
Genetic Heterogeneity, Therapeutic Hurdle Confronting Sorafenib and Immune Checkpoint Inhibitors in Hepatocellular Carcinoma


Abstract
Simple Summary
Abstract
1. Introduction
2. HCC Conventional Therapy
3. Sorafenib as a Frontline Therapy for Advanced HCC
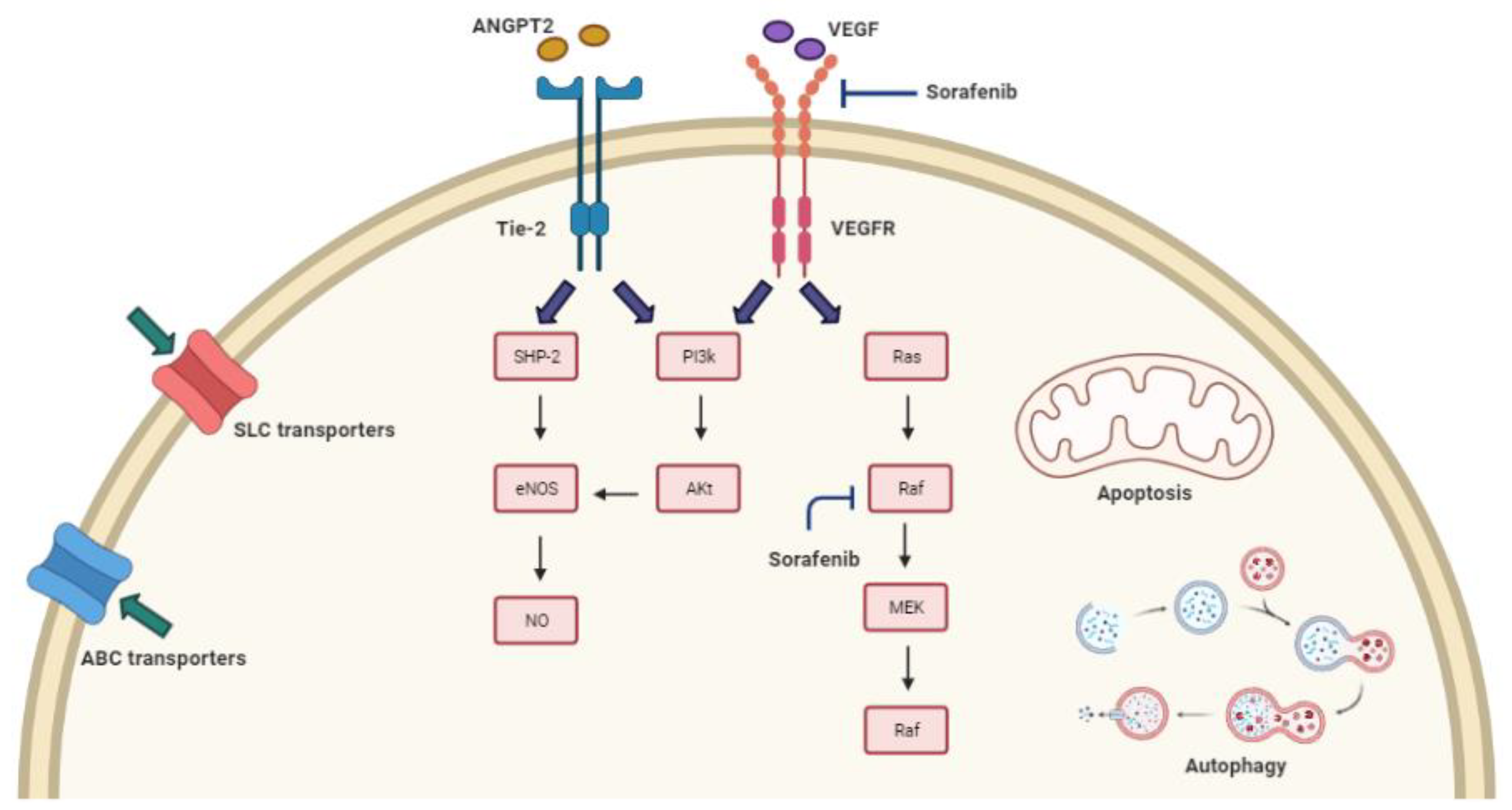
3.1. Mechanisms of Sorafenib Resistance and the Related Genomic Background
3.2. Sorafenib Resistance and Drug Transport across Cell Membrane
3.2.1. ABC Transporters
3.2.2. SLC Transporters
3.3. Sorafenib Resistance and Imbalance in the Regulation of Cell Death
3.3.1. Autophagy
3.3.2. Apoptosis
3.4. Sorafenib Resistance Based on Genetic Alterations of Molecular Targets and Signaling Pathways
3.5. Sorafenib Resistance and Polymorphisms of eNOS and ANGPT-2 Genes
4. Immune Checkpoint Inhibitors; an Update of HCC Therapeutic Armamentarium
Immune Checkpoint Resistance in HCC and Genomic Background
5. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- Torre, L.A.; Bray, F.; Siegel, R.L.; Ferlay, J.; Lortet-Tieulent, J.; Jemal, A. Global cancer statistics, 2012. CA A Cancer J. Clin. 2015, 65, 87–108. [Google Scholar] [CrossRef]
- Llovet, J.M.; Zucman-Rossi, J.; Pikarsky, E.; Sangro, B.; Schwartz, M.; Sherman, M.; Gores, G. Hepatocellular carcinoma. Nat. Rev. Dis. Primers 2016, 2, 16018. [Google Scholar] [CrossRef]
- Cheng, A.L.; Kang, Y.K.; Chen, Z.; Tsao, C.J.; Qin, S.; Kim, J.S.; Luo, R.; Feng, J.; Ye, S.; Yang, T.S.; et al. Efficacy and safety of sorafenib in patients in the Asia-Pacific region with advanced hepatocellular carcinoma: A phase III randomised, double-blind, placebo-controlled trial. Lancet Oncol. 2009, 10, 25–34. [Google Scholar] [CrossRef]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.-F.; de Oliveira, A.C.; Santoro, A.; Raoul, J.-L.; Forner, A.; et al. Sorafenib in Advanced Hepatocellular Carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef]
- Liu, Z.; Lin, Y.; Zhang, J.; Zhang, Y.; Li, Y.; Liu, Z.; Li, Q.; Luo, M.; Liang, R.; Ye, J. Molecular targeted and immune checkpoint therapy for advanced hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 2019, 38, 447. [Google Scholar] [CrossRef]
- Cheng, A.-L.; Finn, R.S.; Qin, S.; Han, K.-H.; Ikeda, K.; Piscaglia, F.; Baron, A.D.; Park, J.-W.; Han, G.; Jassem, J.; et al. Phase III trial of lenvatinib (LEN) vs sorafenib (SOR) in first-line treatment of patients (pts) with unresectable hepatocellular carcinoma (uHCC). J. Clin. Oncol. 2017, 35, 4001. [Google Scholar] [CrossRef]
- Mody, K.; Abou-Alfa, G.K. Systemic Therapy for Advanced Hepatocellular Carcinoma in an Evolving Landscape. Curr. Treat Options Oncol. 2019, 20, 3. [Google Scholar] [CrossRef]
- Kudo, M. Immune Checkpoint Inhibition in Hepatocellular Carcinoma: Basics and Ongoing Clinical Trials. Oncology 2017, 92 (Suppl. S1), 50–62. [Google Scholar] [CrossRef]
- Iñarrairaegui, M.; Melero, I.; Sangro, B. Immunotherapy of Hepatocellular Carcinoma: Facts and Hopes. Clin. Cancer Res. 2018, 24, 1518–1524. [Google Scholar] [CrossRef] [PubMed]
- Shrestha, R.; Prithviraj, P.; Anaka, M.; Bridle, K.R.; Crawford, D.H.G.; Dhungel, B.; Steel, J.C.; Jayachandran, A. Monitoring Immune Checkpoint Regulators as Predictive Biomarkers in Hepatocellular Carcinoma. Front. Oncol. 2018, 8, 269. [Google Scholar] [CrossRef] [PubMed]
- Makarova-Rusher, O.V.; Medina-Echeverz, J.; Duffy, A.G.; Greten, T.F. The yin and yang of evasion and immune activation in HCC. J. Hepatol. 2015, 62, 1420–1429. [Google Scholar] [CrossRef]
- Abd El Aziz, M.A.; Facciorusso, A.; Nayfeh, T.; Saadi, S.; Elnaggar, M.; Cotsoglou, C.; Sacco, R. Immune Checkpoint Inhibitors for Unresectable Hepatocellular Carcinoma. Vaccines 2020, 8, 616. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Dang, H.; Wang, X.W. The significance of intertumor and intratumor heterogeneity in liver cancer. Exp. Mol. Med. 2018, 50, e416. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.D.; Hainaut, P.; Gores, G.J.; Amadou, A.; Plymoth, A.; Roberts, L.R. A global view of hepatocellular carcinoma: Trends, risk, prevention and management. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 589–604. [Google Scholar] [CrossRef]
- Serper, M.; Taddei, T.H.; Mehta, R.; D’Addeo, K.; Dai, F.; Aytaman, A.; Baytarian, M.; Fox, R.; Hunt, K.; Goldberg, D.S.; et al. Association of Provider Specialty and Multidisciplinary Care With Hepatocellular Carcinoma Treatment and Mortality. Gastroenterology 2017, 152, 1954–1964. [Google Scholar] [CrossRef]
- Johnson, T.M.; Overgard, E.B.; Cohen, A.E.; DiBaise, J.K. EASL-EORTC clinical practice guidelines: Management of hepatocellular carcinoma. J. Hepatol. 2012, 56, 908–943. [Google Scholar] [CrossRef]
- Belghiti, J.; Fuks, D. Liver resection and transplantation in hepatocellular carcinoma. Liver Cancer 2012, 1, 71–82. [Google Scholar] [CrossRef]
- Tabrizian, P.; Jibara, G.; Shrager, B.; Schwartz, M.; Roayaie, S. Recurrence of hepatocellular cancer after resection: Patterns, treatments, and prognosis. Ann. Surg. 2015, 261, 947–955. [Google Scholar] [CrossRef]
