
Apoptosis Deregulation and the Development of Cancer Multi-Drug Resistance


 ,
,  and
and 
Abstract
Simple Summary
Abstract
1. Introduction
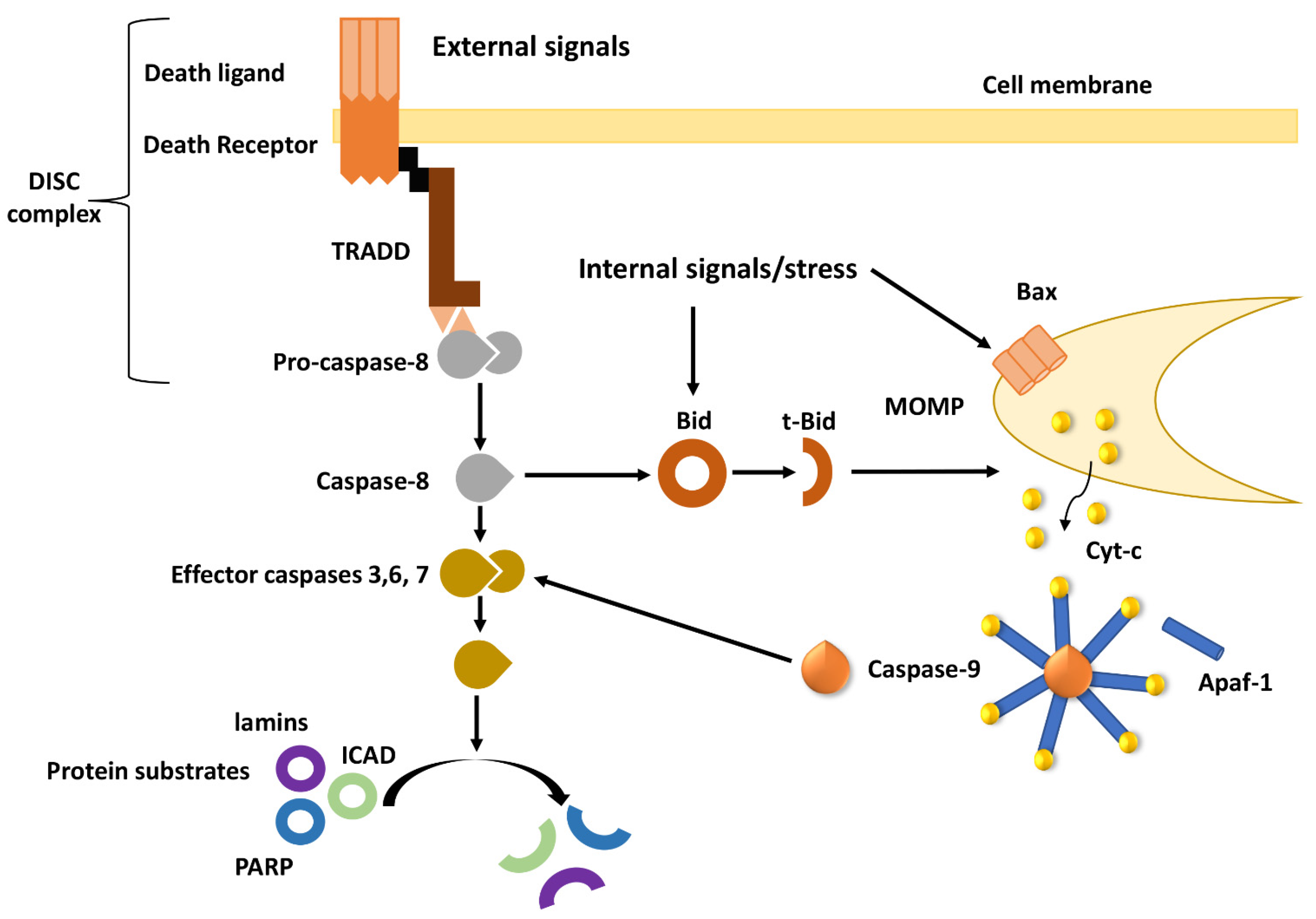
2. Overview of Apoptotic Pathways
3. Deregulation of the Intrinsic Apoptotic Pathway in MDR Tumors
3.1. Bcl-2 Family Deregulation in MDR
3.2. Inhibitors of Apoptosis Proteins (IAPs) and Their Role in MDR
3.3. PI3K/AKT Pathway in Multi-Drug Resistance
4. Implications of the TME in Apoptosis and MDR
4.1. Cancer Associated Fibroblasts in Apoptosis and MDR
4.1.1. CAF-Derived Extracellular Vesicles
4.1.2. Interleukins Secreted by CAFs
4.1.3. Regulation of Sex Determining Region Y-box 2 by CAFs
4.1.4. Growth Promoting Proteins Released by CAFs
5. Therapeutic Approaches to Induce Apoptosis in MDR Cancers

{kind=link}
{kind=link}
| Therapeutic Class | Compound | Observed Effect | Model | Ref |
|---|---|---|---|---|
| Small molecule inhibitors | Venetoclax (Bcl-2 inhibitor) | Directly blocked the wild-type ABCG2 efflux function and inhibited the ATPase activity of ABCG2. | Human embryonic kidney cell line HEK293 overexpressing ABCG2 in vitro. | [231] |
| ABT-737 (BH3-mimetic) | In combination with Fenretinide, synergistically induced cyt-c release, activation of caspases, Bax, t-Bid and Bak. | MDR neuroblastoma cell lines in vitro. | [232] | |
| Nutlin5 (MDM2-p53 antagonist) | Reversed MDR-1-mediated multidrug resistance in a p53-independent manner. | High MDR-1-expressing p53 mutant neuroblastoma cell lines in vitro. | [233] | |
| MI-219 (MDM2 inhibitor) | Sensitized cells to androgen ablation and radiotherapy by inducing DNA damage and apoptosis. | Prostate Cancer Cells in vitro. | [234] | |
| Thiosemicarbazone | Inhibited cell cycle progression at the G1 phase. | MCF7 and MCF7/ADR cells in vitro. | [235] | |
| LY294002 (PI3K inhibitor) | Inhibited the expression of p-Akt and P-gp. | Leukemia cell line K562/DNR and gastric cancer cell line SGC7901/ADR in vitro. | [236] | |
| Metformin (Metabolic inhibitor) | In combination with 2-deoxyglucose selectively enhanced cytotoxicity of Doxorubicin leading to G2/M arrest and apoptosis. | MCF-7/Dox breast cancer cells in vitro. | [237] | |
| BEZ235 (PI3K/mTOR inhibitor) | Caused a dose-dependent decrease in cell viability in combination with Dox, associated with an increase in cleaved PARP. | Ovarian A2780 and pancreatic MiaPaca2 cancer cells in vitro. | [238] | |
| AZ D8055 (mTORC1/2 inhibitor) | Inhibition of mTOR and caspase-3 cleavage in platinum-resistant cells. | Advanced-stage ovarian clear cell carcinoma patient-derived xenograft models. | [239] | |
| Rapamycine (mTOR inhibitor) | Inhibited PI3K/AKT pathway, blocked proliferation, sensitized cells to Tamoxifen and Fulvestrant. | Breast cancer cells resistant to endocrine therapy in vitro. | [240] | |
| YM155 (Survivin inhibitor) | Survivin depletion and p53 activation. | Neuroblastoma cell lines and their sublines with acquired resistance to clinically relevant drugs in vitro. | [241] | |
| Natural agents and derivatives | Wagonin | Promoted TRAIL-induced apoptosis in vitro and downregulated the expression of anti-apoptotic XIAP, cFLIPL, cIAP-1 and cIAP-2. | Non-small cell lung cancer in vivo. | [242] |
| Luteolin | Generated ROS leading to DNA damage and activated the ATR/Chk2/p53 pathway independently of the P-gp efflux pump. | MDR breast cancer cells in vitro. | [243] | |
| Fisetin | Concurrent treatment with chemotherapeutic drugs activated caspases -8 and -3, release of cyt-c and inhibited survival pathways IGF1R and AKT. | Colon cancer cells resistant to both Irinotecan and Oxaliplatin in vitro/in vivo. | [244] | |
| Genistein | Pre-treatment inhibited NFkB activity and led to increased growth inhibition and apoptosis in combination with Cisplatin and Docetaxel. | Prostate and lung cancer cells in vitro/in vivo. | [245] | |
| Resveratrol | - Induced apoptosis by upregulating miR-34c and p53. | - Platinum-resistant colorectal cancer cells, in vivo. | [246] | |
| - Reversed MDR by targeting Survivin and activating caspase-3. | - Non-small cell lung MDR cancer cells, in vivo. | [247] | ||
| Curcumin | - Sensitized cells to capecitabine by inhibiting NFkB, reduced Bcl-2, IAP-1, Survivin, COX-2, MMP-2, ICAM-1, CXCR4 and VEGF | - Colorectal cancer to capecitabine in vivo | [248] | |
| - Difluorinated Curcumin downregulated PTEN inhibitor, miR-21. | - Colorectal cancer cells resistant to 5-FU and oxaliplatin in vitro. | [249] | ||
| - In combination with EGCG led to synergistic effects through activation of the caspase-dependent signaling pathway, and downregulation of Bcl-2 and Survivin. | - Resistant breast cancer cells in vitro. | [250] | ||
| Ellagic acid | In combination with 5-FU increased the Bax/Bcl-2 ratio, caused changes in mitochondrial membrane potential, activated caspase-3 and induced apoptosis. | Colorectal cancer cells in vitro. | [251] | |
| O-methylated coumarin | Inhibited the PI3K/Akt signaling pathway. | Myelogenous leukemia K562/ADM cells in vitro. | [252] | |
| Vitamin E and derivatives | TPGS induced cell cycle arrest and apoptosis selectively in Survivin-overexpressing breast cancer cells. | Breast cancer cells in vitro. | [253] | |
| TME/Immune regulation | Pirfenidone | Induced apoptosis in CAFs at high concentration; at low concentrations induced apoptosis and decreased tumor progression synergistically with Cisplatin. | NSCLC cells in vitro and in vivo. | [254] |
| Combination of anti–CTL-4 plus anti–PD1 therapy | Mediated a switch from expansion of phenotypically exhausted CD8+ T cells to expansion of activated effector CD8+ T cells. | Melanoma patients. | [255] | |
| Combination of EGFR-TKIs and anti-PD-1/PD-L1 antibodies | PD-L1 mediated by EGFR activation could induce the apoptosis of T cells through PD-L1/PD-1 axis in tumor cells. | EGFR-TKIs-resistant NSCLC cells with EGFR mutation in vitro. | [256] | |
| MEDI9447 | Antibody targeting ectoenzyme CD73, increased CD8+ effector cells and activated macrophages. | Mouse syngeneic colorectal tumor growth in vivo. | [257] | |
| Epigenetic drugs | Hydralazine (DNMTi) | In combination with Magnesium Valproate LP improved progression-free survival. | Metastatic Recurrent or Persistent Cervical Cancer patients. | [258] |
| Parthenolide (HDACi) | NFkB and HIF1-α Inhibition. | Brain, breast, colon cancer cell lines in vitro. | [259] | |
| Decitabine (DNMTi) and Panobinostat (HDACi) | In combination with alkylating agent temozolomide showed great improvements in disease stabilization and remission. | Resistant metastatic melanoma patients | [260] | |
| Azacitidine and Valproic acid | In combination with carboplatin demonstrates decreased DR4 methylation and shows modest evidence of antitumor activity | Patients with heavily treated advanced ovarian cancer. | [261] | |
| BRD4i (BRD4 inhibitor) | Induced homologous recombination deficiency and sensitized cells to PARP inhibition. | Multiple tumor lineages regardless of BRCA1/2, TP53, RAS or BRAF mutation status in vitro and in vivo. | [262] |
6. Future Perspectives: The Implication and Therapeutic Exploitation of Epigenetics in MDR
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef]
- Yersal, O.; Barutca, S. Biological subtypes of breast cancer: Prognostic and therapeutic implications. World J. Clin. Oncol. 2014, 5, 412–424. [Google Scholar] [CrossRef] [PubMed]
- Tsaur, I.; Heidegger, I.; Kretschmer, A.; Borgmann, H.; Gandaglia, G.; Briganti, A.; de Visschere, P.; Mathieu, R.; Valerio, M.; van den Bergh, R.; et al. Aggressive variants of prostate cancer—Are we ready to apply specific treatment right now? Cancer Treat. Rev. 2019, 75, 20–26. [Google Scholar] [CrossRef]
- Nguyen, K.S.; Neal, J.W.; Wakelee, H. Review of the current targeted therapies for non-small-cell lung cancer. World J. Clin. Oncol. 2014, 5, 576–587. [Google Scholar] [CrossRef] [PubMed]
- Kozovska, Z.; Gabrisova, V.; Kucerova, L. Colon cancer: Cancer stem cells markers, drug resistance and treatment. Biomed. Pharmacother. Biomed. Pharmacother. 2014, 68, 911–916. [Google Scholar] [CrossRef] [PubMed]
- Assaraf, Y.G.; Brozovic, A.; Goncalves, A.C.; Jurkovicova, D.; Line, A.; Machuqueiro, M.; Saponara, S.; Sarmento-Ribeiro, A.B.; Xavier, C.P.R.; Vasconcelos, M.H. The multi-factorial nature of clinical multidrug resistance in cancer. Drug Resist. Updates Rev. Comment. Antimicrob. Anticancer Chemother. 2019, 46, 100645. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Plati, J.; Bucur, O.; Khosravi-Far, R. Apoptotic cell signaling in cancer progression and therapy. Integr. Biol. 2011, 3, 279–296. [Google Scholar] [CrossRef] [PubMed]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef]
- Savill, J.; Fadok, V. Corpse clearance defines the meaning of cell death. Nature 2000, 407, 784–788. [Google Scholar] [CrossRef]
- Kurosaka, K.; Takahashi, M.; Watanabe, N.; Kobayashi, Y. Silent cleanup of very early apoptotic cells by macrophages. J. Immunol. 2003, 171, 4672–4679. [Google Scholar] [CrossRef] [PubMed]
- Suliman, A.; Lam, A.; Datta, R.; Srivastava, R.K. Intracellular mechanisms of TRAIL: Apoptosis through mitochondrial-dependent and -independent pathways. Oncogene 2001, 20, 2122–2133. [Google Scholar] [CrossRef] [PubMed]
- Ashkenazi, A.; Dixit, V.M. Death receptors: Signaling and modulation. Science 1998, 281, 1305–1308. [Google Scholar] [CrossRef] [PubMed]
- Chicheportiche, Y.; Bourdon, P.R.; Xu, H.; Hsu, Y.M.; Scott, H.; Hession, C.; Garcia, I.; Browning, J.L. TWEAK, a new secreted ligand in the tumor necrosis factor family that weakly induces apoptosis. J. Biol. Chem. 1997, 272, 32401–32410. [Google Scholar] [CrossRef]
