
miR-22 Modulates Lenalidomide Activity by Counteracting MYC Addiction in Multiple Myeloma

 ,
,  , ,
, ,  ,
, 

Abstract
:Simple Summary
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Multiple Myeloma Cell Lines, Primary Cells, and Reagents
2.2. Analysis of Cell Viability and Apoptosis
2.3. Flow Cytometry and Degranulation Assay
2.4. Cytotoxicity Assay
2.5. Transduction of Cells
2.6. RNA Extraction and Quantitative Real-Time PCR
2.7. Gene Expression Profiling
2.8. In Vitro Transfection of MM Cells
2.9. Luciferase Reporter Experiments
2.10. Western Blot Analysis
2.11. ChIP
3. Results
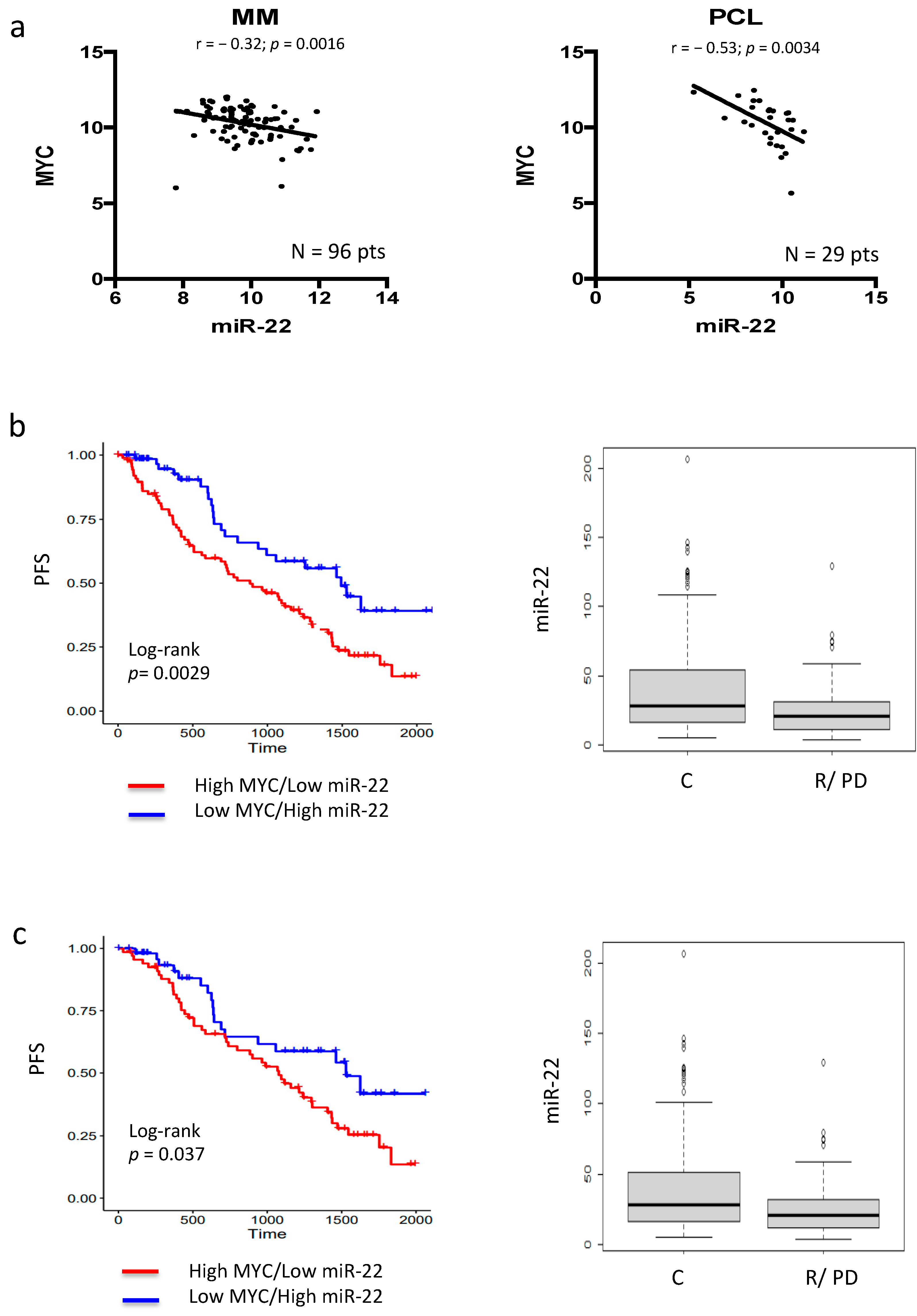
3.1. MYC–miR-22 Inverse Correlation Predicts Poor Response to IMiDs in MM Patients
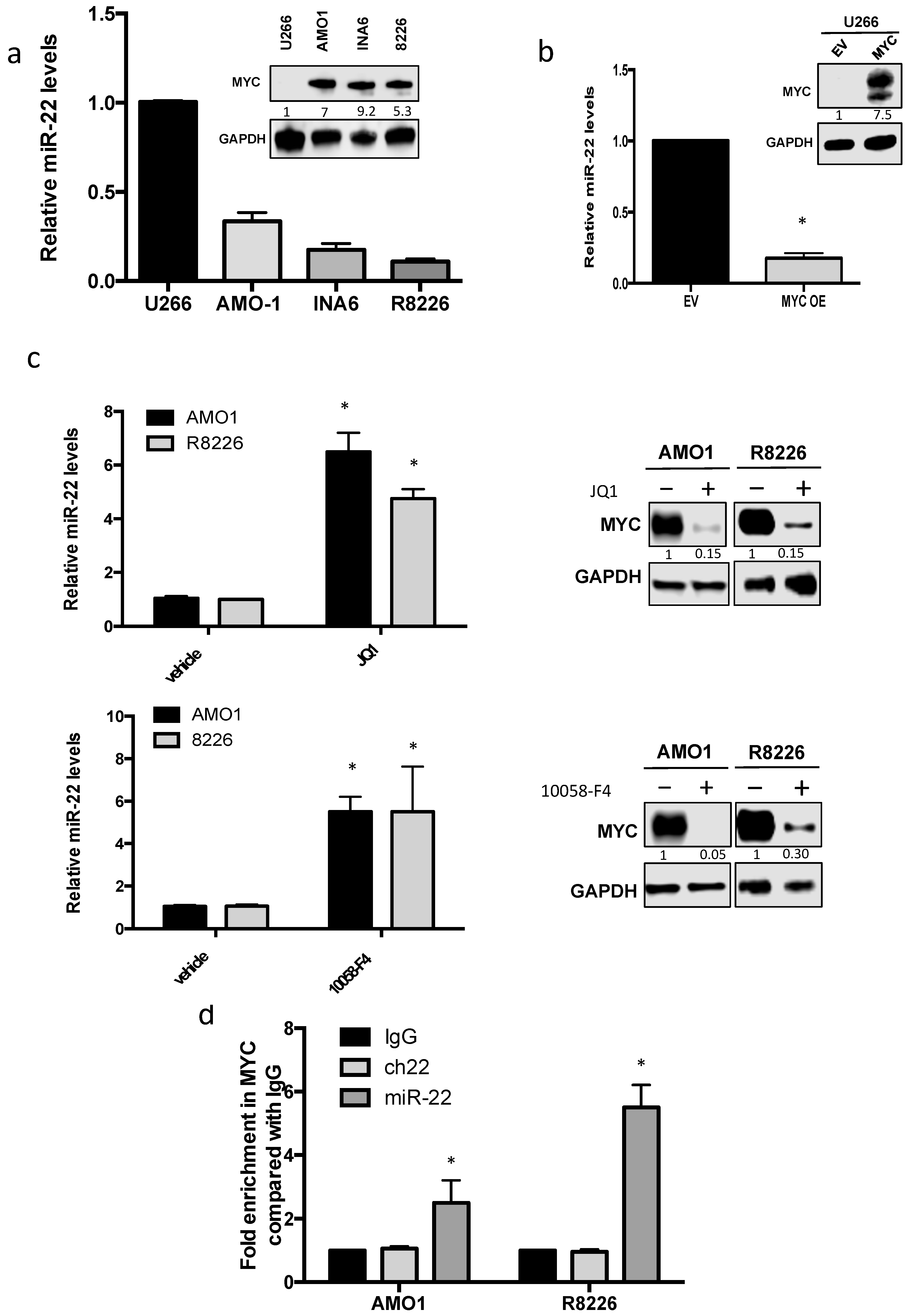
3.2. MYC Represses miR-22 Transcription in MM
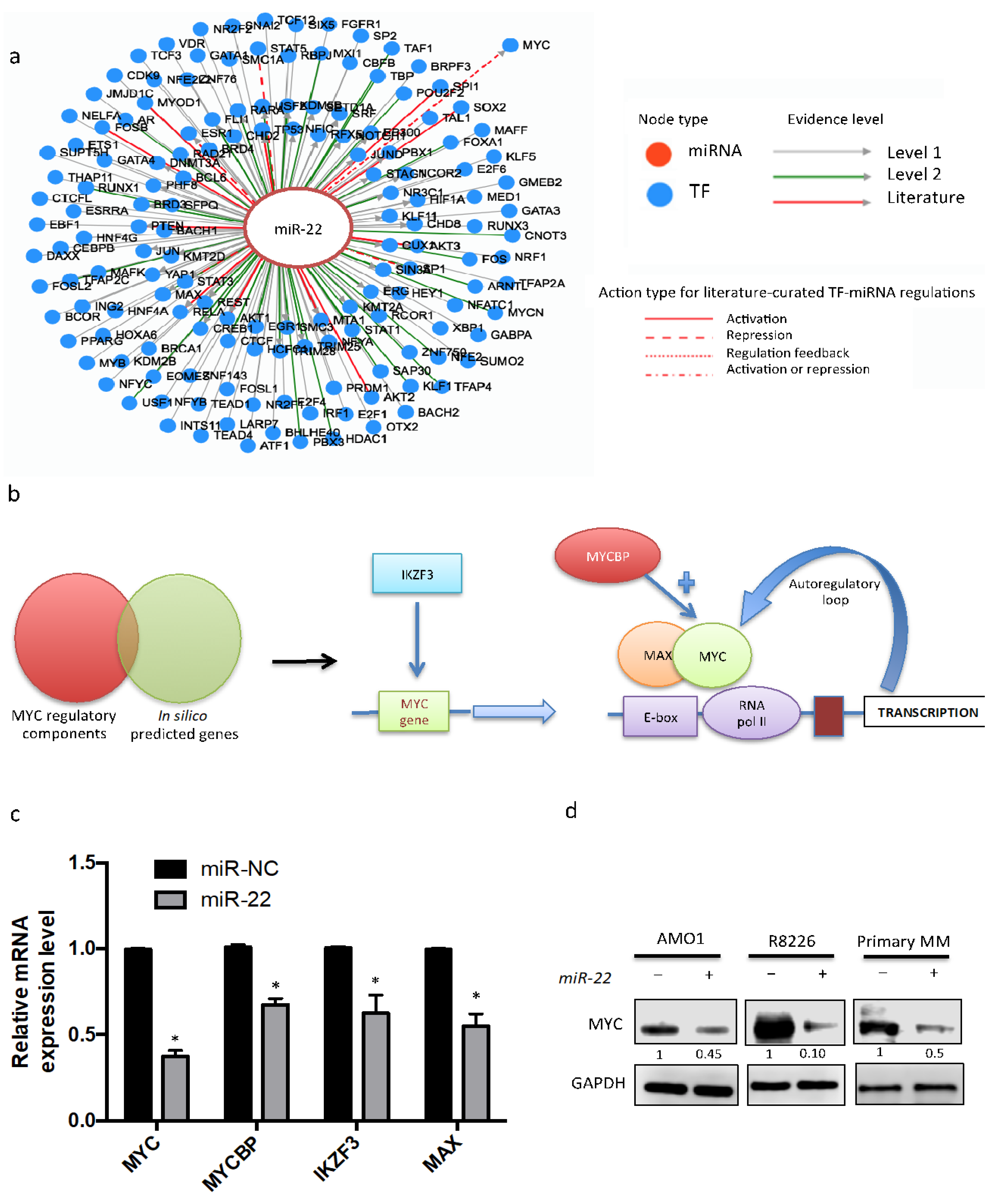
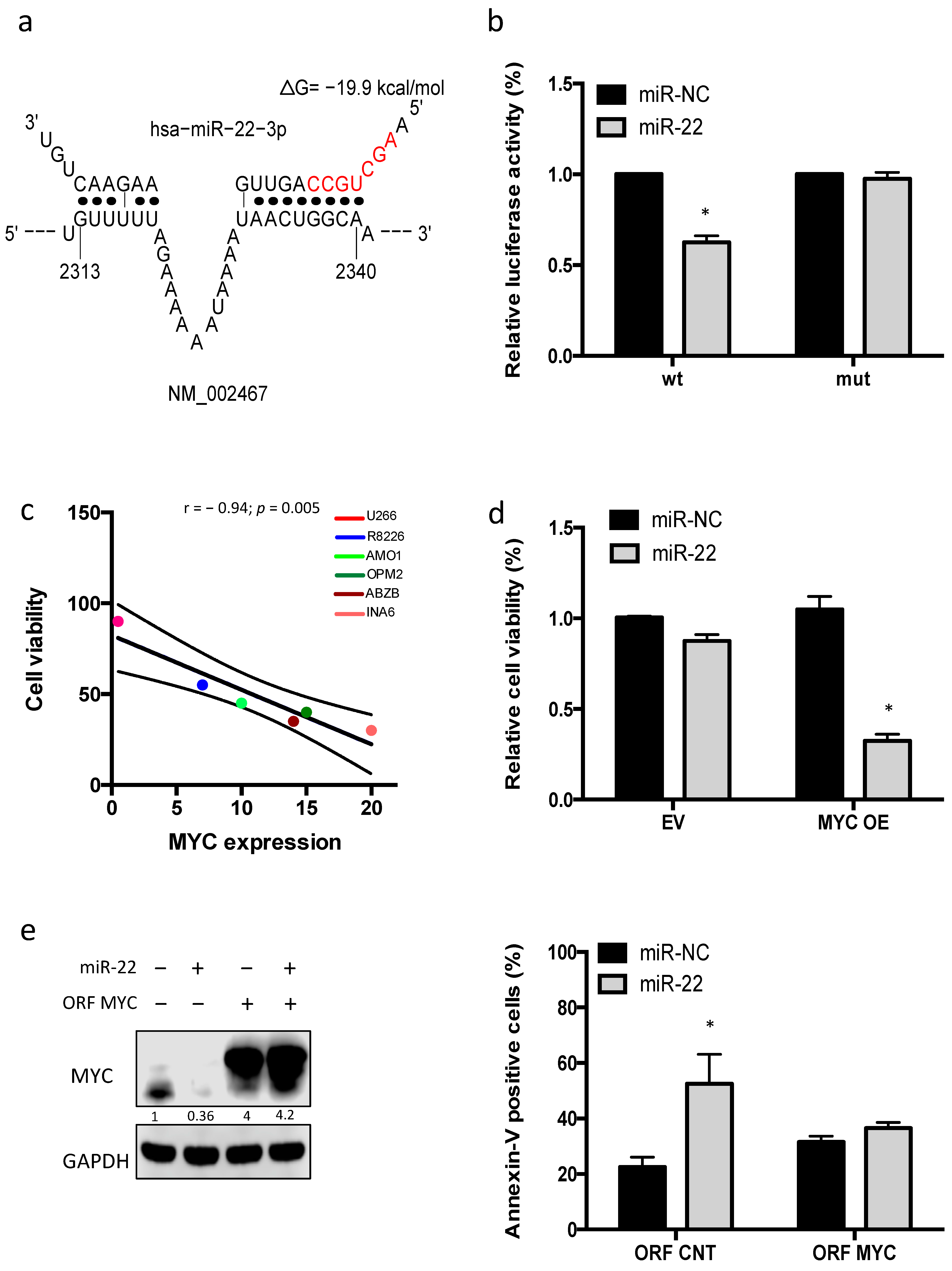
3.3. Enforced Expression of miR-22 Inhibits MYC Expression and Function
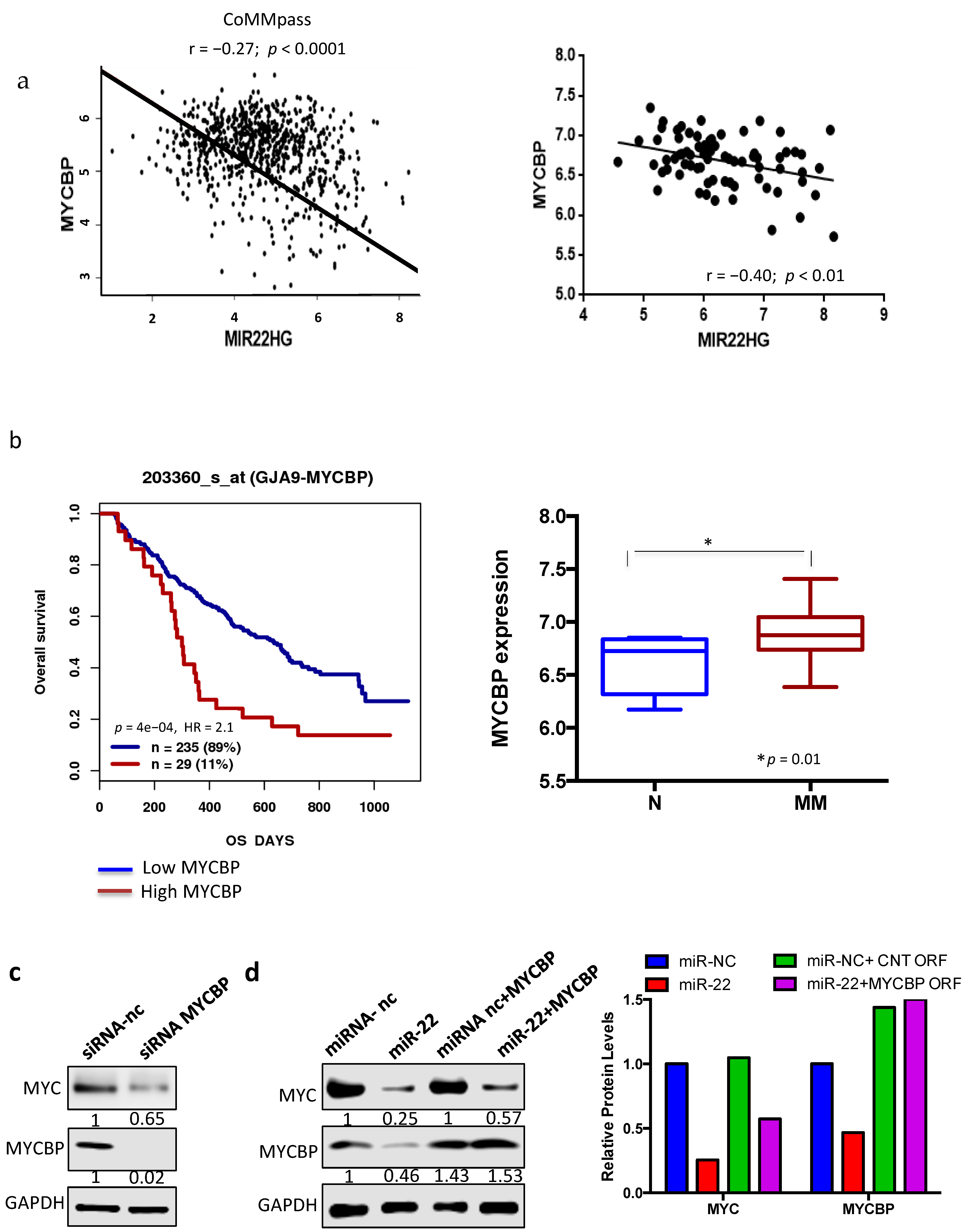
3.4. miR-22-Dependent Regulation of MYC Expression in MM Cells
3.5. miR-22 Triggers MYC-Dependent Synthetic Lethality
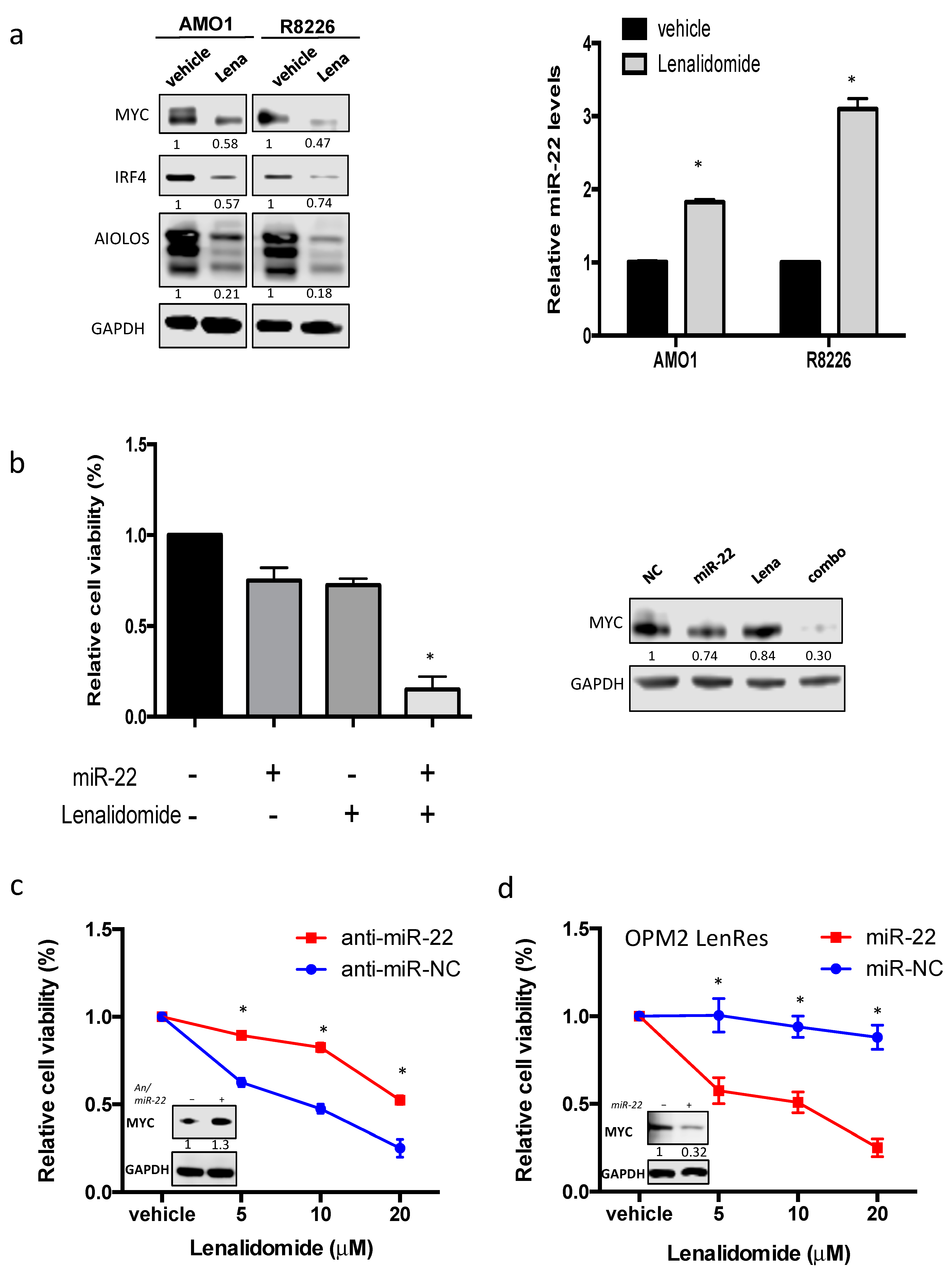
3.6. miR-22 Sensitizes MM Cells to Lenalidomide by Reducing MYC Expression
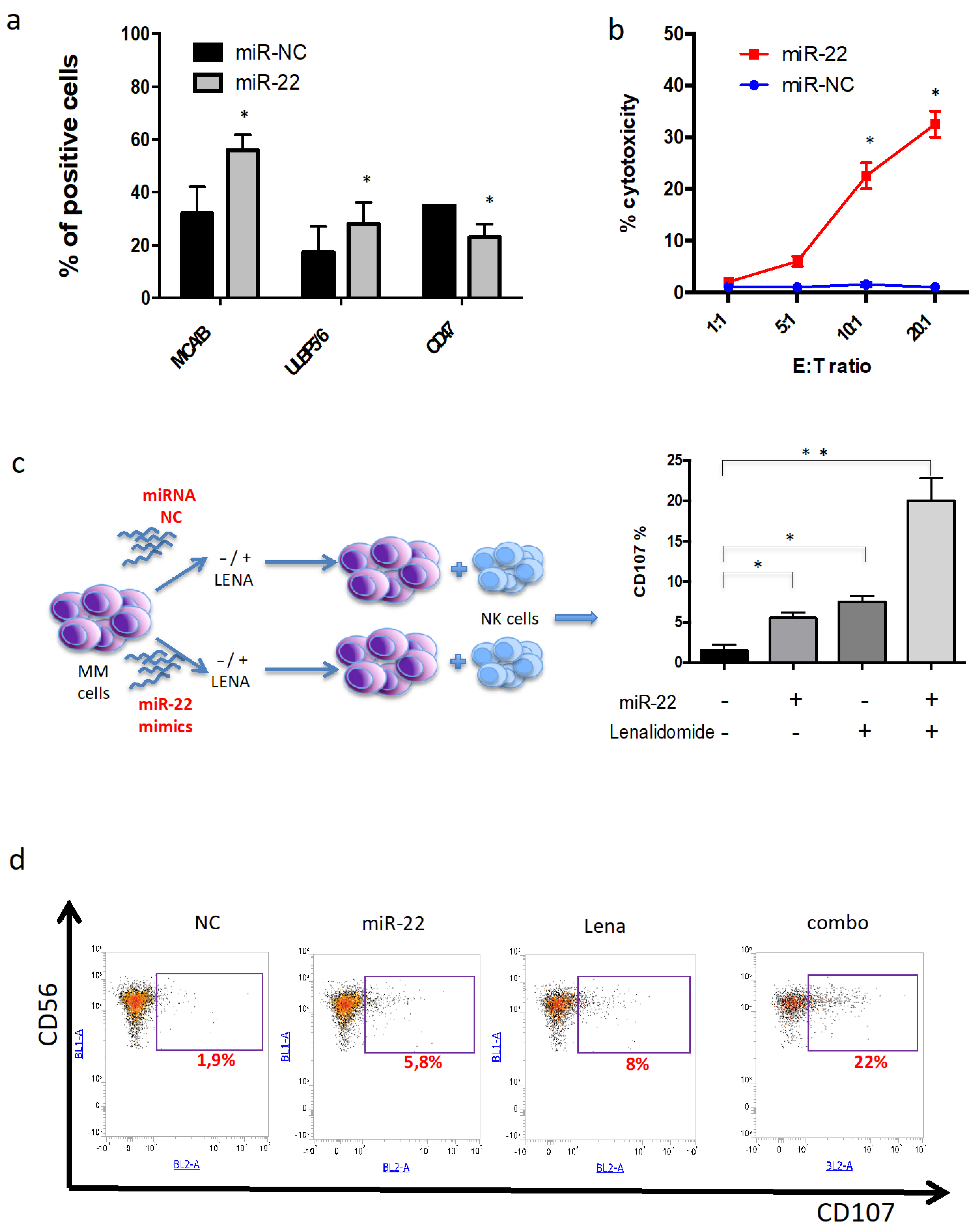
3.7. miR-22 Potentiates NK-Mediated Cytotoxicity Induced by Lenalidomide
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Van de Donk, N.; Pawlyn, C.; Yong, K.L. Multiple myeloma. Lancet 2021, 397, 410–427. [Google Scholar] [CrossRef]
- Offidani, M.; Corvatta, L.; More, S.; Olivieri, A. Novel Experimental Drugs for Treatment of Multiple Myeloma. J. Exp. Pharmacol. 2021, 13, 245–264. [Google Scholar] [CrossRef] [PubMed]
- Dang, C.V.; Reddy, E.P.; Shokat, K.M.; Soucek, L. Drugging the ‘undruggable’ cancer targets. Nat. Rev. Cancer 2017, 17, 502–508. [Google Scholar] [CrossRef] [PubMed]