- Yang, J.D.; Roberts, L.R. Hepatocellular carcinoma: A global view. Nat. Rev. Gastroenterol. Hepatol. 2010, 7, 448–458. [Google Scholar] [CrossRef]
- Takayasu, K.; Arii, S.; Ikai, I.; Omata, M.; Okita, K.; Ichida, T.; Matsuyama, Y.; Nakanuma, Y.; Kojiro, M.; Makuuchi, M.; et al. Prospective Cohort Study of Transarterial Chemoembolization for Unresectable Hepatocellular Carcinoma in 8510 Patients. Gastroenterology 2006, 131, 461–469. [Google Scholar] [CrossRef]
- Lencioni, R.; de Baere, T.; Soulen, M.C.; Rilling, W.S.; Geschwind, J.F. Lipiodol transarterial chemoembolization for hepatocellular carcinoma: A systematic review of efficacy and safety data. Hepatology 2016, 64, 106–116. [Google Scholar] [CrossRef]
- Yao, F.Y.; Hirose, R.; LaBerge, J.M.; Davern, T.J., 3rd; Bass, N.M.; Kerlan, R.K., Jr.; Merriman, R.; Feng, S.; Freise, C.E.; Ascher, N.L.; et al. A prospective study on downstaging of hepatocellular carcinoma prior to liver transplantation. Liver Transpl. 2005, 11, 1505–1514. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, S.; Carter, C.; Lynch, M.; Lowinger, T.; Dumas, J.; Smith, R.A.; Schwartz, B.; Simantov, R.; Kelley, S. Discovery and development of sorafenib: A multikinase inhibitor for treating cancer. Nat. Rev. Drug Discov. 2006, 5, 835–844. [Google Scholar] [CrossRef]
- Abou-Alfa, G.K.; Niedzwieski, D.; Knox, J.J.; Kaubisch, A.; Posey, J.; Tan, B.R.; Kavan, P.; Goel, R.; Murray, J.J.; Bekaii-Saab, T.S. Phase III Randomized Study of Sorafenib Plus Doxorubicin Versus Sorafenib in Patients with Advanced Hepatocellular Carcinoma (HCC): CALGB 80802 (Alliance); American Society of Clinical Oncology: Alexandria, VA, USA, 2016. [Google Scholar]
- Cheng, A.L.; Kang, Y.K.; Lin, D.Y.; Park, J.W.; Kudo, M.; Qin, S.; Chung, H.C.; Song, X.; Xu, J.; Poggi, G.; et al. Sunitinib versus sorafenib in advanced hepatocellular cancer: Results of a randomized phase III trial. J. Clin. Oncol. 2013, 31, 4067–4075. [Google Scholar] [CrossRef]
- Johnson, P.J.; Qin, S.; Park, J.W.; Poon, R.T.; Raoul, J.L.; Philip, P.A.; Hsu, C.H.; Hu, T.H.; Heo, J.; Xu, J.; et al. Brivanib versus sorafenib as first-line therapy in patients with unresectable, advanced hepatocellular carcinoma: Results from the randomized phase III BRISK-FL study. J. Clin. Oncol. 2013, 31, 3517–3524. [Google Scholar] [CrossRef] [PubMed]
- Cainap, C.; Qin, S.; Huang, W.T.; Chung, I.J.; Pan, H.; Cheng, Y.; Kudo, M.; Kang, Y.K.; Chen, P.J.; Toh, H.C.; et al. Linifanib versus Sorafenib in patients with advanced hepatocellular carcinoma: Results of a randomized phase III trial. J. Clin. Oncol. 2015, 33, 172–179. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, S.M.; Carter, C.; Tang, L.; Wilkie, D.; McNabola, A.; Rong, H.; Chen, C.; Zhang, X.; Vincent, P.; McHugh, M. BAY 43-9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res. 2004, 64, 7099–7109. [Google Scholar] [CrossRef]
- Ito, Y.; Sasaki, Y.; Horimoto, M.; Wada, S.; Tanaka, Y.; Kasahara, A.; Ueki, T.; Hirano, T.; Yamamoto, H.; Fujimoto, J. Activation of mitogen-activated protein kinases/extracellular signal-regulated kinases in human hepatocellular carcinoma. Hepatology 1998, 27, 951–958. [Google Scholar] [CrossRef]
- Lord, R.; Suddle, A.; Ross, P.J. Emerging strategies in the treatment of advanced hepatocellular carcinoma: The role of targeted therapies. Int. J. Clin. Pract. 2011, 65, 182–188. [Google Scholar] [CrossRef]
- Llovet, J.M.; Bruix, J. Molecular targeted therapies in hepatocellular carcinoma. Hepatology 2008, 48, 1312–1327. [Google Scholar] [CrossRef]
- Chen, W.; Yang, J.; Zhang, Y.; Cai, H.; Chen, X.; Sun, D. Regorafenib reverses HGF-induced sorafenib resistance by inhibiting epithelial-mesenchymal transition in hepatocellular carcinoma. FEBS Open Bio. 2019, 9, 335–347. [Google Scholar] [CrossRef]
- Beretta, G.L.; Cassinelli, G.; Pennati, M.; Zuco, V.; Gatti, L. Overcoming ABC transporter-mediated multidrug resistance: The dual role of tyrosine kinase inhibitors as multitargeting agents. Eur. J. Med. Chem. 2017, 142, 271–289. [Google Scholar] [CrossRef] [PubMed]
- Tandia, M.; Mhiri, A.; Paule, B.; Saffroy, R.; Cailliez, V.; Noé, G.; Farinotti, R.; Bonhomme-Faivre, L. Correlation between clinical response to sorafenib in hepatocellular carcinoma treatment and polymorphisms of P-glycoprotein (ABCB1) and of breast cancer resistance protein (ABCG2): Monocentric study. Cancer Chemother. Pharmacol. 2017, 79, 759–766. [Google Scholar] [CrossRef] [PubMed]
- Wei, D.; Zhang, H.; Peng, R.; Huang, C.; Bai, R. ABCC2 (1249G > A) polymorphism implicates altered transport activity for sorafenib. Xenobiotica 2017, 47, 1008–1014. [Google Scholar] [CrossRef]
- Herraez, E.; Lozano, E.; Macias, R.I.R.; Vaquero, J.; Bujanda, L.; Banales, J.M.; Marin, J.J.G.; Briz, O. Expression of SLC22A1 variants may affect the response of hepatocellular carcinoma and cholangiocarcinoma to sorafenib. Hepatology 2013, 58, 1065–1073. [Google Scholar] [CrossRef]