- Peter, M.E.; Krammer, P.H. Mechanisms of CD95 (APO-1/Fas)-mediated apoptosis. Curr. Opin. Immunol. 1998, 10, 545–551. [Google Scholar] [CrossRef]
- Rubio-Moscardo, F.; Blesa, D.; Mestre, C.; Siebert, R.; Balasas, T.; Benito, A.; Rosenwald, A.; Climent, J.; Martinez, J.I.; Schilhabel, M.; et al. Characterization of 8p21.3 chromosomal deletions in B-cell lymphoma: TRAIL-R1 and TRAIL-R2 as candidate dosage-dependent tumor suppressor genes. Blood 2005, 106, 3214–3222. [Google Scholar] [CrossRef] [PubMed]
- Guicciardi, M.E.; Gores, G.J. Life and death by death receptors. FASEB J. 2009, 23, 1625–1637. [Google Scholar] [CrossRef]
- Boldin, M.P.; Mett, I.L.; Varfolomeev, E.E.; Chumakov, I.; Shemer-Avni, Y.; Camonis, J.H.; Wallach, D. Self-association of the “death domains” of the p55 tumor necrosis factor (TNF) receptor and Fas/APO1 prompts signaling for TNF and Fas/APO1 effects. J. Biol. Chem. 1995, 270, 387–391. [Google Scholar] [CrossRef]
- Wajant, H. The Fas signaling pathway: More than a paradigm. Science 2002, 296, 1635–1636. [Google Scholar] [CrossRef]
- Hsu, H.; Xiong, J.; Goeddel, D.V. The TNF receptor 1-associated protein TRADD signals cell death and NF-kappa B activation. Cell 1995, 81, 495–504. [Google Scholar] [CrossRef]
- Jin, Z.; El-Deiry, W.S. Overview of cell death signaling pathways. Cancer Biol. Ther. 2005, 4, 139–163. [Google Scholar] [CrossRef] [PubMed]
- Kischkel, F.C.; Hellbardt, S.; Behrmann, I.; Germer, M.; Pawlita, M.; Krammer, P.H.; Peter, M.E. Cytotoxicity-dependent APO-1 (Fas/CD95)-associated proteins form a death-inducing signaling complex (DISC) with the receptor. EMBO J. 1995, 14, 5579–5588. [Google Scholar] [CrossRef]
- Li, J.; Yuan, J. Caspases in apoptosis and beyond. Oncogene 2008, 27, 6194–6206. [Google Scholar] [CrossRef] [PubMed]
- Pop, C.; Salvesen, G.S. Human caspases: Activation, specificity, and regulation. J. Biol. Chem. 2009, 284, 21777–21781. [Google Scholar] [CrossRef]
- Cohen, G.M. Caspases: The executioners of apoptosis. Biochem. J. 1997, 326 Pt 1, 1–16. [Google Scholar] [CrossRef]
- Degterev, A.; Boyce, M.; Yuan, J. A decade of caspases. Oncogene 2003, 22, 8543–8567. [Google Scholar] [CrossRef]
- Saelens, X.; Festjens, N.; Vande Walle, L.; van Gurp, M.; van Loo, G.; Vandenabeele, P. Toxic proteins released from mitochondria in cell death. Oncogene 2004, 23, 2861–2874. [Google Scholar] [CrossRef] [PubMed]
- Chinnaiyan, A.M. The apoptosome: Heart and soul of the cell death machine. Neoplasia 1999, 1, 5–15. [Google Scholar] [CrossRef] [PubMed]
- Hill, M.M.; Adrain, C.; Duriez, P.J.; Creagh, E.M.; Martin, S.J. Analysis of the composition, assembly kinetics and activity of native Apaf-1 apoptosomes. EMBO J. 2004, 23, 2134–2145. [Google Scholar] [CrossRef]
- Van Loo, G.; van Gurp, M.; Depuydt, B.; Srinivasula, S.M.; Rodriguez, I.; Alnemri, E.S.; Gevaert, K.; Vandekerckhove, J.; Declercq, W.; Vandenabeele, P. The serine protease Omi/HtrA2 is released from mitochondria during apoptosis. Omi interacts with caspase-inhibitor XIAP and induces enhanced caspase activity. Cell Death Differ. 2002, 9, 20–26. [Google Scholar] [CrossRef] [PubMed]
- Schimmer, A.D. Inhibitor of apoptosis proteins: Translating basic knowledge into clinical practice. Cancer Res. 2004, 64, 7183–7190. [Google Scholar] [CrossRef] [PubMed]
- Giam, M.; Huang, D.C.; Bouillet, P. BH3-only proteins and their roles in programmed cell death. Oncogene 2008, 27 (Suppl. S1), S128–136. [Google Scholar] [CrossRef] [PubMed]
- Danial, N.N. BCL-2 family proteins: Critical checkpoints of apoptotic cell death. Clin. Cancer Res. 2007, 13, 7254–7263. [Google Scholar] [CrossRef]
- Youle, R.J.; Strasser, A. The BCL-2 protein family: Opposing activities that mediate cell death. Nat. Rev. Mol. Cell Biol. 2008, 9, 47–59. [Google Scholar] [CrossRef] [PubMed]
- Hinds, M.G.; Day, C.L. Regulation of apoptosis: Uncovering the binding determinants. Curr. Opin. Struct. Biol. 2005, 15, 690–699. [Google Scholar] [CrossRef] [PubMed]
- Ghiotto, F.; Fais, F.; Bruno, S. BH3-only proteins: The death-puppeteer’s wires. Cytom. A 2010, 77, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Esposti, M.D. The roles of Bid. Apoptosis 2002, 7, 433–440. [Google Scholar] [CrossRef]
- Igney, F.H.; Krammer, P.H. Death and anti-death: Tumour resistance to apoptosis. Nat. Rev. Cancer 2002, 2, 277–288. [Google Scholar] [CrossRef]
- Cullen, S.P.; Martin, S.J. Caspase activation pathways: Some recent progress. Cell Death Differ. 2009, 16, 935–938. [Google Scholar] [CrossRef]
- Enari, M.; Sakahira, H.; Yokoyama, H.; Okawa, K.; Iwamatsu, A.; Nagata, S. A caspase-activated DNase that degrades DNA during apoptosis, and its inhibitor ICAD. Nature 1998, 391, 43–50. [Google Scholar] [CrossRef]
- Hong, S.J.; Dawson, T.M.; Dawson, V.L. Nuclear and mitochondrial conversations in cell death: PARP-1 and AIF signaling. Trends Pharm. Sci. 2004, 25, 259–264. [Google Scholar] [CrossRef]
- Luthi, A.U.; Martin, S.J. The CASBAH: A searchable database of caspase substrates. Cell Death Differ. 2007, 14, 641–650. [Google Scholar] [CrossRef] [PubMed]
- Khosravi-Far, R.; Esposti, M.D. Death receptor signals to mitochondria. Cancer Biol. Ther. 2004, 3, 1051–1057. [Google Scholar] [CrossRef] [PubMed]
- Housman, G.; Byler, S.; Heerboth, S.; Lapinska, K.; Longacre, M.; Snyder, N.; Sarkar, S. Drug resistance in cancer: An overview. Cancers 2014, 6, 1769–1792. [Google Scholar] [CrossRef] [PubMed]
- Campana, D.; Coustan-Smith, E.; Manabe, A.; Buschle, M.; Raimondi, S.C.; Behm, F.G.; Ashmun, R.; Arico, M.; Biondi, A.; Pui, C.H. Prolonged survival of B-lineage acute lymphoblastic leukemia cells is accompanied by overexpression of bcl-2 protein. Blood 1993, 81, 1025–1031. [Google Scholar] [CrossRef] [PubMed]
- Mason, K.D.; Vandenberg, C.J.; Scott, C.L.; Wei, A.H.; Cory, S.; Huang, D.C.; Roberts, A.W. In vivo efficacy of the Bcl-2 antagonist ABT-737 against aggressive Myc-driven lymphomas. Proc. Natl. Acad. Sci. USA 2008, 105, 17961–17966. [Google Scholar] [CrossRef] [PubMed]
- Othman, R.T.; Kimishi, I.; Bradshaw, T.D.; Storer, L.C.; Korshunov, A.; Pfister, S.M.; Grundy, R.G.; Kerr, I.D.; Coyle, B. Overcoming multiple drug resistance mechanisms in medulloblastoma. Acta Neuropathol. Commun. 2014, 2, 57. [Google Scholar] [CrossRef] [PubMed]
- Beroukhim, R.; Mermel, C.H.; Porter, D.; Wei, G.; Raychaudhuri, S.; Donovan, J.; Barretina, J.; Boehm, J.S.; Dobson, J.; Urashima, M.; et al. The landscape of somatic copy-number alteration across human cancers. Nature 2010, 463, 899–905. [Google Scholar] [CrossRef]
- Cory, S.; Roberts, A.W.; Colman, P.M.; Adams, J.M. Targeting BCL-2-like Proteins to Kill Cancer Cells. Trends Cancer 2016, 2, 443–460. [Google Scholar] [CrossRef] [PubMed]
- McDonnell, T.J.; Deane, N.; Platt, F.M.; Nunez, G.; Jaeger, U.; McKearn, J.P.; Korsmeyer, S.J. bcl-2-immunoglobulin transgenic mice demonstrate extended B cell survival and follicular lymphoproliferation. Cell 1989, 57, 79–88. [Google Scholar] [CrossRef]
- Strasser, A.; Whittingham, S.; Vaux, D.L.; Bath, M.L.; Adams, J.M.; Cory, S.; Harris, A.W. Enforced BCL2 expression in B-lymphoid cells prolongs antibody responses and elicits autoimmune disease. Proc. Natl. Acad. Sci. USA 1991, 88, 8661–8665. [Google Scholar] [CrossRef]
- Sentman, C.L.; Shutter, J.R.; Hockenbery, D.; Kanagawa, O.; Korsmeyer, S.J. bcl-2 inhibits multiple forms of apoptosis but not negative selection in thymocytes. Cell 1991, 67, 879–888. [Google Scholar] [CrossRef]
- Thomas, S.; Quinn, B.A.; Das, S.K.; Dash, R.; Emdad, L.; Dasgupta, S.; Wang, X.Y.; Dent, P.; Reed, J.C.; Pellecchia, M.; et al. Targeting the Bcl-2 family for cancer therapy. Expert Opin. Ther. Targets 2013, 17, 61–75. [Google Scholar] [CrossRef]
- Khoo, K.H.; Verma, C.S.; Lane, D.P. Drugging the p53 pathway: Understanding the route to clinical efficacy. Nat. Rev. Drug Discov. 2014, 13, 217–236. [Google Scholar] [CrossRef]
- Kruiswijk, F.; Labuschagne, C.F.; Vousden, K.H. p53 in survival, death and metabolic health: A lifeguard with a licence to kill. Nat. Rev. Mol. Cell Biol. 2015, 16, 393–405. [Google Scholar] [CrossRef] [PubMed]
- Green, D.R.; Kroemer, G. Cytoplasmic functions of the tumour suppressor p53. Nature 2009, 458, 1127–1130. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.; Ma, D.; Wang, Y. The PROTAC technology in drug development. Cell Biochem. Funct. 2019, 37, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Marino, G.; Niso-Santano, M.; Baehrecke, E.H.; Kroemer, G. Self-consumption: The interplay of autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 2014, 15, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Hientz, K.; Mohr, A.; Bhakta-Guha, D.; Efferth, T. The role of p53 in cancer drug resistance and targeted chemotherapy. Oncotarget 2017, 8, 8921–8946. [Google Scholar] [CrossRef] [PubMed]
- Sanz, G.; Singh, M.; Peuget, S.; Selivanova, G. Inhibition of p53 inhibitors: Progress, challenges and perspectives. J. Mol. Cell Biol. 2019, 11, 586–599. [Google Scholar] [CrossRef]
- Liu, B.; Chen, Y.; St Clair, D.K. ROS and p53: A versatile partnership. Free Radic. Biol. Med. 2008, 44, 1529–1535. [Google Scholar] [CrossRef]
- Mita, A.C.; Mita, M.M.; Nawrocki, S.T.; Giles, F.J. Survivin: Key regulator of mitosis and apoptosis and novel target for cancer therapeutics. Clin. Cancer Res. 2008, 14, 5000–5005. [Google Scholar] [CrossRef]
- Vaux, D.L.; Silke, J. IAPs, RINGs and ubiquitylation. Nat. Rev. Mol. Cell Biol. 2005, 6, 287–297. [Google Scholar] [CrossRef]
- Varfolomeev, E.; Blankenship, J.W.; Wayson, S.M.; Fedorova, A.V.; Kayagaki, N.; Garg, P.; Zobel, K.; Dynek, J.N.; Elliott, L.O.; Wallweber, H.J.; et al. IAP antagonists induce autoubiquitination of c-IAPs, NF-kappaB activation, and TNFalpha-dependent apoptosis. Cell 2007, 131, 669–681. [Google Scholar] [CrossRef]
