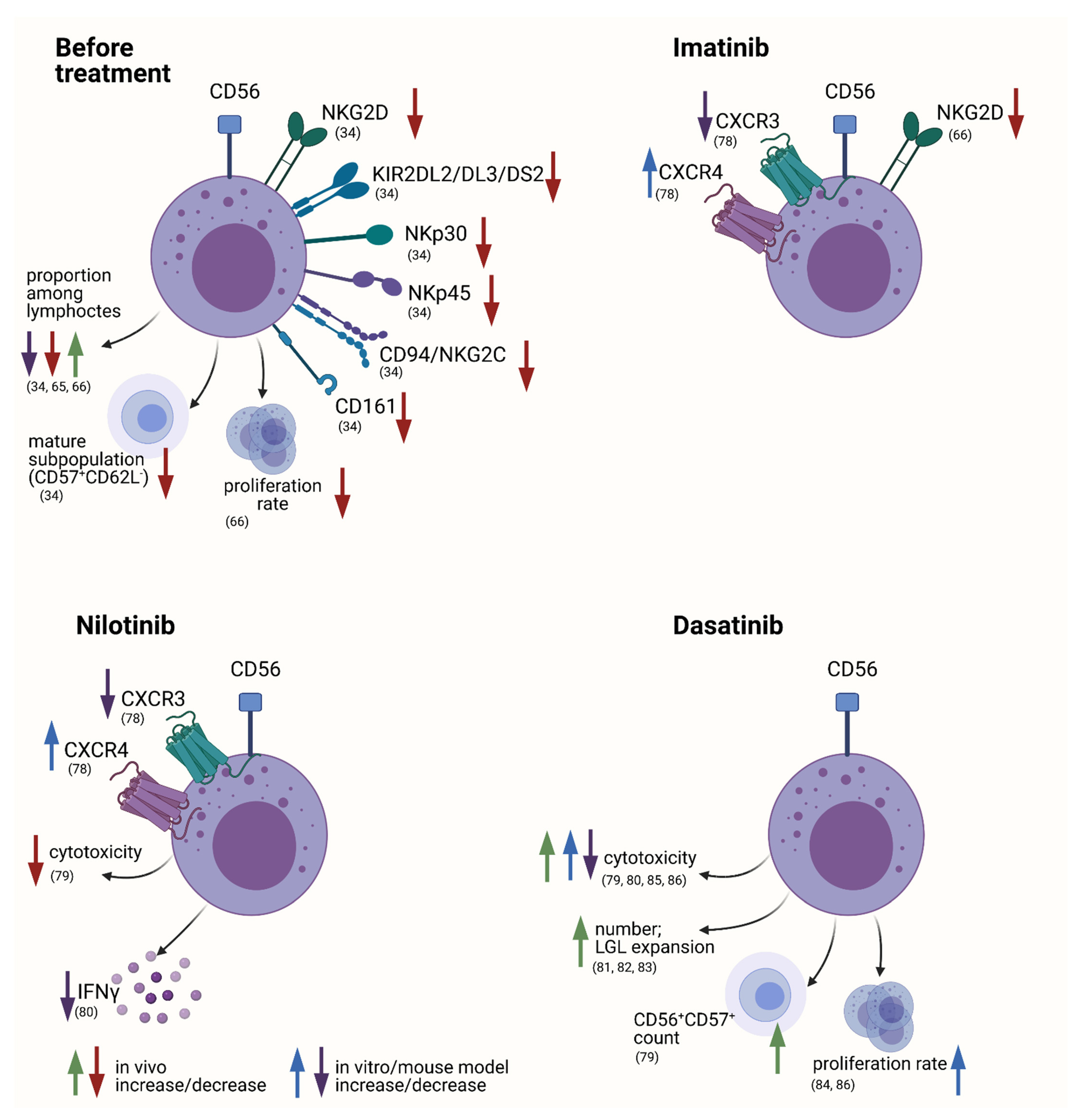
4.1.1. NK Cells in CML
General Alterations in the NK Cell Compartment
Specific Changes during Course of Treatment with Possible Functional Consequences
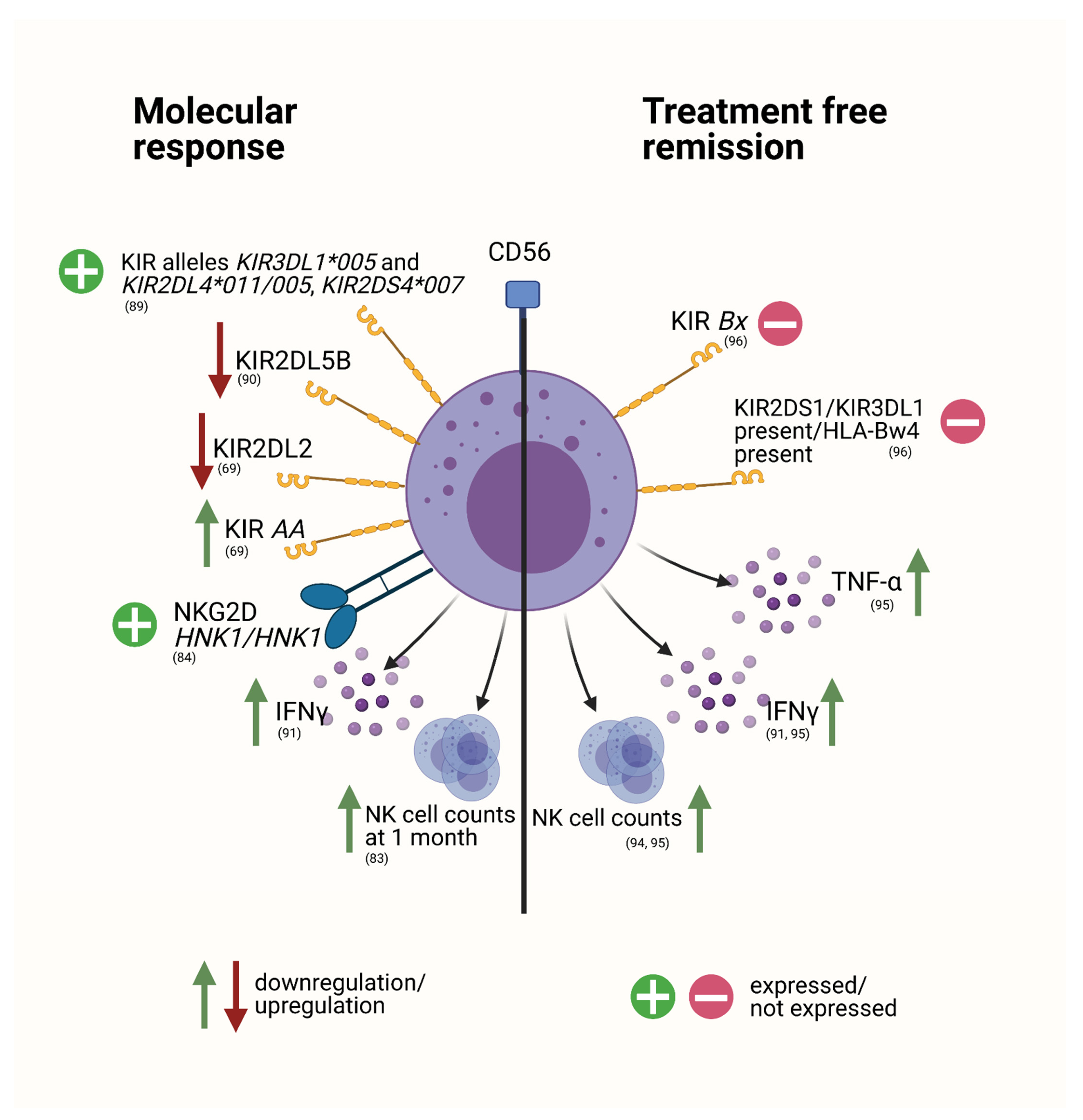
The Role of NK Cells to Disease Progression and Response to Therapy
NK Cells in the Setting of Treatment Discontinuation
4.2. Classical MPNs
4.2.1. Immunological Changes in Classical MPNs
4.2.2. NK Cells in Classical MPNs
4.2.3. The Effect of Treatment on NK Cells
Acetylsalicylic Acid (ASA)
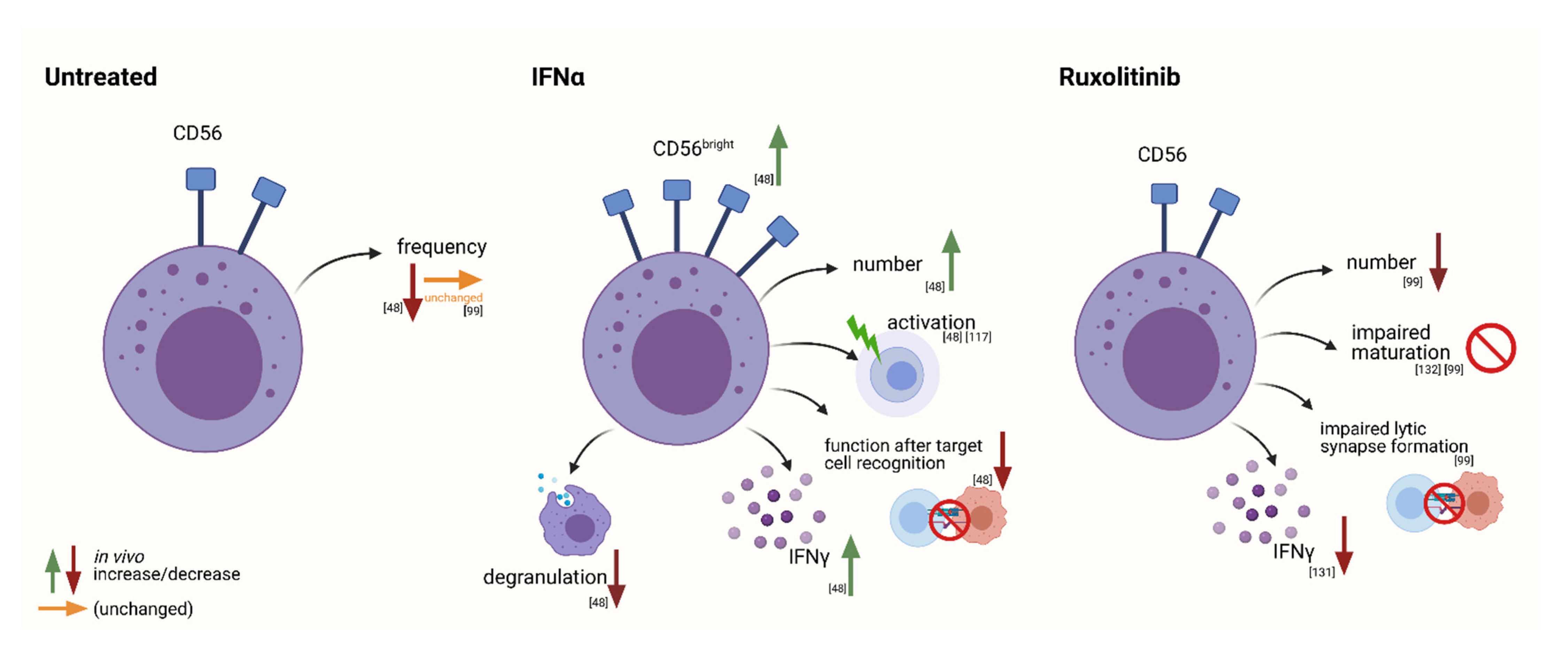
Interferon-Alpha2
Hydroxyurea
JAK Inhibitors
Immunomodulatory Drugs (IMiDs)
5. Discussion
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Grabek, J.; Straube, J.; Bywater, M.; Lane, S.W. MPN: The Molecular Drivers of Disease Initiation, Progression and Transformation and their Effect on Treatment. Cells 2020, 9, 1901. [Google Scholar] [CrossRef]
- Barbui, T.; Thiele, J.; Gisslinger, H.; Kvasnicka, H.M.; Vannucchi, A.M.; Guglielmelli, P.; Orazi, A.; Tefferi, A. The 2016 WHO classification and diagnostic criteria for myeloproliferative neoplasms: Document summary and in-depth discussion. Blood Cancer J. 2018, 8, 1–11. [Google Scholar] [CrossRef]
- Tefferi, A. Myeloproliferative neoplasms: A decade of discoveries and treatment advances. Am. J. Hematol. 2016, 91, 50–58. [Google Scholar] [CrossRef]
- Vainchenker, W.; Kralovics, R. Genetic basis and molecular pathophysiology of classical myeloproliferative neoplasms. Blood 2017, 129, 667–679. [Google Scholar] [CrossRef]
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef]
- Shallis, R.M.; Wang, R.; Davidoff, A.; Ma, X.; Podoltsev, N.A.; Zeidan, A.M. Epidemiology of the classical myeloproliferative neoplasms: The four corners of an expansive and complex map. Blood Rev. 2020, 42, 100706. [Google Scholar] [CrossRef] [PubMed]
- Anderson, L.A.; McMullin, M.F. Epidemiology of MPN: What Do We Know? Curr. Hematol. Malign-Rep. 2014, 9, 340–349. [Google Scholar] [CrossRef] [PubMed]
- Titmarsh, G.J.; Duncombe, A.S.; McMullin, M.F.; O’Rorke, M.; Mesa, R.; De Vocht, F.; Horan, S.; Fritschi, L.; Clarke, M.; Anderson, L.A. How common are myeloproliferative neoplasms? A systematic review and meta-analysis. Am. J. Hematol. 2014, 89, 581–587. [Google Scholar] [CrossRef] [PubMed]
- Helbig, G.; Soja, A.; Bartkowska-Chrobok, A.; Kyrcz-Krzemień, S. Chronic eosinophilic leukemia-not otherwise specified has a poor prognosis with unresponsiveness to conventional treatment and high risk of acute transformation. Am. J. Hematol. 2012, 87, 643–645. [Google Scholar] [CrossRef]
- Szuber, N.; Elliott, M.; Tefferi, A. Chronic neutrophilic leukemia: 2020 update on diagnosis, molecular genetics, prognosis, and management. Am. J. Hematol. 2020, 95, 212–224. [Google Scholar] [CrossRef]
- James, C.; Ugo, V.; Le Couédic, J.-P.; Staerk, J.; Delhommeau, F.; Lacout, C.; Garçon, L.; Raslova, H.; Berger, R.; Griscelli, A.B.; et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nat. Cell Biol. 2005, 434, 1144–1148. [Google Scholar] [CrossRef] [PubMed]
- Levine, R.L.; Wadleigh, M.; Cools, J.; Ebert, B.L.; Wernig, G.; Huntly, B.; Boggon, T.J.; Wlodarska, I.; Clark, J.J.; Moore, S.; et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell 2005, 7, 387–397. [Google Scholar] [CrossRef] [PubMed]
- Chereda, B.; Melo, J.V. Natural course and biology of CML. Ann. Hematol. 2015, 94, 107–121. [Google Scholar] [CrossRef] [PubMed]
- Quintás-Cardama, A.; Cortes, J. Molecular biology of bcr-abl1–positive chronic myeloid leukemia. Blood 2009, 113, 1619–1630. [Google Scholar] [CrossRef] [PubMed]
- Ren, R. Mechanisms of BCR–ABL in the pathogenesis of chronic myelogenous leukaemia. Nat. Rev. Cancer 2005, 5, 172–183. [Google Scholar] [CrossRef]
- Jabbour, E.; Kantarjian, H. Chronic myeloid leukemia: 2018 update on diagnosis, therapy and monitoring. Am. J. Hematol. 2018, 93, 442–459. [Google Scholar] [CrossRef]
- Tefferi, A.; Vannucchi, A.M.; Barbui, T. Polycythemia vera treatment algorithm. Blood Cancer J. 2018, 8, 1–7. [Google Scholar] [CrossRef]
- Spivak, J.L. Polycythemia vera. Curr. Treat. Options Oncol. 2018, 19, 1–14. [Google Scholar] [CrossRef]
- Tefferi, A.; Pardanani, A. Essential thrombocythemia. Engl. J. Med. 2019, 381, 2135–2144. [Google Scholar] [CrossRef]
- Tefferi, A. Primary myelofibrosis: 2021 update on diagnosis, risk-stratification and management. Am. J. Hematol. 2021, 96, 145–162. [Google Scholar] [CrossRef]
- Finazzi, G.; Vannucchi, A.M.; Barbui, T. Prefibrotic myelofibrosis: Treatment algorithm. Blood Cancer J. 2018, 8, 104. [Google Scholar] [CrossRef]