- Alonso-Peña, M.; Sanchez-Martin, A.; Sanchon-Sanchez, P.; Soto-Muñiz, M.; Espinosa-Escudero, R.; Marin, J.J.G. Pharmacogenetics of hepatocellular carcinoma and cholangiocarcinoma. Cancer Drug Resist. 2019, 2, 680–709. [Google Scholar] [CrossRef]
- Lee, Y.S.; Kim, B.H.; Kim, B.C.; Shin, A.; Kim, J.S.; Hong, S.H.; Hwang, J.A.; Lee, J.A.; Nam, S.; Lee, S.H.; et al. SLC15A2 genomic variation is associated with the extraordinary response of sorafenib treatment: Whole-genome analysis in patients with hepatocellular carcinoma. Oncotarget 2015, 6, 16449–16460. [Google Scholar] [CrossRef]
- Brecht, K.; Schäfer, A.M.; Meyer Zu Schwabedissen, H.E. Uptake Transporters of the SLC21, SLC22A, and SLC15A Families in Anticancer Therapy-Modulators of Cellular Entry or Pharmacokinetics? Cancers 2020, 12, 2263. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.-B.; Zhan, M.-X.; Zhao, W.; Liu, B.; Huang, J.-W.; He, X.; Fu, S.-R.; Zhao, Y.; Li, Y.; Hu, B.-S.; et al. The relationship of kinase insert domain receptor gene polymorphisms and clinical outcome in advanced hepatocellular carcinoma patients treated with sorafenib. Med. Oncol. 2014, 31, 209. [Google Scholar] [CrossRef]
- Scartozzi, M.; Faloppi, L.; Svegliati Baroni, G.; Loretelli, C.; Piscaglia, F.; Iavarone, M.; Toniutto, P.; Fava, G.; De Minicis, S.; Mandolesi, A.; et al. VEGF and VEGFR genotyping in the prediction of clinical outcome for HCC patients receiving sorafenib: The ALICE-1 study. Int. J. Cancer 2014, 135, 1247–1256. [Google Scholar] [CrossRef]
- Casadei Gardini, A.; Marisi, G.; Faloppi, L.; Scarpi, E.; Foschi, F.G.; Iavarone, M.; Lauletta, G.; Corbelli, J.; Valgiusti, M.; Facchetti, F.; et al. eNOS polymorphisms and clinical outcome in advanced HCC patients receiving sorafenib: Final results of the ePHAS study. Oncotarget 2016, 7, 27988–27999. [Google Scholar] [CrossRef]
- Marisi, G.; Petracci, E.; Raimondi, F.; Faloppi, L.; Foschi, F.G.; Lauletta, G.; Iavarone, M.; Canale, M.; Valgiusti, M.; Neri, L.M.; et al. ANGPT2 and NOS3 Polymorphisms and Clinical Outcome in Advanced Hepatocellular Carcinoma Patients Receiving Sorafenib. Cancers 2019, 11, 1023. [Google Scholar] [CrossRef] [PubMed]
- Gillet, J.-P.; Gottesman, M.M. Mechanisms of Multidrug Resistance in Cancer. In Multi-Drug Resistance in Cancer; Zhou, J., Ed.; Humana Press: Totowa, NJ, USA, 2010; pp. 47–76. [Google Scholar]
- Chu, X.; Korzekwa, K.; Elsby, R.; Fenner, K.; Galetin, A.; Lai, Y.; Matsson, P.; Moss, A.; Nagar, S.; Rosania, G.R.; et al. Intracellular drug concentrations and transporters: Measurement, modeling, and implications for the liver. Clin Pharm. 2013, 94, 126–141. [Google Scholar] [CrossRef] [PubMed]
- Higgins, C.F. ABC Transporters: From Microorganisms to Man. Annu. Rev. Cell Biol. 1992, 8, 67–113. [Google Scholar] [CrossRef] [PubMed]
- Dean, M.; Allikmets, R. Complete characterization of the human ABC gene family. J. Bioenerg. Biomembr. 2001, 33, 475–479. [Google Scholar] [CrossRef]
- Robey, R.W.; Pluchino, K.M.; Hall, M.D.; Fojo, A.T.; Bates, S.E.; Gottesman, M.M. Revisiting the role of ABC transporters in multidrug-resistant cancer. Nat. Rev. Cancer 2018, 18, 452–464. [Google Scholar] [CrossRef] [PubMed]
- Gatti, L.; Cossa, G.; Beretta, G.L.; Zaffaroni, N.; Perego, P. Novel insights into targeting ATP-binding cassette transporters for antitumor therapy. Curr. Med. Chem. 2011, 18, 4237–4249. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Q.; Zhou, Y.; Lauschke, V.M. Ethnogeographic and inter-individual variability of human ABC transporters. Hum. Genet. 2020, 139, 623–646. [Google Scholar] [CrossRef]
- Lepper, E.R.; Nooter, K.; Verweij, J.; Acharya, M.R.; Figg, W.D.; Sparreboom, A. Mechanisms of resistance to anticancer drugs: The role of the polymorphic ABC transporters ABCB1 and ABCG2. Pharmacogenomics 2005, 6, 115–138. [Google Scholar] [CrossRef] [PubMed]
- Gao, J. Association of MDR1 gene polymorphisms with the risk of hepatocellular carcinoma in the Chinese Han population. Braz. J. Med. Biol. Res. 2013, 46, 311–317. [Google Scholar] [CrossRef][Green Version]
- Li, X.F.; He, H.B.; Zhu, Y.S.; He, J.K.; Ye, W.W.; Chen, Y.X.; Lou, L.Q. Association between the c.3751G>a genetic variant of MDR1 and hepatocellular carcinoma risk in a Chinese Han population. Asian Pac. J. Cancer Prev. 2013, 14, 5361–5365. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ren, Y.Q.; Han, J.Q.; Cao, J.B.; Li, S.X.; Fan, G.R. Association of MDR1 gene polymorphisms with susceptibility to hepatocellular carcinoma in the Chinese population. Asian Pac. J. Cancer Prev. 2012, 13, 5451–5454. [Google Scholar] [CrossRef][Green Version]
- Borel, F.; Han, R.; Visser, A.; Petry, H.; van Deventer, S.J.; Jansen, P.L.; Konstantinova, P. Adenosine triphosphate-binding cassette transporter genes up-regulation in untreated hepatocellular carcinoma is mediated by cellular microRNAs. Hepatology 2012, 55, 821–832. [Google Scholar] [CrossRef]
