
Skin Cancers and the Contribution of Rho GTPase Signaling Networks to Their Progression


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
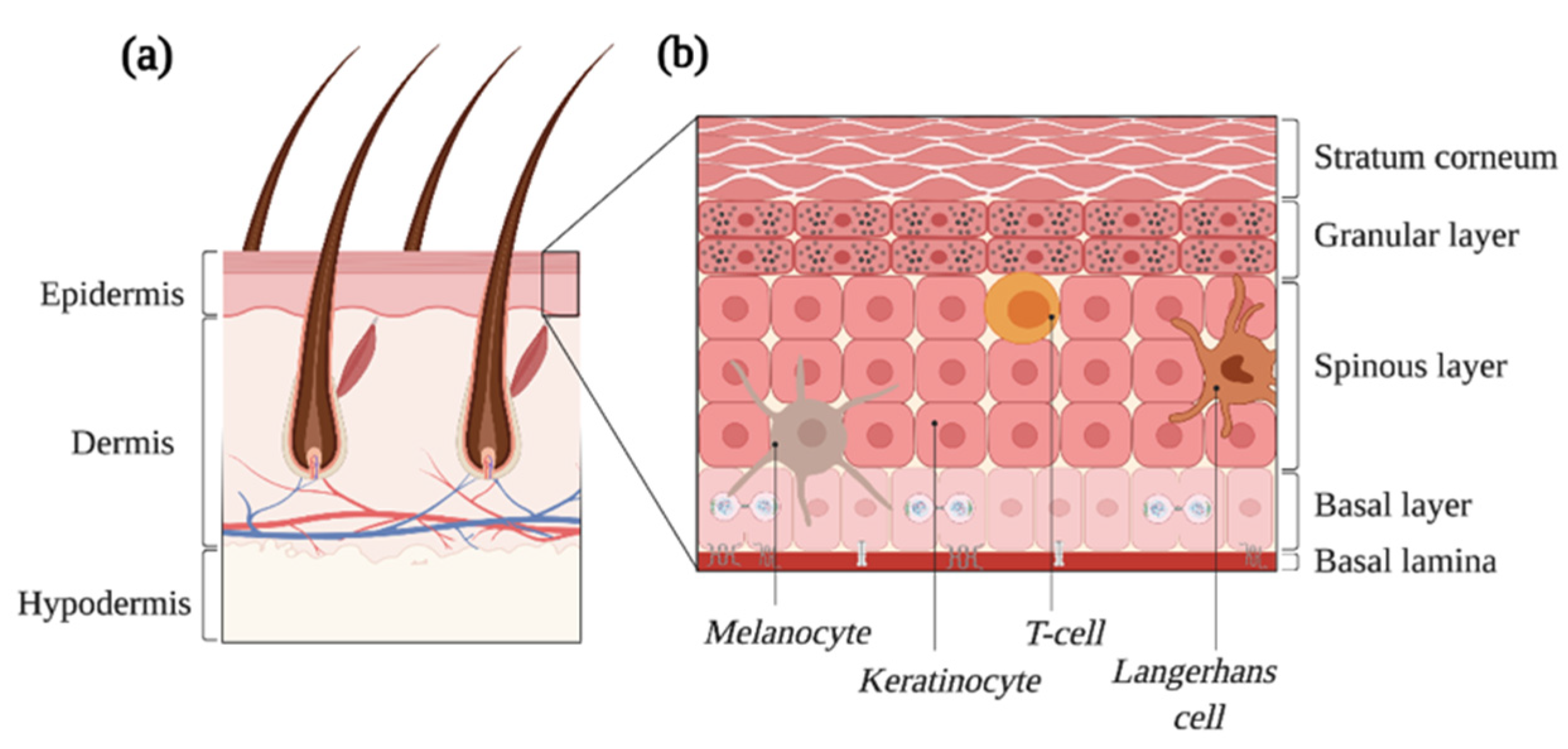
2. The Normal Skin
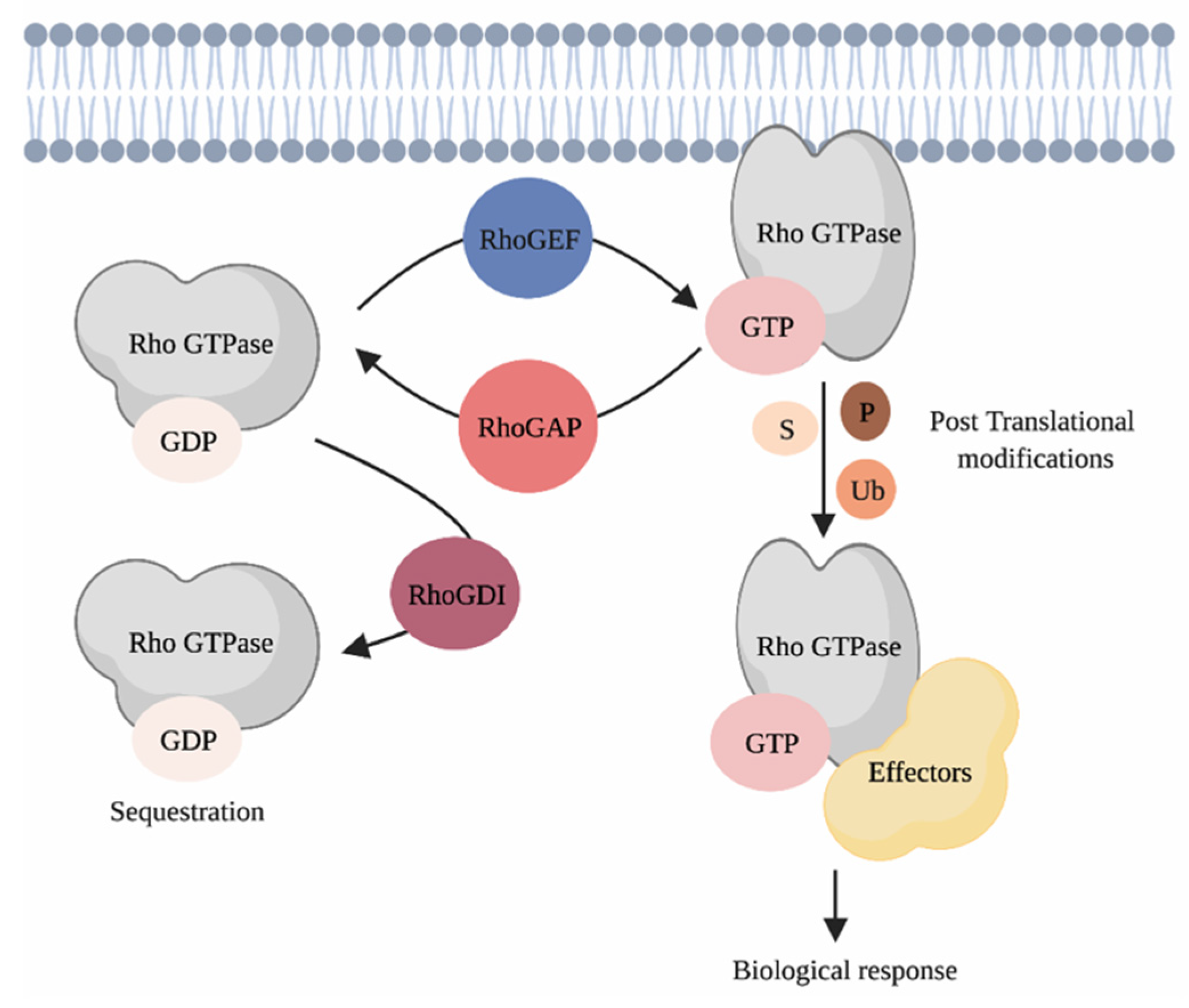
3. Rho GTPases and Their Regulation
4. Cutaneous Squamous Cell Carcinoma
4.1. The Pathogenesis of Squamous Cell Carcinoma
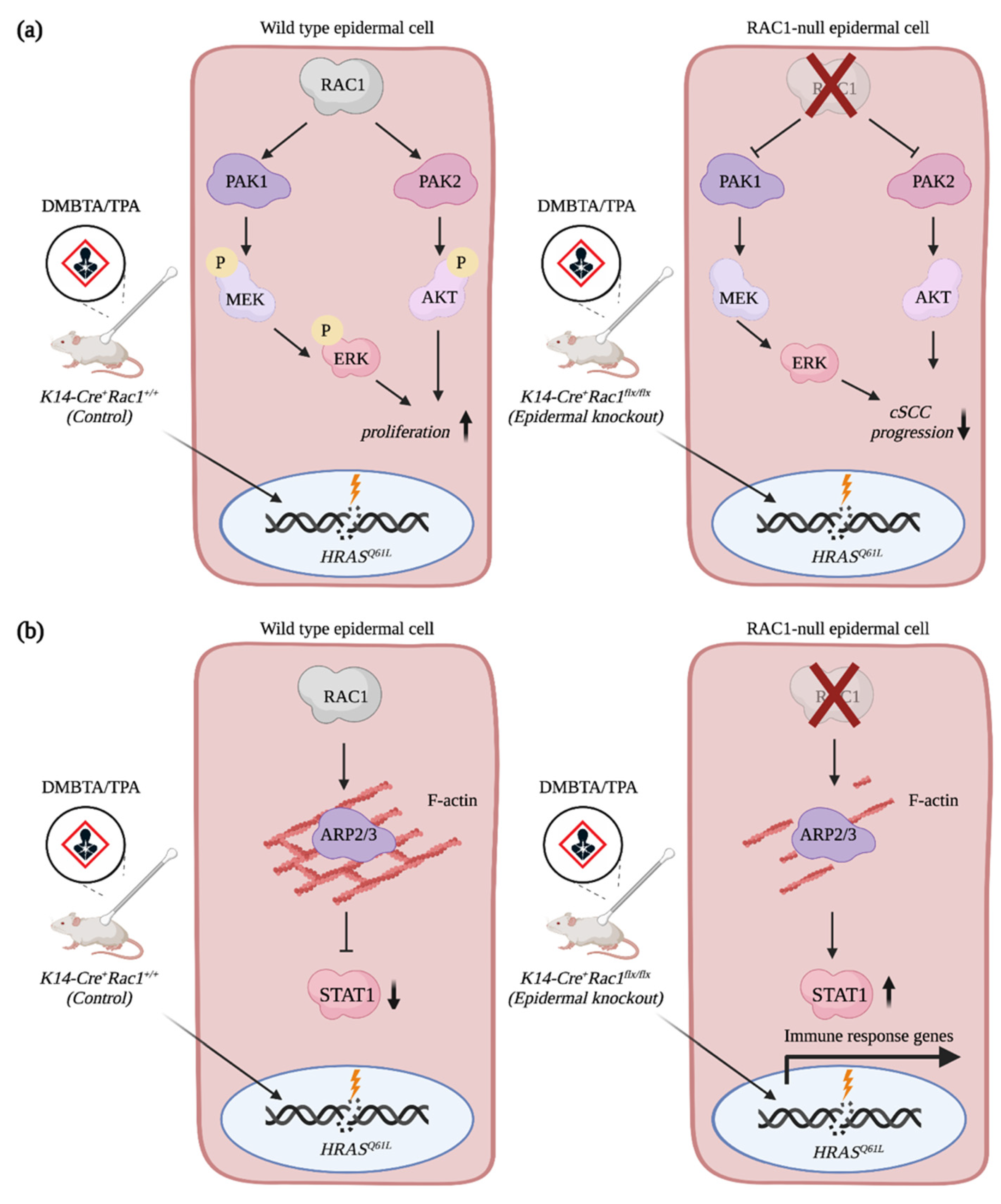
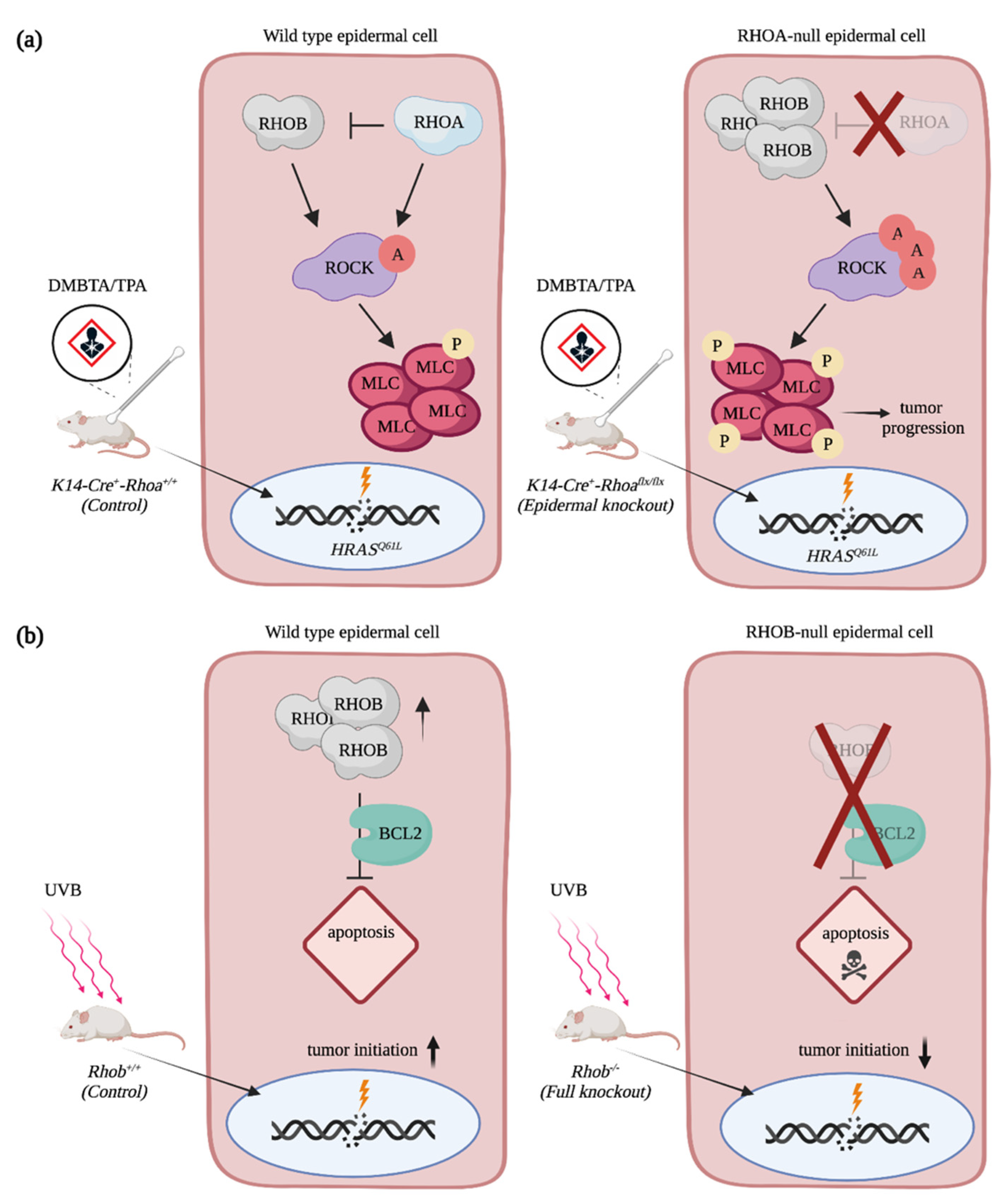
4.2. The Rho GTPases RAC1 and RHOA Act Antagonistically during SCC Progression
4.3. The Contribution of Rho GTPases Regulators to SCC Progression
5. Basal Cell Carcinoma of the Skin, the Most Common Cancer Worldwide
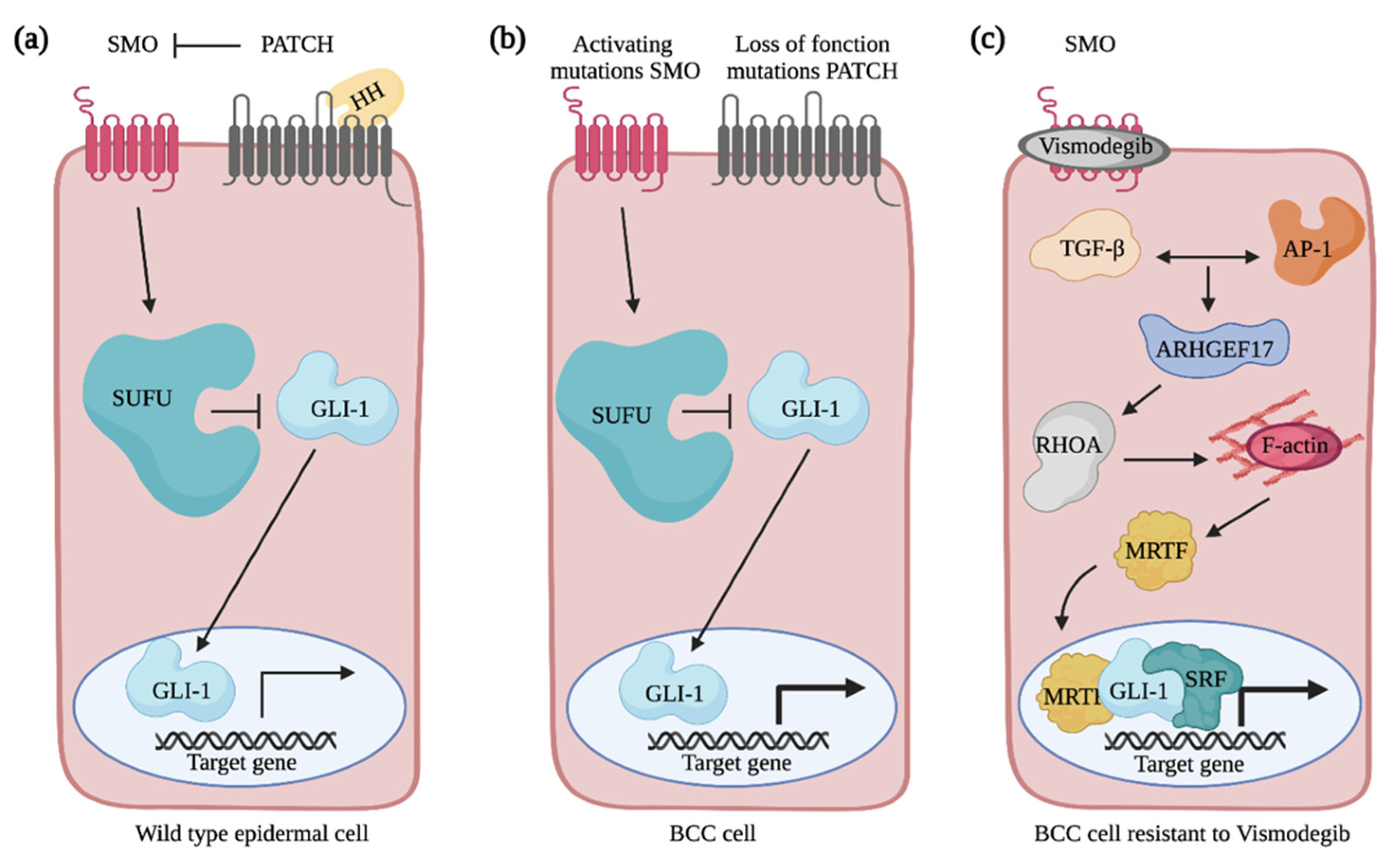
5.1. Basal Cell Carcinoma Is Triggered by Hedgehog Signaling
5.2. Rho GTPases and the Non-Canonical Activation of GLI Transcriptional Activity
6. Melanoma, a Highly Invasive Cancer
6.1. Melanoma, the More Aggressive Skin Cancer
6.2. The Discovery of a Fast-Cycling Rho GTPase
7. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Apalla, Z.; Nashan, D.; Weller, R.B.; Castellsagué, X. Skin Cancer: Epidemiology, Disease Burden, Pathophysiology, Diagnosis, and Therapeutic Approaches. Dermatol. Ther. 2017, 7, 5–19. [Google Scholar] [CrossRef] [PubMed]
- Kosmadaki, M.G.; Gilchrest, B.A. The demographics of aging in the United States: Implications for dermatology. Arch. Dermatol. 2002, 138, 1427–1428. [Google Scholar] [CrossRef]
- Qureshi, A.A.; Wei-Passanese, E.X.; Li, T.; Han, J. Host risk factors for the development of multiple non-melanoma skin cancers. J. Eur. Acad. Dermatol. Venereol. 2013, 27, 565–570. [Google Scholar] [CrossRef]
- Cives, M.; Mannavola, F.; Lospalluti, L.; Sergi, M.C.; Cazzato, G.; Filoni, E.; Cavallo, F.; Giudice, G.; Stucci, L.S.; Porta, C.; et al. Non-melanoma skin cancers: Biological and clinical features. Int. J. Mol. Sci. 2020, 21, 5394. [Google Scholar] [CrossRef] [PubMed]
- Rogers, H.W.; Weinstock, M.A.; Feldman, S.R.; Coldiron, B.M. Incidence estimate of nonmelanoma skin cancer (keratinocyte carcinomas) in the us population, 2012. JAMA Dermatol. 2015, 151, 1081–1086. [Google Scholar] [CrossRef]
- Muzic, J.G.; Schmitt, A.R.; Wright, A.C.; Alniemi, D.T.; Zubair, A.S.; Olazagasti Lourido, J.M.; Sosa Seda, I.M.; Weaver, A.L.; Baum, C.L. Incidence and Trends of Basal Cell Carcinoma and Cutaneous Squamous Cell Carcinoma: A Population-Based Study in Olmsted County, Minnesota, 2000 to 2010. Mayo Clin. Proc. 2017, 92, 890–898. [Google Scholar] [CrossRef] [PubMed]
- Dahmene, M.; Quirion, L.; Laurin, M. High Throughput strategies Aimed at Closing the GAP in Our Knowledge of Rho GTPase Signaling. Cells 2020, 9, 1430. [Google Scholar] [CrossRef]
- Porter, A.P.; Papaioannou, A.; Malliri, A. Deregulation of Rho GTPases in cancer. Small GTPases 2016, 7, 123–138. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.; Yoon, S.R.; Lim, J.; Cho, H.J.; Lee, H.G. Dysregulation of rho gtpases in human cancers. Cancers 2020, 12, 1179. [Google Scholar] [CrossRef]
- Gonzales, K.A.U.; Fuchs, E. Skin and Its Regenerative Powers: An Alliance between Stem Cells and Their Niche. Dev. Cell 2017, 43, 387–401. [Google Scholar] [CrossRef] [PubMed]
- Grice, E.A.; Segre, J.A. The skin microbiome. Nat. Rev. Microbiol. 2011, 9, 244–253. [Google Scholar] [CrossRef] [PubMed]
- Kabashima, K.; Honda, T.; Ginhoux, F.; Egawa, G. The immunological anatomy of the skin. Nat. Rev. Immunol. 2019, 19, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Kanitakis, J. Anatomy, histology and immunohistochemistry of normal human skin. Eur. J. Dermatol. 2002, 12, 390–401. [Google Scholar]
- Simpson, C.L.; Patel, D.M.; Green, K.J. Deconstructing the skin: Cytoarchitectural determinants of epidermal morphogenesis. Nat. Rev. Mol. Cell Biol. 2011, 12, 565–580. [Google Scholar] [CrossRef]
- Kondo, T.; Hearing, V.J. Update on the regulation of mammalian melanocyte function and skin pigmentation. Expert Rev. Dermatol. 2011, 6, 97–108. [Google Scholar] [CrossRef]
- Heasman, S.J.; Ridley, A.J. Mammalian Rho GTPases: New insights into their functions from in vivo studies. Nat. Rev. Mol. Cell Biol. 2008, 9, 690–701. [Google Scholar] [CrossRef] [PubMed]
- Cherfils, J.; Zeghouf, M. Regulation of small GTPases by GEFs, GAPs, and GDIs. Physiol. Rev. 2013, 93, 269–309. [Google Scholar] [CrossRef]
- Sasaki, T.; Kato, M.; Takai, Y. Consequences of weak interaction of rho GDI with the GTP-bound forms of rho p21 and rac p21. J. Biol. Chem. 1993, 268, 23959–23963. [Google Scholar] [CrossRef]
- Cook, D.R.; Rossman, K.L.; Der, C.J. Rho guanine nucleotide exchange factors: Regulators of Rho GTPase activity in development and disease. Oncogene 2014, 33, 4021–4035. [Google Scholar] [CrossRef] [PubMed]
- Laurin, M.; Côté, J.F. Insights into the biological functions of Dock family guanine nucleotide exchange factors. Genes Dev. 2014, 28, 533–547. [Google Scholar] [CrossRef]
- Tcherkezian, J.; Lamarche-Vane, N. Current knowledge of the large RhoGAP family of proteins. Biol. Cell 2007, 99, 67–86. [Google Scholar] [CrossRef]
- Garcia-Mata, R.; Boulter, E.; Burridge, K. The “invisible hand”: Regulation of RHO GTPases by RHOGDIs. Nat. Rev. Mol. Cell Biol. 2011, 12, 493–504. [Google Scholar] [CrossRef]