- Tefferi, A.; Barbui, T. Polycythemia vera and essential thrombocythemia: 2017 update on diagnosis, risk-stratification, and management. Am. J. Hematol. 2017, 92, 94–108. [Google Scholar] [CrossRef] [PubMed]
- Barosi, G. An Immune Dysregulation in MPN. Curr. Hematol. Malign-Rep. 2014, 9, 331–339. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-related inflammation. Nat. Cell Biol. 2008, 454, 436–444. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zuo, X. Cytokines frequently implicated in myeloproliferative neoplasms. Cytokine X 2019, 1, 100005. [Google Scholar] [CrossRef] [PubMed]
- Longhitano, L.; Volti, G.L.; Giallongo, C.; Spampinato, M.; Barbagallo, I.; Di Rosa, M.; Romano, A.; Avola, R.; Tibullo, D.; Palumbo, G.A. The Role of Inflammation and Inflammasome in Myeloproliferative Disease. J. Clin. Med. 2020, 9, 2334. [Google Scholar] [CrossRef]
- Braun, L.M.; Zeiser, R. Immunotherapy in Myeloproliferative Diseases. Cells 2020, 9, 1559. [Google Scholar] [CrossRef]
- Marty, C.; Lacout, C.; Droin, N.; Le Couédic, J.-P.; Ribrag, V.; Solary, E.; Vainchenker, W.; Villeval, J.-L.; Plo, I. A role for reactive oxygen species in JAK2V617F myeloproliferative neoplasm progression. Leukemia 2013, 27, 2187–2195. [Google Scholar] [CrossRef] [PubMed]
- Hasselbalch, H.C.; Thomassen, M.; Riley, C.H.; Kjær, L.; Larsen, T.S.; Jensen, M.K.; Bjerrum, O.W.; Kruse, T.A.; Skov, V. Whole Blood Transcriptional Profiling Reveals Deregulation of Oxidative and Antioxidative Defence Genes in Myelofibrosis and Related Neoplasms. Potential Implications of Downregulation of Nrf2 for Genomic Instability and Disease Progression. PLoS ONE 2014, 9, e112786. [Google Scholar] [CrossRef]
- Bock, O.; Höftmann, J.; Theophile, K.; Hussein, K.; Wiese, B.; Schlué, J.; Kreipe, H. Bone Morphogenetic Proteins Are Overexpressed in the Bone Marrow of Primary Myelofibrosis and Are Apparently Induced by Fibrogenic Cytokines. Am. J. Pathol. 2008, 172, 951–960. [Google Scholar] [CrossRef]
- Murthy, G.S.G.; Atallah, E. Treatment-Free Remission in CML: The US Perspective. Curr. Hematol. Malign-Rep. 2019, 14, 56–61. [Google Scholar] [CrossRef]
- Hughes, A.; Yong, A.S.M. Immune Effector Recovery in Chronic Myeloid Leukemia and Treatment-Free Remission. Front. Immunol. 2017, 8, 469. [Google Scholar] [CrossRef]
- Ureshino, H.; Shindo, T.; Kimura, S. Role of cancer immunology in chronic myelogenous leukemia. Leuk. Res. 2020, 88, 106273. [Google Scholar] [CrossRef] [PubMed]
- Hughes, A.; Clarson, J.; Tang, C.; Vidovic, L.; White, D.; Hughes, T.; Yong, A. CML patients with deep molecular responses to TKI have restored immune effectors and decreased PD-1 and immune suppressors. Blood 2017, 129, 1166–1176. [Google Scholar] [CrossRef]
- Vitale, M.; Della Chiesa, M.; Carlomagno, S.; Pende, D.; Aricò, M.; Moretta, L.; Moretta, A. NK-dependent DC maturation is mediated by TNFα and IFNγ released upon engagement of the NKp30 triggering receptor. Blood 2005, 106, 566–571. [Google Scholar] [CrossRef] [PubMed]
- Di Vito, C.; Mikulak, J.; Mavilio, D. On the Way to Become a Natural Killer Cell. Front. Immunol. 2019, 10, 1812. [Google Scholar] [CrossRef]
- Del Zotto, G.; Marcenaro, E.; Vacca, P.; Sivori, S.; Pende, D.; Della Chiesa, M.; Moretta, F.; Ingegnere, T.; Mingari, M.C.; Moretta, A.; et al. Markers and function of human NK cells in normal and pathological conditions. Cytom. Part B Clin. Cytom. 2017, 92, 100–114. [Google Scholar] [CrossRef] [PubMed]
- Scoville, S.D.; Freud, A.G.; Caligiuri, M.A. Modeling Human Natural Killer Cell Development in the Era of Innate Lymphoid Cells. Front. Immunol. 2017, 8, 360. [Google Scholar] [CrossRef]
- Bonanni, V.; Sciume, G.; Santoni, A.; Bernardini, G. Bone Marrow NK Cells: Origin, Distinctive Features, and Requirements for Tissue Localization. Front. Immunol. 2019, 10, 1569. [Google Scholar] [CrossRef]
- Goh, W.; Huntington, N.D. Regulation of Murine Natural Killer Cell Development. Front. Immunol. 2017, 8, 130. [Google Scholar] [CrossRef]
- Horowitz, A.; Strauss-Albee, D.; Leipold, M.; Kubo, J.; Nemat-Gorgani, N.; Dogan, O.C.; Dekker, C.L.; Mackey, S.; Maecker, H.; Swan, G.E.; et al. Genetic and Environmental Determinants of Human NK Cell Diversity Revealed by Mass Cytometry. Sci. Transl. Med. 2013, 5, 208ra145. [Google Scholar] [CrossRef] [PubMed]
- Moretta, A.; Bottino, C.; Vitale, M.; Pende, D.; Biassoni, R.; Mingari, M.C.; Moretta, L. Receptors for Hla Class-I Molecules in Human Natural Killer Cells. Annu. Rev. Immunol. 1996, 14, 619–648. [Google Scholar] [CrossRef]