- Huang, W.C.; Hsieh, Y.L.; Hung, C.M.; Chien, P.H.; Chien, Y.F.; Chen, L.C.; Tu, C.Y.; Chen, C.H.; Hsu, S.C.; Lin, Y.M.; et al. BCRP/ABCG2 inhibition sensitizes hepatocellular carcinoma cells to sorafenib. PLoS ONE 2013, 8, e83627. [Google Scholar] [CrossRef] [PubMed]
- Timucin, M.; Alagozlu, H.; Ozdemir, S.; Ozdemir, O. Association between ABCB1 (MDR1) Gene polymorphism and unresponsiveness combined therapy in chronic hepatitis C virus. Hepat. Mon. 2013, 13, e7522. [Google Scholar] [CrossRef]
- Fredriksson, R.; Nordström, K.J.; Stephansson, O.; Hägglund, M.G.; Schiöth, H.B. The solute carrier (SLC) complement of the human genome: Phylogenetic classification reveals four major families. FEBS Lett. 2008, 582, 3811–3816. [Google Scholar] [CrossRef]
- Höglund, P.J.; Nordström, K.J.; Schiöth, H.B.; Fredriksson, R. The solute carrier families have a remarkably long evolutionary history with the majority of the human families present before divergence of Bilaterian species. Mol. Biol. Evol. 2011, 28, 1531–1541. [Google Scholar] [CrossRef]
- Schlessinger, A.; Yee, S.W.; Sali, A.; Giacomini, K.M. SLC classification: An update. Clin. Pharmacol. Ther. 2013, 94, 19–23. [Google Scholar] [CrossRef] [PubMed]
- Colas, C.; Ung, P.M.-U.; Schlessinger, A. SLC Transporters: Structure, Function, and Drug Discovery. Medchemcomm 2016, 7, 1069–1081. [Google Scholar] [CrossRef]
- Lin, L.; Yee, S.W.; Kim, R.B.; Giacomini, K.M. SLC transporters as therapeutic targets: Emerging opportunities. Nat. Rev. Drug Discov. 2015, 14, 543–560. [Google Scholar] [CrossRef]
- Schaller, L.; Lauschke, V.M. The genetic landscape of the human solute carrier (SLC) transporter superfamily. Hum. Genet. 2019, 138, 1359–1377. [Google Scholar] [CrossRef] [PubMed]
- Minematsu, T.; Giacomini, K.M. Interactions of tyrosine kinase inhibitors with organic cation transporters and multidrug and toxic compound extrusion proteins. Mol. Cancer Ther. 2011, 10, 531–539. [Google Scholar] [CrossRef]
- Zhao, Q.; Zheng, B.; Meng, S.; Xu, Y.; Guo, J.; Chen, L.-J.; Xiao, J.; Zhang, W.; Tan, Z.-R.; Tang, J.; et al. Increased expression of SLC46A3 to oppose the progression of hepatocellular carcinoma and its effect on sorafenib therapy. Biomed. Pharmacother. 2019, 114, 108864. [Google Scholar] [CrossRef]
- Galimov, E.R.; Lohr, J.N.; Gems, D. When and How Can Death Be an Adaptation? Biochemistry 2019, 84, 1433–1437. [Google Scholar] [CrossRef]
- Lee, Y.; Overholtzer, M. After-Death Functions of Cell Death. Yale J. Biol. Med. 2019, 92, 687–694. [Google Scholar]
- Mizushima, N. Autophagy: Process and function. Genes Dev. 2007, 21, 2861–2873. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.S.; Vats, S.; Chia, A.Y.-Q.; Tan, T.Z.; Deng, S.; Ong, M.S.; Arfuso, F.; Yap, C.T.; Goh, B.C.; Sethi, G.; et al. Dual role of autophagy in hallmarks of cancer. Oncogene 2018, 37, 1142–1158. [Google Scholar] [CrossRef]
- Liu, J.; Fan, L.; Wang, H.; Sun, G. Autophagy, a double-edged sword in anti-angiogenesis therapy. Med. Oncol. 2015, 33, 10. [Google Scholar] [CrossRef]
- Che, N.; Ng, K.Y.; Wong, T.L.; Tong, M.; Kau, P.W.; Chan, L.H.; Lee, T.K.; Huen, M.S.; Yun, J.P.; Ma, S. PRMT6 deficiency induces autophagy in hostile microenvironments of hepatocellular carcinoma tumors by regulating BAG5-associated HSC70 stability. Cancer Lett. 2020. [Google Scholar] [CrossRef]
- Zhai, B.; Hu, F.; Jiang, X.; Xu, J.; Zhao, D.; Liu, B.; Pan, S.; Dong, X.; Tan, G.; Wei, Z.; et al. Inhibition of Akt reverses the acquired resistance to sorafenib by switching protective autophagy to autophagic cell death in hepatocellular carcinoma. Mol. Cancer 2014, 13, 1589–1598. [Google Scholar] [CrossRef]
- Shimizu, S.; Takehara, T.; Hikita, H.; Kodama, T.; Tsunematsu, H.; Miyagi, T.; Hosui, A.; Ishida, H.; Tatsumi, T.; Kanto, T.; et al. Inhibition of autophagy potentiates the antitumor effect of the multikinase inhibitor sorafenib in hepatocellular carcinoma. Int. J. Cancer 2012, 131, 548–557. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.H.; Ding, Z.B.; Zhou, J.; Hui, B.; Shi, G.M.; Ke, A.W.; Wang, X.Y.; Dai, Z.; Peng, Y.F.; Gu, C.Y.; et al. Targeting autophagy enhances sorafenib lethality for hepatocellular carcinoma via ER stress-related apoptosis. Autophagy 2011, 7, 1159–1172. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.; Chen, Z.; Zhang, W.; Cheng, Y.; Zhang, B.; Wu, F.; Wang, Q.; Wang, S.; Rong, D.; Reiter, F.P.; et al. The mechanisms of sorafenib resistance in hepatocellular carcinoma: Theoretical basis and therapeutic aspects. Signal Transduct. Target. Ther. 2020, 5, 87. [Google Scholar] [CrossRef]
- Shen, M.; Lin, L. Functional variants of autophagy-related genes are associated with the development of hepatocellular carcinoma. Life Sci. 2019, 235, 116675. [Google Scholar] [CrossRef]
- Yuan, J.; Zhang, N.; Yin, L.; Zhu, H.; Zhang, L.; Zhou, L.; Yang, M. Clinical implications of the autophagy core gene variations in advanced lung adenocarcinoma treated with Gefitinib. Sci. Rep. 2017, 7, 1–10. [Google Scholar] [CrossRef]
