
Malignant Ascites in Ovarian Cancer: Cellular, Acellular, and Biophysical Determinants of Molecular Characteristics and Therapy Response

 , ,
, ,  , , ,
, , ,
Simple Summary
Abstract
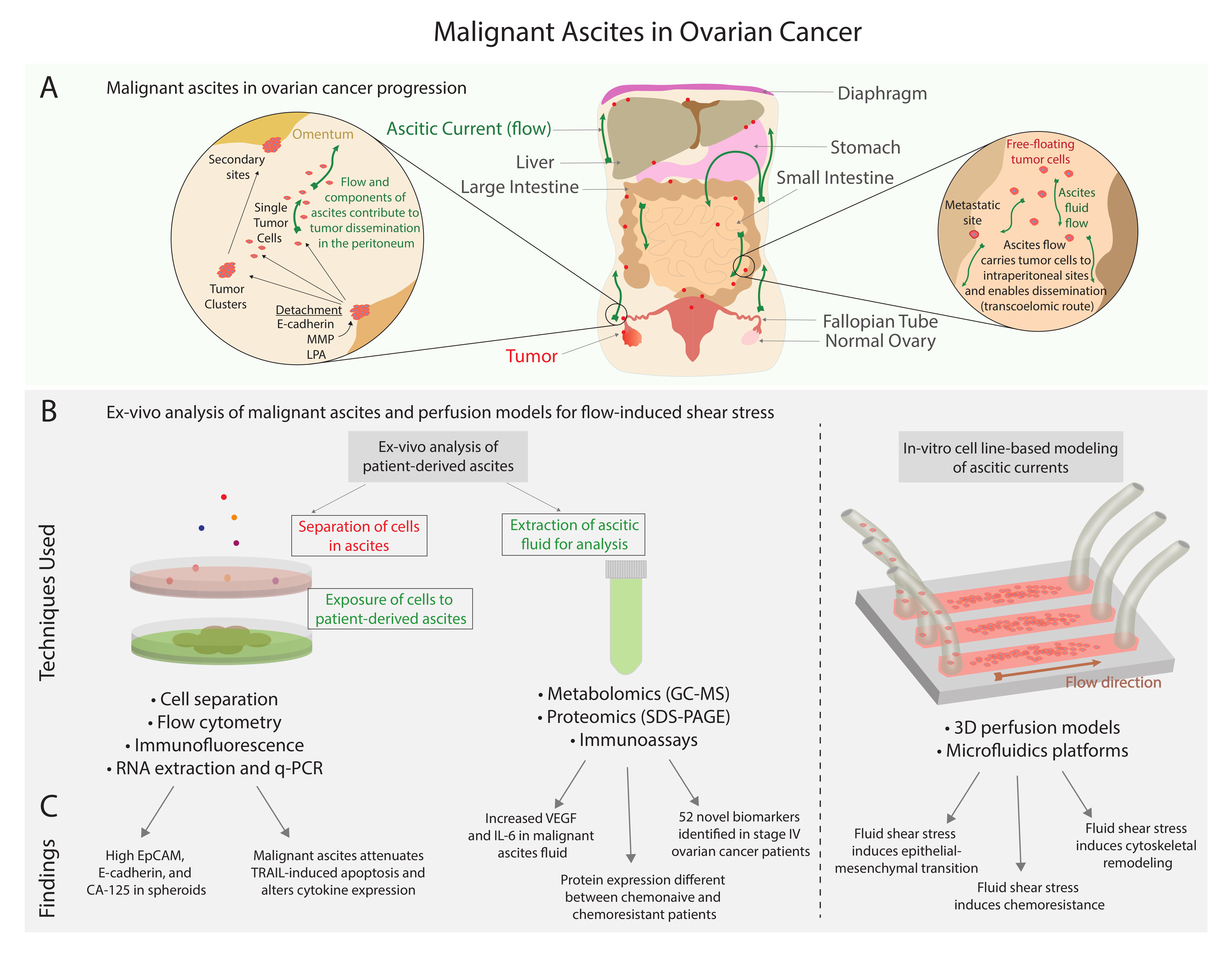
1. Introduction
1.1. Heterogeneity in Ovarian Cancer
1.2. Epithelial-Mesenchymal Transition in Ovarian Cancer
1.3. Prevalence and Symptoms of Ascites
1.4. Pathophysiology of Ascites and Its Role in Ovarian Cancer
2. Malignant Ascites and the Tumor-Promoting Microenvironment
2.1. Angiogenesis-Regulating Factors
2.2. Adhesion-Regulating Factors
2.3. Inflammatory and Immune Response Factors
2.3.1. Innate Cellular Immune Response Factors
Macrophages
Natural Killer (NK) Cells
Dendritic Cells (DCs)
Myeloid-Derived Suppressor Cells (MDSCs)
2.3.2. Adaptive Immune Response Factors
T Helper (Th), Cytotoxic T (Tc), and T Helper 17 (Th17) Cells
Regulatory T Cells (Tregs)
2.4. Lysosomes, Secreted Vesicles, and Ascites
2.5. Proliferation Regulating Factors
2.6. Metabolomic and Proteomic Profiling of Ascites Fluid and Blood in Ovarian Cancer

{kind=link}
{kind=link}
| Reference | Major Findings |
|---|---|
| Kuk et al., 2009. [284] |
|
| Elschenbroich et al., 2011. [298] |
|
| Shender et al., 2014. [285] |
|
| Ahmed et al., 2016. [283] |
|
2.7. Magnitude and Directionality of Ascites
3. Interstitial Fluid Pressure and Fluid Stress
3.1. Interstitial Fluid Pressure in Cancer
3.2. In Vitro Modeling of Fluid Stress in Ovarian Cancer
3.3. In Vivo Modeling of Fluid Stress in Ovarian Cancer
| Cell Line (i.p. Injection) | Species of Origin | Ability to Form Tumor | Ascites Present? |
|---|---|---|---|
| OVCAR-4 | Human | Low-Medium [352,355] | No [352,353] |
| Caov-3 | Human | Low [352] | No [352,353] |
| NIH:OVCAR-3 | Human | Medium-High [352,355] | Yes [354,357] No [352,353] |
| OV-90 | Human | Medium [352] | No [352] |
| OVCAR-8 | Human | High [352] | Yes [353] No [352] |
| HEY A8 | Human | High [352] | Yes [352] |
| SK-OV-3 | Human | High [352,354] | Yes [352,354,355] |
| A2780 | Human | High [352] | Yes [352] |
| IGROV-1 | Human | Low [352] | No [352] |
| OVCAR-5 | Human | Medium-High [352,356] | Yes [356] No [352,353] |
| ID8 | Mouse | High [352] | Yes [352] |
3.4. Compressive Force of Ascites
4. Treatment Options for Ascites
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Ahmed, N.; Stenvers, K.L. Getting to know ovarian cancer ascites: Opportunities for targeted therapy-based translational research. Front. Oncol. 2013, 3, 256. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef]
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 2015, 136, E359–E386. [Google Scholar] [CrossRef] [PubMed]
- Ford, C.E.; Werner, B.; Hacker, N.F.; Warton, K. The untapped potential of ascites in ovarian cancer research and treatment. Br. J. Cancer 2020, 123, 9–16. [Google Scholar] [CrossRef]
- Kim, S.; Kim, B.; Song, Y.S. Ascites modulates cancer cell behavior, contributing to tumor heterogeneity in ovarian cancer. Cancer Sci. 2016, 107, 1173–1178. [Google Scholar] [CrossRef]
- Sangisetty, S.L.; Miner, T.J. Malignant ascites: A review of prognostic factors, pathophysiology and therapeutic measures. World J. Gastrointest. Surg. 2012, 4, 87–95. [Google Scholar] [CrossRef]
- Hudson, L.G.; Zeineldin, R.; Stack, M.S. Phenotypic plasticity of neoplastic ovarian epithelium: Unique cadherin profiles in tumor progression. Clin. Exp. Metastasis 2008, 25, 643–655. [Google Scholar] [CrossRef]
- Rizvi, I.; Gurkan, U.A.; Tasoglu, S.; Alagic, N.; Celli, J.P.; Mensah, L.B.; Mai, Z.; Demirci, U.; Hasan, T. Flow induces epithelial-mesenchymal transition, cellular heterogeneity and biomarker modulation in 3D ovarian cancer nodules. Proc. Natl. Acad. Sci. USA 2013, 110, E1974–E1983. [Google Scholar] [CrossRef]