- Hodge, R.G.; Ridley, A.J. Regulating Rho GTPases and their regulators. Nat. Rev. Mol. Cell Biol. 2016, 17, 496–510. [Google Scholar] [CrossRef]
- Fueller, F.; Kubatzky, K.F. The small GTPase RhoH is an atypical regulator of haematopoietic cells. Cell Commun. Signal. 2008, 6, 6. [Google Scholar] [CrossRef]
- Ridley, A.J. Anne Ridley: Networking with Rho GTPases. Trends Cell Biol. 2016, 26, 465–466. [Google Scholar] [CrossRef] [PubMed]
- Van Aelst, L.; Symons, M. Role of Rho family GTPases in epithelial morphogenesis. Genes Dev. 2002, 16, 1032–1054. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Danés, A.; Blanpain, C. Deciphering the cells of origin of squamous cell carcinomas. Nat. Rev. Cancer 2018, 18, 549–561. [Google Scholar] [CrossRef] [PubMed]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A.; Kinzler, K.W. Cancer genome landscapes. Science 2013, 340, 1546–1558. [Google Scholar] [CrossRef]
- Pickering, C.R.; Zhou, J.H.; Lee, J.J.; Drummond, J.A.; Peng, S.A.; Saade, R.E.; Tsai, K.Y.; Curry, J.L.; Tetzlaff, M.T.; Lai, S.Y.; et al. Mutational landscape of aggressive cutaneous squamous cell carcinoma. Clin. Cancer Res. 2014, 20, 6582–6592. [Google Scholar] [CrossRef]
- Lechner, M.; Frampton, G.M.; Fenton, T.; Feber, A.; Palmer, G.; Jay, A.; Pillay, N.; Forster, M.; Cronin, M.T.; Lipson, D.; et al. Targeted next-generation sequencing of head and neck squamous cell carcinoma identifies novel genetic alterations in HPV+ and HPV- tumors. Genome Med. 2013, 5. [Google Scholar] [CrossRef]
- Pickering, C.R.; Zhang, J.; Yoo, S.Y.; Bengtsson, L.; Moorthy, S.; Neskey, D.M.; Zhao, M.; Ortega Alves, M.V.; Chang, K.; Drummond, J.; et al. Integrative genomic characterization of oral squamous cell carcinoma identifies frequent somatic drivers. Cancer Discov. 2013, 3, 770–781. [Google Scholar] [CrossRef] [PubMed]
- Hammerman, P.S.; Voet, D.; Lawrence, M.S.; Voet, D.; Jing, R.; Cibulskis, K.; Sivachenko, A.; Stojanov, P.; McKenna, A.; Lander, E.S.; et al. Comprehensive genomic characterization of squamous cell lung cancers. Nature 2012, 489, 519–525. [Google Scholar] [CrossRef]
- Kim, Y.; Hammerman, P.S.; Kim, J.; Yoon, J.A.; Lee, Y.; Sun, J.M.; Wilkerson, M.D.; Pedamallu, C.S.; Cibulskis, K.; Yoo, Y.K.; et al. Integrative and comparative genomic analysis of lung squamous cell carcinomas in East Asian patients. J. Clin. Oncol. 2014, 32, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Gao, Z.; Li, F.; Li, X.; Sun, Y.; Wang, M.; Li, D.; Wang, R.; Li, F.; Fang, R.; et al. Whole exome sequencing identifies frequent somatic mutations in cell-cell adhesion genes in Chinese patients with lung squamous cell carcinoma. Sci. Rep. 2015, 5, 14237. [Google Scholar] [CrossRef] [PubMed]
- Burk, R.D.; Chen, Z.; Saller, C.; Tarvin, K.; Carvalho, A.L.; Scapulatempo-Neto, C.; Silveira, H.C.; Fregnani, J.H.; Creighton, C.J.; Anderson, M.L.; et al. Integrated genomic and molecular characterization of cervical cancer. Nature 2017, 543, 378–384. [Google Scholar] [CrossRef]
- Ojesina, A.I.; Lichtenstein, L.; Freeman, S.S.; Pedamallu, C.S.; Imaz-Rosshandler, I.; Pugh, T.J.; Cherniack, A.D.; Ambrogio, L.; Cibulskis, K.; Bertelsen, B.; et al. Landscape of genomic alterations in cervical carcinomas. Nature 2014, 506, 371–375. [Google Scholar] [CrossRef] [PubMed]
- Hoadley, K.A.; Yau, C.; Wolf, D.M.; Cherniack, A.D.; Tamborero, D.; Ng, S.; Leiserson, M.D.M.; Niu, B.; McLellan, M.D.; Uzunangelov, V.; et al. Multiplatform analysis of 12 cancer types reveals molecular classification within and across tissues of origin. Cell 2014, 158, 929–944. [Google Scholar] [CrossRef]
- Chang, D.; Shain, A.H. The landscape of driver mutations in cutaneous squamous cell carcinoma. NPJ Genom. Med. 2021, 6, 61. [Google Scholar] [CrossRef]
- South, A.P.; Purdie, K.J.; Watt, S.A.; Haldenby, S.; Den Breems, N.Y.; Dimon, M.; Arron, S.T.; Kluk, M.J.; Aster, J.C.; McHugh, A.; et al. NOTCH1 mutations occur early during cutaneous squamous cell carcinogenesis. J. Investig. Dermatol. 2014, 134, 2630–2638. [Google Scholar] [CrossRef]
- Kim, J.; Bowlby, R.; Mungall, A.J.; Robertson, A.G.; Odze, R.D.; Cherniack, A.D.; Shih, J.; Pedamallu, C.S.; Cibulskis, C.; Dunford, A.; et al. Integrated genomic characterization of oesophageal carcinoma. Nature 2017, 541, 169–174. [Google Scholar] [CrossRef]
- Song, Y.; Li, L.; Ou, Y.; Gao, Z.; Li, E.; Li, X.; Zhang, W.; Wang, J.; Xu, L.; Zhou, Y.; et al. Identification of genomic alterations in oesophageal squamous cell cancer. Nature 2014, 508, 91–95. [Google Scholar] [CrossRef]
- Lin, D.C.; Hao, J.J.; Nagata, Y.; Xu, L.; Shang, L.; Meng, X.; Sato, Y.; Okuno, Y.; Varela, A.M.; Ding, L.W.; et al. Genomic and molecular characterization of esophageal squamous cell carcinoma. Nat. Genet. 2014, 46, 467–473. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.B.; Chen, Z.L.; Li, J.G.; Hu, X.D.; Shi, X.J.; Sun, Z.M.; Zhang, F.; Zhao, Z.R.; Li, Z.T.; Liu, Z.Y.; et al. Genetic landscape of esophageal squamous cell carcinoma. Nat. Genet. 2014, 46, 1097–1102. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, M.S.; Sougnez, C.; Lichtenstein, L.; Cibulskis, K.; Lander, E.; Gabriel, S.B.; Getz, G.; Ally, A.; Balasundaram, M.; Birol, I.; et al. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature 2015, 517, 576–582. [Google Scholar] [CrossRef]