- Morvan, M.; Lanier, L.L. NK cells and cancer: You can teach innate cells new tricks. Nat. Rev. Cancer 2016, 16, 7–19. [Google Scholar] [CrossRef] [PubMed]
- Raulet, D.H. Missing self recognition and self tolerance of natural killer (NK) cells. Semin. Immunol. 2006, 18, 145–150. [Google Scholar] [CrossRef]
- Gasser, S.; Orsulic, S.; Brown, E.J.; Raulet, D.H. The DNA damage pathway regulates innate immune system ligands of the NKG2D receptor. Nat. Cell Biol. 2005, 436, 1186–1190. [Google Scholar] [CrossRef] [PubMed]
- Pardoll, D.M. Distinct Mechanisms of Tumor Resistance to NK Killing: Of Mice and Men. Immunity 2015, 42, 605–606. [Google Scholar] [CrossRef][Green Version]
- Boissel, N.; Rea, D.; Tieng, V.; Dulphy, N.; Brun, M.; Cayuela, J.-M.; Rousselot, P.; Tamouza, R.; Le Bouteiller, P.; Mahon, F.-X.; et al. BCR/ABL Oncogene Directly Controls MHC Class I Chain-Related Molecule A Expression in Chronic Myelogenous Leukemia. J. Immunol. 2006, 176, 5108–5116. [Google Scholar] [CrossRef] [PubMed]
- Riley, C.H.; Hansen, M.; Brimnes, M.K.; Hasselbalch, H.C.; Bjerrum, O.W.; Straten, P.T.; Svane, I.M.; Jensen, M.K. Expansion of circulating CD56bright natural killer cells in patients with JAK2-positive chronic myeloproliferative neoplasms during treatment with interferon-α. Eur. J. Haematol. 2015, 94, 227–234. [Google Scholar] [CrossRef]
- Van Acker, H.H.; Capsomidis, A.; Smits, E.; Van Tendeloo, V.F. CD56 in the Immune System: More Than a Marker for Cytotoxicity? Front. Immunol. 2017, 8, 892. [Google Scholar] [CrossRef]
- Yeap, W.H.; Wong, K.L.; Shimasaki, N.; Teo, E.C.Y.; Quek, J.K.S.; Yong, H.X.; Diong, C.P.; Bertoletti, A.; Linn, Y.C.; Wong, S.C. CD16 is indispensable for antibody-dependent cellular cytotoxicity by human monocytes. Sci. Rep. 2016, 6, 34310. [Google Scholar] [CrossRef]
- Cooper, M.; Fehniger, T.; Caligiuri, M.A. The biology of human natural killer-cell subsets. Trends Immunol. 2001, 22, 633–640. [Google Scholar] [CrossRef]
- López-Soto, A.; Gonzalez, S.; Smyth, M.J.; Galluzzi, L. Control of Metastasis by NK Cells. Cancer Cell 2017, 32, 135–154. [Google Scholar] [CrossRef] [PubMed]
- Street, S.E.; Hayakawa, Y.; Zhan, Y.; Lew, A.; MacGregor, D.; Jamieson, A.; Diefenbach, A.; Yagita, H.; Godfrey, D.; Smyth, M.J. Innate Immune Surveillance of Spontaneous B Cell Lymphomas by Natural Killer Cells and γδ T Cells. J. Exp. Med. 2004, 199, 879–884. [Google Scholar] [CrossRef]
- Carlsten, M.; Järås, M. Natural Killer Cells in Myeloid Malignancies: Immune Surveillance, NK Cell Dysfunction, and Pharmacological Opportunities to Bolster the Endogenous NK Cells. Front. Immunol. 2019, 10, 2357. [Google Scholar] [CrossRef]
- Ilander, M.; Hekim, C.; Mustjoki, S. Immunology and Immunotherapy of Chronic Myeloid Leukemia. Curr. Hematol. Malign-Rep. 2014, 9, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, N.; Miura, I.; Saitoh, K.; Miura, A.B. Lineage involvement of stem cells bearing the philadelphia chromosome in chronic myeloid leukemia in the chronic phase as shown by a combination of fluorescence-activated cell sorting and fluorescence in situ hybridization. Blood 1998, 92, 4758–4763. [Google Scholar] [CrossRef]
- Min, C.-K.; Yang, I.H.; Kim, D.W.; Lee, J.W.; Han, C.W.; Min, W.S.; Kim, C.C. Detection of the BCR-ABL fusion gene in natural killer cells in patients with chronic myelogenous leukemia. Acta Haematol. 2000, 104, 135–138. [Google Scholar] [CrossRef]
- Cho, E.K.; Heo, D.S.; Seol, J.G.; Seo, E.J.; Chi, H.S.; Kim, E.S.; Lee, Y.Y.; Kim, B.K.; Kim, N.K. Ontogeny of natural killer cells and T cells by analysis of BCR–ABL rearrangement from patients with chronic myelogenous leukaemia. Br. J. Haematol. 2000, 11, 216–222. [Google Scholar]
- Nakajima, H.; Zhao, R.; Lund, T.C.; Ward, J.; Dolan, M.; Hirsch, B.; Miller, J.S. The BCR/ABL transgene causes abnormal NK cell differentiation and can be found in circulating NK cells of advanced phase chronic myelogenous leukemia patients. J. Immunol. 2002, 168, 643–650. [Google Scholar] [CrossRef]
- Pattengale, P.K.; Sundstrom, C.; Yu, A.; Levine, A. Lysis of fresh leukemic blasts by interferon-activated human natural killer cells. Nat. Immun. Cell Growth Regul. 1983, 3, 165–180. [Google Scholar]
- Lotzová, E.; Savary, C.A.; Herberman, R.B. Inhibition of clonogenic growth of fresh leukemia cells by unstimulated and IL-2 stimulated NK cells of normal donors. Leuk. Res. 1987, 11, 1059–1066. [Google Scholar] [CrossRef]
- Cervantes, F.; Pierson, B.A.; McGlave, P.B.; Verfaillie, C.; Miller, J.S. Autologous activated natural killer cells suppress primitive chronic myelogenous leukemia progenitors in long-term culture. Blood 1996, 87, 2476–2485. [Google Scholar] [CrossRef] [PubMed]