- Nath, S.; Pigula, M.; Khan, A.P.; Hanna, W.; Ruhi, M.K.; Dehkordy, F.M.; Pushpavanam, K.; Rege, K.; Moore, K.; Tsujita, Y.; et al. Flow-induced Shear Stress Confers Resistance to Carboplatin in an Adherent Three-Dimensional Model for Ovarian Cancer: A Role for EGFR-Targeted Photoimmunotherapy Informed by Physical Stress. J. Clin. Med. 2020, 9, 924. [Google Scholar] [CrossRef]
- Garrison, R.N.; Galloway, R.H.; Heuser, L.S. Mechanisms of malignant ascites production. J. Surg. Res. 1987, 42, 126–132. [Google Scholar] [CrossRef]
- Parsons, S.L.; Watson, S.A.; Steele, R.J.C. Malignant ascites. Br. J. Surg. 1996, 83, 6–14. [Google Scholar] [CrossRef] [PubMed]
- Shen-Gunther, J.; Mannel, R.S. Ascites as a predictor of ovarian malignancy. Gynecol. Oncol. 2002, 87, 77–83. [Google Scholar] [CrossRef]
- Smith, E.M.; Jayson, G.C. The current and future management of malignant ascites. Clin. Oncol. R. Coll. Radiol. 2003, 15, 59–72. [Google Scholar] [CrossRef] [PubMed]
- Adam, R.A.; Adam, Y.G. Malignant ascites: Past, present, and future. J. Am. Coll. Surg. 2004, 198, 999–1011. [Google Scholar] [CrossRef] [PubMed]
- Burleson, K.M.; Casey, R.C.; Skubitz, K.M.; Pambuccian, S.E.; Oegema, T.R., Jr.; Skubitz, A.P. Ovarian carcinoma ascites spheroids adhere to extracellular matrix components and mesothelial cell monolayers. Gynecol. Oncol. 2004, 93, 170–181. [Google Scholar] [CrossRef]
- Becker, G.; Galandi, D.; Blum, H.E. Malignant ascites: Systematic review and guideline for treatment. Eur. J. Cancer 2006, 42, 589–597. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, C.A.; Maxwell, G.L.; Chernofsky, M.R.; Bernstein, S.A.; Farley, J.H.; Rose, G.S. Intraperitoneal bevacizumab for the palliation of malignant ascites in refractory ovarian cancer. Gynecol. Oncol. 2008, 111, 530–532. [Google Scholar] [CrossRef]
- Huang, H.; Li, Y.J.; Lan, C.Y.; Huang, Q.D.; Feng, Y.L.; Huang, Y.W.; Liu, J.H. Clinical significance of ascites in epithelial ovarian cancer. Neoplasma 2013, 60, 546–552. [Google Scholar] [CrossRef] [PubMed]
- Kipps, E.; Tan, D.S.; Kaye, S.B. Meeting the challenge of ascites in ovarian cancer: New avenues for therapy and research. Nat. Rev. Cancer 2013, 13, 273–282. [Google Scholar] [CrossRef]
- Smolle, E.; Taucher, V.; Haybaeck, J. Malignant ascites in ovarian cancer and the role of targeted therapeutics. Anticancer Res. 2014, 34, 1553–1561. [Google Scholar] [PubMed]
- Hodge, C.; Badgwell, B.D. Palliation of malignant ascites. J. Surg. Oncol. 2019, 120, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Cao, J.; Melamed, A.; Worley, M.; Gockley, A.; Jones, D.; Nia, H.T.; Zhang, Y.; Stylianopoulos, T.; Kumar, A.S.; et al. Losartan treatment enhances chemotherapy efficacy and reduces ascites in ovarian cancer models by normalizing the tumor stroma. Proc. Natl. Acad. Sci. USA 2019, 116, 2210–2219. [Google Scholar] [CrossRef]
- Uruski, P.; Mikula-Pietrasik, J.; Pakula, M.; Budkiewicz, S.; Drzewiecki, M.; Gaiday, A.N.; Wierzowiecka, M.; Naumowicz, E.; Moszynski, R.; Tykarski, A.; et al. Malignant Ascites Promote Adhesion of Ovarian Cancer Cells to Peritoneal Mesothelium and Fibroblasts. Int. J. Mol. Sci. 2021, 22, 4222. [Google Scholar] [CrossRef] [PubMed]
- Lengyel, E. Ovarian cancer development and metastasis. Am. J. Pathol. 2010, 177, 1053–1064. [Google Scholar] [CrossRef]
- Vaughan, S.; Coward, J.I.; Bast, R.C., Jr.; Berchuck, A.; Berek, J.S.; Brenton, J.D.; Coukos, G.; Crum, C.C.; Drapkin, R.; Etemadmoghadam, D.; et al. Rethinking ovarian cancer: Recommendations for improving outcomes. Nat. Rev. Cancer 2011, 11, 719–725. [Google Scholar] [CrossRef]
- Prat, J. Ovarian carcinomas: Five distinct diseases with different origins, genetic alterations, and clinicopathological features. Virchows Arch. Int. J. Pathol. 2012, 460, 237–249. [Google Scholar] [CrossRef]
- Reid, B.M.; Permuth, J.B.; Sellers, T.A. Epidemiology of ovarian cancer: A review. Cancer Biol. Med. 2017, 14, 9–32. [Google Scholar] [CrossRef]
- Kurman, R.J.; Shih, I.-M.M. The dualistic model of ovarian carcinogenesis revisited, revised, and expanded. Am. J. Pathol. 2016, 186, 733–747. [Google Scholar] [CrossRef]