- Chiecchio, L.; Dagrada, G.P.; White, H.E.; Towsend, M.R.; Protheroe, R.K.; Cheung, K.L.; Stockley, D.M.; Orchard, K.H.; Cross, N.C.; Ross, F.M.; et al. Frequent upregulation of MYC in plasma cell leukemia. Genes Chromosomes Cancer 2009, 48, 624–636. [Google Scholar] [CrossRef]
- Misund, K.; Keane, N.; Stein, C.K.; Asmann, Y.W.; Day, G.; Welsh, S.; Van Wier, S.A.; Riggs, D.L.; Ahmann, G.; Chesi, M.; et al. MYC dysregulation in the progression of multiple myeloma. Leukemia 2020, 34, 322–326. [Google Scholar] [CrossRef]
- Greco, C.; D’Agnano, I.; Vitelli, G.; Vona, R.; Marino, M.; Mottolese, M.; Zuppi, C.; Capoluongo, E.; Ameglio, F. c-MYC deregulation is involved in melphalan resistance of multiple myeloma: Role of PDGF-BB. Int. J. Immunopathol. Pharmacol. 2006, 19, 67–79. [Google Scholar] [CrossRef] [Green Version]
- Franssen, L.E.; Nijhof, I.S.; Couto, S.; Levin, M.D.; Bos, G.M.; Broijl, A.; Klein, S.K.; Ren, Y.; Wang, M.; Koene, H.R.; et al. Cereblon loss and up-regulation of c-Myc are associated with lenalidomide resistance in multiple myeloma patients. Haematologica 2018, 103, e368–e371. [Google Scholar] [CrossRef]
- Dang, C.V. MYC on the path to cancer. Cell 2012, 149, 22–35. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, G.J. Emerging roles of Myc in stem cell biology and novel tumor therapies. J. Exp. Clin. Cancer Res. 2018, 37, 173. [Google Scholar] [CrossRef] [Green Version]
- Frenzel, A.; Loven, J.; Henriksson, M.A. Targeting MYC-Regulated miRNAs to Combat Cancer. Genes Cancer 2010, 1, 660–667. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef] [Green Version]
- Garzon, R.; Marcucci, G.; Croce, C.M. Targeting microRNAs in cancer: Rationale, strategies and challenges. Nat. Rev. Drug Discov. 2010, 9, 775–789. [Google Scholar] [CrossRef] [Green Version]