- Agrawal, N.; Frederick, M.J.; Pickering, C.R.; Bettegowda, C.; Chang, K.; Li, R.J.; Fakhry, C.; Xie, T.X.; Zhang, J.; Wang, J.; et al. Exome sequencing of head and neck squamous cell carcinoma reveals inactivating mutations in NOTCH1. Science 2011, 333, 1154–1157. [Google Scholar] [CrossRef]
- Stransky, N.; Egloff, A.M.; Tward, A.D.; Kostic, A.D.; Cibulskis, K.; Sivachenko, A.; Kryukov, G.V.; Lawrence, M.S.; Sougnez, C.; McKenna, A.; et al. The mutational landscape of head and neck squamous cell carcinoma. Science 2011, 333, 1157–1160. [Google Scholar] [CrossRef]
- Fania, L.; Didona, D.; Di Pietro, F.R.; Verkhovskaia, S.; Morese, R.; Paolino, G.; Donati, M.; Ricci, F.; Coco, V.; Ricci, F.; et al. Cutaneous squamous cell carcinoma: From pathophysiology to novel therapeutic approaches. Biomedicines 2021, 9, 171. [Google Scholar] [CrossRef] [PubMed]
- McCullough, A. Comprehensive genomic characterization of squamous cell lung cancers. Yearb. Pathol. Lab. Med. 2013, 2013, 290–291. [Google Scholar] [CrossRef][Green Version]
- Nassar, D.; Latil, M.; Boeckx, B.; Lambrechts, D.; Blanpain, C. Genomic landscape of carcinogen-induced and genetically induced mouse skin squamous cell carcinoma. Nat. Med. 2015, 21, 946–954. [Google Scholar] [CrossRef] [PubMed]
- Martincorena, I.; Roshan, A.; Gerstung, M.; Ellis, P.; Van Loo, P.; McLaren, S.; Wedge, D.C.; Fullam, A.; Alexandrov, L.B.; Tubio, J.M.; et al. High burden and pervasive positive selection of somatic mutations in normal human skin. Science 2015, 348, 880–886. [Google Scholar] [CrossRef] [PubMed]
- Paradisi, A.; Waterboer, T.; Ricci, F.; Sampogna, F.; Pawlita, M.; Abeni, D. Concomitant seropositivity for HPV 16 and cutaneous HPV types increases the risk of recurrent squamous cell carcinoma of the skin. Eur. J. Dermatol. 2020, 30, 493–498. [Google Scholar] [CrossRef]
- Torchia, D.; Massi, D.; Caproni, M.; Fabbri, P. Multiple cutaneous precanceroses and carcinomas from combined iatrogenic/professional exposure to arsenic. Int. J. Dermatol. 2008, 47, 592–593. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.H.; Cohen, D.N.; Rady, P.L.; Tyring, S.K. BRAF inhibitor-associated cutaneous squamous cell carcinoma: New mechanistic insight, emerging evidence for viral involvement and perspectives on clinical management. Br. J. Dermatol. 2017, 177, 914–923. [Google Scholar] [CrossRef]
- zur Hausen, H. Papillomaviruses in the causation of human cancers—A brief historical account. Virology 2009, 384, 260–265. [Google Scholar] [CrossRef]
- Harvey, N.T.; Millward, M.; Wood, B.A. Squamoproliferative lesions arising in the setting of BRAF inhibition. Am. J. Dermatopathol. 2012, 34, 822–826. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Spevak, W.; Zhang, Y.; Burton, E.A.; Ma, Y.; Habets, G.; Zhang, J.; Lin, J.; Ewing, T.; Matusow, B.; et al. RAF inhibitors that evade paradoxical MAPK pathway activation. Nature 2015, 526, 583–586. [Google Scholar] [CrossRef]
- Green, A.C.; Olsen, C.M. Cutaneous squamous cell carcinoma: An epidemiological review. Br. J. Dermatol. 2017, 177, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Jensen, P.; Hansen, S.; Moller, B.; Leivestad, T.; Pfeifer, P.; Geiran, O.; Fauchald, P.; Simonsen, S. Skin cancer in kidney and heart transplant recipients and different long-term immunosuppressive therapy regimens. J. Am. Acad. Dermatol. 1999, 40, 177–186. [Google Scholar] [CrossRef]
- Hartevelt, M.M.; Bouwes Bavinck, J.N.; Kootte, A.M.M.; Vermeer, B.J.; Vandenbroucke, J.P. Incidence of skin cancer after renal transplantation in the netherlands. Transplantation 1990, 49, 506–509. [Google Scholar] [CrossRef]
- Lindelöf, B.; Sigurgeirsson, B.; Gäbel, H.; Stern, R.S. Incidence of skin cancer in 5356 patients following organ transplantation. Br. J. Dermatol. 2000, 143, 513–519. [Google Scholar] [CrossRef]
- Migden, M.R.; Rischin, D.; Schmults, C.D.; Guminski, A.; Hauschild, A.; Lewis, K.D.; Chung, C.H.; Hernandez-Aya, L.; Lim, A.M.; Chang, A.L.S.; et al. PD-1 Blockade with Cemiplimab in Advanced Cutaneous Squamous-Cell Carcinoma. N. Engl. J. Med. 2018. [Google Scholar] [CrossRef]
- Abel, E.L.; Angel, J.M.; Kiguchi, K.; DiGiovanni, J. Multi-stage chemical carcinogenesis in mouse skin: Fundamentals and applications. Nat. Protoc. 2009, 4, 1350–1362. [Google Scholar] [CrossRef] [PubMed]
- Balmain, A.; Ramsden, M.; Bowden, G.T.; Smith, J. Activation of the mouse cellular Harvey-ras gene in chemically induced benign skin papillomas. Nature 1984, 307, 658–660. [Google Scholar] [CrossRef]
- Patel, V.; Rosenfeldt, H.M.; Lyons, R.; Servitja, J.M.; Bustelo, X.R.; Siroff, M.; Gutkind, J.S. Persistent activation of Rac1 in squamous carcinomas of the head and neck: Evidence for an EGFR/Vav2 signaling axis involved in cell invasion. Carcinogenesis 2007, 28, 1145–1152. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Pedersen, E.; Basse, A.; Lefever, T.; Peyrollier, K.; Kapoor, S.; Mei, Q.; Karlsson, R.; Chrostek-Grashoff, A.; Brakebusch, C. Rac1 is crucial for Ras-dependent skin tumor formation by controlling Pak1-Mek-Erk hyperactivation and hyperproliferation in vivo. Oncogene 2010, 29, 3362–3373. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Fu, M.; Zhang, G.; Zhou, Y.; Zhu, S.; Liu, J.; Wang, D.; Deng, A.; Wang, Z. Rac1 regulates skin tumors by regulation of keratin 17 through recruitment and interaction with CD11b+Gr1+ cells. Oncotarget 2014, 5, 4406–4417. [Google Scholar] [CrossRef][Green Version]
