
Whole-Genome Analysis of De Novo Somatic Point Mutations Reveals Novel Mutational Biomarkers in Pancreatic Cancer

 , , , , and
, , , , and
Abstract
:Simple Summary
Abstract

1. Introduction
2. Materials and Methods
2.1. Data
2.2. Data Cleaning and Filtration
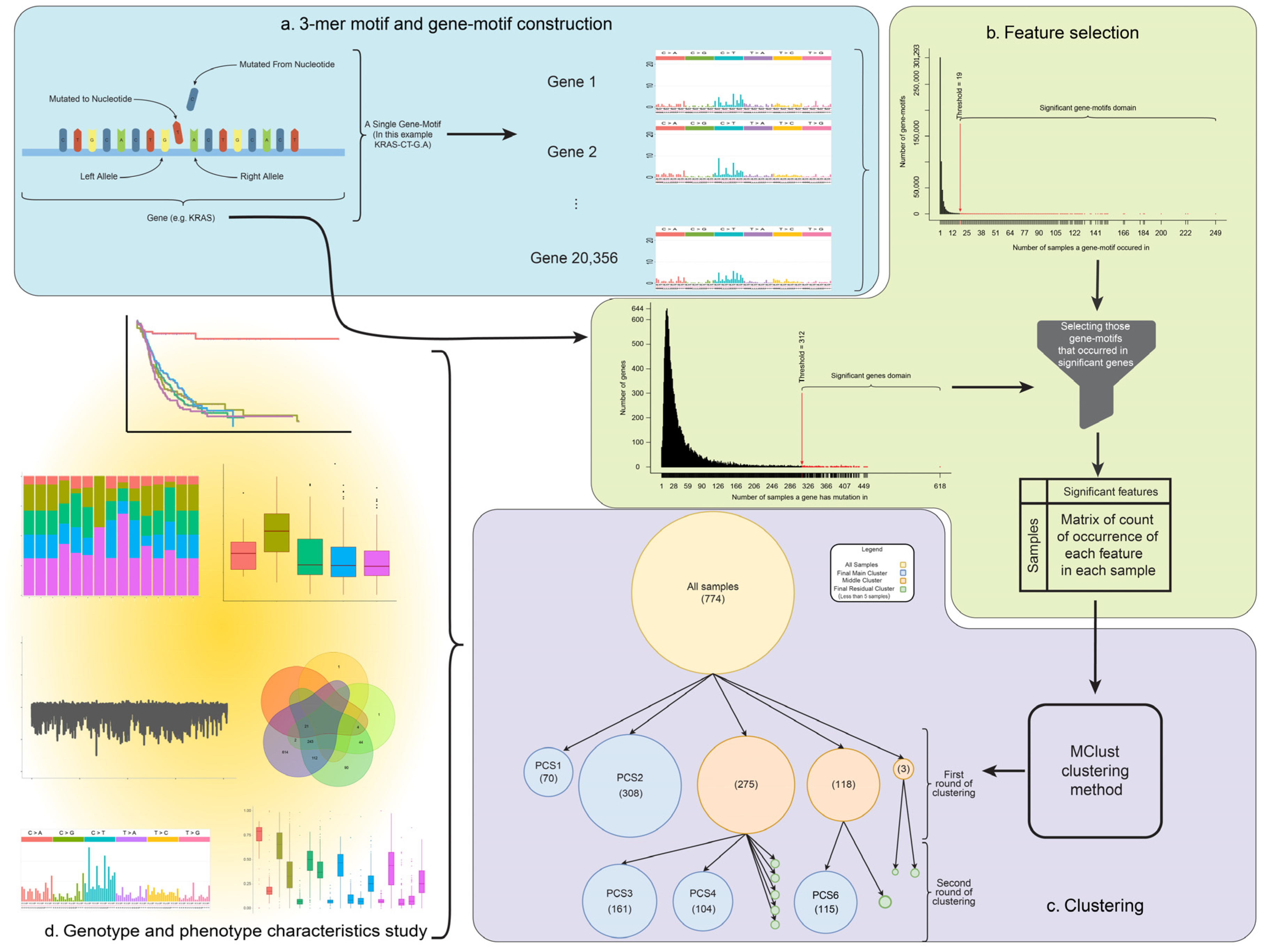
2.3. Feature Selection
2.4. Clustering Method
2.5. Differential Analysis
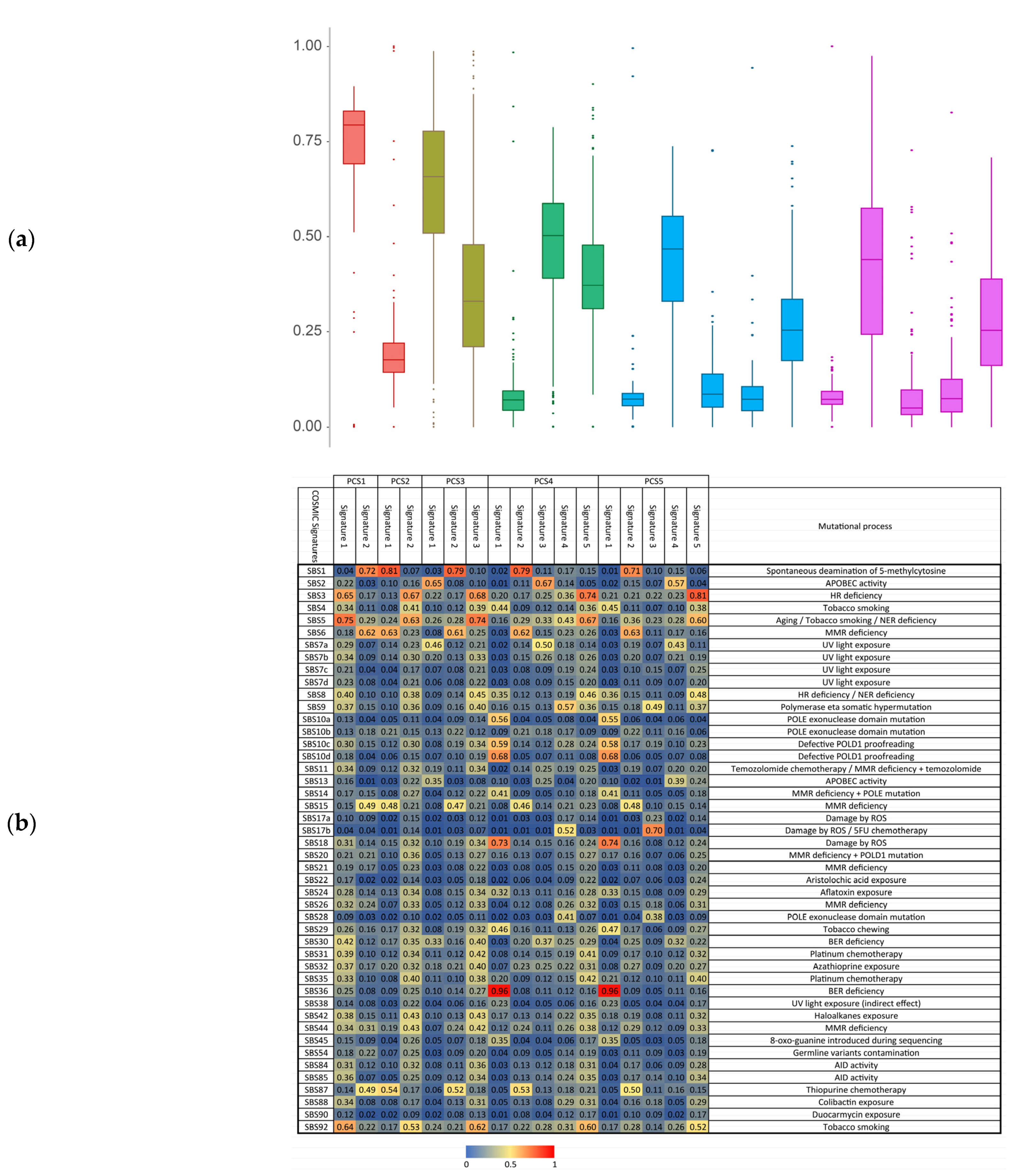
2.6. Mutational Signature Analysis
2.7. Motif Analysis
2.8. Transcript Type Analysis
2.9. Gene Association
2.10. Gene Expression Analysis
2.11. Gene Ontology and Pathway
2.12. Gender and Project Code Analysis
2.13. Literature Search for Non-Coding Interacting Genes
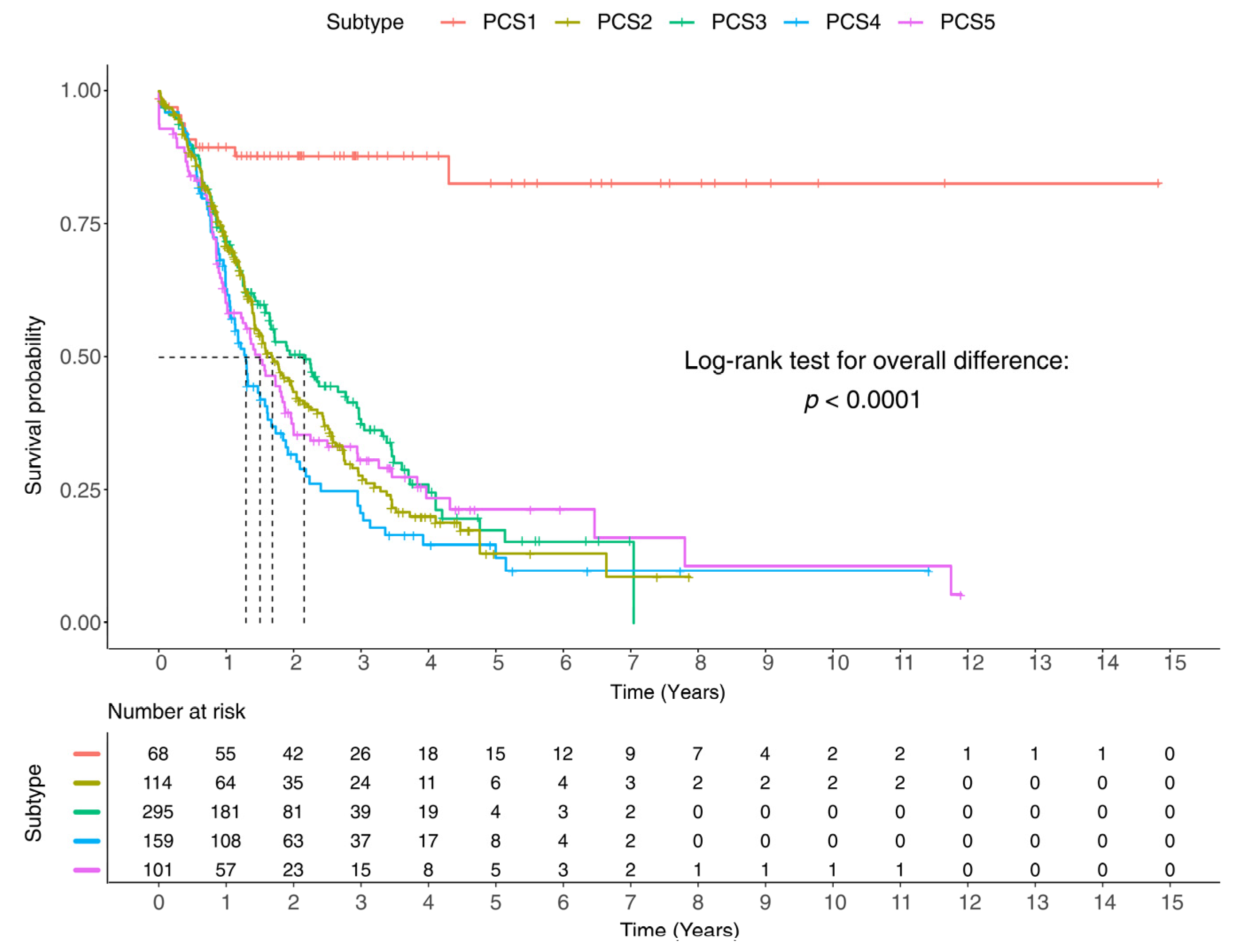
2.14. Survival Analysis
2.15. Comparing the Overall Mutation Rate in Subtypes
3. Results
3.1. Background Model to Identify Subtypes in Pancreatic Cancer
3.2. Relative Frequency of Mutations in the PC Subtypes
3.3. Biological Characterization of Each Subtype
3.4. Motif Rates and Signatures Analysis
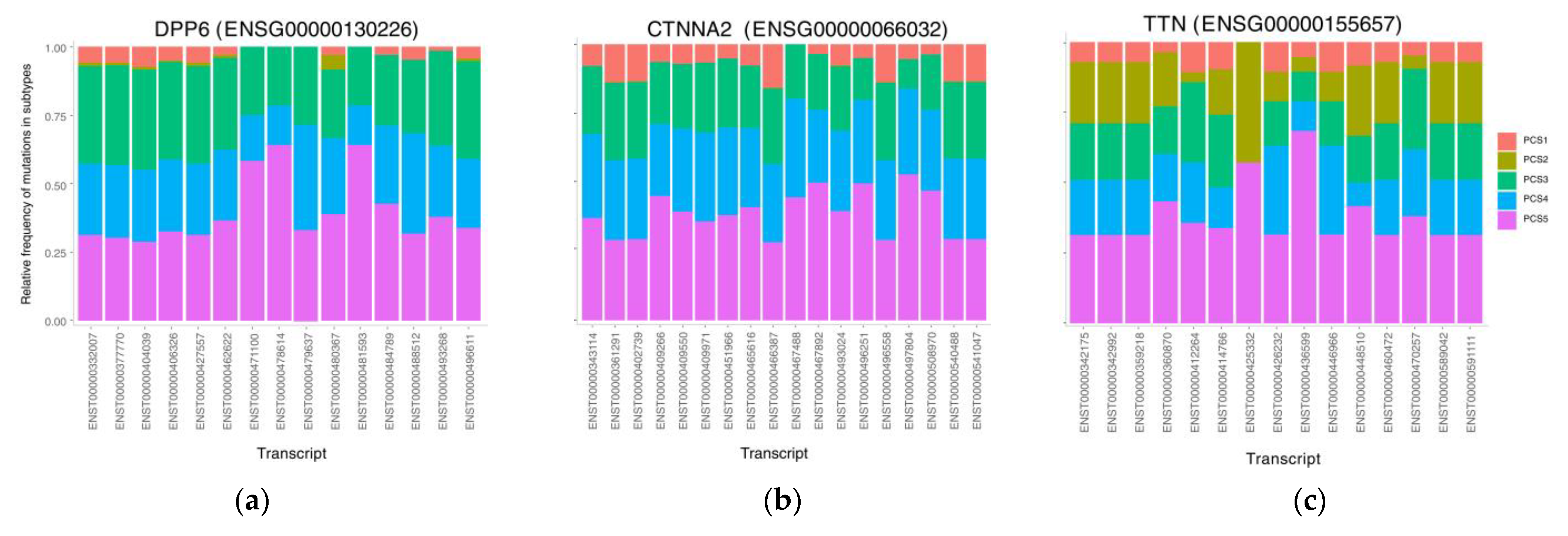
3.5. The Mutational Rate in Transcripts
3.6. Gene Expression Analysis and Finding Differentially Expressed Genes