- Li, N.; Fan, X.; Wang, X.; Deng, H.; Zhang, K.; Zhang, X.; Han, Q.; Lv, Y.; Liu, Z. Autophagy-Related 5 Gene rs510432 Polymorphism Is Associated with Hepatocellular Carcinoma in Patients with Chronic Hepatitis B Virus Infection. Immunol. Investig. 2019, 48, 378–391. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Fan, X.; Wang, X.; Zhang, X.; Zhang, K.; Han, Q.; Lv, Y.; Liu, Z. Genetic association of polymorphisms at the intergenic region between PRDM1 and ATG5 with hepatitis B virus infection in Han Chinese patients. J. Med. Virol. 2020, 92, 1198–1205. [Google Scholar] [CrossRef]
- Mariño, G.; Niso-Santano, M.; Baehrecke, E.H.; Kroemer, G. Self-consumption: The interplay of autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 2014, 15, 81–94. [Google Scholar] [CrossRef]
- Maiuri, M.C.; Zalckvar, E.; Kimchi, A.; Kroemer, G. Self-eating and self-killing: Crosstalk between autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 2007, 8, 741–752. [Google Scholar] [CrossRef]
- Moreno-Càceres, J.; Fabregat, I. Apoptosis in liver carcinogenesis and chemotherapy. Hepat. Oncol. 2015, 2, 381–397. [Google Scholar] [CrossRef]
- Niu, L.; Liu, L.; Yang, S.; Ren, J.; Lai, P.B.S.; Chen, G.G. New insights into sorafenib resistance in hepatocellular carcinoma: Responsible mechanisms and promising strategies. Biochim. Biophys. Acta Rev. Cancer 2017, 1868, 564–570. [Google Scholar] [CrossRef]
- Shimizu, S.; Takehara, T.; Hikita, H.; Kodama, T.; Miyagi, T.; Hosui, A.; Tatsumi, T.; Ishida, H.; Noda, T.; Nagano, H.; et al. The let-7 family of microRNAs inhibits Bcl-xL expression and potentiates sorafenib-induced apoptosis in human hepatocellular carcinoma. J. Hepatol. 2010, 52, 698–704. [Google Scholar] [CrossRef] [PubMed]
- Tutusaus, A.; Stefanovic, M.; Boix, L.; Cucarull, B.; Zamora, A.; Blasco, L.; de Frutos, P.G.; Reig, M.; Fernandez-Checa, J.C.; Marí, M.; et al. Antiapoptotic BCL-2 proteins determine sorafenib/regorafenib resistance and BH3-mimetic efficacy in hepatocellular carcinoma. Oncotarget 2018, 9, 16701–16717. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.; Viatour, P. Hepatocellular carcinoma: Old friends and new tricks. Exp. Mol. Med. 2020. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Hernández, M.A.; Chapresto-Garzón, R.; Cadenas, M.; Navarro-Villarán, E.; Negrete, M.; Gómez-Bravo, M.A.; Victor, V.M.; Padillo, F.J.; Muntané, J. Differential effectiveness of tyrosine kinase inhibitors in 2D/3D culture according to cell differentiation, p53 status and mitochondrial respiration in liver cancer cells. Cell Death Dis. 2020, 11, 339. [Google Scholar] [CrossRef]
- Hussain, S.P.; Schwank, J.; Staib, F.; Wang, X.W.; Harris, C.C. TP53 mutations and hepatocellular carcinoma: Insights into the etiology and pathogenesis of liver cancer. Oncogene 2007, 26, 2166–2176. [Google Scholar] [CrossRef]
- Wei, J.-C.; Qu, K.; Wang, Z.-X.; Wu, Q.-F.; Zhang, L.-Q.; Pang, Q.; Liu, C. Sorafenib inhibits proliferation and invasion of human hepatocellular carcinoma cells via up-regulation of p53 and suppressing FoxM1. Acta Pharmacol. Sin. 2015, 36, 241–251. [Google Scholar] [CrossRef] [PubMed]
- Omar, H.A.; Tolba, M.F.; Hung, J.-H.; Al-Tel, T.H. OSU-2S/Sorafenib synergistic antitumor combination against hepatocellular carcinoma: The role of PKCδ/p53. Front. Pharmacol. 2016, 7, 463. [Google Scholar] [CrossRef]
- Pang, R.W.; Joh, J.W.; Johnson, P.J.; Monden, M.; Pawlik, T.M.; Poon, R.T. Biology of hepatocellular carcinoma. Ann. Surg. Oncol. 2008, 15, 962–971. [Google Scholar] [CrossRef]
- Poon, R.T.; Lau, C.; Pang, R.; Ng, K.K.; Yuen, J.; Fan, S.T. High serum vascular endothelial growth factor levels predict poor prognosis after radiofrequency ablation of hepatocellular carcinoma: Importance of tumor biomarker in ablative therapies. Ann. Surg. Oncol. 2007, 14, 1835–1845. [Google Scholar] [CrossRef]
- Marin, J.J.G.; Serrano, M.A.; Monte, M.J.; Sanchez-Martin, A.; Temprano, A.G.; Briz, O.; Romero, M.R. Role of Genetic Variations in the Hepatic Handling of Drugs. Int. J. Mol. Sci. 2020, 21, 2884. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zheng, Y.; Zhang, W.; Yu, H.; Lou, K.; Zhang, Y.; Qin, Q.; Zhao, B.; Yang, Y.; Hui, R. Polymorphisms of KDRGene are associated with coronary heart disease. J. Am. Coll. Cardiol. 2007, 50, 760–767. [Google Scholar] [CrossRef]
- Pinyol, R.; Montal, R.; Bassaganyas, L.; Sia, D.; Takayama, T.; Chau, G.Y.; Mazzaferro, V.; Roayaie, S.; Lee, H.C.; Kokudo, N.; et al. Molecular predictors of prevention of recurrence in HCC with sorafenib as adjuvant treatment and prognostic factors in the phase 3 STORM trial. Gut 2019, 68, 1065–1075. [Google Scholar] [CrossRef] [PubMed]
- Harding, J.J.; Nandakumar, S.; Armenia, J.; Khalil, D.N.; Albano, M.; Ly, M.; Shia, J.; Hechtman, J.F.; Kundra, R.; El Dika, I.; et al. Prospective Genotyping of Hepatocellular Carcinoma: Clinical Implications of Next-Generation Sequencing for Matching Patients to Targeted and Immune Therapies. Clin. Cancer Res. 2019, 25, 2116–2126. [Google Scholar] [CrossRef]