- Pedersen, E.; Wang, Z.; Stanley, A.; Peyrollier, K.; Rösner, L.M.; Werfel, T.; Quondamatteo, F.; Brakebusch, C. RAC1 in keratinocytes regulates crosstalk to immune cells by Arp2/3-dependent control of STAT1. J. Cell Sci. 2012, 125, 5379–5390. [Google Scholar] [CrossRef]
- Winge, M.C.G.; Marinkovich, M.P. Epidermal activation of the small GTPase Rac1 in psoriasis pathogenesis. Small GTPases 2019, 10, 163–168. [Google Scholar] [CrossRef]
- Winge, M.C.G.; Ohyama, B.; Dey, C.N.; Boxer, L.M.; Li, W.; Ehsani-Chimeh, N.; Truong, A.K.; Wu, D.; Armstrong, A.W.; Makino, T.; et al. RAC1 activation drives pathologic interactions between the epidermis and immune cells. J. Clin. Investig. 2016, 126, 2661–2677. [Google Scholar] [CrossRef]
- Deshmukh, J.; Pofahl, R.; Haase, I. Epidermal rac1 regulates the DNA damage response and protects from UV-light-induced keratinocyte apoptosis and skin carcinogenesis. Cell Death Dis. 2017, 8, e2664. [Google Scholar] [CrossRef]
- McCauley, H.A.; Chevrier, V.; Birnbaum, D.; Guasch, G. De-repression of the RAC activator ELMO1 in cancer stem cells drives progression of TGFβ-deficient squamous cell carcinoma from transition zones. eLife 2017, 6, e22914. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, M.; Maruyama, S.; Abé, T.; Tsuneki, M.; Kato, H.; Izumi, K.; Tanuma, J.-I.; Cheng, J.; Saku, T. Rac1-dependent phagocytosis of apoptotic cells by oral squamous cell carcinoma cells: A possible driving force for tumor progression. Exp. Cell Res. 2020, 392, 112013. [Google Scholar] [CrossRef]
- Skvortsov, S.; Dudás, J.; Eichberger, P.; Witsch-Baumgartner, M.; Loeffler-Ragg, J.; Pritz, C.; Schartinger, V.H.; Maier, H.; Hall, J.; Debbage, P.; et al. Rac1 as a potential therapeutic target for chemo-radioresistant head and neck squamous cell carcinomas (HNSCC). Br. J. Cancer 2014, 110, 2677–2687. [Google Scholar] [CrossRef]
- Skvortsov, S.; Jimenez, C.R.; Knol, J.C.; Eichberger, P.; Schiestl, B.; Debbage, P.; Skvortsova, I.; Lukas, P. Radioresistant head and neck squamous cell carcinoma cells: Intracellular signaling, putative biomarkers for tumor recurrences and possible therapeutic targets. Radiother. Oncol. 2011, 101, 177–182. [Google Scholar] [CrossRef] [PubMed]
- García-Mariscal, A.; Li, H.; Pedersen, E.; Peyrollier, K.; Ryan, K.M.; Stanley, A.; Quondamatteo, F.; Brakebusch, C. Loss of RhoA promotes skin tumor formation and invasion by upregulation of RhoB. Oncogene 2018, 37, 847–860. [Google Scholar] [CrossRef] [PubMed]
- Liu, A.-X.; Rane, N.; Liu, J.-P.; Prendergast, G.C. RhoB Is Dispensable for Mouse Development, but It Modifies Susceptibility to Tumor Formation as Well as Cell Adhesion and Growth Factor Signaling in Transformed Cells. Mol. Cell. Biol. 2001, 21, 6906–6912. [Google Scholar] [CrossRef]
- Meyer, N.; Peyret-Lacombe, A.; Canguilhem, B.; Médale-Giamarchi, C.; Mamouni, K.; Cristini, A.; Monferran, S.; Lamant, L.; Filleron, T.; Pradines, A.; et al. RhoB promotes cancer initiation by protecting keratinocytes from UVB-induced apoptosis but limits tumor aggressiveness. J. Investig. Dermatol. 2014, 134, 203–212. [Google Scholar] [CrossRef]
- Canguilhem, B.; Pradines, A.; Baudouin, C.; Boby, C.; Lajoie-Mazenc, I.; Charveron, M.; Favre, G. RhoB protects human keratinocytes from UVB-induced apoptosis through epidermal growth factor receptor signaling. J. Biol. Chem. 2005, 280, 43257–43263. [Google Scholar] [CrossRef]
- Jiang, L.; Liu, X.; Kolokythas, A.; Yu, J.; Wang, A.; Heidbreder, C.E.; Shi, F.; Zhou, X. Downregulation of the Rho GTPase signaling pathway is involved in the microRNA-138-mediated inhibition of cell migration and invasion in tongue squamous cell carcinoma. Int. J. Cancer 2010, 127, 505–512. [Google Scholar] [CrossRef]
- Wang, H.; Guo, W.; Jian, Q.; Xue, K.; Huang, M.; Chi, S.; Li, C.; Li, C. MicroRNA-340 inhibits squamous cell carcinoma cell proliferation, migration and invasion by downregulating RhoA. J. Dermatol. Sci. 2018, 92, 197–206. [Google Scholar] [CrossRef]
- Graziano, V.; De Laurenzi, V. Role of p63 in cancer development. Biochim. Biophys. Acta Rev. Cancer 2011, 1816, 57–66. [Google Scholar] [CrossRef]
- Lawrence, M.S.; Stojanov, P.; Mermel, C.H.; Robinson, J.T.; Garraway, L.A.; Golub, T.R.; Meyerson, M.; Gabriel, S.B.; Lander, E.S.; Getz, G. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature 2014, 505, 495–501. [Google Scholar] [CrossRef]
- Abraham, C.G.; Ludwig, M.P.; Andrysik, Z.; Pandey, A.; Joshi, M.; Galbraith, M.D.; Sullivan, K.D.; Espinosa, J.M. ΔNp63α Suppresses TGFB2 Expression and RHOA Activity to Drive Cell Proliferation in Squamous Cell Carcinomas. Cell Rep. 2018, 24, 3224–3236. [Google Scholar] [CrossRef] [PubMed]
- Menacho-Márquez, M.; García-Escudero, R.; Ojeda, V.; Abad, A.; Delgado, P.; Costa, C.; Ruiz, S.; Alarcón, B.; Paramio, J.M.; Bustelo, X.R. The Rho Exchange Factors Vav2 and Vav3 Favor Skin Tumor Initiation and Promotion by Engaging Extracellular Signaling Loops. PLoS Biol. 2013, 11, e1001615. [Google Scholar] [CrossRef] [PubMed]
- Malliri, A.; Van der Kammen, R.A.; Clark, K.; Van der Valk, M.; Michiels, F.; Collard, J.G. Mice deficient in the Rac activator Tiam1 are resistant to Ras-induced skin tumours. Nature 2002, 417, 867–871. [Google Scholar] [CrossRef] [PubMed]