- Morelli, E.; Biamonte, L.; Federico, C.; Amodio, N.; Di Martino, M.T.; Gallo Cantafio, M.E.; Manzoni, M.; Scionti, F.; Samur, M.K.; Gullà, A.; et al. Therapeutic vulnerability of multiple myeloma to MIR17PTi, a first-in-class inhibitor of pri-miR-17-92. Blood 2018, 132, 1050–1063. [Google Scholar] [CrossRef]
- Di Martino, M.T.; Gullà, A.; Gallo Cantafio, M.E.; Altomare, E.; Amodio, N.; Leone, E.; Morelli, E.; Lio, S.G.; Caracciolo, D.; Rossi, M.; et al. In vitro and in vivo activity of a novel locked nucleic acid (LNA)-inhibitor-miR-221 against multiple myeloma cells. PLoS ONE 2014, 9, e89659. [Google Scholar] [CrossRef] [PubMed]
- Stamato, M.A.; Juli, G.; Romeo, E.; Ronchetti, D.; Arbitrio, M.; Caracciolo, D.; Neri, A.; Tagliaferri, P.; Tassone, P.; Amodio, N.; et al. Inhibition of EZH2 triggers the tumor suppressive miR-29b network in multiple myeloma. Oncotarget 2017, 8, 106527–106537. [Google Scholar] [CrossRef] [Green Version]
- Rossi, M.; Amodio, N.; Di Martino, M.T.; Caracciolo, D.; Tagliaferri, P.; Tassone, P. From target therapy to miRNA therapeutics of human multiple myeloma: Theoretical and technological issues in the evolving scenario. Curr. Drug Targ. 2013, 14, 1144–1149. [Google Scholar] [CrossRef]
- Beg, M.S.; Brenner, A.J.; Sachdev, J.; Borad, M.; Kang, Y.K.; Stoudemire, J.; Smith, S.; Bader, A.G.; Kim, S.; Hong, D.S.; et al. Phase I study of MRX34, a liposomal miR-34a mimic, administered twice weekly in patients with advanced solid tumors. Investig. New Drugs 2017, 35, 180–188. [Google Scholar] [CrossRef]
- Van Zandwijk, N.; Pavlakis, N.; Kao, S.C.; Linton, A.; Boyer, M.J.; Clarke, S.; Huynh, Y.; Chrzanowska, A.; Fulham, M.J.; Bailey, D.L.; et al. Safety and activity of microRNA-loaded minicells in patients with recurrent malignant pleural mesothelioma: A first-in-man, phase 1, open-label, dose-escalation study. Lancet Oncol. 2017, 18, 1386–1396. [Google Scholar] [CrossRef]