- Gnoni, A.; Licchetta, A.; Memeo, R.; Argentiero, A.; Solimando, A.G.; Longo, V.; Delcuratolo, S.; Brunetti, O. Role of BRAF in Hepatocellular Carcinoma: A Rationale for Future Targeted Cancer Therapies. Medicine 2019, 55, 754. [Google Scholar] [CrossRef] [PubMed]
- Sereno, M.; Moreno, V.; Moreno Rubio, J.; Gómez-Raposo, C.; García Sánchez, S.; Hernández Jusdado, R.; Falagan, S.; Zambrana Tébar, F.; Casado Sáenz, E. A significant response to sorafenib in a woman with advanced lung adenocarcinoma and a BRAF non-V600 mutation. Anticancer Drugs 2015, 26, 1004–1007. [Google Scholar] [CrossRef] [PubMed]
- Casadei Gardini, A.; Chiadini, E.; Faloppi, L.; Marisi, G.; Delmonte, A.; Scartozzi, M.; Loretelli, C.; Lucchesi, A.; Oboldi, D.; Dubini, A.; et al. Efficacy of sorafenib in BRAF-mutated non-small-cell lung cancer (NSCLC) and no response in synchronous BRAF wild type-hepatocellular carcinoma: A case report. BMC Cancer 2016, 16, 429. [Google Scholar] [CrossRef]
- Moawad, A.W.; Szklaruk, J.; Lall, C.; Blair, K.J.; Kaseb, A.O.; Kamath, A.; Rohren, S.A.; Elsayes, K.M. Angiogenesis in Hepatocellular Carcinoma; Pathophysiology, Targeted Therapy, and Role of Imaging. J. Hepatocell Carcinoma 2020, 7, 77–89. [Google Scholar] [CrossRef]
- Huang, H.; Bhat, A.; Woodnutt, G.; Lappe, R. Targeting the ANGPT–TIE2 pathway in malignancy. Nat. Rev. Cancer 2010, 10, 575–585. [Google Scholar] [CrossRef] [PubMed]
- Ziche, M.; Morbidelli, L. Molecular regulation of tumour angiogenesis by nitric oxide. Eur. Cytokine Netw. 2009, 20, 164–170. [Google Scholar] [CrossRef]
- Llovet, J.M.; Peña, C.E.; Lathia, C.D.; Shan, M.; Meinhardt, G.; Bruix, J. Plasma biomarkers as predictors of outcome in patients with advanced hepatocellular carcinoma. Clin. Cancer Res. 2012, 18, 2290–2300. [Google Scholar] [CrossRef]
- Miyahara, K.; Nouso, K.; Tomoda, T.; Kobayashi, S.; Hagihara, H.; Kuwaki, K.; Toshimori, J.; Onishi, H.; Ikeda, F.; Miyake, Y.; et al. Predicting the treatment effect of sorafenib using serum angiogenesis markers in patients with hepatocellular carcinoma. J. Gastroenterol. Hepatol. 2011, 26, 1604–1611. [Google Scholar] [CrossRef] [PubMed]
- Fares, C.M.; Van Allen, E.M.; Drake, C.G.; Allison, J.P.; Hu-Lieskovan, S. Mechanisms of Resistance to Immune Checkpoint Blockade: Why Does Checkpoint Inhibitor Immunotherapy Not Work for All Patients? Am. Soc. Clin. Oncol. Educ. Book 2019, 39, 147–164. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.C.; Sharma, R.; Anang, N.A.S.; Levine, J.H.; Zhao, Y.; Mancuso, J.J.; Setty, M.; Sharma, P.; Wang, J.; Pe’er, D.; et al. Negative Co-stimulation Constrains T Cell Differentiation by Imposing Boundaries on Possible Cell States. Immunity 2019, 50, 1084–1098.e1010. [Google Scholar] [CrossRef] [PubMed]
- Zheng, C.; Zheng, L.; Yoo, J.K.; Guo, H.; Zhang, Y.; Guo, X.; Kang, B.; Hu, R.; Huang, J.Y.; Zhang, Q.; et al. Landscape of Infiltrating T Cells in Liver Cancer Revealed by Single-Cell Sequencing. Cell 2017, 169, 1342–1356.e1316. [Google Scholar] [CrossRef]
- Sharma, P.; Allison, J.P. Dissecting the mechanisms of immune checkpoint therapy. Nat. Rev. Immunol. 2020, 20, 75–76. [Google Scholar] [CrossRef]
- Dal Bo, M.; De Mattia, E.; Baboci, L.; Mezzalira, S.; Cecchin, E.; Assaraf, Y.G.; Toffoli, G. New insights into the pharmacological, immunological, and CAR-T-cell approaches in the treatment of hepatocellular carcinoma. Drug Resist. Updates 2020, 51, 100702. [Google Scholar] [CrossRef] [PubMed]
- Collins, A.V.; Brodie, D.W.; Gilbert, R.J.C.; Iaboni, A.; Manso-Sancho, R.; Walse, B.; Stuart, D.I.; van der Merwe, P.A.; Davis, S.J. The Interaction Properties of Costimulatory Molecules Revisited. Immunity 2002, 17, 201–210. [Google Scholar] [CrossRef]
- Masteller, E.L.; Chuang, E.; Mullen, A.C.; Reiner, S.L.; Thompson, C.B. Structural Analysis of CTLA-4 Function In Vivo. J. Immunol. 2000, 164, 5319–5327. [Google Scholar] [CrossRef] [PubMed]
- Fallarino, F.; Fields, P.E.; Gajewski, T.F. B7-1 engagement of cytotoxic T lymphocyte antigen 4 inhibits T cell activation in the absence of CD28. J. Exp. Med. 1998, 188, 205–210. [Google Scholar] [CrossRef] [PubMed]
- Baumeister, S.H.; Freeman, G.J.; Dranoff, G.; Sharpe, A.H. Coinhibitory Pathways in Immunotherapy for Cancer. Annu. Rev. Immunol. 2016, 34, 539–573. [Google Scholar] [CrossRef]
- Keir, M.E.; Butte, M.J.; Freeman, G.J.; Sharpe, A.H. PD-1 and Its Ligands in Tolerance and Immunity. Annu. Rev. Immunol. 2008, 26, 677–704. [Google Scholar] [CrossRef] [PubMed]
- Fife, B.T.; Bluestone, J.A. Control of peripheral T-cell tolerance and autoimmunity via the CTLA-4 and PD-1 pathways. Immunol. Rev. 2008, 224, 166–182. [Google Scholar] [CrossRef] [PubMed]
- Pinato, D.J.; Guerra, N.; Fessas, P.; Murphy, R.; Mineo, T.; Mauri, F.A.; Mukherjee, S.K.; Thursz, M.; Wong, C.N.; Sharma, R.; et al. Immune-based therapies for hepatocellular carcinoma. Oncogene 2020, 39, 3620–3637. [Google Scholar] [CrossRef]
- Pinter, M.; Jain, R.K.; Duda, D.G. The Current Landscape of Immune Checkpoint Blockade in Hepatocellular Carcinoma: A Review. JAMA Oncol. 2020. [Google Scholar] [CrossRef]