3.7. Gene Ontology and Pathway Analyses
3.8. Clinical Report and Survival Analysis of the Identified Subtypes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [Green Version]
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting Cancer Incidence and Deaths to 2030: The Unexpected Burden of Thyroid, Liver, and Pancreas Cancers in the United States. Cancer Res. 2014, 74, 2913–2921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collisson, E.A.; Bailey, P.; Chang, D.K.; Biankin, A.V. Molecular subtypes of pancreatic cancer. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 207–220. [Google Scholar] [CrossRef] [PubMed]
- Slamon, D.J.; Leyland-Jones, B.; Shak, S.; Fuchs, H.; Paton, V.; Bajamonde, A.; Fleming, T.; Eiermann, W.; Wolter, J.; Pegram, M.; et al. Use of Chemotherapy plus a Monoclonal Antibody against HER2 for Metastatic Breast Cancer That Overexpresses HER2. N. Engl. J. Med. 2001, 344, 783–792. [Google Scholar] [CrossRef] [PubMed]
- Lynch, T.J.; Bell, D.W.; Sordella, R.; Gurubhagavatula, S.; Okimoto, R.A.; Brannigan, B.W.; Harris, P.L.; Haserlat, S.M.; Supko, J.G.; Haluska, F.G.; et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 2004, 350, 2129–2139. [Google Scholar] [CrossRef]