- Salvesen, G.S.; Duckett, C.S. IAP proteins: Blocking the road to death’s door. Nat. Rev. Mol. Cell Biol. 2002, 3, 401–410. [Google Scholar] [CrossRef]
- Eckelman, B.P.; Salvesen, G.S.; Scott, F.L. Human inhibitor of apoptosis proteins: Why XIAP is the black sheep of the family. EMBO Rep. 2006, 7, 988–994. [Google Scholar] [CrossRef] [PubMed]
- LaCasse, E.C.; Mahoney, D.J.; Cheung, H.H.; Plenchette, S.; Baird, S.; Korneluk, R.G. IAP-targeted therapies for cancer. Oncogene 2008, 27, 6252–6275. [Google Scholar] [CrossRef] [PubMed]
- Hunter, A.M.; LaCasse, E.C.; Korneluk, R.G. The inhibitors of apoptosis (IAPs) as cancer targets. Apoptosis 2007, 12, 1543–1568. [Google Scholar] [CrossRef] [PubMed]
- Peng, X.H.; Karna, P.; O’Regan, R.M.; Liu, X.; Naithani, R.; Moriarty, R.M.; Wood, W.C.; Lee, H.Y.; Yang, L. Down-regulation of inhibitor of apoptosis proteins by deguelin selectively induces apoptosis in breast cancer cells. Mol. Pharm. 2007, 71, 101–111. [Google Scholar] [CrossRef]
- Esposito, I.; Kleeff, J.; Abiatari, I.; Shi, X.; Giese, N.; Bergmann, F.; Roth, W.; Friess, H.; Schirmacher, P. Overexpression of cellular inhibitor of apoptosis protein 2 is an early event in the progression of pancreatic cancer. J. Clin. Pathol. 2007, 60, 885–895. [Google Scholar] [CrossRef]
- Zhang, S.; Ding, F.; Luo, A.; Chen, A.; Yu, Z.; Ren, S.; Liu, Z.; Zhang, L. XIAP is highly expressed in esophageal cancer and its downregulation by RNAi sensitizes esophageal carcinoma cell lines to chemotherapeutics. Cancer Biol. Ther. 2007, 6, 973–980. [Google Scholar] [CrossRef]
- Rathore, R.; McCallum, J.E.; Varghese, E.; Florea, A.M.; Busselberg, D. Overcoming chemotherapy drug resistance by targeting inhibitors of apoptosis proteins (IAPs). Apoptosis 2017, 22, 898–919. [Google Scholar] [CrossRef]
- Shiozaki, A.; Kataoka, K.; Fujimura, M.; Yuki, H.; Sakai, M.; Saito, S. Survivin inhibits apoptosis in cytotrophoblasts. Placenta 2003, 24, 65–76. [Google Scholar] [CrossRef] [PubMed]
- Lehner, R.; Bobak, J.; Kim, N.W.; Shroyer, A.L.; Shroyer, K.R. Localization of telomerase hTERT protein and survivin in placenta: Relation to placental development and hydatidiform mole. Obstet. Gynecol. 2001, 97, 965–970. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, D.S.; Grossman, D.; Plescia, J.; Li, F.; Zhang, H.; Villa, A.; Tognin, S.; Marchisio, P.C.; Altieri, D.C. Regulation of apoptosis at cell division by p34cdc2 phosphorylation of survivin. Proc. Natl. Acad. Sci. USA 2000, 97, 13103–13107. [Google Scholar] [CrossRef] [PubMed]
- Altieri, D.C. Survivin and IAP proteins in cell-death mechanisms. Biochem. J. 2010, 430, 199–205. [Google Scholar] [CrossRef]
- Waligorska-Stachura, J.; Jankowska, A.; Wasko, R.; Liebert, W.; Biczysko, M.; Czarnywojtek, A.; Baszko-Blaszyk, D.; Shimek, V.; Ruchala, M. Survivin--prognostic tumor biomarker in human neoplasms—Review. Ginekol. Pol. 2012, 83, 537–540. [Google Scholar]
- Xu, C.; Yamamoto-Ibusuki, M.; Yamamoto, Y.; Yamamoto, S.; Fujiwara, S.; Murakami, K.; Okumura, Y.; Yamaguchi, L.; Fujiki, Y.; Iwase, H. High survivin mRNA expression is a predictor of poor prognosis in breast cancer: A comparative study at the mRNA and protein level. Breast Cancer 2012. [Google Scholar] [CrossRef]
- Kelly, R.J.; Lopez-Chavez, A.; Citrin, D.; Janik, J.E.; Morris, J.C. Impacting tumor cell-fate by targeting the inhibitor of apoptosis protein survivin. Mol. Cancer 2011, 10, 35. [Google Scholar] [CrossRef]
- Neophytou, C.M.; Constantinou, A.C. C. Survivin: Transcriptional Regulation and Protein Function in Cancer. J. Immunol. 2017. [Google Scholar]
- Richmond, A. Nf-kappa B, chemokine gene transcription and tumour growth. Nat. Rev. Immunol. 2002, 2, 664–674. [Google Scholar] [CrossRef]
- Dohi, T.; Okada, K.; Xia, F.; Wilford, C.E.; Samuel, T.; Welsh, K.; Marusawa, H.; Zou, H.; Armstrong, R.; Matsuzawa, S.; et al. An IAP-IAP complex inhibits apoptosis. J. Biol. Chem. 2004, 279, 34087–34090. [Google Scholar] [CrossRef]
- Song, Z.; Yao, X.; Wu, M. Direct interaction between survivin and Smac/DIABLO is essential for the anti-apoptotic activity of survivin during taxol-induced apoptosis. J. Biol. Chem. 2003, 278, 23130–23140. [Google Scholar] [CrossRef] [PubMed]
- Ceballos-Cancino, G.; Espinosa, M.; Maldonado, V.; Melendez-Zajgla, J. Regulation of mitochondrial Smac/DIABLO-selective release by survivin. Oncogene 2007, 26, 7569–7575. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Aljahdali, I.; Ling, X. Cancer therapeutics using survivin BIRC5 as a target: What can we do after over two decades of study? J. Exp. Clin. Cancer Res. 2019, 38, 368. [Google Scholar] [CrossRef]
- Hay, N. The Akt-mTOR tango and its relevance to cancer. Cancer Cell 2005, 8, 179–183. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wang, T.; Du, J.; Li, Y.; Wang, X.; Zhou, Y.; Yu, X.; Fan, W.; Zhu, Q.; Tong, X.; et al. The Critical Role of PTEN/PI3K/AKT Signaling Pathway in Shikonin-Induced Apoptosis and Proliferation Inhibition of Chronic Myeloid Leukemia. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2018, 47, 981–993. [Google Scholar] [CrossRef]
- Rahmani, F.; Ziaeemehr, A.; Shahidsales, S.; Gharib, M.; Khazaei, M.; Ferns, G.A.; Ryzhikov, M.; Avan, A.; Hassanian, S.M. Role of regulatory miRNAs of the PI3K/AKT/mTOR signaling in the pathogenesis of hepatocellular carcinoma. J. Cell. Physiol. 2020, 235, 4146–4152. [Google Scholar] [CrossRef]
- Soltani, A.; Torki, S.; Ghahfarokhi, M.S.; Jami, M.S.; Ghatrehsamani, M. Targeting the phosphoinositide 3-kinase/AKT pathways by small molecules and natural compounds as a therapeutic approach for breast cancer cells. Mol. Biol. Rep. 2019, 46, 4809–4816. [Google Scholar] [CrossRef] [PubMed]
- Ediriweera, M.K.; Tennekoon, K.H.; Samarakoon, S.R. Role of the PI3K/AKT/mTOR signaling pathway in ovarian cancer: Biological and therapeutic significance. Semin. Cancer Biol. 2019, 59, 147–160. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.M.; Zhang, T.; Liu, Y.B.; Deng, S.H.; Han, R.; Liu, T.; Li, J.; Xu, Y. The PAX6-ZEB2 axis promotes metastasis and cisplatin resistance in non-small cell lung cancer through PI3K/AKT signaling. Cell Death Dis. 2019, 10, 349. [Google Scholar] [CrossRef]
- Rittler, D.; Baranyi, M.; Molnar, E.; Garay, T.; Jalsovszky, I.; Varga, I.K.; Hegedus, L.; Aigner, C.; Tovari, J.; Timar, J.; et al. The Antitumor Effect of Lipophilic Bisphosphonate BPH1222 in Melanoma Models: The Role of the PI3K/Akt Pathway and the Small G Protein Rheb. Int. J. Mol. Sci. 2019, 20, 4917. [Google Scholar] [CrossRef] [PubMed]
- Fayard, E.; Tintignac, L.A.; Baudry, A.; Hemmings, B.A. Protein kinase B/Akt at a glance. J. Cell Sci. 2005, 118, 5675–5678. [Google Scholar] [CrossRef] [PubMed]
- Stambolic, V.; Suzuki, A.; de la Pompa, J.L.; Brothers, G.M.; Mirtsos, C.; Sasaki, T.; Ruland, J.; Penninger, J.M.; Siderovski, D.P.; Mak, T.W. Negative regulation of PKB/Akt-dependent cell survival by the tumor suppressor PTEN. Cell 1998, 95, 29–39. [Google Scholar] [CrossRef]
- Vazquez, F.; Ramaswamy, S.; Nakamura, N.; Sellers, W.R. Phosphorylation of the PTEN tail regulates protein stability and function. Mol. Cell Biol. 2000, 20, 5010–5018. [Google Scholar] [CrossRef]
- Stephens, L.; Anderson, K.; Stokoe, D.; Erdjument-Bromage, H.; Painter, G.F.; Holmes, A.B.; Gaffney, P.R.; Reese, C.B.; McCormick, F.; Tempst, P.; et al. Protein kinase B kinases that mediate phosphatidylinositol 3,4,5-trisphosphate-dependent activation of protein kinase B. Science 1998, 279, 710–714. [Google Scholar] [CrossRef]
- Sarbassov, D.D.; Guertin, D.A.; Ali, S.M.; Sabatini, D.M. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 2005, 307, 1098–1101. [Google Scholar] [CrossRef]
- Jacinto, E.; Facchinetti, V.; Liu, D.; Soto, N.; Wei, S.; Jung, S.Y.; Huang, Q.; Qin, J.; Su, B. SIN1/MIP1 maintains rictor-mTOR complex integrity and regulates Akt phosphorylation and substrate specificity. Cell 2006, 127, 125–137. [Google Scholar] [CrossRef] [PubMed]
- Alessi, D.R.; Andjelkovic, M.; Caudwell, B.; Cron, P.; Morrice, N.; Cohen, P.; Hemmings, B.A. Mechanism of activation of protein kinase B by insulin and IGF-1. EMBO J. 1996, 15, 6541–6551. [Google Scholar] [CrossRef]
- Carnero, A.; Blanco-Aparicio, C.; Renner, O.; Link, W.; Leal, J.F. The PTEN/PI3K/AKT signalling pathway in cancer, therapeutic implications. Curr. Cancer Drug Targets 2008, 8, 187–198. [Google Scholar] [CrossRef]
- Sale, E.M.; Sale, G.J. Protein kinase B: Signalling roles and therapeutic targeting. Cell Mol. Life Sci. 2008, 65, 113–127. [Google Scholar] [CrossRef]
- Asanuma, H.; Torigoe, T.; Kamiguchi, K.; Hirohashi, Y.; Ohmura, T.; Hirata, K.; Sato, M.; Sato, N. Survivin expression is regulated by coexpression of human epidermal growth factor receptor 2 and epidermal growth factor receptor via phosphatidylinositol 3-kinase/AKT signaling pathway in breast cancer cells. Cancer Res. 2005, 65, 11018–11025. [Google Scholar] [CrossRef] [PubMed]
- Crawford, A.; Nahta, R. Targeting Bcl-2 in Herceptin-Resistant Breast Cancer Cell Lines. Curr. Pharm. Pers. Med. 2011, 9, 184–190. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Du, K.; Montminy, M. CREB is a regulatory target for the protein kinase Akt/PKB. J. Biol. Chem. 1998, 273, 32377–32379. [Google Scholar] [CrossRef] [PubMed]
- Song, G.; Ouyang, G.; Bao, S. The activation of Akt/PKB signaling pathway and cell survival. J. Cell Mol. Med. 2005, 9, 59–71. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Chen, Y.; Liu, G.; Li, C.; Song, Y.; Cao, Z.; Li, W.; Hu, J.; Lu, C.; Liu, Y. PI3K/AKT pathway as a key link modulates the multidrug resistance of cancers. Cell Death Dis. 2020, 11, 797. [Google Scholar] [CrossRef]