- Cebo, C.; Da Rocha, S.; Wittnebel, S.; Turhan, A.G.; Abdelali, J.; Caillat-Zucman, S.; Bourhis, J.H.; Chouaib, S.; Caignard, A. The Decreased Susceptibility of Bcr/Abl Targets to NK Cell-Mediated Lysis in Response to Imatinib Mesylate Involves Modulation of NKG2D Ligands, GM1 Expression, and Synapse Formation. J. Immunol. 2006, 176, 864–872. [Google Scholar] [CrossRef]
- Yong, A.S.M.; Keyvanfar, K.; Hensel, N.; Eniafe, R.; Savani, B.N.; Berg, M.; Lundqvist, A.; Adams, S.; Sloand, E.M.; Goldman, J.M.; et al. Primitive quiescent CD34+ cells in chronic myeloid leukemia are targeted by in vitro expanded natural killer cells, which are functionally enhanced by bortezomib. Blood 2009, 113, 875–882. [Google Scholar] [CrossRef]
- Sopper, S.; Mustjoki, S.; Gjertsen, B.T.; Giles, F.; Hochhaus, A.; Janssen, J.J.W.M.; Porkka, K.; Wolf, D. NK cell dynamics and association with molecular response in early chronic phase chronic myelogenous leukemia (CML-CP) patients treated with nilotinib. Leukemia 2017, 31, 2264–2267. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.I.-U.; Koschmieder, S.; Kerstiens, L.; Schemionek, M.; Altvater, B.; Pscherer, S.; Gerss, J.; Maecker, H.T.; Berdel, W.E.; Juergens, H.; et al. NK cells are dysfunctional in human chronic myelogenous leukemia before and on imatinib treatment and in BCR–ABL-positive mice. Leukemia 2011, 26, 465–474. [Google Scholar] [CrossRef]
- Kumagai, T.; Matsuki, E.; Inokuchi, K.; Ohashi, K.; Shinagawa, A.; Takeuchi, J.; Yoshida, C.; Okamoto, S.; Wakita, H.; Kozai, Y.; et al. Relative increase in lymphocytes from as early as 1 month predicts improved response to dasatinib in chronic-phase chronic myelogenous leukemia. Int. J. Hematol. 2013, 99, 41–52. [Google Scholar] [CrossRef]
- Toubert, A.; Turhan, A.; Guerci-Bresler, A.; Dulphy, N.; Réa, D. Lymphocytes NK: Un rôle majeur dans le contrôle immunologique de la leucémie myéloïde chronique. J. Sci. 2018, 34, 540–546. [Google Scholar] [CrossRef]
- La Nasa, G.; Caocci, G.; Littera, R.; Atzeni, S.; Vacca, A.; Mulas, O.; Langiu, M.; Greco, M.; Orru, S.; Orrù, N.; et al. Homozygosity for killer immunoglobin-like receptor haplotype A predicts complete molecular response to treatment with tyrosine kinase inhibitors in chronic myeloid leukemia patients. Exp. Hematol. 2013, 41, 424–431. [Google Scholar] [CrossRef] [PubMed]
- Danier, A.C.A.; De Melo, R.P.; Napimoga, M.H.; Laguna-Abreu, M.T.C. The role of natural killer cells in chronic myeloid leukemia. Rev. Bras. Hematol. Hemoter. 2011, 33, 216–220. [Google Scholar] [CrossRef][Green Version]
- Bumbea, H.; Vladareanu, A.-M.; Voican, I.; Cisleanu, D.; Barsan, L.; Onisai, M. Chronic myeloid leukemia therapy in the era of tyrosine kinase inhibitors. The first molecular targeted treatment. J. Med. Life 2010, 3, 162–166. [Google Scholar]
- Hochhaus, A.; Baccarani, M.; Silver, R.T.; Schiffer, C.; Apperley, J.F.; Cervantes, F.; Clark, R.E.; Cortes, J.E.; Deininger, M.W.; Guilhot, F.; et al. European LeukemiaNet 2020 recommendations for treating chronic myeloid leukemia. Leukemia 2020, 34, 966–984. [Google Scholar] [CrossRef]
- Kreutzman, A.; Rohon, P.; Faber, E.; Indrák, K.; Juvonen, V.; Kairisto, V.; Voglova, J.; Sinisalo, M.; Flochová, E.; Vakkila, J.; et al. Chronic Myeloid Leukemia Patients in Prolonged Remission following Interferon-α Monotherapy Have Distinct Cytokine and Oligoclonal Lymphocyte Profile. PLoS ONE 2011, 6, e23022. [Google Scholar] [CrossRef]
- Ilander, M.; Kreutzman, A.; Rohon, P.; Melo, T.; Faber, E.; Porkka, K.; Vakkila, J.; Mustjoki, S. Enlarged Memory T-Cell Pool and Enhanced Th1-Type Responses in Chronic Myeloid Leukemia Patients Who Have Successfully Discontinued IFN-α Monotherapy. PLoS ONE 2014, 9, e87794. [Google Scholar] [CrossRef] [PubMed]
- de Castro, F.A.; Palma, P.V.B.; Morais, F.R.; Simões, B.P.; Carvalho, P.V.B.; Ismael, S.J.; Voltarelli, C.P.; Morais, J.C. Immunological effects of interferon-alpha on chronic myelogenous leukemia. Leuk. Lymphoma 2003, 44, 2061–2067. [Google Scholar] [CrossRef] [PubMed]
- Alves, R.; McArdle, S.E.B.; Vadakekolathu, J.; Gonçalves, A.C.; Tavares, P.; Pereira, A.; Almeida, A.M.; Sarmento-Ribeiro, A.B.; Rutella, S. Flow cytometry and targeted immune transcriptomics identify distinct profiles in patients with chronic myeloid leukemia receiving tyrosine kinase inhibitors with or without interferon-α. J. Transl. Med. 2020, 18, 2–15. [Google Scholar] [CrossRef]