- Lambert, J.M.; Lambert, Q.T.; Reuther, G.W.; Malliri, A.; Siderovski, D.P.; Sondek, J.; Collard, J.G.; Der, C.J. Tiam1 mediates Ras activation of Rac by a PI(3)K-independent mechanism. Nat. Cell Biol. 2002, 4, 621–625. [Google Scholar] [CrossRef] [PubMed]
- Lorenzo-Martín, L.F.; Rodríguez-Fdez, S.; Fabbiano, S.; Abad, A.; García-Macías, M.C.; Dosil, M.; Cuadrado, M.; Robles-Valero, J.; Bustelo, X.R. Vav2 pharmaco-mimetic mice reveal the therapeutic value and caveats of the catalytic inactivation of a Rho exchange factor. Oncogene 2020, 39, 5098–5111. [Google Scholar] [CrossRef]
- Lorenzo-Martín, L.F.; Fernández-Parejo, N.; Menacho-Márquez, M.; Rodríguez-Fdez, S.; Robles-Valero, J.; Zumalave, S.; Fabbiano, S.; Pascual, G.; García-Pedrero, J.M.; Abad, A.; et al. VAV2 signaling promotes regenerative proliferation in both cutaneous and head and neck squamous cell carcinoma. Nat. Commun. 2020, 11, 4788. [Google Scholar] [CrossRef]
- Wang, H.; Wang, Y.; Liang, B.; He, F.; Li, Y.; Che, J.; Li, X.; Zhao, H.; Shi, G. The Rho GTPase RhoE exerts tumor-suppressing effects in human esophageal squamous cell carcinoma via negatively regulating epidermal growth factor receptor. J. Cancer Res. Ther. 2016, 12, C60–C63. [Google Scholar] [CrossRef]
- Zhao, H.; Yang, J.; Fan, T.; Li, S.; Ren, X. RhoE functions as a tumor suppressor in esophageal squamous cell carcinoma and modulates the PTEN/PI3K/Akt signaling pathway. Tumor Biol. 2012, 33, 1363–1374. [Google Scholar] [CrossRef]
- Sreekantaswamy, S.; Endo, J.; Chen, A.; Butler, D.; Morrison, L.; Linos, E. Aging and the treatment of basal cell carcinoma. Clin. Dermatol. 2019, 37, 373–378. [Google Scholar] [CrossRef]
- Ikehata, H.; Ono, T. The mechanisms of UV mutagenesis. J. Radiat. Res. 2011, 52, 115–125. [Google Scholar] [CrossRef]
- Miller, D.L.; Weinstock, M.A. Nonmelanoma skin cancer in the United States: Incidence. J. Am. Acad. Dermatol. 1994, 30, 774–778. [Google Scholar] [CrossRef]
- Epstein, E.H. Basal cell carcinomas: Attack of the hedgehog. Nat. Rev. Cancer 2008, 8, 743–754. [Google Scholar] [CrossRef]
- Fania, L.; Didona, D.; Morese, R.; Campana, I.; Coco, V.; Di Pietro, F.R.; Ricci, F.; Pallotta, S.; Candi, E.; Abeni, D.; et al. Basal cell carcinoma: From pathophysiology to novel therapeutic approaches. Biomedicines 2020, 8, 449. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.K.; Mays, R.R.; Abramovits, W.; Vincent, K.D. Odomzo® (Sonidegib). Skinmed 2018, 16, 35–38. [Google Scholar] [PubMed]
- Shord, S.S.; Casey, D.; Zhao, H.; Demko, S.; Keegan, P.; Pazdur, R. FDA Approval summary: Sonidegib—Response. Clin. Cancer Res. 2017, 23, 5994. [Google Scholar] [CrossRef][Green Version]
- Basset-Seguin, N.; Sharpe, H.J.; De Sauvage, F.J. Efficacy of Hedgehog pathway inhibitors in basal cell carcinoma. Mol. Cancer Ther. 2015, 14, 633–641. [Google Scholar] [CrossRef] [PubMed]
- Sekulic, A.; Migden, M.R.; Oro, A.E.; Dirix, L.; Lewis, K.D.; Hainsworth, J.D.; Solomon, J.A.; Yoo, S.; Arron, S.T.; Friedlander, P.A.; et al. Efficacy and Safety of Vismodegib in Advanced Basal-Cell Carcinoma. N. Engl. J. Med. 2012, 366, 2171–2179. [Google Scholar] [CrossRef]
- Chang, A.L.S.; Oro, A.E. Initial assessment of tumor regrowth after vismodegib in advanced basal cell carcinoma. Arch. Dermatol. 2012, 148, 1324–1325. [Google Scholar] [CrossRef]
- Axelson, M.; Liu, K.; Jiang, X.; He, K.; Wang, J.; Zhao, H.; Kufrin, D.; Palmby, T.; Dong, Z.; Russell, A.M.; et al. U.S. Food and Drug Administration approval: Vismodegib for recurrent, locally advanced, or metastatic basal cell carcinoma. Clin. Cancer Res. 2013, 19, 2289–2293. [Google Scholar] [CrossRef] [PubMed]
- Atwood, S.X.; Sarin, K.Y.; Li, J.R.; Yao, C.Y.; Urman, N.M.; Chang, A.L.S.; Tang, J.Y.; Oro, A.E. Rolling the Genetic Dice: Neutral and Deleterious Smoothened Mutations in Drug-Resistant Basal Cell Carcinoma. J. Investig. Dermatol. 2015, 135, 2138–2141. [Google Scholar] [CrossRef] [PubMed]
- Youssef, K.K.; Lapouge, G.; Bouvrée, K.; Rorive, S.; Brohée, S.; Appelstein, O.; Larsimont, J.C.; Sukumaran, V.; Van De Sande, B.; Pucci, D.; et al. Adult interfollicular tumour-initiating cells are reprogrammed into an embryonic hair follicle progenitor-like fate during basal cell carcinoma initiation. Nat. Cell Biol. 2012, 14, 1282–1294. [Google Scholar] [CrossRef] [PubMed]
- Larsimont, J.C.; Youssef, K.K.; Sánchez-Danés, A.; Sukumaran, V.; Defrance, M.; Delatte, B.; Liagre, M.; Baatsen, P.; Marine, J.C.; Lippens, S.; et al. Sox9 Controls Self-Renewal of Oncogene Targeted Cells and Links Tumor Initiation and Invasion. Cell Stem Cell 2015, 17, 60–73. [Google Scholar] [CrossRef] [PubMed]
- Fiore, V.F.; Krajnc, M.; Quiroz, F.G.; Levorse, J.; Pasolli, H.A.; Shvartsman, S.Y.; Fuchs, E. Mechanics of a multilayer epithelium instruct tumour architecture and function. Nature 2020, 585, 433–439. [Google Scholar] [CrossRef]
- Tucci, M.G.; Lucarini, G.; Zizzi, A.; Rocchetti, R.; Brancorsini, D.; Di Primio, R.; Ricotti, F.; Ricotti, G. Cdc42 is involved in basal cell carcinoma carcinogenesis. Arch. Dermatol. Res. 2013, 305, 835–840. [Google Scholar] [CrossRef]