- Welsh, G.I.; Wilson, C.; Proud, C.G. GSK3: A SHAGGY frog story. Trends Cell Biol. 1996, 6, 274–279. [Google Scholar] [CrossRef]
- Srivastava, A.K.; Pandey, S.K. Potential mechanism(s) involved in the regulation of glycogen synthesis by insulin. Mol. Cell Biochem. 1998, 182, 135–141. [Google Scholar] [CrossRef]
- Diehl, J.A.; Cheng, M.; Roussel, M.F.; Sherr, C.J. Glycogen synthase kinase-3beta regulates cyclin D1 proteolysis and subcellular localization. Genes Dev. 1998, 12, 3499–3511. [Google Scholar] [CrossRef]
- Somanath, P.R.; Vijai, J.; Kichina, J.V.; Byzova, T.; Kandel, E.S. The role of PAK-1 in activation of MAP kinase cascade and oncogenic transformation by Akt. Oncogene 2009, 28, 2365–2369. [Google Scholar] [CrossRef]
- Manning, B.D.; Cantley, L.C. AKT/PKB signaling: Navigating downstream. Cell 2007, 129, 1261–1274. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Tang, N.; Hadden, T.J.; Rishi, A.K. Akt, FoxO and regulation of apoptosis. Biochim. Biophys. Acta 2011, 1813, 1978–1986. [Google Scholar] [CrossRef] [PubMed]
- Farhan, M.; Silva, M.; Xingan, X.; Huang, Y.; Zheng, W. Role of FOXO Transcription Factors in Cancer Metabolism and Angiogenesis. Cells 2020, 9, 1586. [Google Scholar] [CrossRef]
- Slamon, D.J.; Clark, G.M.; Wong, S.G.; Levin, W.J.; Ullrich, A.; McGuire, W.L. Human breast cancer: Correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science 1987, 235, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.P.; Hu, M.C.; Miller, S.A.; Yu, Z.; Xia, W.; Lin, S.Y.; Hung, M.C. HER-2/neu blocks tumor necrosis factor-induced apoptosis via the Akt/NF-kappaB pathway. J. Biol. Chem. 2000, 275, 8027–8031. [Google Scholar] [CrossRef]
- Salomon, D.S.; Brandt, R.; Ciardiello, F.; Normanno, N. Epidermal growth factor-related peptides and their receptors in human malignancies. Crit. Rev. Oncol. Hematol. 1995, 19, 183–232. [Google Scholar] [CrossRef]
- Shayesteh, L.; Lu, Y.; Kuo, W.L.; Baldocchi, R.; Godfrey, T.; Collins, C.; Pinkel, D.; Powell, B.; Mills, G.B.; Gray, J.W. PIK3CA is implicated as an oncogene in ovarian cancer. Nat. Genet. 1999, 21, 99–102. [Google Scholar] [CrossRef]
- Byun, D.S.; Cho, K.; Ryu, B.K.; Lee, M.G.; Park, J.I.; Chae, K.S.; Kim, H.J.; Chi, S.G. Frequent monoallelic deletion of PTEN and its reciprocal associatioin with PIK3CA amplification in gastric carcinoma. Int. J. Cancer 2003, 104, 318–327. [Google Scholar] [CrossRef]
- Samuels, Y.; Diaz, L.A., Jr.; Schmidt-Kittler, O.; Cummins, J.M.; Delong, L.; Cheong, I.; Rago, C.; Huso, D.L.; Lengauer, C.; Kinzler, K.W.; et al. Mutant PIK3CA promotes cell growth and invasion of human cancer cells. Cancer Cell 2005, 7, 561–573. [Google Scholar] [CrossRef]
- Stal, O.; Perez-Tenorio, G.; Akerberg, L.; Olsson, B.; Nordenskjold, B.; Skoog, L.; Rutqvist, L.E. Akt kinases in breast cancer and the results of adjuvant therapy. Breast Cancer Res. 2003, 5, R37–R44. [Google Scholar] [CrossRef]
- Roy, H.K.; Olusola, B.F.; Clemens, D.L.; Karolski, W.J.; Ratashak, A.; Lynch, H.T.; Smyrk, T.C. AKT proto-oncogene overexpression is an early event during sporadic colon carcinogenesis. Carcinogenesis 2002, 23, 201–205. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Zhu, J.; Du, W.; Ning, W.; Zhang, Y.; Zeng, Y.; Liu, Z.; Huang, J.A. AKT2 drives cancer progression and is negatively modulated by miR-124 in human lung adenocarcinoma. Respir. Res. 2020, 21, 227. [Google Scholar] [CrossRef] [PubMed]
- Rychahou, P.G.; Kang, J.; Gulhati, P.; Doan, H.Q.; Chen, L.A.; Xiao, S.Y.; Chung, D.H.; Evers, B.M. Akt2 overexpression plays a critical role in the establishment of colorectal cancer metastasis. Proc. Natl. Acad. Sci. USA 2008, 105, 20315–20320. [Google Scholar] [CrossRef]
- Arboleda, M.J.; Lyons, J.F.; Kabbinavar, F.F.; Bray, M.R.; Snow, B.E.; Ayala, R.; Danino, M.; Karlan, B.Y.; Slamon, D.J. Overexpression of AKT2/protein kinase Bbeta leads to up-regulation of beta1 integrins, increased invasion, and metastasis of human breast and ovarian cancer cells. Cancer Res. 2003, 63, 196–206. [Google Scholar] [PubMed]
- Ozes, O.N.; Akca, H.; Mayo, L.D.; Gustin, J.A.; Maehama, T.; Dixon, J.E.; Donner, D.B. A phosphatidylinositol 3-kinase/Akt/mTOR pathway mediates and PTEN antagonizes tumor necrosis factor inhibition of insulin signaling through insulin receptor substrate-1. Proc. Natl. Acad. Sci. USA 2001, 98, 4640–4645. [Google Scholar] [CrossRef] [PubMed]
- Osaki, M.; Oshimura, M.; Ito, H. PI3K-Akt pathway: Its functions and alterations in human cancer. Apoptosis 2004, 9, 667–676. [Google Scholar] [CrossRef] [PubMed]
- Neophytou, C.M.; Panagi, M.; Stylianopoulos, T.; Papageorgis, P. The Role of Tumor Microenvironment in Cancer Metastasis: Molecular Mechanisms and Therapeutic Opportunities. Cancers 2021, 13, 2053. [Google Scholar] [CrossRef] [PubMed]
- Neophytou, C.M.; Pierides, C.; Christodoulou, M.I.; Costeas, P.; Kyriakou, T.C.; Papageorgis, P. The Role of Tumor-Associated Myeloid Cells in Modulating Cancer Therapy. Front. Oncol. 2020, 10, 899. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Lostao, L.; Anel, A.; Pardo, J. How Do Cytotoxic Lymphocytes Kill Cancer Cells? Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2015, 21, 5047–5056. [Google Scholar] [CrossRef]
- Anel, A.; Buferne, M.; Boyer, C.; Schmitt-Verhulst, A.M.; Golstein, P. T cell receptor-induced Fas ligand expression in cytotoxic T lymphocyte clones is blocked by protein tyrosine kinase inhibitors and cyclosporin A. Eur. J. Immunol. 1994, 24, 2469–2476. [Google Scholar] [CrossRef] [PubMed]
- Bossi, G.; Griffiths, G.M. CTL secretory lysosomes: Biogenesis and secretion of a harmful organelle. Semin. Immunol. 2005, 17, 87–94. [Google Scholar] [CrossRef]
- De Saint Basile, G.; Menasche, G.; Fischer, A. Molecular mechanisms of biogenesis and exocytosis of cytotoxic granules. Nat. Rev. Immunol. 2010, 10, 568–579. [Google Scholar] [CrossRef] [PubMed]
- Tecchio, C.; Huber, V.; Scapini, P.; Calzetti, F.; Margotto, D.; Todeschini, G.; Pilla, L.; Martinelli, G.; Pizzolo, G.; Rivoltini, L.; et al. IFNalpha-stimulated neutrophils and monocytes release a soluble form of TNF-related apoptosis-inducing ligand (TRAIL/Apo-2 ligand) displaying apoptotic activity on leukemic cells. Blood 2004, 103, 3837–3844. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.T.; Kim, J.H.; Kwon, S.J.; Stein, M.N.; Hong, J.H.; Nagaya, N.; Billakanti, S.; Kim, M.M.; Kim, W.J.; Kim, I.Y. Dihydrotestosterone Increases Cytotoxic Activity of Macrophages on Prostate Cancer Cells via TRAIL. Endocrinology 2019, 160, 2049–2060. [Google Scholar] [CrossRef]
- Freeman, Z.T.; Nirschl, T.R.; Hovelson, D.H.; Johnston, R.J.; Engelhardt, J.J.; Selby, M.J.; Kochel, C.M.; Lan, R.Y.; Zhai, J.; Ghasemzadeh, A.; et al. A conserved intratumoral regulatory T cell signature identifies 4-1BB as a pan-cancer target. J. Clin. Investig. 2020, 130, 1405–1416. [Google Scholar] [CrossRef]
- Hu, B.S.; Tang, T.; Jia, J.L.; Xie, B.C.; Wu, T.L.; Sheng, Y.Y.; Xue, Y.Z.; Tang, H.M. CD137 agonist induces gastric cancer cell apoptosis by enhancing the functions of CD8(+) T cells via NF-kappaB signaling. Cancer Cell Int. 2020, 20, 513. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.; Zhong, Y.; Li, X.; Li, Y.; Li, X.; Cao, J.; Fan, H.; Yuan, Y.; Ji, Z.; Qiao, B.; et al. ILs-3, 6 and 11 increase, but ILs-10 and 24 decrease stemness of human prostate cancer cells in vitro. Oncotarget 2015, 6, 42687–42703. [Google Scholar] [CrossRef]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef]
- Bejarano, L.; Jordao, M.J.C.; Joyce, J.A. Therapeutic Targeting of the Tumor Microenvironment. Cancer Discov. 2021, 11, 933–959. [Google Scholar] [CrossRef] [PubMed]
- Khalaf, K.; Hana, D.; Chou, J.T.; Singh, C.; Mackiewicz, A.; Kaczmarek, M. Aspects of the Tumor Microenvironment Involved in Immune Resistance and Drug Resistance. Front. Immunol. 2021, 12, 656364. [Google Scholar] [CrossRef]
- Jain, R.K.; Martin, J.D.; Stylianopoulos, T. The role of mechanical forces in tumor growth and therapy. Annu. Rev. Biomed. Eng. 2014, 16, 321–346. [Google Scholar] [CrossRef]
- Muz, B.; de la Puente, P.; Azab, F.; Azab, A.K. The role of hypoxia in cancer progression, angiogenesis, metastasis, and resistance to therapy. Hypoxia 2015, 3, 83–92. [Google Scholar] [CrossRef]
- Gerweck, L.E.; Vijayappa, S.; Kozin, S. Tumor pH controls the in vivo efficacy of weak acid and base chemotherapeutics. Mol. Cancer Ther. 2006, 5, 1275–1279. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, H.; Sakai, R. Direct Interaction between Carcinoma Cells and Cancer Associated Fibroblasts for the Regulation of Cancer Invasion. Cancers 2015, 7, 2054–2062. [Google Scholar] [CrossRef]
- Joshi, R.S.; Kanugula, S.S.; Sudhir, S.; Pereira, M.P.; Jain, S.; Aghi, M.K. The Role of Cancer-Associated Fibroblasts in Tumor Progression. Cancers 2021, 13, 1399. [Google Scholar] [CrossRef]
- Augsten, M. Cancer-associated fibroblasts as another polarized cell type of the tumor microenvironment. Front. Oncol. 2014, 4, 62. [Google Scholar] [CrossRef]
- Guo, L.; Li, B.; Yang, J.; Shen, J.; Ji, J.; Miao, M. Fibroblastderived exosomal microRNA369 potentiates migration and invasion of lung squamous cell carcinoma cells via NF1mediated MAPK signaling pathway. Int. J. Mol. Med. 2020, 46, 595–608. [Google Scholar] [CrossRef] [PubMed]
- Valcz, G.; Sipos, F.; Tulassay, Z.; Molnar, B.; Yagi, Y. Importance of carcinoma-associated fibroblast-derived proteins in clinical oncology. J. Clin. Pathol. 2014, 67, 1026–1031. [Google Scholar] [CrossRef]