- Rix, U.; Hantschel, O.; Dürnberger, G.; Rix, L.L.R.; Planyavsky, M.; Fernbach, N.V.; Kaupe, I.; Bennett, K.L.; Valent, P.; Colinge, J.; et al. Chemical proteomic profiles of the BCR-ABL inhibitors imatinib, nilotinib, and dasatinib reveal novel kinase and nonkinase targets. Blood 2007, 110, 4055–4063. [Google Scholar] [CrossRef] [PubMed]
- Bellora, F.; Dondero, A.; Corrias, M.V.; Casu, B.; Regis, S.; Caliendo, F.; Moretta, A.; Cazzola, M.; Elena, C.; Vinti, L.; et al. Imatinib and Nilotinib Off-Target Effects on Human NK Cells, Monocytes, and M2 Macrophages. J. Immunol. 2017, 199, 1516–1525. [Google Scholar] [CrossRef]
- Hayashi, Y.; Nakamae, H.; Katayama, T.; Nakane, T.; Koh, H.; Nakamae, M.; Hirose, A.; Hagihara, K.; Terada, Y.; Nakao, Y.; et al. Different immunoprofiles in patients with chronic myeloid leukemia treated with imatinib, nilotinib or dasatinib. Leuk. Lymphoma 2012, 53, 1084–1089. [Google Scholar] [CrossRef] [PubMed]
- Salih, J.; Hilpert, J.; Placke, T.; Grünebach, F.; Steinle, A.; Salih, H.R.; Krusch, M. The BCR/ABL-inhibitors imatinib, nilotinib and dasatinib differentially affect NK cell reactivity. Int. J. Cancer 2010, 127, 2119–2128. [Google Scholar] [CrossRef]
- Qiu, Z.-Y.; Xu, W.; Li, J.-Y. Large granular lymphocytosis during dasatinib therapy. Cancer Biol. Ther. 2013, 15, 247–255. [Google Scholar] [CrossRef]
- Kreutzman, A.; Juvonen, V.; Kairisto, V.; Ekblom, M.; Stenke, L.; Seggewiss, R.; Porkka, K.; Mustjoki, S. Mono/oligoclonal T and NK cells are common in chronic myeloid leukemia patients at diagnosis and expand during dasatinib therapy. Blood 2010, 116, 772–782. [Google Scholar] [CrossRef]
- Iriyama, N.; Fujisawa, S.; Yoshida, C.; Wakita, H.; Chiba, S.; Okamoto, S.; Kawakami, K.; Takezako, N.; Kumagai, T.; Inokuchi, K.; et al. Early cytotoxic lymphocyte expansion contributes to a deep molecular response to dasatinib in patients with newly diagnosed chronic myeloid leukemia in the chronic phase: Results of the D-first study. Am. J. Hematol. 2015, 90, 819–824. [Google Scholar] [CrossRef]
- Hara, R.; Onizuka, M.; Matsusita, E.; Kikkawa, E.; Nakamura, Y.; Matsushita, H.; Ohgiya, D.; Murayama, H.; Machida, S.; Ohmachi, K.; et al. NKG2D gene polymorphisms are associated with disease control of chronic myeloid leukemia by dasatinib. Int. J. Hematol. 2017, 106, 666–674. [Google Scholar] [CrossRef]
- Hassold, N.; Seystahl, K.; Kempf, K.; Urlaub, D.; Zekl, M.; Einsele, H.; Watzl, C.; Wischhusen, J.; Seggewiss-Bernhardt, R. Enhancement of natural killer cell effector functions against selected lymphoma and leukemia cell lines by dasatinib. Int. J. Cancer 2012, 131, E916–E927. [Google Scholar] [CrossRef] [PubMed]
- Uchiyama, T.; Sato, N.; Narita, M.; Yamahira, A.; Iwabuchi, M.; Furukawa, T.; Sone, H.; Takahashi, M. Direct effect of dasatinib on proliferation and cytotoxicity of natural killer cells in in vitro study. Hematol. Oncol. 2013, 31, 156–163. [Google Scholar] [CrossRef]
- Breccia, M.; Abruzzese, E.; Bocchia, M.; Bonifacio, M.; Castagnetti, F.; Fava, C.; Galimberti, S.; Gozzini, A.; Gugliotta, G.; Iurlo, A.; et al. Chronic myeloid leukemia management at the time of the COVID-19 pandemic in Italy. A campus CML survey. Leukemia 2020, 34, 2260–2261. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, F.; Jo, D.-H.; Lee, S.-H. Can Natural Killer Cells Be a Principal Player in Anti-SARS-CoV-2 Immunity? Front. Immunol. 2020, 11, 3246. [Google Scholar] [CrossRef]
- Ureshino, H.; Shindo, T.; Kojima, H.; Kusunoki, Y.; Miyazaki, Y.; Tanaka, H.; Saji, H.; Kawaguchi, A.; Kimura, S. Allelic Polymorphisms of KIRs and HLAs Predict Favorable Responses to Tyrosine Kinase Inhibitors in CML. Cancer Immunol. Res. 2018, 6, 745–754. [Google Scholar] [CrossRef] [PubMed]
- Yeung, D.T.; Tang, C.; Vidovic, L.; White, D.L.; Branford, S.; Hughes, T.P.; Yong, A.S. KIR2DL5B genotype predicts outcomes in CML patients treated with response-directed sequential imatinib/nilotinib strategy. Blood 2015, 126, 2720–2723. [Google Scholar] [CrossRef]
- Mizoguchi, I.; Yoshimoto, T.; Katagiri, S.; Mizuguchi, J.; Tauchi, T.; Kimura, Y.; Inokuchi, K.; Ohyashiki, J.H.; Ohyashiki, K. Sustained upregulation of effector natural killer cells in chronic myeloid leukemia after discontinuation of imatinib. Cancer Sci. 2013, 104, 1146–1153. [Google Scholar] [CrossRef]
- Molica, M.; Noguera, N.I.; Trawinska, M.M.; Martinelli, G.; Cerchione, C.; Abruzzese, E. Treatment-free remission in chronic myeloid leukemia: Lights and shadows. Hematol. Rep. 2020, 12 (Suppl. S1), 8950. [Google Scholar] [CrossRef]
- Rea, D.; Henry, G.; Khaznadar, Z.; Etienne, G.; Guilhot, F.; Nicolini, F.; Guilhot, J.; Rousselot, P.; Huguet, F.; Legros, L.; et al. Natural killer-cell counts are associated with molecular relapse-free survival after imatinib discontinuation in chronic myeloid leukemia: The IMMUNOSTIM study. Haematology 2017, 102, 1368–1377. [Google Scholar] [CrossRef] [PubMed]