- Kurman, R.J.; Shih, I.-M. Seromucinous Tumors of the Ovary. What’s in a Name? Int. J. Gynecol. Pathol. Off. J. Int. Soc. Gynecol. Pathol. 2016, 35, 78–81. [Google Scholar] [CrossRef]
- Bowtell, D.D.; Böhm, S.; Ahmed, A.A.; Aspuria, P.-J.J.; Bast, R.C.; Beral, V.; Berek, J.S.; Birrer, M.J.; Blagden, S.; Bookman, M.A.; et al. Rethinking ovarian cancer II: Reducing mortality from high-grade serous ovarian cancer. Nat. Rev. Cancer 2015, 15, 668–679. [Google Scholar] [CrossRef]
- Auersperg, N. The origin of ovarian cancers—Hypotheses and controversies. Front. Biosci. 2013, 5, 709–719. [Google Scholar] [CrossRef] [PubMed]
- Kroeger, P.T., Jr.; Drapkin, R. Pathogenesis and heterogeneity of ovarian cancer. Curr. Opin. Obstet. Gynecol. 2017, 29, 26–34. [Google Scholar] [CrossRef]
- Kurman, R.J.; Shih, I.-M. Molecular pathogenesis and extraovarian origin of epithelial ovarian cancer—Shifting the paradigm. Hum. Pathol. 2011, 42, 918–931. [Google Scholar] [CrossRef] [PubMed]
- Patch, A.-M.; Christie, E.L.; Etemadmoghadam, D.; Garsed, D.W.; George, J.; Fereday, S.; Nones, K.; Cowin, P.; Alsop, K.; Bailey, P.J.; et al. Whole-genome characterization of chemoresistant ovarian cancer. Nature 2015, 521, 489–494. [Google Scholar] [CrossRef]
- Tan, D.S.; Agarwal, R.; Kaye, S.B. Mechanisms of transcoelomic metastasis in ovarian cancer. Lancet Oncol. 2006, 7, 925–934. [Google Scholar] [CrossRef]
- Bjørge, L.; Junnikkala, S.; Kristoffersen, E.K.; Hakulinen, J.; Matre, R.; Meri, S. Resistance of ovarian teratocarcinoma cell spheroids to complement-mediated lysis. Br. J. Cancer 1997, 75, 1247–1255. [Google Scholar] [CrossRef][Green Version]
- Aslam, N.; Marino, C.R. Malignant ascites: New concepts in pathophysiology, diagnosis, and management. Arch. Intern. Med. 2001, 161, 2733–2737. [Google Scholar] [CrossRef]
- du Bois, A.; Reuss, A.; Pujade-Lauraine, E.; Harter, P.; Ray-Coquard, I.; Pfisterer, J. Role of surgical outcome as prognostic factor in advanced epithelial ovarian cancer: A combined exploratory analysis of 3 prospectively randomized phase 3 multicenter trials: By the Arbeitsgemeinschaft Gynaekologische Onkologie Studiengruppe Ovarialkarzinom (AGO-OVAR) and the Groupe d’Investigateurs Nationaux Pour les Etudes des Cancers de l’Ovaire (GINECO). Cancer 2009, 115, 1234–1244. [Google Scholar] [CrossRef] [PubMed]
- Anuradha, S.; Webb, P.M.; Blomfield, P.; Brand, A.H.; Friedlander, M.; Leung, Y.; Obermair, A.; Oehler, M.K.; Quinn, M.; Steer, C.; et al. Survival of Australian women with invasive epithelial ovarian cancer: A population-based study. Med. J. Aust. 2014, 201, 283–288. [Google Scholar] [CrossRef]
- Cannistra, S.A. Cancer of the Ovary. N. Engl. J. Med. 2004, 351, 2519–2529. [Google Scholar] [CrossRef] [PubMed]
- Manning-Geist, B.L.; Hicks-Courant, K.; Gockley, A.A.; Clark, R.M.; del Carmen, M.G.; Growdon, W.B.; Horowitz, N.S.; Berkowitz, R.S.; Muto, M.G.; Worley, M.J. Moving beyond “complete surgical resection” and “optimal”: Is low-volume residual disease another option for primary debulking surgery? Gynecol. Oncol. 2018, 150, 233–238. [Google Scholar] [CrossRef]
- Vergote, I.; Tropé, C.G.; Amant, F.; Kristensen, G.B.; Ehlen, T.; Johnson, N.; Verheijen, R.H.M.; van der Burg, M.E.L.; Lacave, A.J.; Panici, P.B.; et al. Neoadjuvant chemotherapy or primary surgery in stage IIIC or IV ovarian cancer. N. Engl. J. Med. 2010, 363, 943–953. [Google Scholar] [CrossRef]
- Kehoe, S.; Hook, J.; Nankivell, M.; Jayson, G.C.; Kitchener, H.; Lopes, T.; Luesley, D.; Perren, T.; Bannoo, S.; Mascarenhas, M.; et al. Primary chemotherapy versus primary surgery for newly diagnosed advanced ovarian cancer (CHORUS): An open-label, randomised, controlled, non-inferiority trial. Lancet 2015, 386, 249–257. [Google Scholar] [CrossRef]