- Paraiso, K.H.; Smalley, K.S. Fibroblast-mediated drug resistance in cancer. Biochem. Pharmacol. 2013, 85, 1033–1041. [Google Scholar] [CrossRef] [PubMed]
- Choe, C.; Shin, Y.S.; Kim, S.H.; Jeon, M.J.; Choi, S.J.; Lee, J.; Kim, J. Tumor-stromal interactions with direct cell contacts enhance motility of non-small cell lung cancer cells through the hedgehog signaling pathway. Anticancer Res. 2013, 33, 3715–3723. [Google Scholar]
- Ren, J.; Ding, L.; Zhang, D.; Shi, G.; Xu, Q.; Shen, S.; Wang, Y.; Wang, T.; Hou, Y. Carcinoma-associated fibroblasts promote the stemness and chemoresistance of colorectal cancer by transferring exosomal lncRNA H19. Theranostics 2018, 8, 3932–3948. [Google Scholar] [CrossRef]
- Chen, W.J.; Ho, C.C.; Chang, Y.L.; Chen, H.Y.; Lin, C.A.; Ling, T.Y.; Yu, S.L.; Yuan, S.S.; Chen, Y.J.; Lin, C.Y.; et al. Cancer-associated fibroblasts regulate the plasticity of lung cancer stemness via paracrine signalling. Nat. Commun. 2014, 5, 3472. [Google Scholar] [CrossRef] [PubMed]
- Lacina, L.; Plzak, J.; Kodet, O.; Szabo, P.; Chovanec, M.; Dvorankova, B.; Smetana, K., Jr. Cancer Microenvironment: What Can We Learn from the Stem Cell Niche. Int. J. Mol. Sci. 2015, 16, 24094–24110. [Google Scholar] [CrossRef]
- Xi, C.; Wang, J.; Sun, H.; Zhang, X.; Kang, H. Loss of microRNA-30e induced by extracellular vesicles from cancer-associated fibroblasts promotes breast cancer progression by binding to CTHRC1. Exp. Mol. Pathol. 2021, 118, 104586. [Google Scholar] [CrossRef]
- Eichelmann, A.K.; Matuszcak, C.; Hummel, R.; Haier, J. Role of miRNAs in cell signaling of cancer associated fibroblasts. Int. J. Biochem. Cell Biol. 2018, 101, 94–102. [Google Scholar] [CrossRef]
- Boomgarden, A.C.; Sheehan, C.; D’Souza-Schorey, C. Extracellular Vesicles in the Tumor Microenvironment: Various Implications in Tumor Progression. Adv. Exp. Med. Biol. 2020, 1259, 155–170. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wei, H.; Wang, J.; Li, L.; Chen, A.; Li, Z. MicroRNA-181d-5p-Containing Exosomes Derived from CAFs Promote EMT by Regulating CDX2/HOXA5 in Breast Cancer. Mol. Ther. Nucleic Acids 2020, 19, 654–667. [Google Scholar] [CrossRef]
- Beermann, J.; Piccoli, M.T.; Viereck, J.; Thum, T. Non-coding RNAs in Development and Disease: Background, Mechanisms, and Therapeutic Approaches. Physiol. Rev. 2016, 96, 1297–1325. [Google Scholar] [CrossRef] [PubMed]
- Di Martino, M.T.; Riillo, C.; Scionti, F.; Grillone, K.; Polera, N.; Caracciolo, D.; Arbitrio, M.; Tagliaferri, P.; Tassone, P. miRNAs and lncRNAs as Novel Therapeutic Targets to Improve Cancer Immunotherapy. Cancers 2021, 13, 1587. [Google Scholar] [CrossRef] [PubMed]
- Cui, H.; Onyango, P.; Brandenburg, S.; Wu, Y.; Hsieh, C.L.; Feinberg, A.P. Loss of imprinting in colorectal cancer linked to hypomethylation of H19 and IGF2. Cancer Res. 2002, 62, 6442–6446. [Google Scholar]
- Berteaux, N.; Lottin, S.; Monte, D.; Pinte, S.; Quatannens, B.; Coll, J.; Hondermarck, H.; Curgy, J.J.; Dugimont, T.; Adriaenssens, E. H19 mRNA-like noncoding RNA promotes breast cancer cell proliferation through positive control by E2F1. J. Biol. Chem. 2005, 280, 29625–29636. [Google Scholar] [CrossRef]
- Lecerf, C.; Peperstraete, E.; Le Bourhis, X.; Adriaenssens, E. Propagation and Maintenance of Cancer Stem Cells: A Major Influence of the Long Non-Coding RNA H19. Cells 2020, 9, 2613. [Google Scholar] [CrossRef] [PubMed]
- Vermeulen, L.; De Sousa, E.M.F.; van der Heijden, M.; Cameron, K.; de Jong, J.H.; Borovski, T.; Tuynman, J.B.; Todaro, M.; Merz, C.; Rodermond, H.; et al. Wnt activity defines colon cancer stem cells and is regulated by the microenvironment. Nat. Cell Biol. 2010, 12, 468–476. [Google Scholar] [CrossRef]
- Gao, Z.; Wang, Q.; Ji, M.; Guo, X.; Li, L.; Su, X. Exosomal lncRNA UCA1 modulates cervical cancer stem cell self-renewal and differentiation through microRNA-122-5p/SOX2 axis. J. Transl. Med. 2021, 19, 229. [Google Scholar] [CrossRef]
- Lv, X.; Lian, Y.; Liu, Z.; Xiao, J.; Zhang, D.; Yin, X. Exosomal long non-coding RNA LINC00662 promotes non-small cell lung cancer progression by miR-320d/E2F1 axis. Aging 2021, 13, 6010–6024. [Google Scholar] [CrossRef]
- Liu, K.; Gao, L.; Ma, X.; Huang, J.J.; Chen, J.; Zeng, L.; Ashby, C.R., Jr.; Zou, C.; Chen, Z.S. Long non-coding RNAs regulate drug resistance in cancer. Mol. Cancer 2020, 19, 54. [Google Scholar] [CrossRef] [PubMed]
- Ying, L.; Zhu, Z.; Xu, Z.; He, T.; Li, E.; Guo, Z.; Liu, F.; Jiang, C.; Wang, Q. Cancer Associated Fibroblast-Derived Hepatocyte Growth Factor Inhibits the Paclitaxel-Induced Apoptosis of Lung Cancer A549 Cells by Up-Regulating the PI3K/Akt and GRP78 Signaling on a Microfluidic Platform. PLoS ONE 2015, 10, e0129593. [Google Scholar] [CrossRef]
- Chen, C.; Hou, J.; Yu, S.; Li, W.; Wang, X.; Sun, H.; Qin, T.; Claret, F.X.; Guo, H.; Liu, Z. Role of cancer-associated fibroblasts in the resistance to antitumor therapy, and their potential therapeutic mechanisms in non-small cell lung cancer. Oncol. Lett. 2021, 21, 413. [Google Scholar] [CrossRef] [PubMed]
- Tao, L.; Huang, G.; Wang, R.; Pan, Y.; He, Z.; Chu, X.; Song, H.; Chen, L. Cancer-associated fibroblasts treated with cisplatin facilitates chemoresistance of lung adenocarcinoma through IL-11/IL-11R/STAT3 signaling pathway. Sci. Rep. 2016, 6, 38408. [Google Scholar] [CrossRef]
- Bockhorn, J.; Dalton, R.; Nwachukwu, C.; Huang, S.; Prat, A.; Yee, K.; Chang, Y.F.; Huo, D.; Wen, Y.; Swanson, K.E.; et al. MicroRNA-30c inhibits human breast tumour chemotherapy resistance by regulating TWF1 and IL-11. Nat. Commun. 2013, 4, 1393. [Google Scholar] [CrossRef]
- Lokau, J.; Agthe, M.; Garbers, C. Generation of Soluble Interleukin-11 and Interleukin-6 Receptors: A Crucial Function for Proteases during Inflammation. Mediat. Inflamm. 2016, 2016, 1785021. [Google Scholar] [CrossRef] [PubMed]
- Delbridge, A.R.; Grabow, S.; Strasser, A.; Vaux, D.L. Thirty years of BCL-2: Translating cell death discoveries into novel cancer therapies. Nat. Rev. Cancer 2016, 16, 99–109. [Google Scholar] [CrossRef]
- Real, P.J.; Sierra, A.; De Juan, A.; Segovia, J.C.; Lopez-Vega, J.M.; Fernandez-Luna, J.L. Resistance to chemotherapy via Stat3-dependent overexpression of Bcl-2 in metastatic breast cancer cells. Oncogene 2002, 21, 7611–7618. [Google Scholar] [CrossRef] [PubMed]
- Borsellino, N.; Belldegrun, A.; Bonavida, B. Endogenous interleukin 6 is a resistance factor for cis-diamminedichloroplatinum and etoposide-mediated cytotoxicity of human prostate carcinoma cell lines. Cancer Res. 1995, 55, 4633–4639. [Google Scholar]
- Garcia-Tunon, I.; Ricote, M.; Ruiz, A.; Fraile, B.; Paniagua, R.; Royuela, M. IL-6, its receptors and its relationship with bcl-2 and bax proteins in infiltrating and in situ human breast carcinoma. Histopathology 2005, 47, 82–89. [Google Scholar] [CrossRef] [PubMed]
- Gritsko, T.; Williams, A.; Turkson, J.; Kaneko, S.; Bowman, T.; Huang, M.; Nam, S.; Eweis, I.; Diaz, N.; Sullivan, D.; et al. Persistent activation of stat3 signaling induces survivin gene expression and confers resistance to apoptosis in human breast cancer cells. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2006, 12, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Kumari, N.; Dwarakanath, B.S.; Das, A.; Bhatt, A.N. Role of interleukin-6 in cancer progression and therapeutic resistance. Tumour Biol. J. Int. Soc. Oncodev. Biol. Med. 2016, 37, 11553–11572. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.P.; Yang, D.C.; Elliott, R.L.; Head, J.F. Down-regulation of expression of interleukin-6 and its receptor results in growth inhibition of MCF-7 breast cancer cells. Anticancer Res. 2011, 31, 2899–2906. [Google Scholar] [PubMed]
- So, K.A.; Min, K.J.; Hong, J.H.; Lee, J.K. Interleukin-6 expression by interactions between gynecologic cancer cells and human mesenchymal stem cells promotes epithelial-mesenchymal transition. Int. J. Oncol. 2015, 47, 1451–1459. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, H.; Hirata, Y.; Nakagawa, H.; Sakamoto, K.; Hayakawa, Y.; Takahashi, R.; Nakata, W.; Sakitani, K.; Serizawa, T.; Hikiba, Y.; et al. Interleukin-6 mediates epithelial-stromal interactions and promotes gastric tumorigenesis. PLoS ONE 2013, 8, e60914. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Tao, P.; Zhou, Q.; Li, J.; Yu, Z.; Wang, X.; Li, J.; Li, C.; Yan, M.; Zhu, Z.; et al. IL-6 secreted by cancer-associated fibroblasts promotes epithelial-mesenchymal transition and metastasis of gastric cancer via JAK2/STAT3 signaling pathway. Oncotarget 2017, 8, 20741–20750. [Google Scholar] [CrossRef] [PubMed]
- Ham, I.H.; Oh, H.J.; Jin, H.; Bae, C.A.; Jeon, S.M.; Choi, K.S.; Son, S.Y.; Han, S.U.; Brekken, R.A.; Lee, D.; et al. Targeting interleukin-6 as a strategy to overcome stroma-induced resistance to chemotherapy in gastric cancer. Mol. Cancer 2019, 18, 68. [Google Scholar] [CrossRef]
- Ma, Y.; Zhu, J.; Chen, S.; Li, T.; Ma, J.; Guo, S.; Hu, J.; Yue, T.; Zhang, J.; Wang, P.; et al. Activated gastric cancer-associated fibroblasts contribute to the malignant phenotype and 5-FU resistance via paracrine action in gastric cancer. Cancer Cell Int. 2018, 18, 104. [Google Scholar] [CrossRef] [PubMed]
- Hwang, R.F.; Moore, T.; Arumugam, T.; Ramachandran, V.; Amos, K.D.; Rivera, A.; Ji, B.; Evans, D.B.; Logsdon, C.D. Cancer-associated stromal fibroblasts promote pancreatic tumor progression. Cancer Res. 2008, 68, 918–926. [Google Scholar] [CrossRef]
- Muerkoster, S.; Wegehenkel, K.; Arlt, A.; Witt, M.; Sipos, B.; Kruse, M.L.; Sebens, T.; Kloppel, G.; Kalthoff, H.; Folsch, U.R.; et al. Tumor stroma interactions induce chemoresistance in pancreatic ductal carcinoma cells involving increased secretion and paracrine effects of nitric oxide and interleukin-1beta. Cancer Res. 2004, 64, 1331–1337. [Google Scholar] [CrossRef] [PubMed]