- Zhang, L.; Liao, Y.; Tang, L. MicroRNA-34 family: A potential tumor suppressor and therapeutic candidate in cancer. J. Exp. Clin. Cancer Res. 2019, 38, 53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, T.C.; Yu, D.; Lee, Y.S.; Wentzel, E.A.; Arking, D.E.; West, K.M.; Dang, C.V.; Thomas-Tikhonenko, A.; Mendell, J.T. Widespread microRNA repression by Myc contributes to tumorigenesis. Nat. Genet. 2008, 40, 43–50. [Google Scholar] [CrossRef] [Green Version]
- Amodio, N.; Di Martino, M.T.; Foresta, U.; Leone, E.; Lionetti, M.; Leotta, M.; Gulla, A.M.; Pitari, M.R.; Conforti, F.; Rossi, M.; et al. miR-29b sensitizes multiple myeloma cells to bortezomib-induced apoptosis through the activation of a feedback loop with the transcription factor Sp1. Cell Death Dis. 2012, 3, e436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gullà, A.; Di Martino, M.T.; Cantafio, M.E.G.; Morelli, E.; Amodio, N.; Botta, C.; Pitari, M.R.; Lio, S.G.; Britti, D.; Stamato, M.A.; et al. A 13 mer LNA-i-miR-221 Inhibitor Restores Drug Sensitivity in Melphalan-Refractory Multiple Myeloma Cells. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2016, 22, 1222–1233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murray, M.Y.; Rushworth, S.A.; Zaitseva, L.; Bowles, K.M.; Macewan, D.J. Attenuation of dexamethasone-induced cell death in multiple myeloma is mediated by miR-125b expression. Cell Cycle 2013, 12, 2144–2153. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.H.; Lee, S.E.; Lee, M.; Kim, S.H.; Yim, S.H.; Kim, T.W.; Min, C.K.; Chung, Y.J. Circulating microRNA expressions can predict the outcome of lenalidomide plus low-dose dexamethasone treatment in patients with refractory/relapsed multiple myeloma. Haematologica 2017, 102, e456–e459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caracciolo, D.; Riillo, C.; Ballerini, A.; Gaipa, G.; Lhermitte, L.; Rossi, M.; Botta, C.; Duroyon, E.; Grillone, K.; Cantafio, M.E.G.; et al. Therapeutic afucosylated monoclonal antibody and bispecific T-cell engagers for T-cell acute lymphoblastic leukemia. J. Immunother. Cancer 2021, 9, e002026. [Google Scholar] [CrossRef]