- Manning-Geist, B.L.; Hicks-Courant, K.; Gockley, A.A.; Clark, R.M.; Del Carmen, M.G.; Growdon, W.B.; Horowitz, N.S.; Berkowitz, R.S.; Muto, M.G.; Worley, M.J. A novel classification of residual disease after interval debulking surgery for advanced-stage ovarian cancer to better distinguish oncologic outcome. Am. J. Obstet. Gynecol. 2019, 221, 326–e1. [Google Scholar] [CrossRef]
- Trinidad, C.V.; Tetlow, A.L.; Bantis, L.E.; Godwin, A.K. Reducing Ovarian Cancer Mortality Through Early Detection: Approaches Using Circulating Biomarkers. Cancer Prev. Res. (Phila. Pa.) 2020, 13, 241–252. [Google Scholar] [CrossRef]
- Marusyk, A.; Polyak, K. Tumor heterogeneity: Causes and consequences. Biochim. Biophys. Acta 2010, 1805, 105–117. [Google Scholar] [CrossRef] [PubMed]
- Blagden, S.P. Harnessing pandemonium: The clinical implications of tumor heterogeneity in ovarian cancer. Front. Oncol. 2015, 5, 149. [Google Scholar] [CrossRef]
- Tothill, R.W.; Tinker, A.V.; George, J.; Brown, R.; Fox, S.B.; Lade, S.; Johnson, D.S.; Trivett, M.K.; Etemadmoghadam, D.; Locandro, B.; et al. Novel Molecular Subtypes of Serous and Endometrioid Ovarian Cancer Linked to Clinical Outcome. Clin. Cancer Res. 2008, 14, 5198–5208. [Google Scholar] [CrossRef]
- Loret, N.; Denys, H.; Tummers, P.; Berx, G. The role of epithelial-to-mesenchymal plasticity in ovarian cancer progression and therapy resistance. Cancers 2019, 11, 838. [Google Scholar] [CrossRef]
- Ahmed, N.; Thompson, E.W.; Quinn, M.A. Epithelial–mesenchymal interconversions in normal ovarian surface epithelium and ovarian carcinomas: An exception to the norm. J. Cell. Physiol. 2007, 213, 581–588. [Google Scholar] [CrossRef]
- Salamanca, C.M.; Maines-Bandiera, S.L.; Leung, P.C.K.; Hu, Y.-L.; Auersperg, N. Effects of epidermal growth factor/hydrocortisone on the growth and differentiation of human ovarian surface epithelium. J. Soc. Gynecol. Investig. 2004, 11, 241–251. [Google Scholar] [CrossRef]
- Ahmed, N.; Maines-Bandiera, S.; Quinn, M.A.; Unger, W.G.; Dedhar, S.; Auersperg, N. Molecular pathways regulating EGF-induced epithelio-mesenchymal transition in human ovarian surface epithelium. Am. J. Physiology.-Cell Physiol. 2006, 290, C1532–C1542. [Google Scholar] [CrossRef]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.J.J.; Nieto, M.A. Epithelial-Mesenchymal Transitions in Development and Disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Sennino, B.; Ishiguro-Oonuma, T.; Wei, Y.; Naylor, R.M.; Williamson, C.W.; Bhagwandin, V.; Tabruyn, S.P.; You, W.-K.; Chapman, H.A.; Christensen, J.G.; et al. Suppression of tumor invasion and metastasis by concurrent inhibition of c-Met and VEGF signaling in pancreatic neuroendocrine tumors. Cancer Discov. 2012, 2, 270–287. [Google Scholar] [CrossRef]
- Du, F.; Wu, X.; Liu, Y.; Wang, T.; Qi, X.; Mao, Y.; Jiang, L.; Zhu, Y.; Chen, Y.; Zhu, R.; et al. Acquisition of paclitaxel resistance via PI3K-dependent epithelial-mesenchymal transition in A2780 human ovarian cancer cells. Oncol. Rep. 2013, 30, 1113–1118. [Google Scholar] [CrossRef]
- Haslehurst, A.M.; Koti, M.; Dharsee, M.; Nuin, P.; Evans, K.; Geraci, J.; Childs, T.; Chen, J.; Li, J.; Weberpals, J.; et al. EMT transcription factors snail and slug directly contribute to cisplatin resistance in ovarian cancer. BMC Cancer 2012, 12, 91. [Google Scholar] [CrossRef]
- Izumiya, M.; Kabashima, A.; Higuchi, H.; Igarashi, T.; Sakai, G.; Iizuka, H.; Nakamura, S.; Adachi, M.; Hamamoto, Y.; Funakoshi, S.; et al. Chemoresistance is associated with cancer stem cell-like properties and epithelial-to-mesenchymal transition in pancreatic cancer cells. Anticancer Res. 2012, 32, 3847–3853. [Google Scholar]
- Thomson, S.; Buck, E.; Petti, F.; Griffin, G.; Brown, E.; Ramnarine, N.; Iwata, K.K.; Gibson, N.; Haley, J.D. Epithelial to mesenchymal transition is a determinant of sensitivity of non-small-cell lung carcinoma cell lines and xenografts to epidermal growth factor receptor inhibition. Cancer Res. 2005, 65, 9455–9462. [Google Scholar] [CrossRef]