- Hoseong Yang, S.; Andl, T.; Grachtchouk, V.; Wang, A.; Liu, J.; Syu, L.J.; Ferris, J.; Wang, T.S.; Glick, A.B.; Millar, S.E.; et al. Pathological responses to oncogenic Hedgehog signaling in skin are dependent on canonical Wnt/β-catenin signaling. Nat. Genet. 2008, 40, 1130–1135. [Google Scholar] [CrossRef] [PubMed]
- Whitson, R.J.; Lee, A.; Urman, N.M.; Mirza, A.; Yao, C.Y.; Brown, A.S.; Li, J.R.; Shankar, G.; Fry, M.A.; Atwood, S.X.; et al. Noncanonical hedgehog pathway activation through SRF-MKL1 promotes drug resistance in basal cell carcinomas. Nat. Med. 2018, 24, 271–281. [Google Scholar] [CrossRef]
- Miralles, F.; Posern, G.; Zaromytidou, A.I.; Treisman, R. Actin dynamics control SRF activity by regulation of its coactivator MAL. Cell 2003, 113, 329–342. [Google Scholar] [CrossRef]
- Yao, C.D.; Haensel, D.; Gaddam, S.; Patel, T.; Atwood, S.X.; Sarin, K.Y.; Whitson, R.J.; McKellar, S.; Shankar, G.; Aasi, S.; et al. AP-1 and TGFß cooperativity drives non-canonical Hedgehog signaling in resistant basal cell carcinoma. Nat. Commun. 2020, 11. [Google Scholar] [CrossRef]
- DasGupta, R.; Fuchs, E. Multiple roles for activated LEF/TCF transcription complexes during hair follicle development and differentiation. Development 1999, 126, 4557–4568. [Google Scholar] [CrossRef]
- Saxena, N.; Mok, K.W.; Rendl, M. An updated classification of hair follicle morphogenesis. Exp. Dermatol. 2019, 28, 332–344. [Google Scholar] [CrossRef]
- Millar, S.E. Molecular mechanisms regulating hair follicle development. J. Investig. Dermatol. 2002, 118, 216–225. [Google Scholar] [CrossRef] [PubMed]
- Veltri, A.; Lang, C.; Lien, W.H. Concise Review: Wnt Signaling Pathways in Skin Development and Epidermal Stem Cells. Stem Cells 2018, 36, 22–35. [Google Scholar] [CrossRef] [PubMed]
- Chiang, C.; Swan, R.Z.; Grachtchouk, M.; Bolinger, M.; Litingtung, Y.; Robertson, E.K.; Cooper, M.K.; Gaffield, W.; Westphal, H.; Beachy, P.A.; et al. Essential role for Sonic hedgehog during hair follicle morphogenesis. Dev. Biol. 1999, 205, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Luxenburg, C.; Zaidel-Bar, R. From cell shape to cell fate via the cytoskeleton—Insights from the epidermis. Exp. Cell Res. 2019, 378, 232–237. [Google Scholar] [CrossRef]
- Devenport, D.; Fuchs, E. Planar polarization in embryonic epidermis orchestrates global asymmetric morphogenesis of hair follicles. Nat. Cell Biol. 2008, 10, 1257–1268. [Google Scholar] [CrossRef]
- Ahtiainen, L.; Lefebvre, S.; Lindfors, P.H.; Renvoisé, E.; Shirokova, V.; Vartiainen, M.K.; Thesleff, I.; Mikkola, M.L. Directional Cell Migration, but Not Proliferation, Drives Hair Placode Morphogenesis. Dev. Cell 2014, 28, 588–602. [Google Scholar] [CrossRef]
- Le, H.Q.; Ghatak, S.; Yeung, C.Y.C.; Tellkamp, F.; Günschmann, C.; Dieterich, C.; Yeroslaviz, A.; Habermann, B.; Pombo, A.; Niessen, C.M.; et al. Mechanical regulation of transcription controls Polycomb-mediated gene silencing during lineage commitment. Nat. Cell Biol. 2016, 18, 864–875. [Google Scholar] [CrossRef]
- Luxenburg, C.; Heller, E.; Pasolli, H.A.; Chai, S.; Nikolova, M.; Stokes, N.; Fuchs, E. Wdr1-mediated cell shape dynamics and cortical tension are essential for epidermal planar cell polarity. Nat. Cell Biol. 2015, 17, 592–604. [Google Scholar] [CrossRef]
- Rogers, G.E. Hair follicle differentiation and regulation. Int. J. Dev. Biol. 2004, 48, 163–170. [Google Scholar] [CrossRef]
- Laurin, M.; Gomez, N.C.; Levorse, J.; Sendoel, A.; Sribour, M.; Fuchs, E. An RNAi screen unravels the complexities of rho GTPase networks in skin morphogenesis. eLife 2019, 8, e50226. [Google Scholar] [CrossRef] [PubMed]
- Hawryluk, E.B.; Tsao, H. Melanoma: Clinical features and genomic insights. Cold Spring Harb. Perspect. Med. 2014, 4, a015388. [Google Scholar] [CrossRef]
- Scolyer, R.A.; Long, G.V.; Thompson, J.F. Evolving concepts in melanoma classification and their relevance to multidisciplinary melanoma patient care. Mol. Oncol. 2011, 5, 124–136. [Google Scholar] [CrossRef] [PubMed]
- Bertolotto, C. Melanoma: From Melanocyte to Genetic Alterations and Clinical Options. Scientifica 2013, 2013, 635203. [Google Scholar] [CrossRef] [PubMed]
- Akbani, R.; Akdemir, K.C.; Aksoy, B.A.; Albert, M.; Ally, A.; Amin, S.B.; Arachchi, H.; Arora, A.; Auman, J.T.; Ayala, B.; et al. Genomic Classification of Cutaneous Melanoma. Cell 2015, 161, 1681–1696. [Google Scholar] [CrossRef]
- Curtin, J.A.; Fridlyand, J.; Kageshita, T.; Patel, H.N.; Busam, K.J.; Kutzner, H.; Cho, K.-H.; Aiba, S.; Bröcker, E.-B.; LeBoit, P.E.; et al. Distinct Sets of Genetic Alterations in Melanoma. N. Engl. J. Med. 2005, 353, 2135–2147. [Google Scholar] [CrossRef] [PubMed]
- Smalley, K.S.M. Understanding melanoma signaling networks as the basis for molecular targeted therapy. J. Investig. Dermatol. 2010, 130, 28–37. [Google Scholar] [CrossRef]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef]
- Pollock, P.M.; Harper, U.L.; Hansen, K.S.; Yudt, L.M.; Stark, M.; Robbins, C.M.; Moses, T.Y.; Hostetter, G.; Wagner, U.; Kakareka, J.; et al. High frequency of BRAF mutations in nevi. Nat. Genet. 2003, 33, 19–20. [Google Scholar] [CrossRef]
- Moreira, A.; Heinzerling, L.; Bhardwaj, N.; Friedlander, P. Current melanoma treatments: Where do we stand? Cancers 2021, 13, 221. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, R.W.; Fisher, D.E. Treatment of Advanced Melanoma in 2020 and Beyond. J. Investig. Dermatol. 2021, 141, 23–31. [Google Scholar] [CrossRef]