- Amodio, N.; Gallo Cantafio, M.E.; Botta, C.; Agosti, V.; Federico, C.; Caracciolo, D.; Ronchetti, D.; Rossi, M.; Driessen, C.; Neri, A.; et al. Replacement of miR-155 Elicits Tumor Suppressive Activity and Antagonizes Bortezomib Resistance in Multiple Myeloma. Cancers 2019, 11, 236. [Google Scholar] [CrossRef] [Green Version]
- Lionetti, M.; Barbieri, M.; Todoerti, K.; Agnelli, L.; Marzorati, S.; Fabris, S.; Ciceri, G.; Galletti, S.; Milesi, G.; Mazzoni, M.; et al. Molecular spectrum of BRAF, NRAS and KRAS gene mutations in plasma cell dyscrasias: Implication for MEK-ERK pathway activation. Oncotarget 2015, 6, 24205–24217. [Google Scholar] [CrossRef] [Green Version]
- Gallo Cantafio, M.E.; Grillone, K.; Caracciolo, D.; Scionti, F.; Arbitrio, M.; Barbieri, V.; Pensabene, L.; Guzzi, P.H.; Di Martino, M.T. From Single Level Analysis to Multi-Omics Integrative Approaches: A Powerful Strategy towards the Precision Oncology. High Throughput 2018, 7, 33. [Google Scholar] [CrossRef] [Green Version]
- Dib, A.; Gabrea, A.; Glebov, O.K.; Bergsagel, P.L.; Kuehl, W.M. Characterization of MYC translocations in multiple myeloma cell lines. J. Natl. Cancer Inst. Monographs 2008, 2008, 25–31. [Google Scholar] [CrossRef]
- Rennie, W.; Liu, C.; Carmack, C.S.; Wolenc, A.; Kanoria, S.; Lu, J.; Long, D.; Ding, Y. STarMir: A web server for prediction of microRNA binding sites. Nucl. Acids Res. 2014, 42, W114–W118. [Google Scholar] [CrossRef] [Green Version]