- Garcea, G.; Neal, C.; Pattenden, C.; Steward, W.; Berry, D. Molecular prognostic markers in pancreatic cancer: A systematic review. Eur. J. Cancer 2005, 41, 2213–2236. [Google Scholar] [CrossRef] [PubMed]
- Collisson, E.A.; Sadanandam, A.; Olson, P.; Gibb, W.J.; Truitt, M.; Gu, S.; Cooc, J.; Weinkle, J.; Kim, G.E.; Jakkula, L.; et al. Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy. Nat. Med. 2011, 17, 500–503. [Google Scholar] [CrossRef] [PubMed]
- Moffitt, R.; Marayati, R.; Flate, E.L.; Volmar, K.E.; Loeza, S.G.H.; Hoadley, K.; Rashid, N.U.; Williams, L.A.; Eaton, S.C.; Chung, A.H.; et al. Virtual microdissection identifies distinct tumor- and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nat. Genet. 2015, 47, 1168–1178. [Google Scholar] [CrossRef] [PubMed]
- Bailey, P.; Initiative, A.P.C.G.; Chang, D.K.; Nones, K.; Johns, A.L.; Patch, A.-M.; Gingras, M.-C.; Miller, D.K.; Christ, A.N.; Bruxner, T.J.C.; et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nat. Cell Biol. 2016, 531, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Sivakumar, S.; de Santiago, I.; Chlon, L.; Markowetz, F. Master