- Kim, G.; McKee, A.E.; Ning, Y.M.; Hazarika, M.; Theoret, M.; Johnson, J.R.; Xu, Q.C.; Tang, S.; Sridhara, R.; Jiang, X.; et al. FDA approval summary: Vemurafenib for treatment of unresectable or metastatic melanoma with the BRAFV600E mutation mutation. Clin. Cancer Res. 2014, 20, 4994–5000. [Google Scholar] [CrossRef]
- Namikawa, K.; Yamazaki, N. Targeted Therapy and Immunotherapy for Melanoma in Japan. Curr. Treat. Options Oncol. 2019, 20, 7. [Google Scholar] [CrossRef]
- Robert, C.; Schachter, J.; Long, G.V.; Arance, A.; Grob, J.J.; Mortier, L.; Daud, A.; Carlino, M.S.; McNeil, C.; Lotem, M.; et al. Pembrolizumab versus Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2015, 372, 2521–2532. [Google Scholar] [CrossRef]
- Hodi, F.S.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Rutkowski, P.; Cowey, C.L.; Lao, C.D.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Nivolumab plus ipilimumab or nivolumab alone versus ipilimumab alone in advanced melanoma (CheckMate 067): 4-year outcomes of a multicentre, randomised, phase 3 trial. Lancet Oncol. 2018, 19, 1480–1492. [Google Scholar] [CrossRef]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.-J.; Rutkowski, P.; Lao, C.D.; Cowey, C.L.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Five-Year Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2019, 381, 1535–1546. [Google Scholar] [CrossRef]
- Arozarena, I.; Wellbrock, C. Targeting invasive properties of melanoma cells. FEBS J. 2017, 284, 2148–2162. [Google Scholar] [CrossRef]
- Sanz-Moreno, V.; Gadea, G.; Ahn, J.; Paterson, H.; Marra, P.; Pinner, S.; Sahai, E.; Marshall, C.J. Rac Activation and Inactivation Control Plasticity of Tumor Cell Movement. Cell 2008, 135, 510–523. [Google Scholar] [CrossRef] [PubMed]
- Sahai, E.; Marshall, C.J. Differing modes for tumour cell invasion have distinct requirements for Rho/ROCK signalling and extracellular proteolysis. Nat. Cell Biol. 2003, 5, 711–719. [Google Scholar] [CrossRef] [PubMed]
- Gadea, G.; Sanz-Moreno, V.; Self, A.; Godi, A.; Marshall, C.J. DOCK10-Mediated Cdc42 Activation Is Necessary for Amoeboid Invasion of Melanoma Cells. Curr. Biol. 2008, 18, 1456–1465. [Google Scholar] [CrossRef] [PubMed]
- Hodis, E.; Watson, I.R.; Kryukov, G.V.; Arold, S.T.; Imielinski, M.; Theurillat, J.P.; Nickerson, E.; Auclair, D.; Li, L.; Place, C.; et al. A landscape of driver mutations in melanoma. Cell 2012, 150, 251–263. [Google Scholar] [CrossRef] [PubMed]
- Krauthammer, M.; Kong, Y.; Ha, B.H.; Evans, P.; Bacchiocchi, A.; McCusker, J.P.; Cheng, E.; Davis, M.J.; Goh, G.; Choi, M.; et al. Exome sequencing identifies recurrent somatic RAC1 mutations in melanoma. Nat. Genet. 2012, 44, 1006–1014. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.J.; Ha, B.H.; Holman, E.C.; Halaban, R.; Schlessinger, J.; Boggon, T.J. RAC1P29S is a spontaneously activating cancer-associated GTPase. Proc. Natl. Acad. Sci. USA 2013, 110, 912–917. [Google Scholar] [CrossRef]
- Watson, I.R.; Li, L.; Cabeceiras, P.K.; Mahdavi, M.; Gutschner, T.; Genovese, G.; Wang, G.; Fang, Z.; Tepper, J.M.; Stemke-Hale, K.; et al. The RAC1 P29S hotspot mutation in melanoma confers resistance to pharmacological inhibition of RAF. Cancer Res. 2014, 74, 4845–4852. [Google Scholar] [CrossRef] [PubMed]
- Araiza-Olivera, D.; Feng, Y.; Semenova, G.; Prudnikova, T.Y.; Rhodes, J.; Chernoff, J. Suppression of RAC1-driven malignant melanoma by group A PAK inhibitors. Oncogene 2018, 37, 944–952. [Google Scholar] [CrossRef]
- Dalton, L.E.; Kamarashev, J.; Barinaga-Rementeria Ramirez, I.; White, G.; Malliri, A.; Hurlstone, A. Constitutive rac activation is not sufficient to initiate melanocyte neoplasia but accelerates malignant progression. J. Investig. Dermatol. 2013, 133, 1572–1581. [Google Scholar] [CrossRef]
- Li, A.; Ma, Y.; Jin, M.; Mason, S.; Mort, R.L.; Blyth, K.; Larue, L.; Sansom, O.J.; MacHesky, L.M. Activated mutant NRas Q61K drives aberrant melanocyte signaling, survival, and invasiveness via a Rac1-dependent mechanism. J. Investig. Dermatol. 2012, 132, 2610–2621. [Google Scholar] [CrossRef]
- Berger, M.F.; Hodis, E.; Heffernan, T.P.; Deribe, Y.L.; Lawrence, M.S.; Protopopov, A.; Ivanova, E.; Watson, I.R.; Nickerson, E.; Ghosh, P.; et al. Melanoma genome sequencing reveals frequent PREX2 mutations. Nature 2012, 485, 502–506. [Google Scholar] [CrossRef]
- Lindsay, C.R.; Lawn, S.; Campbell, A.D.; Faller, W.J.; Rambow, F.; Mort, R.L.; Timpson, P.; Li, A.; Cammareri, P.; Ridgway, R.A.; et al. P-Rex1 is required for efficient melanoblast migration and melanoma metastasis. Nat. Commun. 2011, 2, 555. [Google Scholar] [CrossRef]
- Monaghan-Benson, E.; Burridge, K. Mutant B-RAF regulates a Rac-dependent cadherin switch in melanoma. Oncogene 2013, 32, 4836–4844. [Google Scholar] [CrossRef]
- Kaczorowski, M.; Biecek, P.; Donizy, P.; Pieniazek, M.; Matkowski, R.; Halon, A. Low rhoa expression is associated with adverse outcome in melanoma patients: A clinicopathological analysis. Am. J. Transl. Res. 2019, 11, 4524–4532. [Google Scholar]
- Espinha, G.; Osaki, J.H.; Costa, E.T.; Forti, F.L. Inhibition of the RhoA GTPase activity increases sensitivity of melanoma cells to UV radiation effects. Oxid. Med. Cell. Longev. 2016, 2016, 2696952. [Google Scholar] [CrossRef]