- Caracciolo, D.; Scionti, F.; Juli, G.; Altomare, E.; Golino, G.; Todoerti, K.; Grillone, K.; Riillo, C.; Arbitrio, M.; Iannone, M.; et al. Exploiting MYC-induced PARPness to target genomic instability in multiple myeloma. Haematologica 2020, 106, 185–195. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.X.; Shi, C.X.; Bruins, L.A.; Wang, X.; Riggs, D.L.; Porter, B.; Ahmann, J.M.; de Campos, C.B.; Braggio, E.; Bergsagel, P.L.; et al. Identification of lenalidomide resistance pathways in myeloma and targeted resensitization using cereblon replacement, inhibition of STAT3 or targeting of IRF4. Blood Cancer J. 2019, 9, 19. [Google Scholar] [CrossRef] [Green Version]
- Quach, H.; Ritchie, D.; Stewart, A.K.; Neeson, P.; Harrison, S.; Smyth, M.J.; Prince, H.M. Mechanism of action of immunomodulatory drugs (IMiDS) in multiple myeloma. Leukemia 2010, 24, 22–32. [Google Scholar] [CrossRef] [Green Version]
- Fionda, C.; Abruzzese, M.P.; Zingoni, A.; Cecere, F.; Vulpis, E.; Peruzzi, G.; Soriani, A.; Molfetta, R.; Paolini, R.; Ricciardi, M.R.; et al. The IMiDs targets IKZF-1/3 and IRF4 as novel negative regulators of NK cell-activating ligands expression in multiple myeloma. Oncotarget 2015, 6, 23609–23630. [Google Scholar] [CrossRef] [Green Version]
- Abruzzese, M.P.; Bilotta, M.T.; Fionda, C.; Zingoni, A.; Soriani, A.; Vulpis, E.; Borrelli, C.; Zitti, B.; Petrucci, M.T.; Ricciardi, M.R.; et al. Inhibition of bromodomain and extra-terminal (BET) proteins increases NKG2D ligand MICA expression and sensitivity to NK cell-mediated cytotoxicity in multiple myeloma cells: Role of cMYC-IRF4-miR-125b interplay. J. Hematol. Oncol. 2016, 9, 134. [Google Scholar] [CrossRef] [PubMed]
- Sessa, M.; Cavazzini, F.; Cavallari, M.; Rigolin, G.M.; Cuneo, A. A Tangle of Genomic Aberrations Drives Multiple Myeloma and Correlates with Clinical Aggressiveness of the Disease: A Comprehensive Review from a Biological Perspective to Clinical Trial Results. Genes 2020, 11, 1453. [Google Scholar] [CrossRef]