- Setrerrahmane, S.; Xu, H. Tumor-related interleukins: Old validated targets for new anti-cancer drug development. Mol. Cancer 2017, 16, 153. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, S.C.; Fahrmann, J.F.; Celiktas, M.; Aguilar, M.; Marini, K.D.; Jolly, M.K.; Katayama, H.; Wang, H.; Murage, E.N.; Dennison, J.B.; et al. MCAM Mediates Chemoresistance in Small-Cell Lung Cancer via the PI3K/AKT/SOX2 Signaling Pathway. Cancer Res. 2017, 77, 4414–4425. [Google Scholar] [CrossRef] [PubMed]
- Jung, K.; Wang, P.; Gupta, N.; Gopal, K.; Wu, F.; Ye, X.; Alshareef, A.; Bigras, G.; McMullen, T.P.; Abdulkarim, B.S.; et al. Profiling gene promoter occupancy of Sox2 in two phenotypically distinct breast cancer cell subsets using chromatin immunoprecipitation and genome-wide promoter microarrays. Breast Cancer Res. 2014, 16, 470. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, D.; Senbanjo, L.; Majumdar, S.; Franklin, R.B.; Chellaiah, M.A. Androgen receptor expression reduces stemness characteristics of prostate cancer cells (PC3) by repression of CD44 and SOX2. J. Cell. Biochem. 2018. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Teng, C.; Huang, W.; Peng, J.; Wang, C. SOX2 Promotes the Epithelial to Mesenchymal Transition of Esophageal Squamous Cells by Modulating Slug Expression through the Activation of STAT3/HIF-alpha Signaling. Int. J. Mol. Sci. 2015, 16, 21643–21657. [Google Scholar] [CrossRef] [PubMed]
- Piva, M.; Domenici, G.; Iriondo, O.; Rabano, M.; Simoes, B.M.; Comaills, V.; Barredo, I.; Lopez-Ruiz, J.A.; Zabalza, I.; Kypta, R.; et al. Sox2 promotes tamoxifen resistance in breast cancer cells. EMBO Mol. Med. 2014, 6, 66–79. [Google Scholar] [CrossRef] [PubMed]
- Finicelli, M.; Benedetti, G.; Squillaro, T.; Pistilli, B.; Marcellusi, A.; Mariani, P.; Santinelli, A.; Latini, L.; Galderisi, U.; Giordano, A. Expression of stemness genes in primary breast cancer tissues: The role of SOX2 as a prognostic marker for detection of early recurrence. Oncotarget 2014, 5, 9678–9688. [Google Scholar] [CrossRef] [PubMed]
- Dittmer, A.; Dittmer, J. Carcinoma-Associated Fibroblasts Promote Growth of Sox2-Expressing Breast Cancer Cells. Cancers 2020, 12, 3435. [Google Scholar] [CrossRef] [PubMed]
- Shekhar, M.P.; Santner, S.; Carolin, K.A.; Tait, L. Direct involvement of breast tumor fibroblasts in the modulation of tamoxifen sensitivity. Am. J. Pathol. 2007, 170, 1546–1560. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Sun, Y. Targeting oncogenic SOX2 in human cancer cells: Therapeutic application. Protein Cell 2020, 11, 82–84. [Google Scholar] [CrossRef]
- Deying, W.; Feng, G.; Shumei, L.; Hui, Z.; Ming, L.; Hongqing, W. CAF-derived HGF promotes cell proliferation and drug resistance by up-regulating the c-Met/PI3K/Akt and GRP78 signalling in ovarian cancer cells. Biosci. Rep. 2017, 37. [Google Scholar] [CrossRef] [PubMed]
- Moran-Jones, K. The Therapeutic Potential of Targeting the HGF/cMET Axis in Ovarian Cancer. Mol. Diagn. Ther. 2016, 20, 199–212. [Google Scholar] [CrossRef] [PubMed]
- Otte, J.M.; Schmitz, F.; Kiehne, K.; Stechele, H.U.; Banasiewicz, T.; Krokowicz, P.; Nakamura, T.; Folsch, U.R.; Herzig, K. Functional expression of HGF and its receptor in human colorectal cancer. Digestion 2000, 61, 237–246. [Google Scholar] [CrossRef] [PubMed]
- Yamada, T.; Matsumoto, K.; Wang, W.; Li, Q.; Nishioka, Y.; Sekido, Y.; Sone, S.; Yano, S. Hepatocyte growth factor reduces susceptibility to an irreversible epidermal growth factor receptor inhibitor in EGFR-T790M mutant lung cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2010, 16, 174–183. [Google Scholar] [CrossRef] [PubMed]
- Meng, Q.; Xia, C.; Fang, J.; Rojanasakul, Y.; Jiang, B.H. Role of PI3K and AKT specific isoforms in ovarian cancer cell migration, invasion and proliferation through the p70S6K1 pathway. Cell. Signal. 2006, 18, 2262–2271. [Google Scholar] [CrossRef] [PubMed]
- Yao, R.; Cooper, G.M. Requirement for phosphatidylinositol-3 kinase in the prevention of apoptosis by nerve growth factor. Science 1995, 267, 2003–2006. [Google Scholar] [CrossRef]
- Shin, B.K.; Wang, H.; Yim, A.M.; Le Naour, F.; Brichory, F.; Jang, J.H.; Zhao, R.; Puravs, E.; Tra, J.; Michael, C.W.; et al. Global profiling of the cell surface proteome of cancer cells uncovers an abundance of proteins with chaperone function. J. Biol. Chem. 2003, 278, 7607–7616. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Lee, A.S. Glucose regulated proteins in cancer progression, drug resistance and immunotherapy. Cancer Biol. Ther. 2006, 5, 741–744. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Jiang, Y.; Jia, Z.; Li, Q.; Gong, W.; Wang, L.; Wei, D.; Yao, J.; Fang, S.; Xie, K. Association of elevated GRP78 expression with increased lymph node metastasis and poor prognosis in patients with gastric cancer. Clin. Exp. Metastasis 2006, 23, 401–410. [Google Scholar] [CrossRef]
- Davidson, D.J.; Haskell, C.; Majest, S.; Kherzai, A.; Egan, D.A.; Walter, K.A.; Schneider, A.; Gubbins, E.F.; Solomon, L.; Chen, Z.; et al. Kringle 5 of human plasminogen induces apoptosis of endothelial and tumor cells through surface-expressed glucose-regulated protein 78. Cancer Res. 2005, 65, 4663–4672. [Google Scholar] [CrossRef] [PubMed]
- Arnaudeau, S.; Arboit, P.; Bischof, P.; Shin-ya, K.; Tomida, A.; Tsuruo, T.; Irion, O.; Cohen, M. Glucose-regulated protein 78: A new partner of p53 in trophoblast. Proteomics 2009, 9, 5316–5327. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Li, J.; Lee, A.S. GRP78/BiP inhibits endoplasmic reticulum BIK and protects human breast cancer cells against estrogen starvation-induced apoptosis. Cancer Res. 2007, 67, 3734–3740. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Zhang, Y.; Fu, Y.; Chan, L.; Lee, A.S. Novel mechanism of anti-apoptotic function of 78-kDa glucose-regulated protein (GRP78): Endocrine resistance factor in breast cancer, through release of B-cell lymphoma 2 (BCL-2) from BCL-2-interacting killer (BIK). J. Biol. Chem. 2011, 286, 25687–25696. [Google Scholar] [CrossRef]
- Hao, Y.; Zhang, L.; He, J.; Guo, Z.; Ying, L.; Xu, Z.; Zhang, J.; Lu, J.; Wang, Q. Functional investigation of NCI-H460-inducible myofibroblasts on the chemoresistance to VP-16 with a microfluidic 3D co-culture device. PLoS ONE 2013, 8, e61754. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Sun, C.; Li, N.; Shan, W.; Lu, H.; Guo, L.; Guo, E.; Xia, M.; Weng, D.; Meng, L.; et al. Cisplatin-induced CCL5 secretion from CAFs promotes cisplatin-resistance in ovarian cancer via regulation of the STAT3 and PI3K/Akt signaling pathways. Int. J. Oncol. 2016, 48, 2087–2097. [Google Scholar] [CrossRef] [PubMed]
- Mehlen, P.; Delloye-Bourgeois, C.; Chedotal, A. Novel roles for Slits and netrins: Axon guidance cues as anticancer targets? Nat. Rev. Cancer 2011, 11, 188–197. [Google Scholar] [CrossRef]
- Kefeli, U.; Ucuncu Kefeli, A.; Cabuk, D.; Isik, U.; Sonkaya, A.; Acikgoz, O.; Ozden, E.; Uygun, K. Netrin-1 in cancer: Potential biomarker and therapeutic target? Tumour Biol. 2017, 39, 1010428317698388. [Google Scholar] [CrossRef] [PubMed]
- Sung, P.J.; Rama, N.; Imbach, J.; Fiore, S.; Ducarouge, B.; Neves, D.; Chen, H.W.; Bernard, D.; Yang, P.C.; Bernet, A.; et al. Cancer-Associated Fibroblasts Produce Netrin-1 to Control Cancer Cell Plasticity. Cancer Res. 2019, 79, 3651–3661. [Google Scholar] [CrossRef]
- Adams, C.; Cazzanelli, G.; Rasul, S.; Hitchinson, B.; Hu, Y.; Coombes, R.C.; Raguz, S.; Yague, E. Apoptosis inhibitor TRIAP1 is a novel effector of drug resistance. Oncol. Rep. 2015, 34, 415–422. [Google Scholar] [CrossRef] [PubMed]
- Park, W.R.; Nakamura, Y. p53CSV, a novel p53-inducible gene involved in the p53-dependent cell-survival pathway. Cancer Res. 2005, 65, 1197–1206. [Google Scholar] [CrossRef] [PubMed]
- Ketteler, J.; Panic, A.; Reis, H.; Wittka, A.; Maier, P.; Herskind, C.; Yague, E.; Jendrossek, V.; Klein, D. Progression-Related Loss of Stromal Caveolin 1 Levels Mediates Radiation Resistance in Prostate Carcinoma via the Apoptosis Inhibitor TRIAP1. J. Clin. Med. 2019, 8, 348. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Tang, X.; He, Q.; Yang, X.; Ren, X.; Wen, X.; Zhang, J.; Wang, Y.; Liu, N.; Ma, J. Overexpression of Mitochondria Mediator Gene TRIAP1 by miR-320b Loss Is Associated with Progression in Nasopharyngeal Carcinoma. PLoS Genet. 2016, 12, e1006183. [Google Scholar] [CrossRef] [PubMed]
- Kunou, S.; Shimada, K.; Takai, M.; Sakamoto, A.; Aoki, T.; Hikita, T.; Kagaya, Y.; Iwamoto, E.; Sanada, M.; Shimada, S.; et al. Exosomes secreted from cancer-associated fibroblasts elicit anti-pyrimidine drug resistance through modulation of its transporter in malignant lymphoma. Oncogene 2021, 40, 3989–4003. [Google Scholar] [CrossRef]
- Staiger, A.M.; Duppel, J.; Dengler, M.A.; van der Kuip, H.; Vohringer, M.C.; Aulitzky, W.E.; Rosenwald, A.; Ott, G.; Horn, H. An analysis of the role of follicular lymphoma-associated fibroblasts to promote tumor cell viability following drug-induced apoptosis. Leuk. Lymphoma 2017, 58, 1922–1930. [Google Scholar] [CrossRef] [PubMed]
- Brien, G.; Trescol-Biemont, M.C.; Bonnefoy-Berard, N. Downregulation of Bfl-1 protein expression sensitizes malignant B cells to apoptosis. Oncogene 2007, 26, 5828–5832. [Google Scholar] [CrossRef]
- Scarfo, L.; Ghia, P. Reprogramming cell death: BCL2 family inhibition in hematological malignancies. Immunol. Lett. 2013, 155, 36–39. [Google Scholar] [CrossRef]