- Okada, M.; Imagawa, J.; Tanaka, H.; Nakamae, H.; Hino, M.; Murai, K.; Ishida, Y.; Kumagai, T.; Sato, S.; Ohashi, K.; et al. Final 3-year Results of the Dasatinib Discontinuation Trial in Patients with Chronic Myeloid Leukemia Who Received Dasatinib as a Second-line Treatment. Clin. Lymphoma Myeloma Leuk. 2018, 18, 353–360.e1. [Google Scholar] [CrossRef] [PubMed]
- Ilander, M.; Olsson-Strömberg, U.; Schlums, H.; Guilhot, J.; Brück, O.; Lähteenmäki, H.; Kasanen, T.; Koskenvesa, P.; Söderlund, S.; Hoglund, M.; et al. Increased proportion of mature NK cells is associated with successful imatinib discontinuation in chronic myeloid leukemia. Leukemia 2017, 31, 1108–1116. [Google Scholar] [CrossRef]
- Caocci, G.; Martino, B.; Greco, M.; Abruzzese, E.; Trawinska, M.M.; Lai, S.; Ragatzu, P.; Galimberti, S.; Baratè, C.; Mulas, O.; et al. Killer immunoglobulin-like receptors can predict TKI treatment-free remission in chronic myeloid leukemia patients. Exp. Hematol. 2015, 43, 1015–1018.e1. [Google Scholar] [CrossRef]
- Ye, X.-P.; Bao, S.; Gao, H.-M.; Guo, Y.; Wei, Y.-P. A case of myeloproliferative neoplasm with a normal complete blood cell count: A novel problem of the JAK2 era. Oncol. Lett. 2016, 11, 2134–2136. [Google Scholar] [CrossRef]
- Ghanima, W.; Cooper, N.; Rodeghiero, F.; Godeau, B.; Bussel, J.B. Thrombopoietin receptor agonists: Ten years later. Haematology 2019, 104, 1112–1123. [Google Scholar] [CrossRef]
- Schönberg, K.; Rudolph, J.; Vonnahme, M.; Yajnanarayana, S.P.; Cornez, I.; Hejazi, M.; Manser, A.R.; Uhrberg, M.; Verbeek, W.; Koschmieder, S.; et al. JAK Inhibition Impairs NK Cell Function in Myeloproliferative Neoplasms. Cancer Res. 2015, 75, 2187–2199. [Google Scholar] [CrossRef]
- Marchioli, R.; Finazzi, G.; Specchia, G.; Cacciola, R.; Cavazzina, R.; Cilloni, D.; De Stefano, V.; Guglielmelli, P.; Iurlo, A.; Latagliata, R.; et al. Cardiovascular Events and Intensity of Treatment in Polycythemia Vera. New Engl. J. Med. 2013, 368, 22–33. [Google Scholar] [CrossRef] [PubMed]
- Bönnemann, V.; Claus, M.; Butzeck, B.; Collette, D.; Bröde, P.; Golka, K.; Watzl, C. Analysis of Natural Killer cell functions in patients with hereditary hemochromatosis. EXCLI J. 2020, 19, 430–441. [Google Scholar]
- De Stefano, V.; Finazzi, G.; Barbui, T. Antithrombotic therapy for venous thromboembolism in myeloproliferative neoplasms. Blood Cancer J. 2018, 8, 65. [Google Scholar] [CrossRef]
- Landolfi, R.; Marchioli, R.; Kutti, J.; Gisslinger, H.; Tognoni, G.; Patrono, C.; Barbui, T. Efficacy and Safety of Low-Dose Aspirin in Polycythemia Vera. N. Engl. J. Med. 2004, 350, 114–124. [Google Scholar] [CrossRef]
- Tefferi, A.; Vannucchi, A.M.; Barbui, T. Essential thrombocythemia treatment algorithm. Blood Cancer J. 2018, 8, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Tefferi, A.; Barbui, T. Polycythemia vera and essential thrombocythemia: 2019 update on diagnosis, risk-stratification and management. Am. J. Hematol. 2019, 94, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Antonioli, E.; Guglielmelli, P.; Pieri, L.; Finazzi, M.; Rumi, E.; Martinelli, V.; Vianelli, N.; Randi, M.L.; Bertozzi, I.; De Stefano, V.; et al. Hydroxyurea-related toxicity in 3411 patients with Ph’-negative MPN. Am. J. Hematol. 2012, 87, 552–554. [Google Scholar] [CrossRef] [PubMed]
- Michiels, J.J. Myeloproliferative and thrombotic burden and treatment outcome of thrombocythemia and polycythemia patients. World J. Crit. Care Med. 2015, 4, 230–239. [Google Scholar] [CrossRef]
- Singh, A.; Xu, Y.-J. The Cell Killing Mechanisms of Hydroxyurea. Genes 2016, 7, 99. [Google Scholar] [CrossRef]
- Freissmuth, M. Chemotherapie von Tumorerkrankungen; Springer Science and Business Media LLC: Berlin/Heidelberg, Germany, 2020; pp. 837–916. [Google Scholar]
- Economides, M.P.; Verstovsek, S.; Pemmaraju, N. Novel Therapies in Myeloproliferative Neoplasms (MPN): Beyond JAK Inhibitors. Curr. Hematol. Malign-Rep. 2019, 14, 460–468. [Google Scholar] [CrossRef]
- Daver, N.; Shastri, A.; Kadia, T.; Quintás-Cardama, A.; Jabbour, E.; Konopleva, M.; O’Brien, S.; Pierce, S.; Zhou, L.; Cortes, J.; et al. Modest activity of pomalidomide in patients with myelofibrosis and significant anemia. Leuk. Res. 2013, 37, 1440–1444. [Google Scholar] [CrossRef]