- Tao, J.; Zhao, X.; Tao, J. c-MYC-miRNA circuitry: A central regulator of aggressive B-cell malignancies. Cell Cycle 2014, 13, 191–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caracciolo, D.; Di Martino, M.T.; Amodio, N.; Morelli, E.; Montesano, M.; Botta, C.; Scionti, F.; Talarico, D.; Altomare, E.; Cantafio, M.E.G.; et al. miR-22 suppresses DNA ligase III addiction in multiple myeloma. Leukemia 2018, 33, 487–498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, J.; Du, Q.; Liang, Z. Tumor-suppressive microRNA-22 inhibits the transcription of E-box-containing c-Myc target genes by silencing c-Myc binding protein. Oncogene 2010, 29, 4980–4988. [Google Scholar] [CrossRef] [Green Version]
- Ting, Y.; Medina, D.J.; Strair, R.K.; Schaar, D.G. Differentiation-associated miR-22 represses Max expression and inhibits cell cycle progression. Biochem. Biophys. Res. Commun. 2010, 394, 606–611. [Google Scholar] [CrossRef]
- Hirano, M.; Imai, Y.; Kaito, Y.; Murayama, T.; Sato, K.; Ishida, T.; Yamamoto, J.; Ito, T.; Futami, M.; Ri, M.; et al. Small-molecule HDAC and Akt inhibitors suppress tumor growth and enhance immunotherapy in multiple myeloma. J. Exp. Clin. Cancer Res. 2021, 40, 110. [Google Scholar] [CrossRef] [PubMed]
- Moros, A.; Rodriguez, V.; Saborit-Villarroya, I.; Montraveta, A.; Balsas, P.; Sandy, P.; Martinez, A.; Wiestner, A.; Normant, E.; Campo, E.; et al. Synergistic antitumor activity of lenalidomide with the BET bromodomain inhibitor CPI203 in bortezomib-resistant mantle cell lymphoma. Leukemia 2014, 28, 2049–2059. [Google Scholar] [CrossRef] [PubMed]
- Díaz, T.; Rodríguez, V.; Lozano, E.; Mena, M.P.; Calderón, M.; Rosiñol, L.; Martínez, A.; Tovar, N.; Pérez-Galán, P.; Bladé, J.; et al. The BET bromodomain inhibitor CPI203 improves lenalidomide and dexamethasone activity in in vitro and in vivo models of multiple myeloma by blockade