- Goundiam, O.; Nagel, M.; Vayssade, M. Akt and RhoA inhibition promotes anoikis of aggregated B16F10 melanoma cells. Cell Biol. Int. 2012, 36, 311–319. [Google Scholar] [CrossRef] [PubMed]
- Dua, P.; Gude, R.P. Pentoxifylline impedes migration in B16F10 melanoma by modulating Rho GTPase activity and actin organisation. Eur. J. Cancer 2008, 44, 1587–1595. [Google Scholar] [CrossRef] [PubMed]
- Sarrabayrouse, G.; Synaeve, C.; Leveque, K.; Favre, G.; Tilkin-Mariamé, A.F. Statins stimulate in vitro membrane FasL expression and lymphocyte apoptosis through RhoA/ROCK pathway in murine melanoma cells. Neoplasia 2007, 9, 1078–1090. [Google Scholar] [CrossRef]
- Díaz-Núñez, M.; Díez-Torre, A.; De Wever, O.; Andrade, R.; Arluzea, J.; Silió, M.; Aréchaga, J. Histone deacetylase inhibitors induce invasion of human melanoma cells in vitro via differential regulation of N-cadherin expression and RhoA activity. BMC Cancer 2016, 16, 667. [Google Scholar] [CrossRef]
- Yamamura, S.; Hakomori, S.; Wada, A.; Igarashi, Y. Sphingosine-1-phosphate inhibits haptotactic motility by overproduction of focal adhesion sites in B16 melanoma cells through EDG-induced activation of Rho. Ann. N. Y. Acad. Sci. 2000, 905, 301–307. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, M.; Hayashi, K.; Egi, Y.; Katayama, K.I.; Amano, Y.; Uehata, M.; Ohtsuki, M.; Fujii, A.; Oshita, K.I.; Kataoka, H.; et al. Effect of Wf-536, a novel ROCK inhibitor, against metastasis of B16 melanoma. Cancer Chemother. Pharmacol. 2003, 52, 319–324. [Google Scholar] [CrossRef] [PubMed]
- Klein, R.M.; Higgins, P.J. A switch in RND3-RHOA signaling is critical for melanoma cell invasion following mutant-BRAF inhibition. Mol. Cancer 2011, 10, 114. [Google Scholar] [CrossRef] [PubMed]
- Routhier, A.; Astuccio, M.; Lahey, D.; Monfredo, N.; Johnson, A.; Callahan, W.; Partington, A.; Fellows, K.; Ouellette, L.; Zhidro, S.; et al. Pharmacological inhibition of Rho-kinase signaling with Y-27632 blocks melanoma tumor growth. Oncol. Rep. 2010, 23, 861–867. [Google Scholar] [CrossRef] [PubMed]
- McHardy, T.; Caldwell, J.J.; Cheung, K.M.; Hunter, L.J.; Taylor, K.; Rowlands, M.; Ruddle, R.; Henley, A.; De Brandon, A.H.; Valenti, M.; et al. Discovery of 4-amino-l-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)piperidine-4- carboxamides as selective, orally active inhibitors of protein kinase B (Akt). J. Med. Chem. 2010, 53, 2239–2249. [Google Scholar] [CrossRef]
- Yap, T.A.; Walton, M.I.; Grimshaw, K.M.; Te Poele, R.H.; Eve, P.D.; Valenti, M.R.; De Haven Brandon, A.K.; Martins, V.; Zetterlund, A.; Heaton, S.P.; et al. AT13148 is a novel, oral multi-AGC kinase inhibitor with potent pharmacodynamic and antitumor activity. Clin. Cancer Res. 2012, 18, 3912–3923. [Google Scholar] [CrossRef]
- Sadok, A.; McCarthy, A.; Caldwell, J.; Collins, I.; Garrett, M.D.; Yeo, M.; Hooper, S.; Sahai, E.; Kuemper, S.; Mardakheh, F.K.; et al. Rho kinase inhibitors block melanoma cell migration and inhibit metastasis. Cancer Res. 2015, 75, 2272–2284. [Google Scholar] [CrossRef]
- Chang, F.; Zhang, Y.; Mi, J.; Zhou, Q.; Bai, F.; Xu, X.; Fisher, D.E.; Sun, Q.; Wu, X. ROCK inhibitor enhances the growth and migration of BRAF-mutant skin melanoma cells. Cancer Sci. 2018, 109, 3428–3437. [Google Scholar] [CrossRef]
- Ekström, E.J.; Bergenfelz, C.; von Bülow, V.; Serifler, F.; Carlemalm, E.; Jönsson, G.; Andersson, T.; Leandersson, K. WNT5A induces release of exosomes containing pro-angiogenic and immunosuppressive factors from malignant melanoma cells. Mol. Cancer 2014, 13, 88. [Google Scholar] [CrossRef] [PubMed]
- Ho, H.; Aruri, J.; Kapadia, R.; Mehr, H.; White, M.A.; Ganesan, A.K. RhoJ regulates melanoma chemoresistance by suppressing pathways that sense DNA damage. Cancer Res. 2012, 72, 5516–5528. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, R.; Jahid, S.; Harris, M.; Marzese, D.M.; Espitia, F.; Vasudeva, P.; Chen, C.F.; de Feraudy, S.; Wu, J.; Gillen, D.L.; et al. The RhoJ-BAD signaling network: An Achilles’ heel for BRAF mutant melanomas. PLoS Genet. 2017, 13, e1006913. [Google Scholar] [CrossRef]
- Ho, H.; Soto Hopkin, A.; Kapadia, R.; Vasudeva, P.; Schilling, J.; Ganesan, A.K. RhoJ modulates melanoma invasion by altering actin cytoskeletal dynamics. Pigment Cell Melanoma Res. 2013, 26, 218–225. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pecora, A.; Laprise, J.; Dahmene, M.; Laurin, M. Skin Cancers and the Contribution of Rho GTPase Signaling Networks to Their Progression. Cancers 2021, 13, 4362. https://doi.org/10.3390/cancers13174362
Pecora A, Laprise J, Dahmene M, Laurin M. Skin Cancers and the Contribution of Rho GTPase Signaling Networks to Their Progression. Cancers. 2021; 13(17):4362. https://doi.org/10.3390/cancers13174362
Chicago/Turabian StylePecora, Alessandra, Justine Laprise, Manel Dahmene, and Mélanie Laurin. 2021. "Skin Cancers and the Contribution of Rho GTPase Signaling Networks to Their Progression" Cancers 13, no. 17: 4362. https://doi.org/10.3390/cancers13174362
APA StylePecora, A., Laprise, J., Dahmene, M., & Laurin, M. (2021). Skin Cancers and the Contribution of Rho GTPase Signaling Networks to Their Progression. Cancers, 13(17), 4362. https://doi.org/10.3390/cancers13174362