Regulators of Oncogenic KRAS Response in Pancreatic Cancer: An Integrative Network Biology Analysis. PLoS Med. 2017, 14, e1002223. [Google Scholar] [CrossRef] [Green Version]
- Puleo, F.; Nicolle, R.; Blum, Y.; Cros, J.; Marisa, L.; Demetter, P.; Quertinmont, E.; Svrcek, M.; Elarouci, N.; Iovanna, J.; et al. Stratification of Pancreatic Ductal Adenocarcinomas Based on Tumor and Microenvironment Features. Gastroenterology 2018, 155, 1999–2013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Androulakis, I.P.; Yang, E.; Almon, R.R. Analysis of Time-Series Gene Expression Data: Methods, Challenges, and Opportunities. Annu. Rev. Biomed. Eng. 2007, 9, 205–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res. 2018, 47, D941–D947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuijjer, M.L.; Paulson, J.N.; Salzman, P.; Ding, W.; Quackenbush, J. Cancer subtype identification using somatic mutation data. Br. J. Cancer 2018, 118, 1492–1501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuipers, J.; Thurnherr, T.; Moffa, G.; Suter, P.; Behr, J.; Goosen, R.; Christofori, G.; Beerenwinkel, N. Mutational interactions define novel cancer subgroups. Nat. Commun. 