- Sharma, P.; Allison, J.P. The future of immune checkpoint therapy. Science 2015, 348, 56–61. [Google Scholar] [CrossRef]
- El-Khoueiry, A.B.; Sangro, B.; Yau, T.; Crocenzi, T.S.; Kudo, M.; Hsu, C.; Kim, T.Y.; Choo, S.P.; Trojan, J.; Welling, T.H.R.; et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): An open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet 2017, 389, 2492–2502. [Google Scholar] [CrossRef]
- Yau, T.; Park, J.W.; Finn, R.S.; Cheng, A.L.; Mathurin, P.; Edeline, J.; Kudo, M.; Han, K.H.; Harding, J.J.; Merle, P.; et al. CheckMate 459: A randomized, multi-center phase III study of nivolumab (NIVO) vs sorafenib (SOR) as first-line (1L) treatment in patients (pts) with advanced hepatocellular carcinoma (aHCC). Ann. Oncol. 2019, 30, v874–v875. [Google Scholar] [CrossRef]
- Kudo, M.; Matilla, A.; Santoro, A.; Melero, I.; Gracian, A.C.; Acosta-Rivera, M.; Choo, S.P.; El-Khoueiry, A.B.; Kuromatsu, R.; El-Rayes, B.F.; et al. Checkmate-040: Nivolumab (NIVO) in patients (pts) with advanced hepatocellular carcinoma (aHCC) and Child-Pugh B (CPB) status. J. Clin. Oncol. 2019, 37, 327. [Google Scholar] [CrossRef]
- Zhu, A.X.; Finn, R.S.; Edeline, J.; Cattan, S.; Ogasawara, S.; Palmer, D.; Verslype, C.; Zagonel, V.; Fartoux, L.; Vogel, A.; et al. Pembrolizumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib (KEYNOTE-224): A non-randomised, open-label phase 2 trial. Lancet Oncol. 2018, 19, 940–952. [Google Scholar] [CrossRef]
- Finn, R.S.; Ryoo, B.Y.; Merle, P.; Kudo, M.; Bouattour, M.; Lim, H.Y.; Breder, V.; Edeline, J.; Chao, Y.; Ogasawara, S.; et al. Pembrolizumab As Second-Line Therapy in Patients With Advanced Hepatocellular Carcinoma in KEYNOTE-240: A Randomized, Double-Blind, Phase III Trial. J. Clin. Oncol. 2020, 38, 193–202. [Google Scholar] [CrossRef] [PubMed]
- Deva, S.; Lee, J.-S.; Lin, C.-C.; Yen, C.-J.; Millward, M.; Chao, Y.; Keam, B.; Jameson, M.; Hou, M.-M.; Kang, Y.-K. A phase Ia/Ib trial of tislelizumab, an anti-PD-1 antibody (ab), in patients (pts) with advanced solid tumors. Ann. Oncol. 2018, 29, x24–x25. [Google Scholar] [CrossRef]
- Qin, S.; Ren, Z.; Meng, Z.; Chen, Z.; Chai, X.; Xiong, J.; Bai, Y.; Yang, L.; Zhu, H.; Fang, W. Camrelizumab in patients with previously treated advanced hepatocellular carcinoma: A multicentre, open-label, parallel-group, randomised, phase 2 trial. Lancet Oncol. 2020, 21, 571–580. [Google Scholar] [CrossRef]
- Sangro, B.; Gomez-Martin, C.; de la Mata, M.; Iñarrairaegui, M.; Garralda, E.; Barrera, P.; Riezu-Boj, J.I.; Larrea, E.; Alfaro, C.; Sarobe, P. A clinical trial of CTLA-4 blockade with tremelimumab in patients with hepatocellular carcinoma and chronic hepatitis C. J. Hepatol. 2013, 59, 81–88. [Google Scholar] [CrossRef]
- Powles, T.; O’Donnell, P.H.; Massard, C.; Arkenau, H.-T.; Friedlander, T.W.; Hoimes, C.J.; Lee, J.L.; Ong, M.; Sridhar, S.S.; Vogelzang, N.J. Efficacy and safety of durvalumab in locally advanced or metastatic urothelial carcinoma: Updated results from a phase 1/2 open-label study. JAMA Oncol. 2017, 3, e172411. [Google Scholar] [CrossRef]
- Lee, M.S.; Ryoo, B.-Y.; Hsu, C.-H.; Numata, K.; Stein, S.; Verret, W.; Hack, S.P.; Spahn, J.; Liu, B.; Abdullah, H. Atezolizumab with or without bevacizumab in unresectable hepatocellular carcinoma (GO30140): An open-label, multicentre, phase 1b study. Lancet Oncol. 2020, 21, 808–820. [Google Scholar] [CrossRef]
- Sia, D.; Jiao, Y.; Martinez-Quetglas, I.; Kuchuk, O.; Villacorta-Martin, C.; de Moura, M.C.; Putra, J.; Camprecios, G.; Bassaganyas, L.; Akers, N. Identification of an immune-specific class of hepatocellular carcinoma, based on molecular features. Gastroenterology 2017, 153, 812–826. [Google Scholar] [CrossRef]
- Fujita, M.; Yamaguchi, R.; Hasegawa, T.; Shimada, S.; Arihiro, K.; Hayashi, S.; Maejima, K.; Nakano, K.; Fujimoto, A.; Ono, A. Classification of primary liver cancer with immunosuppression mechanisms and correlation with genomic alterations. EBioMedicine 2020, 53, 102659. [Google Scholar] [CrossRef]
- Shimada, S.; Mogushi, K.; Akiyama, Y.; Furuyama, T.; Watanabe, S.; Ogura, T.; Ogawa, K.; Ono, H.; Mitsunori, Y.; Ban, D. Comprehensive molecular and immunological characterization of hepatocellular carcinoma. EBioMedicine 2019, 40, 457–470. [Google Scholar] [CrossRef]
- Spahn, S.; Roessler, D.; Pompilia, R.; Gabernet, G.; Gladstone, B.P.; Horger, M.; Biskup, S.; Feldhahn, M.; Nahnsen, S.; Hilke, F.J.; et al. Clinical and Genetic Tumor Characteristics of Responding and Non-Responding Patients to PD-1 Inhibition in Hepatocellular Carcinoma. Cancers 2020, 12, 3830. [Google Scholar] [CrossRef]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D. PD-1 blockade in tumors with mismatch-repair deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S. Mutational landscape determines sensitivity to PD-1 blockade in non–small cell lung cancer. Science 2015, 348, 124–128. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.B.; Frampton, G.M.; Rioth, M.J.; Yusko, E.; Xu, Y.; Guo, X.; Ennis, R.C.; Fabrizio, D.; Chalmers, Z.R.; Greenbowe, J. Targeted next generation sequencing identifies markers of response to PD-1 blockade. Cancer Immunol. Res. 2016, 4, 959–967. [Google Scholar] [CrossRef]