- Zhang, H.; Okamoto, M.; Panzhinskiy, E.; Zawada, W.M.; Das, M. PKCdelta/midkine pathway drives hypoxia-induced proliferation and differentiation of human lung epithelial cells. Am. J. Physiol. Cell Physiol. 2014, 306, C648–C658. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wang, Q.L.; Wang, H.; Zhao, S.L.; Huang, Y.H.; Hou, Y.Y. Over-expressed and truncated midkines promote proliferation of BGC823 cells in vitro and tumor growth in vivo. World J. Gastroenterol. 2008, 14, 1858–1865. [Google Scholar] [CrossRef]
- Huang, Y.; Hoque, M.O.; Wu, F.; Trink, B.; Sidransky, D.; Ratovitski, E.A. Midkine induces epithelial-mesenchymal transition through Notch2/Jak2-Stat3 signaling in human keratinocytes. Cell Cycle 2008, 7, 1613–1622. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.; Nie, Y.; Lv, M.; He, L.; Wang, T.; Hou, Y. ERbeta-mediated estradiol enhances epithelial mesenchymal transition of lung adenocarcinoma through increasing transcription of midkine. Mol. Endocrinol. 2012, 26, 1304–1315. [Google Scholar] [CrossRef]
- Zhang, D.; Ding, L.; Li, Y.; Ren, J.; Shi, G.; Wang, Y.; Zhao, S.; Ni, Y.; Hou, Y. Midkine derived from cancer-associated fibroblasts promotes cisplatin-resistance via up-regulation of the expression of lncRNA ANRIL in tumour cells. Sci. Rep. 2017, 7, 16231. [Google Scholar] [CrossRef] [PubMed]
- Lorente, M.; Torres, S.; Salazar, M.; Carracedo, A.; Hernandez-Tiedra, S.; Rodriguez-Fornes, F.; Garcia-Taboada, E.; Melendez, B.; Mollejo, M.; Campos-Martin, Y.; et al. Stimulation of the midkine/ALK axis renders glioma cells resistant to cannabinoid antitumoral action. Cell Death Differ. 2011, 18, 959–973. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, S.M.; Cunningham, C.C.; Golenkov, A.K.; Turkina, A.G.; Novick, S.C.; Rai, K.R. Phase I to II multicenter study of oblimersen sodium, a Bcl-2 antisense oligonucleotide, in patients with advanced chronic lymphocytic leukemia. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2005, 23, 7697–7702. [Google Scholar] [CrossRef]
- O’Brien, S.; Moore, J.O.; Boyd, T.E.; Larratt, L.M.; Skotnicki, A.; Koziner, B.; Chanan-Khan, A.A.; Seymour, J.F.; Bociek, R.G.; Pavletic, S.; et al. Randomized phase III trial of fludarabine plus cyclophosphamide with or without oblimersen sodium (Bcl-2 antisense) in patients with relapsed or refractory chronic lymphocytic leukemia. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2007, 25, 1114–1120. [Google Scholar] [CrossRef]
- Soderquist, R.S.; Eastman, A. BCL2 Inhibitors as Anticancer Drugs: A Plethora of Misleading BH3 Mimetics. Mol. Cancer Ther. 2016, 15, 2011–2017. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.Q.; Li, J.Y.; Teng, Q.X.; Lei, Z.N.; Ji, N.; Cui, Q.; Zeng, L.; Pan, Y.; Yang, D.H.; Chen, Z.S. Venetoclax, a BCL-2 Inhibitor, Enhances the Efficacy of Chemotherapeutic Agents in Wild-Type ABCG2-Overexpression-Mediated MDR Cancer Cells. Cancers 2020, 12, 466. [Google Scholar] [CrossRef]
- Fang, H.; Harned, T.M.; Kalous, O.; Maldonado, V.; DeClerck, Y.A.; Reynolds, C.P. Synergistic activity of fenretinide and the Bcl-2 family protein inhibitor ABT-737 against human neuroblastoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2011, 17, 7093–7104. [Google Scholar] [CrossRef]
- Chen, L.; Zhao, Y.; Halliday, G.C.; Berry, P.; Rousseau, R.F.; Middleton, S.A.; Nichols, G.L.; Del Bello, F.; Piergentili, A.; Newell, D.R.; et al. Structurally diverse MDM2-p53 antagonists act as modulators of MDR-1 function in neuroblastoma. Br. J. Cancer 2014, 111, 716–725. [Google Scholar] [CrossRef][Green Version]
- Feng, F.Y.; Zhang, Y.; Kothari, V.; Evans, J.R.; Jackson, W.C.; Chen, W.; Johnson, S.B.; Luczak, C.; Wang, S.; Hamstra, D.A. MDM2 Inhibition Sensitizes Prostate Cancer Cells to Androgen Ablation and Radiotherapy in a p53-Dependent Manner. Neoplasia 2016, 18, 213–222. [Google Scholar] [CrossRef]
- Bai, J.; Wang, R.-H.; Qiao, Y.; Wang, A.; Fang, C.-J. Schiff base derived from thiosemicarbazone and anthracene showed high potential in overcoming multidrug resistance in vitro with low drug resistance index. Drug Des. Devel. Ther. 2017, 11, 2227–2237. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Qu, X.J.; Liu, Y.P.; Yang, X.H.; Hou, K.Z.; Teng, Y.E.; Zhang, J.D. Reversal effect of PI3-K inhibitor LY294002 on P-glycoprotein-mediated multidrug resistance of human leukemia cell line K562/DNR and gastric cancer cell line SGC7901/ADR. Ai Zheng Aizheng Chin. J. Cancer 2009, 28, 97–99. [Google Scholar]
- Xue, C.; Wang, C.; Sun, Y.; Meng, Q.; Liu, Z.; Huo, X.; Sun, P.; Sun, H.; Ma, X.; Ma, X.; et al. Targeting P-glycoprotein function, p53 and energy metabolism: Combination of metformin and 2-deoxyglucose reverses the multidrug resistance of MCF-7/Dox cells to doxorubicin. Oncotarget 2017, 8, 8622–8632. [Google Scholar] [CrossRef]
- Durrant, D.E.; Das, A.; Dyer, S.; Kukreja, R.C. A dual PI3 kinase/mTOR inhibitor BEZ235 reverses doxorubicin resistance in ABCB1 overexpressing ovarian and pancreatic cancer cell lines. Biochim. Biophys. Acta Gen. Subj. 2020, 1864, 129556. [Google Scholar] [CrossRef]
- Caumanns, J.J.; Berns, K.; Wisman, G.B.A.; Fehrmann, R.S.N.; Tomar, T.; Klip, H.; Meersma, G.J.; Hijmans, E.M.; Gennissen, A.M.C.; Duiker, E.W.; et al. Integrative Kinome Profiling Identifies mTORC1/2 Inhibition as Treatment Strategy in Ovarian Clear Cell Carcinoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2018, 24, 3928–3940. [Google Scholar] [CrossRef]
- Ghayad, S.E.; Bieche, I.; Vendrell, J.A.; Keime, C.; Lidereau, R.; Dumontet, C.; Cohen, P.A. mTOR inhibition reverses acquired endocrine therapy resistance of breast cancer cells at the cell proliferation and gene-expression levels. Cancer Sci. 2008, 99, 1992–2003. [Google Scholar] [CrossRef]
- Voges, Y.; Michaelis, M.; Rothweiler, F.; Schaller, T.; Schneider, C.; Politt, K.; Mernberger, M.; Nist, A.; Stiewe, T.; Wass, M.N.; et al. Effects of YM155 on survivin levels and viability in neuroblastoma cells with acquired drug resistance. Cell Death Dis. 2016, 7, e2410. [Google Scholar] [CrossRef]
- Yang, L.; Wang, Q.; Li, D.; Zhou, Y.; Zheng, X.; Sun, H.; Yan, J.; Zhang, L.; Lin, Y.; Wang, X. Wogonin enhances antitumor activity of tumor necrosis factor-related apoptosis-inducing ligand in vivo through ROS-mediated downregulation of cFLIPL and IAP proteins. Apoptosis 2013, 18, 618–626. [Google Scholar] [CrossRef] [PubMed]
- Rao, P.S.; Satelli, A.; Moridani, M.; Jenkins, M.; Rao, U.S. Luteolin induces apoptosis in multidrug resistant cancer cells without affecting the drug transporter function: Involvement of cell line-specific apoptotic mechanisms. Int. J. Cancer 2012, 130, 2703–2714. [Google Scholar] [CrossRef] [PubMed]
- Jeng, L.B.; Kumar Velmurugan, B.; Chen, M.C.; Hsu, H.H.; Ho, T.J.; Day, C.H.; Lin, Y.M.; Padma, V.V.; Tu, C.C.; Huang, C.Y. Fisetin mediated apoptotic cell death in parental and Oxaliplatin/irinotecan resistant colorectal cancer cells in vitro and in vivo. J. Cell. Physiol. 2018, 233, 7134–7142. [Google Scholar] [CrossRef]
- Li, Y.; Ahmed, F.; Ali, S.; Philip, P.A.; Kucuk, O.; Sarkar, F.H. Inactivation of nuclear factor kappaB by soy isoflavone genistein contributes to increased apoptosis induced by chemotherapeutic agents in human cancer cells. Cancer Res. 2005, 65, 6934–6942. [Google Scholar] [CrossRef]
- Yang, S.; Li, W.; Sun, H.; Wu, B.; Ji, F.; Sun, T.; Chang, H.; Shen, P.; Wang, Y.; Zhou, D. Resveratrol elicits anti-colorectal cancer effect by activating miR-34c-KITLG in vitro and in vivo. BMC Cancer 2015, 15, 969. [Google Scholar] [CrossRef]
- Zhao, W.; Bao, P.; Qi, H.; You, H. Resveratrol down-regulates survivin and induces apoptosis in human multidrug-resistant SPC-A-1/CDDP cells. Oncol. Rep. 2010, 23, 279–286. [Google Scholar] [CrossRef]
- Kunnumakkara, A.B.; Diagaradjane, P.; Anand, P.; Harikumar, K.B.; Deorukhkar, A.; Gelovani, J.; Guha, S.; Krishnan, S.; Aggarwal, B.B. Curcumin sensitizes human colorectal cancer to capecitabine by modulation of cyclin D1, COX-2, MMP-9, VEGF and CXCR4 expression in an orthotopic mouse model. Int. J. Cancer 2009, 125, 2187–2197. [Google Scholar] [CrossRef]
- Roy, S.; Yu, Y.; Padhye, S.B.; Sarkar, F.H.; Majumdar, A.P. Difluorinated-curcumin (CDF) restores PTEN expression in colon cancer cells by down-regulating miR-21. PLoS ONE 2013, 8, e68543. [Google Scholar] [CrossRef]
- Wang, S.; Chen, R.; Zhong, Z.; Shi, Z.; Chen, M.; Wang, Y. Epigallocatechin-3-gallate potentiates the effect of curcumin in inducing growth inhibition and apoptosis of resistant breast cancer cells. Am. J. Chin. Med. 2014, 42, 1279–1300. [Google Scholar] [CrossRef]
- Kao, T.Y.; Chung, Y.C.; Hou, Y.C.; Tsai, Y.W.; Chen, C.H.; Chang, H.P.; Chou, J.L.; Hsu, C.P. Effects of ellagic acid on chemosensitivity to 5-fluorouracil in colorectal carcinoma cells. Anticancer Res. 2012, 32, 4413–4418. [Google Scholar]
- Wang, H.; Jia, X.H.; Chen, J.R.; Wang, J.Y.; Li, Y.J. Osthole shows the potential to overcome P-glycoproteinmediated multidrug resistance in human myelogenous leukemia K562/ADM cells by inhibiting the PI3K/Akt signaling pathway. Oncol. Rep. 2016, 35, 3659–3668. [Google Scholar] [CrossRef]
- Neophytou, C.M.; Constantinou, C.; Papageorgis, P.; Constantinou, A.I. D-alpha-tocopheryl polyethylene glycol succinate (TPGS) induces cell cycle arrest and apoptosis selectively in Survivin-overexpressing breast cancer cells. Biochem. Pharm. 2014, 89, 31–42. [Google Scholar] [CrossRef]
- Mediavilla-Varela, M.; Boateng, K.; Noyes, D.; Antonia, S.J. The anti-fibrotic agent pirfenidone synergizes with cisplatin in killing tumor cells and cancer-associated fibroblasts. BMC Cancer 2016, 16, 176. [Google Scholar] [CrossRef]