2018, 9, 4353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waddell, N.; Pajic, M.; Patch, A.M.; Chang, D.K.; Kassahn, K.S.; Bailey, P.; Johns, A.L.; Miller, D.; Nones, K.; Quek, K.; et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature. 2015, 518, 495–501. [Google Scholar] [CrossRef] [Green Version]
- Alexandrov, L.B.; Kim, J.; Haradhvala, N.J.; Huang, M.N.; Ng, A.W.T.; Wu, Y.; Boot, A.; Covington, K.R.; Gordenin, D.A.; Bergstrom, E.N.; et al. The repertoire of mutational signatures in human cancer. Nat. Cell Biol. 2020, 578, 94–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parvin, H.; Minaei, B.; Alizadeh, H.; Beigi, A. A Novel Classifier Ensemble Method Based on Class Weightening in Huge Dataset. In International Symposium on Neural Networks; Springer: Berlin/Heidelberg, Germany, 2011; pp. 144–150. [Google Scholar] [CrossRef]
- All Codes Related to This Research. Available online: https://github.com/bcb-sut/Pancreatic-Cancer-Subtype-Identification (accessed on 6 January 2021).
- International Cancer Genome Consortium Data Portal. Available online: https://dcc.icgc.org/ (accessed on 1 November 2017).