- Latifi, A.; Luwor, R.B.; Bilandzic, M.; Nazaretian, S.; Stenvers, K.; Pyman, J.; Zhu, H.; Thompson, E.W.; Quinn, M.A.; Findlay, J.K.; et al. Isolation and characterization of tumor cells from the ascites of ovarian cancer patients: Molecular phenotype of chemoresistant ovarian tumors. PLoS ONE 2012, 7, e46858. [Google Scholar] [CrossRef]
- Veatch, A.L.; Carson, L.F.; Ramakrishnan, S. Differential expression of the cell-cell adhesion molecule E-cadherin in ascites and solid human ovarian tumor cells. Int. J. Cancer 1994, 58, 393–399. [Google Scholar] [CrossRef]
- Moreno-Bueno, G.; Peinado, H.; Molina, P.; Olmeda, D.; Cubillo, E.; Santos, V.; Palacios, J.; Portillo, F.; Cano, A. The morphological and molecular features of the epithelial-to-mesenchymal transition. Nat. Protoc. 2009, 4, 1591–1613. [Google Scholar] [CrossRef]
- Sawada, K.; Mitra, A.K.; Radjabi, A.R.; Bhaskar, V.; Kistner, E.O.; Tretiakova, M.; Jagadeeswaran, S.; Montag, A.; Becker, A.; Kenny, H.A.; et al. Loss of E-cadherin promotes ovarian cancer metastasis via alpha 5-integrin, which is a therapeutic target. Cancer Res. 2008, 68, 2329–2339. [Google Scholar] [CrossRef]
- Cowden Dahl, K.D.; Symowicz, J.; Ning, Y.; Gutierrez, E.; Fishman, D.A.; Adley, B.P.; Stack, M.S.; Hudson, L.G. Matrix metalloproteinase 9 is a mediator of epidermal growth factor-dependent e-cadherin loss in ovarian carcinoma cells. Cancer Res. 2008, 68, 4606–4613. [Google Scholar] [CrossRef] [PubMed]
- Chiejina, M.; Kudaravalli, P.; Samant, H. Ascites. In StatPearls; StatPearls Publishing LLC.: Treasure Island, FL, USA, 2021. [Google Scholar]
- Ayantunde, A.A.; Parsons, S.L. Pattern and prognostic factors in patients with malignant ascites: A retrospective study. Ann. Oncol. 2007, 18, 945–949. [Google Scholar] [CrossRef] [PubMed]
- Ayhan, A.; Gultekin, M.; Taskiran, C.; Dursun, P.; Firat, P.; Bozdag, G.; Celik, N.Y.; Yuce, K. Ascites and epithelial ovarian cancers: A reappraisal with respect to different aspects. Int. J. Gynecol. Cancer 2007, 17, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Holm-Nielsen, P. Pathogenesis of ascites in peritoneal carcinomatosis1. Acta Pathol. Microbiol. Scand. 1953, 33, 10–21. [Google Scholar] [CrossRef]
- Belotti, D.; Paganoni, P.; Manenti, L.; Garofalo, A.; Marchini, S.; Taraboletti, G.; Giavazzi, R. Matrix metalloproteinases (MMP9 and MMP2) induce the release of vascular endothelial growth factor (VEGF) by ovarian carcinoma cells: Implications for ascites formation. Cancer Res. 2003, 63, 5224–5229. [Google Scholar]
- Fang, X.; Yu, S.; Bast, R.C.; Liu, S.; Xu, H.-J.; Hu, S.-X.; LaPushin, R.; Claret, F.X.; Aggarwal, B.B.; Lu, Y.; et al. Mechanisms for lysophosphatidic acid-induced cytokine production in ovarian cancer cells. J. Biol. Chem. 2004, 279, 9653–9661. [Google Scholar] [CrossRef]
- Lee, H.K.; Chae, H.S.; Kim, J.S.; Kim, H.K.; Cho, Y.S.; Rho, S.Y.; Kang, J.-H.; Cho, S.G.; Jang, H.S.; Han, K. Vascular Endothelial Growth Factor Levels in Ascites Between Chemonaive and Chemotreated Patients. Yonsei Med. J. 2008, 49, 429–435. [Google Scholar] [CrossRef]
- Hirabayashi, K.; Graham, J. Genesis of ascites in ovarian cancer. Am. J. Obstet. Gynecol. 1970, 106, 492–497. [Google Scholar] [CrossRef]
- Urrunaga, N.H.; Singal, A.G.; Cuthbert, J.A.; Rockey, D.C. Hemorrhagic ascites. Clinical presentation and outcomes in patients with cirrhosis. J. Hepatol. 2013, 58, 1113–1118. [Google Scholar] [CrossRef]
- Akriviadis, E.A. Hemoperitoneum in patients with ascites. Am. J. Gastroenterol 1997, 92, 567–575. [Google Scholar] [PubMed]
- DeSitter, L.; Rector, W.G., Jr. The significance of bloody ascites in patients with cirrhosis. Am. J. Gastroenterol 1984, 79, 136–138. [Google Scholar]
- Thériault, C.; Pinard, M.; Comamala, M.; Migneault, M.; Beaudin, J.; Matte, I.; Boivin, M.; Piché, A.; Rancourt, C. MUC16 (CA125) regulates epithelial ovarian cancer cell growth, tumorigenesis and metastasis. Gynecol. Oncol. 2011, 121, 434–443. [Google Scholar] [CrossRef] [PubMed]