- Xie, F.; Bai, Y.; Yang, X.; Long, J.; Mao, J.; Lin, J.; Wang, D.; Song, Y.; Xun, Z.; Huang, H.; et al. Comprehensive analysis of tumour mutation burden and the immune microenvironment in hepatocellular carcinoma. Int. Immunopharmacol. 2020, 89, 107135. [Google Scholar] [CrossRef]


{kind=link}
| Gene | Protein | Genotype | Reference SNP | Consequences | Reference | |
|---|---|---|---|---|---|---|
| ABCB1 | Export Pumps | MDR1 | 3435 C > T | rs2032582 | Reduced sorafenib plasma levels | [35] |
| ABCC2 | MRP2 | 1249G > A | rs2273697 | Reduced sensitivity | [36] | |
| BCRP | ABCG2 | 34 G > A | rs2,231,137 | Reduced sorafenib plasma levels | [35] | |
| 1143 C > T | rs2,622,604T | Reduced sorafenib plasma levels | ||||
| SLC22A1 | Uptake carriers | OCT1 | R61S fs *10 | novel | Reduced sensitivity | [37] |
| C88A fs *16 | novel | Reduced sensitivity | ||||
| c.262T > C | rs1001179 | Reduced sensitivity | [38] | |||
| c.566C > T | rs34104736 | Reduced sensitivity | ||||
| c.659G > T | rs36103319 | Reduced sensitivity | ||||
| c.859C > G | rs4646278 | Reduced sensitivity | ||||
| SLC15A2 | PEPT2 | 1048 T/T & C/T | rs2257212 | Prolonged PFS | [39,40] | |
| KDR | Drug target | VEGFR2 | AA genotype | rs1870377 | Improved response to sorafenib and longer TTP | [41] |
| CC genotype | rs2071559 | Shorter OS | ||||
| VEGF | VEGF-A | C allele | rs2010963 | Reduced OS and PFS | [42] | |
| VEGF-C | T allele | rs4604006 | Reduced OS and PFS | |||
| NOS3 | eNOS | eNOS−786 TT | rs2070744 | Reduced OS and PFS | [43] | |
| eNOS + 894 GG | rs1799983 | Reduced OS and PFS | [44] | |||
| ANGPT2 | ANGPT2 | TT/GT | rs55633437 | Reduced OS and PFS | [44] | |
| Haplotype (HT2) | Reduced OS and PFS | |||||
| T T G | rs3739392 rs3739391 rs3739390 | |||||
| Target. | PD-1 | CTLA-4 | PD-L1 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Drug | Nivolumab | Pembrolizumab | Tislelizumab | Camrelizumab | Tremelimumab | Durvalumab | Atezolizumab | |||
| Versus | single-arm | single-arm | sorafenib | single-arm | single-arm | sorafenib | single-arm | single arm | single arm | single arm |
| Trial name | CheckMate-040 (Dose-escalation arm) | CheckMate-040 (Dose-expansion arm) | Checkmate-459 | KEYNOTE- 224 | KEYNOTE-240 | / | / | / | / | GO30140 |
| NCT number | NCT01658878 | NCT01658878 | NCT02576509 | NCT02702414 | NCT02702401 | NCT02407990 | NCT02989922 | NCT01008358 | NCT01693562 | NCT02715531 |
| Treatment line | First/second | First/second | First/second | Second | Second | First | Second | First/second | First/second | Second |
| Study phase | I/II | I/II | III | II | III | IA/B | II | II | I/II | IB |
| Study design | randomized | randomized | randomized | non-randomized | randomized | non-randomized | randomized | non-randomized | N/A | randomized |
| Primary end points | Safety and tolerability | ORR | OS | ORR | OS / PFS | Safety | ORR/OS at 6 months | Tumor response | Safety | PFS |
| ORR | 15 | 20 | 17.6 | 18 | 18.3 | 12.2 | 32 | 17.6 | 10 | 17 |
| PFS (months) | 4.1 | 4 | N/A | N/A | 3 | 2.1 | 2.1 | N/A | 2.7 | 3.4 |
| TTP (months) | 3.4 | / | 7.4 | N/A | N/A | N/A | N/A | 6.5 | N/A | N/A |
| OS (months) | 28.6 | 15 | 12.3 | 12.9 | 13.9 | 13.6 (IA) 9.3 (IB) | 13.8 | 8.2 | 13.2 | N/A |
| DOR (months) | 17 | 9.9 | N/A | N/A | 13.8 | N/A | N/A | N/A | N/A | N/A |
| Result | accepted safety/tolerability | positive | OS did not reach statistical significance | positive | OS did not reach statistical significance | accepted safety/tolerability | positive | Need further investigation | accepted safety/tolerability | Not effective as monotherapy |
| Reference | [120] | [122] | [121] | [123] | [124] | [125] | [126] | [127] | [128] | [129] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Atwa, S.M.; Odenthal, M.; El Tayebi, H.M. Genetic Heterogeneity, Therapeutic Hurdle Confronting Sorafenib and Immune Checkpoint Inhibitors in Hepatocellular Carcinoma. Cancers 2021, 13, 4343. https://doi.org/10.3390/cancers13174343
Atwa SM, Odenthal M, El Tayebi HM. Genetic Heterogeneity, Therapeutic Hurdle Confronting Sorafenib and Immune Checkpoint Inhibitors in Hepatocellular Carcinoma. Cancers. 2021; 13(17):4343. https://doi.org/10.3390/cancers13174343
Chicago/Turabian StyleAtwa, Sara M., Margarete Odenthal, and Hend M. El Tayebi. 2021. "Genetic Heterogeneity, Therapeutic Hurdle Confronting Sorafenib and Immune Checkpoint Inhibitors in Hepatocellular Carcinoma" Cancers 13, no. 17: 4343. https://doi.org/10.3390/cancers13174343
APA StyleAtwa, S. M., Odenthal, M., & El Tayebi, H. M. (2021). Genetic Heterogeneity, Therapeutic Hurdle Confronting Sorafenib and Immune Checkpoint Inhibitors in Hepatocellular Carcinoma. Cancers, 13(17), 4343. https://doi.org/10.3390/cancers13174343