- Lawrence, M.; Huber, W.; Pagès, H.; Aboyoun, P.; Carlson, M.; Gentleman, R.; Morgan, M.; Carey, V.J. Software for Computing and Annotating Genomic Ranges. PLoS Comput. Biol. 2013, 9, e1003118. [Google Scholar] [CrossRef]
- Gehring, J.S.; Fischer, B.; Lawrence, M.F.; Huber, W. SomaticSignatures: Inferring mutational signatures from single-nucleotide variants: Figure 1. Bioinformatics 2015, 31, 3673–3675. [Google Scholar] [CrossRef] [Green Version]
- Dashti, H.; Dehzangi, A.; Bayati, M.; Breen, J.; Lovell, N.; Ebrahimi, D.; Rabiee, R.H.; Alinejad-Rokny, H. Integrative analysis of mutated genes and mutational processes reveals seven colorectal cancer subtypes. bioRxiv 2020. [Google Scholar] [CrossRef]
- Scrucca, L.; Fop, M.; Murphy, T.B.; Adrian, E.R. mclust 5: Clustering, Classification and Density Estimation Using Gaussian Finite Mixture Models. R J. 2016, 8, 289–317. [Google Scholar] [CrossRef] [Green Version]
- Fraley, C.; Raftery, A.E. Model-based methods of classification: Using the mclust software in chemometrics. J. Stat. Softw. 2007, 18, 1–13. [Google Scholar] [CrossRef]
- Fraley, C.; Raftery, A.E. How Many Clusters? Which Clustering Method? Answers Via Model-Based Cluster Analysis. Comput. J. 1998, 41, 578–588. [Google Scholar] [CrossRef]
- Bayati, M.; Rabiee, H.R.; Mehrbod, M.; Vafaee, F.; Ebrahimi, D.; Forrest, A.; Alinejad-Rokny, H. CANCERSIGN: A user-friendly and robust tool for identification and classification of mutational signatures and patterns in cancer genomes. Sci. Rep. 2020, 10, 1286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Børresen-Dale, A.L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shamshirband, S.; Fathi, M.; Dehzangi, A.; Chronopoulose, A.T. A review on deep learning approaches in healthcare systems: Taxonomies, challenges, and open issues. J. Biomed. Inf. 2020, 113, 103627. [Google Scholar] [CrossRef]
- Ebrahimi, D.; Alinejad-Rokny, H.; Davenport, M.P. Insights into the Motif Preference of APOBEC3 Enzymes. PLoS ONE 2014, 9, e87679. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Enrichr. Available online: https://amp.pharm.mssm.edu/Enrichr/ (accessed on 6 January 2020).
- Javanmard, R.; Jeddisaravi, K.; Alinejad-Rokny, H. Proposed a New Method for Rules Extraction Using Artificial Neural Network and Artificial Immune System in Cancer Diagnosis. J. Bionanoscience 2013, 7, 665–672. [Google Scholar] [CrossRef]
- Rad, M.P.; Pourshaikh, R. Conceptual Information Retrieval in Cross-Language Searches. Res. J. Appl. Sci. Eng. Technol. 2012, 4, 1714–1720. [Google Scholar]
- Alinejad-Rokny, H.; Parvin, S. Divide and Conquer Classification. Int. J. Sci. Basic Appl. Res. 2011, 5, 2446–2452. [Google Scholar]
- Hasanzadeh, E.; Rokny, H.A.; Poyan, M. Text clustering on latent semantic indexing with particle swarm optimization (PSO) algorithm. Int. J. Phys. Sci. 2012, 7, 16–120. [Google Scholar] [CrossRef]
- Esmaeili, L.; Minaei-Bidgoli, B.; Alinejad-Rokny, H.; Nasiri, M. Hybrid recommender system for joining virtual communities. Res. J. Appl. Sci. Eng. Technol. 2012, 4, 500–509. [Google Scholar]
- Alinejad-Rokny, H.; Anwar, F.; Waters, S.; Davenport, M.; Ebrahimi, D. Source of CpG Depletion in the HIV-1 Genome. Mol. Biol. Evol. 2016, 33, 3205–3212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parvin, H.; MirnabiBaboli, M.; Alinejad-Rokny, H. Proposing a classifier ensemble framework based on classifier selection and decision tree. Eng. Appl. Artif. Intell. 2015, 37, 34–42. [Google Scholar] [CrossRef]
- Woolson, R.F. Rank Tests and a One-Sample Logrank Test for Comparing Observed Survival Data to a Standard Population. Biometrics 1981, 37, 687. [Google Scholar] [CrossRef]