- Gubbels, J.A.A.; Felder, M.; Horibata, S.; Belisle, J.A.; Kapur, A.; Holden, H.; Petrie, S.; Migneault, M.; Rancourt, C.; Connor, J.P.; et al. MUC16 provides immune protection by inhibiting synapse formation between NK and ovarian tumor cells. Mol. Cancer 2010, 9, 11. [Google Scholar] [CrossRef]
- Avraham, R.; Yarden, Y. Feedback regulation of EGFR signalling: Decision making by early and delayed loops. Nat. Rev. Mol. Cell Biol. 2011, 12, 104–117. [Google Scholar] [CrossRef]
- del Carmen, M.G.; Rizvi, I.; Chang, Y.; Moor, A.C.; Oliva, E.; Sherwood, M.; Pogue, B.; Hasan, T. Synergism of epidermal growth factor receptor-targeted immunotherapy with photodynamic treatment of ovarian cancer in vivo. J. Natl. Cancer Inst. 2005, 97, 1516–1524. [Google Scholar] [CrossRef] [PubMed]
- Psyrri, A.; Kassar, M.; Yu, Z.; Bamias, A.; Weinberger, P.M.; Markakis, S.; Kowalski, D.; Camp, R.L.; Rimm, D.L.; Dimopoulos, M.A. Effect of epidermal growth factor receptor expression level on survival in patients with epithelial ovarian cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2005, 11, 8637–8643. [Google Scholar] [CrossRef] [PubMed]
- Alper, O.; Bergmann-Leitner, E.S.; Bennett, T.A.; Hacker, N.F.; Stromberg, K.; Stetler-Stevenson, W.G. Epidermal growth factor receptor signaling and the invasive phenotype of ovarian carcinoma cells. J. Natl. Cancer Inst. 2001, 93, 1375–1384. [Google Scholar] [CrossRef] [PubMed]
- Posadas, E.M.; Liel, M.S.; Kwitkowski, V.; Minasian, L.; Godwin, A.K.; Hussain, M.M.; Espina, V.; Wood, B.J.; Steinberg, S.M.; Kohn, E.C. A phase II and pharmacodynamic study of gefitinib in patients with refractory or recurrent epithelial ovarian cancer. Cancer 2007, 109, 1323–1330. [Google Scholar] [CrossRef]
- Zeineldin, R.; Muller, C.Y.; Stack, M.S.; Hudson, L.G. Targeting the EGF receptor for ovarian cancer therapy. J. Oncol. 2010, 2010, 414676. [Google Scholar] [CrossRef]
- Lane, D.; Matte, I.; Rancourt, C.; Piché, A. Prognostic significance of IL-6 and IL-8 ascites levels in ovarian cancer patients. BMC Cancer 2011, 11, 210. [Google Scholar] [CrossRef]
- Tempfer, C.; Zeisler, H.; Sliutz, G.; Haeusler, G.; Hanzal, E.; Kainz, C. Serum evaluation of interleukin 6 in ovarian cancer patients. Gynecol. Oncol. 1997, 66, 27–30. [Google Scholar] [CrossRef]
- Obata, N.H.; Tamakoshi, K.; Shibata, K.; Kikkawa, F.; Tomoda, Y. Effects of interleukin-6 on in vitro cell attachment, migration and invasion of human ovarian carcinoma. Anticancer Res. 1997, 17, 337–342. [Google Scholar]
- Penson, R.T.; Kronish, K.; Duan, Z.; Feller, A.J.; Stark, P.; Cook, S.E.; Duska, L.R.; Fuller, A.F.; Goodman, A.K.; Nikrui, N.; et al. Cytokines IL-1beta, IL-2, IL-6, IL-8, MCP-1, GM-CSF and TNFalpha in patients with epithelial ovarian cancer and their relationship to treatment with paclitaxel. Int. J. Gynecol. Cancer Off. J. Int. Gynecol. Cancer Soc. 2000, 10, 33–41. [Google Scholar] [CrossRef]
- Nilsson, M.B.; Langley, R.R.; Fidler, I.J. Interleukin-6, secreted by human ovarian carcinoma cells, is a potent proangiogenic cytokine. Cancer Res. 2005, 65, 10794–10800. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xu, R.C.; Zhang, X.L.; Niu, X.L.; Qu, Y.; Li, L.Z.; Meng, X.Y. Interleukin-8 secretion by ovarian cancer cells increases anchorage-independent growth, proliferation, angiogenic potential, adhesion and invasion. Cytokine 2012, 59, 145–155. [Google Scholar] [CrossRef]
- Huang, S.; Robinson, J.B.; DeGuzman, A.; Bucana, C.D.; Fidler, I.J. Blockade of Nuclear Factor-κB Signaling Inhibits Angiogenesis and Tumorigenicity of Human Ovarian Cancer Cells by Suppressing Expression of Vascular Endothelial Growth Factor and Interleukin 8. Cancer Res. 2000, 60, 5334–5339. [Google Scholar] [PubMed]