- Chihara, D.; Masarova, L.; Newberry, K.J.; Maeng, H.; Ravandi, F.; Garcia-Manero, G.; Ferrajoli, A.; Cortes, J.; Kantarjian, H.; Verstovsek, S. Long-term results of a phase II trial of lenalidomide plus prednisone therapy for patients with myelofibrosis. Leuk. Res. 2016, 48, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Xu, Z.; Li, B.; Qin, T.; Zhang, P.; Zhang, H.; Fang, L.; Pan, L.; Hu, N.; Qu, S.; et al. Thalidomide plus prednisone with or without danazol therapy in myelofibrosis: A retrospective analysis of incidence and durability of anemia response. Blood Cancer J. 2018, 8, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Dredge, K.; Marriott, J.B.; Macdonald, C.D.; Man, H.-W.; Chen, R.; Muller, G.W.; Stirling, D.; Dalgleish, A.G. Novel thalidomide analogues display anti-angiogenic activity independently of immunomodulatory effects. Br. J. Cancer 2002, 87, 1166–1172. [Google Scholar] [CrossRef] [PubMed][Green Version]
- De Stefano, V.; Rocca, B.; Tosetto, A.; Soldati, D.; Petrucci, G.; Beggiato, E.; Bertozzi, I.; Betti, S.; Carli, G.; Carpenedo, M.; et al. The Aspirin Regimens in Essential Thrombocythemia (ARES) phase II randomized trial design: Implementation of the serum thromboxane B2 assay as an evaluation tool of different aspirin dosing regimens in the clinical setting. Blood Cancer J. 2018, 8, 49. [Google Scholar] [CrossRef] [PubMed]
- Periayah, M.H.; Halim, A.S.; Saad, A.Z.M. Mechanism Action of Platelets and Crucial Blood Coagulation Pathways in Hemostasis. Int. J. Hematol. Oncol. Stem Cell Res. 2017, 11, 319–327. [Google Scholar]
- Hasselbalch, H.C.; Holmström, M.O. Perspectives on interferon-alpha in the treatment of polycythemia vera and related myeloproliferative neoplasms: Minimal residual disease and cure? Semin. Immunopathol. 2019, 41, 5–19. [Google Scholar] [CrossRef]
- Michallet, M.; Maloisel, F.; Delain, M.; Hellmann, A.; Rosas, A.; Silver, R.T.; Tendler, C. Pegylated recombinant interferon alpha-2b vs recombinant interferon alpha-2b for the initial treatment of chronic-phase chronic myelogenous leukemia: A phase III study. Leukemia 2003, 18, 309–315. [Google Scholar] [CrossRef][Green Version]
- Schneider, W.M.; Chevillotte, M.D.; Rice, C.M. Interferon-Stimulated Genes: A Complex Web of Host Defenses. Annu. Rev. Immunol. 2014, 32, 513–545. [Google Scholar] [CrossRef]
- Kiladjian, J.-J.; Giraudier, S.; Cassinat, B. Interferon-alpha for the therapy of myeloproliferative neoplasms: Targeting the malignant clone. Leukemia 2015, 30, 776–781. [Google Scholar] [CrossRef]
- Riley, C.H.; Brimnes, M.K.; Hansen, M.; Jensen, M.K.; Hasselbalch, H.C.; Kjaer, L.; Straten, P.T.; Svane, I.M. Interferon-α induces marked alterations in circulating regulatory T cells, NK cell subsets, and dendritic cells in patients with JAK2V617F-positive essential thrombocythemia and polycythemia vera. Eur. J. Haematol. 2015, 97, 83–92. [Google Scholar] [CrossRef]
- Kuvibidila, S.; Warrier, R.P.; Haynes, J.; Baliga, S.B. Hydroxyurea and Zileuton Differentially Modulate Cell Proliferation and Interleukin-2 Secretion by Murine Spleen Cells: Possible Implication on the Immune Function and Risk of Pain Crisis in Patients with Sickle Cell Disease. Ochsner. J. 2015, 15, 241–247. [Google Scholar]
- Weinberg, A. In Vitro Hydroxyurea Decreases Th1 Cell-Mediated Immunity. Clin. Diagn. Lab. Immunol. 2001, 8, 702–705. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lu, X.; Ohata, K.; Kondo, Y.; Espinoza, J.L.; Qi, Z.; Nakao, S. Hydroxyurea upregulates NKG2D ligand expression in myeloid leukemia cells synergistically with valproic acid and potentially enhances susceptibility of leukemic cells to natural killer cell-mediated cytolysis. Cancer Sci. 2010, 101, 609–615. [Google Scholar] [CrossRef]
- Kovacsovics-Bankowski, M.; Kelley, T.W.; Efimova, O.; Kim, S.J.; Wilson, A.; Swierczek, S.; Prchal, J. Changes in peripheral blood lymphocytes in polycythemia vera and essential thrombocythemia patients treated with pegylated-interferon alpha and correlation with JAK2 V617F allelic burden. Exp. Hematol. Oncol. 2015, 5, 28. [Google Scholar] [CrossRef] [PubMed]
- Linder, B.; Pinilla-Ibarz, J.; Sweet, K.; Corrales-Yepez, G.; Komrokji, R. Role of tyrosine-kinase inhibitors in myeloproliferative neoplasms: Comparative lessons learned. Onco. Targets Ther. 2016, 9, 4937–4957. [Google Scholar] [CrossRef] [PubMed]
- Verstovsek, S.; Mesa, R.A.; Gotlib, J.; Levy, R.S.; Gupta, V.; DiPersio, J.F.; Catalano, J.V.; Deininger, M.W.; Miller, C.B.; Silver, R.T.; et al. Efficacy, safety and survival with ruxolitinib in patients with myelofibrosis: Results of a median 2-year follow-up of COMFORT-I. Haematology 2013, 98, 1865–1871. [Google Scholar] [CrossRef]