- Karadeniz, P.G.; Ercan, I. Examining Tests for Comparing Survival Curves with Right Censored Data. Stat. Transit. New Ser. 2017, 18, 311–328. [Google Scholar] [CrossRef]
- Zhu, Y.; Neeman, T.; Yap, V.B.; Huttley, G.A. Statistical Methods for Identifying Sequence Motifs Affecting Point Mutations. Genetics 2017, 205, 843–856. [Google Scholar] [CrossRef] [Green Version]
- Alexandrov, L.B.; Kim, J.; Haradhvala, N.J.; Huang, M.N.; Ng, A.W.; Wu, Y.; Boot, A.; Covington, K.R.; Gordenin, D.A.; Bergstrom, E.N.; et al. The repertoire of mutational signatures in human cancer. BioRxiv 2019, 322859. [Google Scholar] [CrossRef]
- Zuo, H.; Han, B.; Poppinga, W.J.; Ringnalda, L.; Kistemaker, L.E.M.; Halayko, A.J.; Gosens, R.; Nikolaev, O.V.; Schmidt, M. Cigarette smoke up-regulates PDE3 and PDE4 to decrease cAMP in airway cells. Br. J. Pharmacol. 2017, 175, 2988–3006. [Google Scholar] [CrossRef]
- Park, S.L.; Carmella, S.G.; Chen, M.; Patel, Y.; Stram, D.O.; Haiman, C.A.; Le Marchand, L.; Hecht, S.S. Mercapturic Acids Derived from the Toxicants Acrolein and Crotonaldehyde in the Urine of Cigarette Smokers from Five Ethnic Groups with Differing Risks for Lung Cancer. PLoS ONE 2015, 10, e0124841. [Google Scholar] [CrossRef]
- Luce, L.N.; Abbate, M.; Cotignola, J.; Giliberto, F. Non-myogenic tumors display altered expression of dystrophin (DMD) and a high frequency of genetic alterations. Oncotarget 2016, 8, 145–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The Gene Ontology Consortium, Expansion of the Gene Ontology knowledgebase and resources. Nucleic Acids Res. 2016, 45, D331–D338. [CrossRef] [Green Version]
- Demir, I.E.; Friess, H.; Ceyhan, G.O. Neural plasticity in pancreatitis and pancreatic cancer. Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 649–659. [Google Scholar] [CrossRef]
- Moh, M.C.; Shen, S. The roles of cell adhesion molecules in tumor suppression and cell migration: A new paradox. Cell Adhes. Migr. 2009, 3, 334–336. [Google Scholar] [CrossRef] [PubMed]
- Bassagañas, S.; Carvalho, S.; Dias, A.; Pérez-Garay, M.; Ortiz, R.; Figueras, J.; Reis, C.; Pinho, S.S.; Peracaula, R. Pancreatic Cancer Cell Glycosylation Regulates Cell Adhesion and Invasion through the Modulation of α2β1 Integrin and E-Cadherin Function. PLoS ONE 2014, 9, e98595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farahani, E.; Patra, H.K.; Jangamreddy, J.R.; Rashedi, I.; Kawalec, M.; Pariti, R.K.R.; Batakis, P.; Wiechec, E. Cell adhesion molecules and their relation to (cancer) cell stemness. Carcinog. 2014, 35, 747–759. [Google Scholar] [CrossRef] [Green Version]
- Biankin, A.V.; Initiative, A.P.C.G.; Waddell, N.; Kassahn, K.S.; Gingras, M.-C.; Muthuswamy, L.B.; Johns, A.L.; Miller, D.K.; Wilson, P.J.; Patch, A.-M.; et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nat. Cell Biol. 2012, 491, 399–405. [Google Scholar] [CrossRef]
- Halfdanarson, T.R.; Rubin, J.; Farnell, M.B.; Grant, C.S.; Petersen, G.M. Pancreatic endocrine neoplasms: Epidemiology and prognosis of pancreatic endocrine tumors. Endocr.-Relat. Cancer 2008, 15, 409–427. [Google Scholar] [CrossRef]
- Ilic, M.; Ilic, I. Epidemiology of pancreatic cancer. World J. Gastroenterol. 2016, 22, 9694–9705. [Google Scholar] [CrossRef]
- Dietlein, F.; Weghorn, D.; Taylor-Weiner, A.; Richters, A.; Reardon, B.; Liu, D.; Lander, E.S.; Van Allen, E.M.; Sunyaev, S.R. Identification of cancer driver genes based on nucleotide context. Nat. Genet. 2020, 52, 208–218. [Google Scholar] [CrossRef] [PubMed]