- Matte, I.; Lane, D.; Laplante, C.; Rancourt, C.; Piché, A. Profiling of cytokines in human epithelial ovarian cancer ascites. Am. J. Cancer Res. 2012, 2, 566–580. [Google Scholar]
- Mocellin, S.; Wang, E.; Marincola, F.M. Cytokines and immune response in the tumor microenvironment. J. Immunother. 2001, 24, 392–407. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.W.; de Waal Malefyt, R.; Coffman, R.L.; O’Garra, A. Interleukin-10 and the interleukin-10 receptor. Annu. Rev. Immunol. 2001, 19, 683–765. [Google Scholar] [CrossRef] [PubMed]
- Moser, M. Dendritic cells in immunity and tolerance-do they display opposite functions? Immunity 2003, 19, 5–8. [Google Scholar] [CrossRef]
- Hu, Y.L.; Tee, M.K.; Goetzl, E.J.; Auersperg, N.; Mills, G.B.; Ferrara, N.; Jaffe, R.B. Lysophosphatidic acid induction of vascular endothelial growth factor expression in human ovarian cancer cells. J. Natl. Cancer Inst. 2001, 93, 762–768. [Google Scholar] [CrossRef]
- Liu, Y.; Burkhalter, R.; Symowicz, J.; Chaffin, K.; Ellerbroek, S.; Stack, M.S. Lysophosphatidic Acid disrupts junctional integrity and epithelial cohesion in ovarian cancer cells. J. Oncol. 2012, 2012, 501492. [Google Scholar] [CrossRef] [PubMed]
- Carreiras, F.; Cruet, S.; Staedel, C.; Sichel, F.; Gauduchon, P. Human ovarian adenocarcinoma cells synthesize vitronectin and use It to organize their adhesion. Gynecol. Oncol. 1999, 72, 312–322. [Google Scholar] [CrossRef]
- Lessan, K.; Aguiar, D.J.; Oegema, T.; Siebenson, L.; Skubitz, A.P. CD44 and beta1 integrin mediate ovarian carcinoma cell adhesion to peritoneal mesothelial cells. Am. J. Pathol. 1999, 154, 1525–1537. [Google Scholar] [CrossRef]
- Carreiras, F.; Rigot, V.; Cruet, S.; Andre, F.; Gauduchon, P.; Marvaldi, J. Migration properties of the human ovarian adenocarcinoma cell line IGROV1: Importance of alpha(v)beta3 integrins and vitronectin. Int. J. Cancer 1999, 80, 285–294. [Google Scholar] [CrossRef]
- Blasi, F.; Sidenius, N. The urokinase receptor: Focused cell surface proteolysis, cell adhesion and signaling. FEBS Lett. 2010, 584, 1923–1930. [Google Scholar] [CrossRef] [PubMed]
- Tecimer, C.; Doering, D.L.; Goldsmith, L.J.; Meyer, J.S.; Abdulhay, G.; Wittliff, J.L. Clinical relevance of urokinase-type plasminogen activator, its receptor and inhibitor type 1 in ovarian cancer. Int. J. Gynecol. Cancer Off. J. Int. Gynecol. Cancer Soc. 2000, 10, 372–381. [Google Scholar] [CrossRef] [PubMed]
- Zhan, N.; Dong, W.G.; Wang, J. The clinical significance of vascular endothelial growth factor in malignant ascites. Tumor Biol. 2016, 37, 3719–3725. [Google Scholar] [CrossRef]
- Masoumi Moghaddam, S.; Amini, A.; Morris, D.L.; Pourgholami, M.H. Significance of vascular endothelial growth factor in growth and peritoneal dissemination of ovarian cancer. Cancer Metastasis Rev. 2012, 31, 143–162. [Google Scholar] [CrossRef] [PubMed]
- Yabushita, H.; Shimazu, M.; Noguchi, M.; Kishida, T.; Narumiya, H.; Sawaguchi, K.; Noguchi, M. Vascular endothelial growth factor activating matrix metalloproteinase in ascitic fluid during peritoneal dissemination of ovarian cancer. Oncol. Rep. 2003, 10, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Herr, D.; Sallmann, A.; Bekes, I.; Konrad, R.; Holzheu, I.; Kreienberg, R.; Wulff, C. VEGF induces ascites in ovarian cancer patients via increasing peritoneal permeability by downregulation of Claudin 5. Gynecol. Oncol. 2012, 127, 210–216. [Google Scholar] [CrossRef] [PubMed]
- Gawrychowski, K.; Szewczyk, G.; Skopińska-Różewska, E.; Małecki, M.; Barcz, E.; Kamiński, P.; Miedzińska-Maciejewska, M.; Śmiertka, W.; Szukiewicz, D.; Skopiński, P. The angiogenic activity of ascites in the course of ovarian cancer as a marker of disease progression. Dis. Markers 2014, 2014, 683757. [Google Scholar] [CrossRef] [PubMed]
- Nishida, N.; Yano, H.; Nishida, T.; Kamura, T.; Kojiro, M. Angiogenesis in cancer. Vasc. Health Risk Manag. 2006, 2, 213–219. [Google Scholar] [CrossRef]