- Kvasnicka, H.M.; Thiele, J.; Bueso-Ramos, C.E.; Sun, W.; Cortes, J.; Kantarjian, H.M.; Verstovsek, S. Long-term effects of ruxolitinib versus best available therapy on bone marrow fibrosis in patients with myelofibrosis. J. Hematol. Oncol. 2018, 11, 1–10. [Google Scholar] [CrossRef]
- Elli, E.M.; Baratè, C.; Mendicino, F.; Palandri, F.; Palumbo, G.A. Mechanisms Underlying the Anti-inflammatory and Immunosuppressive Activity of Ruxolitinib. Front. Oncol. 2019, 9, 1186. [Google Scholar] [CrossRef]
- Heine, A.; Held, S.A.E.; Daecke, S.N.; Wallner, S.; Yajnanarayana, S.P.; Kurts, C.; Wolf, D.; Brossart, P. The JAK-inhibitor ruxolitinib impairs dendritic cell function in vitro and in vivo. Blood 2013, 122, 1192–1202. [Google Scholar] [CrossRef]
- Curran, S.A.; Shyer, J.A.; Angelo, E.T.S.; Talbot, L.R.; Sharma, S.; Chung, D.J.; Heller, G.; Hsu, K.C.; Betts, B.C.; Young, J.W. Human dendritic cells mitigate NK-cell dysfunction mediated by nonselective JAK1/2 blockade. Cancer Immunol. Res. 2017, 5, 52–60. [Google Scholar] [CrossRef]
- Munegowda, M.A.; Hu, J. Transient blocking of NK cell function with small molecule inhibitors for helper dependant adenoviral vector-mediated gene delivery. Cell Biosci. 2015, 5, 1–10. [Google Scholar] [CrossRef]
- Hofmann, S.; Babiak, A.; Greiner, J. Immunotherapy for myeloproliferative neoplasms (MPN). Curr. Cancer Drug Targets 2011, 11, 72–84. [Google Scholar] [CrossRef]
- Chang, X.; Zhu, Y.; Shi, C.; Stewart, A.K. Mechanism of immunomodulatory drugs’ action in the treatment of multiple myeloma. Acta Biochim. Biophys. Sin. 2013, 46, 240–253. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Shen, Y.; Li, S.; Lv, M.; Zhang, X.; Yang, J.; Wang, F.; Yang, J. Importance of the interaction between immune cells and tumor vasculature mediated by thalidomide in cancer treatment (Review). Int. J. Mol. Med. 2016, 38, 1021–1029. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Wolf, D.; Sopper, S. Molecular response prediction in CML: Novel ideas? Oncotarget 2017, 8, 80105–80106. [Google Scholar] [CrossRef] [PubMed]
- Markasz, L.; Stuber, G.; Vanherberghen, B.; Flaberg, E.; Olah, E.; Carbone, E.; Eksborg, S.; Klein, E.; Skribek, H.; Szekely, L. Effect of frequently used chemotherapeutic drugs on the cytotoxic activity of human natural killer cells. Mol. Cancer Ther. 2007, 6, 644–654. [Google Scholar] [CrossRef]
- Abraham, A.A.; Lang, H.; Meier, E.R.; Nickel, R.S.; Dean, M.; Lawal, N.; Speller-Brown, B.; Wang, Y.; Kean, L.; Bollard, C.M. Characterization of natural killer cells expressing markers associated with maturity and cytotoxicity in children and young adults with sickle cell disease. Pediatr. Blood Cancer 2019, 66, e27601. [Google Scholar] [CrossRef] [PubMed]
- McLornan, D.P.; Khan, A.A.; Harrison, C.N. Immunological Consequences of JAK Inhibition: Friend or Foe? Curr. Hematol. Malign-Rep. 2015, 10, 370–379. [Google Scholar] [CrossRef]
- Vargas-Hernández, A.; Mace, E.; Zimmerman, O.; Zerbe, C.S.; Freeman, A.F.; Rosenzweig, S.; Leiding, J.W.; Torgerson, T.; Altman, M.C.; Schussler, E.; et al. Ruxolitinib partially reverses functional natural killer cell deficiency in patients with signal transducer and activator of transcription 1 (STAT1) gain-of-function mutations. J. Allergy Clin. Immunol. 2018, 141, 2142–2155. [Google Scholar] [CrossRef]
- Cao, Y.; Wang, X.; Jin, T.; Tian, Y.; Dai, C.; Widarma, C.; Song, R.; Xu, F. Immune checkpoint molecules in natural killer cells as potential targets for cancer immunotherapy. Signal. Transduct. Target. Ther. 2020, 5, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Cayssials, E.; Guilhot, F. Chronic Myeloid Leukemia: Immunobiology and Novel Immunotherapeutic Approaches. BioDrugs 2017, 31, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Swatler, J.; Turos-Korgul, L.; Kozlowska, E.; Piwocka, K. Immunosuppressive Cell Subsets and Factors in Myeloid Leukemias. Cancers 2021, 13, 1203. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Naismith, E.; Steichen, J.; Sopper, S.; Wolf, D. NK Cells in Myeloproliferative Neoplasms (MPN). Cancers 2021, 13, 4400. https://doi.org/10.3390/cancers13174400
Naismith E, Steichen J, Sopper S, Wolf D. NK Cells in Myeloproliferative Neoplasms (MPN). Cancers. 2021; 13(17):4400. https://doi.org/10.3390/cancers13174400
Chicago/Turabian StyleNaismith, Erin, Janine Steichen, Sieghart Sopper, and Dominik Wolf. 2021. "NK Cells in Myeloproliferative Neoplasms (MPN)" Cancers 13, no. 17: 4400. https://doi.org/10.3390/cancers13174400
APA StyleNaismith, E., Steichen, J., Sopper, S., & Wolf, D. (2021). NK Cells in Myeloproliferative Neoplasms (MPN). Cancers, 13(17), 4400. https://doi.org/10.3390/cancers13174400