- Veeriah, S.; Brennan, C.; Meng, S.; Singh, B.; Fagin, J.A.; Solit, D.B.; Paty, P.B.; Rohle, D.; Vivanco, I.; Chmielecki, J.; et al. The tyrosine phosphatase PTPRD is a tumor suppressor that is frequently inactivated and mutated in glioblastoma and other human cancers. Proc. Natl. Acad. Sci. USA 2009, 106, 9435–9440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Funato, K.; Yamazumi, Y.; Oda, T.; Akiyama, T. Tyrosine phosphatase PTPRD suppresses colon cancer cell migration in coordination with CD44. Exp. Ther. Med. 2011, 2, 457–463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waters, A.M.; Der, C.J. KRAS: The Critical Driver and Therapeutic Target for Pancreatic cancer. Cold Spring Harb. Perspect. Med. 2017, 8, a031435. [Google Scholar] [CrossRef]
- Canon, J.; Rex, K.; Saiki, A.; Mohr, C.; Cooke, K.; Bagal, D.; Gaida, K.; Holt, T.; Knutson, C.G.; Koppada, N.; et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nat. Cell Biol. 2019, 575, 217–223. [Google Scholar] [CrossRef]
- Guo, S.-W.; Zheng, Y.; Lu, Y.; Liu, X.; Geng, J.-G. Slit2 overexpression results in increased microvessel density and lesion size in mice with induced endometriosis. Reprod. Sci. 2013, 20, 285–298. [Google Scholar] [CrossRef]
- Rama, N.; Dubrac, A.; Mathivet, T.; Chárthaigh, R.-A.N.; Genet, G.; Cristofaro, B.; Pibouin-Fragner, L.; Ma, L.; Eichmann, A.; Chédotal, A. Slit2 signaling through Robo1 and Robo2 is required for retinal neovascularization. Nat. Med. 2015, 21, 483–491. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Huang, L.; Sun, Y.; Bai, Y.; Yang, F.; Yu, W.; Li, F.; Zhang, Q.; Wang, B.; Geng, J.; et al. Slit2 Promotes Angiogenic Activity Via the Robo1-VEGFR2-ERK1/2 Pathway in Both In Vivo and In Vitro Studies. Investig. Ophthalmol. Vis. Sci. 2015, 56, 5210–5217. [Google Scholar] [CrossRef] [Green Version]
- Chaturvedi, S.; Yuen, D.A.; Bajwa, A.; Huang, Y.-W.; Sokollik, C.; Huang, L.; Lam, G.Y.; Tole, S.; Liu, G.-Y.; Pan, J.; et al. Slit2 Prevents Neutrophil Recruitment and Renal Ischemia-Reperfusion Injury. J. Am. Soc. Nephrol. 2013, 24, 1274–1287. [Google Scholar] [CrossRef] [Green Version]
- Pinho, A.V.; Van Bulck, M.; Chantrill, L.; Arshi, M.; Sklyarova, T.; Herrmann, D.; Vennin, C.; Gallego-Ortega, D.; Mawson, A.; Giry-Laterriere, M.; et al. ROBO2 is a stroma suppressor gene in the pancreas and acts via TGF-β signalling. Nat. Commun. 2018, 9, 5083. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ghareyazi, A.; Mohseni, A.; Dashti, H.; Beheshti, A.; Dehzangi, A.; Rabiee, H.R.; Alinejad-Rokny, H. Whole-Genome Analysis of De Novo Somatic Point Mutations Reveals Novel Mutational Biomarkers in Pancreatic Cancer. Cancers 2021, 13, 4376. https://doi.org/10.3390/cancers13174376
Ghareyazi A, Mohseni A, Dashti H, Beheshti A, Dehzangi A, Rabiee HR, Alinejad-Rokny H. Whole-Genome Analysis of De Novo Somatic Point Mutations Reveals Novel Mutational Biomarkers in Pancreatic Cancer. Cancers. 2021; 13(17):4376. https://doi.org/10.3390/cancers13174376
Chicago/Turabian StyleGhareyazi, Amin, Amir Mohseni, Hamed Dashti, Amin Beheshti, Abdollah Dehzangi, Hamid R. Rabiee, and Hamid Alinejad-Rokny. 2021. "Whole-Genome Analysis of De Novo Somatic Point Mutations Reveals Novel Mutational Biomarkers in Pancreatic Cancer" Cancers 13, no. 17: 4376. https://doi.org/10.3390/cancers13174376
APA StyleGhareyazi, A., Mohseni, A., Dashti, H., Beheshti, A., Dehzangi, A., Rabiee, H. R., & Alinejad-Rokny, H. (2021). Whole-Genome Analysis of De Novo Somatic Point Mutations Reveals Novel Mutational Biomarkers in Pancreatic Cancer. Cancers, 13(17), 4376. https://doi.org/10.3390/cancers13174376