of Ikaros and MYC signaling. Haematologica 2017, 102, 1776–1784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casey, S.C.; Tong, L.; Li, Y.; Do, R.; Walz, S.; Fitzgerald, K.N.; Gouw, A.M.; Baylot, V.; Gütgemann, I.; Eilers, M.; et al. MYC regulates the antitumor immune response through CD47 and PD-L1. Science 2016, 352, 227–231. [Google Scholar] [CrossRef] [Green Version]
- Casey, S.C.; Baylot, V.; Felsher, D.W. The MYC oncogene is a global regulator of the immune response. Blood 2018, 131, 2007–2015. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Caracciolo, D.; Riillo, C.; Juli, G.; Scionti, F.; Todoerti, K.; Polerà, N.; Grillone, K.; Fiorillo, L.; Arbitrio, M.; Di Martino, M.T.; et al. miR-22 Modulates Lenalidomide Activity by Counteracting MYC Addiction in Multiple Myeloma. Cancers 2021, 13, 4365. https://doi.org/10.3390/cancers13174365
Caracciolo D, Riillo C, Juli G, Scionti F, Todoerti K, Polerà N, Grillone K, Fiorillo L, Arbitrio M, Di Martino MT, et al. miR-22 Modulates Lenalidomide Activity by Counteracting MYC Addiction in Multiple Myeloma. Cancers. 2021; 13(17):4365. https://doi.org/10.3390/cancers13174365
Chicago/Turabian StyleCaracciolo, Daniele, Caterina Riillo, Giada Juli, Francesca Scionti, Katia Todoerti, Nicoletta Polerà, Katia Grillone, Lucia Fiorillo, Mariamena Arbitrio, Maria Teresa Di Martino, and et al. 2021. "miR-22 Modulates Lenalidomide Activity by Counteracting MYC Addiction in Multiple Myeloma" Cancers 13, no. 17: 4365. https://doi.org/10.3390/cancers13174365
APA StyleCaracciolo, D., Riillo, C., Juli, G., Scionti, F., Todoerti, K., Polerà, N., Grillone, K., Fiorillo, L., Arbitrio, M., Di Martino, M. T., Neri, A., Tagliaferri, P., & Tassone, P. (2021). miR-22 Modulates Lenalidomide Activity by Counteracting MYC Addiction in Multiple Myeloma. Cancers, 13(17), 4365. https://doi.org/10.3390/cancers13174365