- Wei, S.C.; Anang, N.A.S.; Sharma, R.; Andrews, M.C.; Reuben, A.; Levine, J.H.; Cogdill, A.P.; Mancuso, J.J.; Wargo, J.A.; Pe’er, D.; et al. Combination anti-CTLA-4 plus anti-PD-1 checkpoint blockade utilizes cellular mechanisms partially distinct from monotherapies. Proc. Natl. Acad. Sci. USA 2019, 116, 22699–22709. [Google Scholar] [CrossRef]
- Chen, N.; Fang, W.; Zhan, J.; Hong, S.; Tang, Y.; Kang, S.; Zhang, Y.; He, X.; Zhou, T.; Qin, T.; et al. Upregulation of PD-L1 by EGFR Activation Mediates the Immune Escape in EGFR-Driven NSCLC: Implication for Optional Immune Targeted Therapy for NSCLC Patients with EGFR Mutation. J. Thorac. Oncol. Off. Publ. Int. Assoc. Study Lung Cancer 2015, 10, 910–923. [Google Scholar] [CrossRef]
- Hay, C.M.; Sult, E.; Huang, Q.; Mulgrew, K.; Fuhrmann, S.R.; McGlinchey, K.A.; Hammond, S.A.; Rothstein, R.; Rios-Doria, J.; Poon, E.; et al. Targeting CD73 in the tumor microenvironment with MEDI9447. Oncoimmunology 2016, 5, e1208875. [Google Scholar] [CrossRef]
- Soto, H.; Sanchez, K.; Escobar, J.Y.; Constanzo, A.; Fernandez, Z.; Melendez, C. Cost-Effectiveness Analysis of Hydralazine and Magnesium Valproate LP Associated with Treatment for Adult Patients with Metastatic Recurrent or Persistent Cervical Cancer in Mexico. Value Health J. Int. Soc. Pharm. Outcomes Res. 2014, 17, A639. [Google Scholar] [CrossRef]
- Dawood, M.; Ooko, E.; Efferth, T. Collateral Sensitivity of Parthenolide via NF-kappaB and HIF-alpha Inhibition and Epigenetic Changes in Drug-Resistant Cancer Cell Lines. Front. Pharm. 2019, 10, 542. [Google Scholar] [CrossRef]
- Xia, C.; Leon-Ferre, R.; Laux, D.; Deutsch, J.; Smith, B.J.; Frees, M.; Milhem, M. Treatment of resistant metastatic melanoma using sequential epigenetic therapy (decitabine and panobinostat) combined with chemotherapy (temozolomide). Cancer Chemother. Pharmacol. 2014, 74, 691–697. [Google Scholar] [CrossRef]
- Falchook, G.S.; Fu, S.; Naing, A.; Hong, D.S.; Hu, W.; Moulder, S.; Wheler, J.J.; Sood, A.K.; Bustinza-Linares, E.; Parkhurst, K.L.; et al. Methylation and histone deacetylase inhibition in combination with platinum treatment in patients with advanced malignancies. Invest. New Drugs 2013, 31, 1192–1200. [Google Scholar] [CrossRef]
- Sun, C.; Yin, J.; Fang, Y.; Chen, J.; Jeong, K.J.; Chen, X.; Vellano, C.P.; Ju, Z.; Zhao, W.; Zhang, D.; et al. BRD4 Inhibition Is Synthetic Lethal with PARP Inhibitors through the Induction of Homologous Recombination Deficiency. Cancer Cell 2018, 33, 401–416.e408. [Google Scholar] [CrossRef]
- Shariati, M.; Meric-Bernstam, F. Targeting AKT for cancer therapy. Expert Opin. Investig. Drugs 2019, 28, 977–988. [Google Scholar] [CrossRef]
- Nitulescu, G.M.; Margina, D.; Juzenas, P.; Peng, Q.; Olaru, O.T.; Saloustros, E.; Fenga, C.; Spandidos, D.; Libra, M.; Tsatsakis, A.M. Akt inhibitors in cancer treatment: The long journey from drug discovery to clinical use (Review). Int. J. Oncol. 2016, 48, 869–885. [Google Scholar] [CrossRef]
- Yang, H.; Villani, R.M.; Wang, H.; Simpson, M.J.; Roberts, M.S.; Tang, M.; Liang, X. The role of cellular reactive oxygen species in cancer chemotherapy. J. Exp. Clin. Cancer Res. 2018, 37, 266. [Google Scholar] [CrossRef] [PubMed]
- Minassian, L.M.; Cotechini, T.; Huitema, E.; Graham, C.H. Hypoxia-Induced Resistance to Chemotherapy in Cancer. Adv. Exp. Med. Biol. 2019, 1136, 123–139. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Xu, J.; Min, T.; Huang, W. Up-regulation of P-glycoprotein expression by catalase via JNK activation in HepG2 cells. Redox Rep. Commun. Free Radic. Res. 2006, 11, 173–178. [Google Scholar] [CrossRef]
- Kotecha, R.; Takami, A.; Espinoza, J.L. Dietary phytochemicals and cancer chemoprevention: A review of the clinical evidence. Oncotarget 2016, 7, 52517–52529. [Google Scholar] [CrossRef]
- Neophytou, C.M.; Constantinou, G.Y. Pro-apoptotic properties of chemopreventive agents. In Natural Products for Cancer Chemoprevention: Single Compounds and Combinations; Pezzuto, J.M., Vang, O., Eds.; Springer International Publishing: Berlin/Heidelberg, Germany, 2020; pp. 517–559. [Google Scholar]
- Hu-Lieskovan, S.; Malouf, G.G.; Jacobs, I.; Chou, J.; Liu, L.; Johnson, M.L. Addressing resistance to immune checkpoint inhibitor therapy: An urgent unmet need. Future Oncol. 2021, 17, 1401–1439. [Google Scholar] [CrossRef]
- Wilting, R.H.; Dannenberg, J.H. Epigenetic mechanisms in tumorigenesis, tumor cell heterogeneity and drug resistance. Drug Resist. Updates Rev. Comment. Antimicrob. Anticancer Chemother. 2012, 15, 21–38. [Google Scholar] [CrossRef]
- Talukdar, S.; Bhoopathi, P.; Emdad, L.; Das, S.; Sarkar, D.; Fisher, P.B. Dormancy and cancer stem cells: An enigma for cancer therapeutic targeting. Adv. Cancer Res. 2019, 141, 43–84. [Google Scholar] [CrossRef]
- Shipitsin, M.; Campbell, L.L.; Argani, P.; Weremowicz, S.; Bloushtain-Qimron, N.; Yao, J.; Nikolskaya, T.; Serebryiskaya, T.; Beroukhim, R.; Hu, M.; et al. Molecular definition of breast tumor heterogeneity. Cancer Cell 2007, 11, 259–273. [Google Scholar] [CrossRef] [PubMed]
- Issa, M.E.; Takhsha, F.S.; Chirumamilla, C.S.; Perez-Novo, C.; Vanden Berghe, W.; Cuendet, M. Epigenetic strategies to reverse drug resistance in heterogeneous multiple myeloma. Clin. Epigenet. 2017, 9, 17. [Google Scholar] [CrossRef]
- Lu, Y.; Chan, Y.T.; Tan, H.Y.; Li, S.; Wang, N.; Feng, Y. Epigenetic regulation in human cancer: The potential role of epi-drug in cancer therapy. Mol. Cancer 2020, 19, 79. [Google Scholar] [CrossRef] [PubMed]
- Ganesan, A.; Arimondo, P.B.; Rots, M.G.; Jeronimo, C.; Berdasco, M. The timeline of epigenetic drug discovery: From reality to dreams. Clin. Epigenet. 2019, 11, 174. [Google Scholar] [CrossRef]
- Guo, M.; Peng, Y.; Gao, A.; Du, C.; Herman, J.G. Epigenetic heterogeneity in cancer. Biomark. Res. 2019, 7, 23. [Google Scholar] [CrossRef]
- Dawson, M.A. The cancer epigenome: Concepts, challenges, and therapeutic opportunities. Science 2017, 355, 1147–1152. [Google Scholar] [CrossRef]
- Mohammad, H.P.; Barbash, O.; Creasy, C.L. Targeting epigenetic modifications in cancer therapy: Erasing the roadmap to cancer. Nat. Med. 2019, 25, 403–418. [Google Scholar] [CrossRef]
- Guo, L.; Lee, Y.T.; Zhou, Y.; Huang, Y. Targeting epigenetic regulatory machinery to overcome cancer therapy resistance. Semin. Cancer Biol. 2021. [Google Scholar] [CrossRef]
- Brown, R.; Curry, E.; Magnani, L.; Wilhelm-Benartzi, C.S.; Borley, J. Poised epigenetic states and acquired drug resistance in cancer. Nat. Rev. Cancer 2014, 14, 747–753. [Google Scholar] [CrossRef]
- Bukowski, K.; Kciuk, M.; Kontek, R. Mechanisms of Multidrug Resistance in Cancer Chemotherapy. Int. J. Mol. Sci. 2020, 21, 3233. [Google Scholar] [CrossRef] [PubMed]
- Juergens, R.A.; Wrangle, J.; Vendetti, F.P.; Murphy, S.C.; Zhao, M.; Coleman, B.; Sebree, R.; Rodgers, K.; Hooker, C.M.; Franco, N.; et al. Combination epigenetic therapy has efficacy in patients with refractory advanced non-small cell lung cancer. Cancer Discov. 2011, 1, 598–607. [Google Scholar] [CrossRef] [PubMed]
- Fuller, M.; Klein, M.; Schmidt, E.; Rohde, C.; Gollner, S.; Schulze, I.; Qianli, J.; Berdel, W.E.; Edemir, B.; Muller-Tidow, C.; et al. 5-azacytidine enhances efficacy of multiple chemotherapy drugs in AML and lung cancer with modulation of CpG methylation. Int. J. Oncol. 2015, 46, 1192–1204. [Google Scholar] [CrossRef]
- Hammerlindl, H.; Schaider, H. Tumor cell-intrinsic phenotypic plasticity facilitates adaptive cellular reprogramming driving acquired drug resistance. J. Cell Commun. Signal. 2018, 12, 133–141. [Google Scholar] [CrossRef]
- Guha, M. HDAC inhibitors still need a home run, despite recent approval. Nat. Rev. Drug Discov. 2015, 14, 225–226. [Google Scholar] [CrossRef]
- Imamura, Y.; Mukohara, T.; Shimono, Y.; Funakoshi, Y.; Chayahara, N.; Toyoda, M.; Kiyota, N.; Takao, S.; Kono, S.; Nakatsura, T.; et al. Comparison of 2D- and 3D-culture models as drug-testing platforms in breast cancer. Oncol. Rep. 2015, 33, 1837–1843. [Google Scholar] [CrossRef] [PubMed]
- Gengenbacher, N.; Singhal, M.; Augustin, H.G. Preclinical mouse solid tumour models: Status quo, challenges and perspectives. Nat. Rev. Cancer 2017, 17, 751–765. [Google Scholar] [CrossRef]
- Ramirez, M.; Rajaram, S.; Steininger, R.J.; Osipchuk, D.; Roth, M.A.; Morinishi, L.S.; Evans, L.; Ji, W.; Hsu, C.H.; Thurley, K.; et al. Diverse drug-resistance mechanisms can emerge from drug-tolerant cancer persister cells. Nat. Commun. 2016, 7, 10690. [Google Scholar] [CrossRef]
- Boumahdi, S.; de Sauvage, F.J. The great escape: Tumour cell plasticity in resistance to targeted therapy. Nat. Rev. Drug Discov. 2020, 19, 39–56. [Google Scholar] [CrossRef]
- Morel, D.; Jeffery, D.; Aspeslagh, S.; Almouzni, G.; Postel-Vinay, S. Combining epigenetic drugs with other therapies for solid tumours—Past lessons and future promise. Nat. Rev. Clin. Oncol. 2020, 17, 91–107. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Neophytou, C.M.; Trougakos, I.P.; Erin, N.; Papageorgis, P. Apoptosis Deregulation and the Development of Cancer Multi-Drug Resistance. Cancers 2021, 13, 4363. https://doi.org/10.3390/cancers13174363
Neophytou CM, Trougakos IP, Erin N, Papageorgis P. Apoptosis Deregulation and the Development of Cancer Multi-Drug Resistance. Cancers. 2021; 13(17):4363. https://doi.org/10.3390/cancers13174363
Chicago/Turabian StyleNeophytou, Christiana M., Ioannis P. Trougakos, Nuray Erin, and Panagiotis Papageorgis. 2021. "Apoptosis Deregulation and the Development of Cancer Multi-Drug Resistance" Cancers 13, no. 17: 4363. https://doi.org/10.3390/cancers13174363
APA StyleNeophytou, C. M., Trougakos, I. P., Erin, N., & Papageorgis, P. (2021). Apoptosis Deregulation and the Development of Cancer Multi-Drug Resistance. Cancers, 13(17), 4363. https://doi.org/10.3390/cancers13174363