- Santin, A.D.; Hermonat, P.L.; Ravaggi, A.; Cannon, M.J.; Pecorelli, S.; Parham, G.P. Secretion of vascular endothelial growth factor in ovarian cancer. Eur. J. Gynaecol. Oncol. 1999, 20, 177–181. [Google Scholar]
- Balcan, E.; Demirkiran, F.; Aydin, Y.; Sanioglu, C.; Bese, T.; Arvas, M.; Akçay, T.; Cift, T. Serum levels of epidermal growth factor, transforming growth factor, and c-erbB2 in ovarian cancer. Int. J. Gynecol. Cancer 2012, 22, 1138–1142. [Google Scholar] [CrossRef]
- Ellis, L.M. Epidermal growth factor receptor in tumor angiogenesis. Hematol./Oncol. Clin. N. Am. 2004, 18, 1007–1021. [Google Scholar] [CrossRef] [PubMed]
- Lassus, H.; Sihto, H.; Leminen, A.; Joensuu, H.; Isola, J.; Nupponen, N.N.; Butzow, R. Gene amplification, mutation, and protein expression of EGFR and mutations of ERBB2 in serous ovarian carcinoma. J. Mol. Med. 2006, 84, 671–681. [Google Scholar] [CrossRef] [PubMed]
- Caswell, P.T.; Chan, M.; Lindsay, A.J.; McCaffrey, M.W.; Boettiger, D.; Norman, J.C. Rab-coupling protein coordinates recycling of alpha5beta1 integrin and EGFR1 to promote cell migration in 3D microenvironments. J. Cell Biol. 2008, 183, 143–155. [Google Scholar] [CrossRef] [PubMed]
- Guarino, M.; Rubino, B.; Ballabio, G. The role of epithelial-mesenchymal transition in cancer pathology. Pathology 2007, 39, 305–318. [Google Scholar] [CrossRef]
- Hoxhaj, G.; Manning, B.D. The PI3K–AKT network at the interface of oncogenic signalling and cancer metabolism. Nat. Rev. Cancer 2020, 20, 74–88. [Google Scholar] [CrossRef]
- Ghoneum, A.; Said, N. PI3K-AKT-mTOR and NFκB Pathways in Ovarian Cancer: Implications for Targeted Therapeutics. Cancers 2019, 11, 949. [Google Scholar] [CrossRef]
- Morales-Ruiz, M.; Tugues, S.; Cejudo-Martín, P.; Ros, J.; Melgar-Lesmes, P.; Fernández-Llama, P.; Arroyo, V.; Rodés, J.; Jiménez, W. Ascites from cirrhotic patients induces angiogenesis through the phosphoinositide 3-kinase/Akt signaling pathway. J. Hepatol 2005, 43, 85–91. [Google Scholar] [CrossRef]
- Takada, Y.; Ye, X.; Simon, S. The integrins. Genome Biol. 2007, 8, 215. [Google Scholar] [CrossRef]
- Ginsberg, M.H.; Partridge, A.; Shattil, S.J. Integrin regulation. Curr. Opin. Cell Biol. 2005, 17, 509–516. [Google Scholar] [CrossRef]
- Hynes, R.O. Integrins: Bidirectional, Allosteric Signaling Machines. Cell 2002, 110, 673–687. [Google Scholar] [CrossRef]
- Schwartz, M.A. Integrins: Emerging Paradigms of Signal Transduction. Annu. Rev. Cell Dev. Biol. 1995, 11, 549–599. [Google Scholar] [CrossRef] [PubMed]
- Harburger, D.S.; Calderwood, D.A. Integrin signalling at a glance. J. Cell Sci. 2009, 122, 159–163. [Google Scholar] [CrossRef]
- Schoenwaelder, S.M.; Burridge, K. Bidirectional signaling between the cytoskeleton and integrins. Curr. Opin. Cell Biol. 1999, 11, 274–286. [Google Scholar] [CrossRef]
- Hamidi, H.; Ivaska, J. Every step of the way: Integrins in cancer progression and metastasis. Nat. Rev. Cancer 2018, 18, 533–548. [Google Scholar] [CrossRef]
- Kobayashi, M.; Sawada, K.; Kimura, T. Potential of Integrin Inhibitors for Treating Ovarian Cancer: A Literature Review. Cancers 2017, 9, 83. [Google Scholar] [CrossRef]
- Fishman, D.A.; Kearns, A.; Chilukuri, K.; Bafetti, L.M.; O’Toole, E.A.; Georgacopoulos, J.; Ravosa, M.J.; Stack, M.S. Metastatic Dissemination of Human Ovarian Epithelial Carcinoma Is Promoted by α2β1-Integrin-Mediated Interaction with Type I Collagen. Invasion Metastasis 1998, 18, 15–26. [Google Scholar] [CrossRef]
- Moser, T.L.; Pizzo, S.V.; Bafetti, L.M.; Fishman, D.A.; Stack, M.S. Evidence for preferential adhesion of ovarian epithelial carcinoma cells to type I collagen mediated by the α2β1 integrin. Int. J. Cancer 1996, 67, 695–701. [Google Scholar] [CrossRef]
- Hu, Z.; Gao, S.; Gao, J.; Hou, R.; Liu, C.; Liu, J.; Li, B.; Liu, D.; Zhang, S.; Lin, B. Elevated levels of Lewis Y and integrin α5β1 correlate with chemotherapeutic drug resistance in epithelial ovarian carcinoma. Int. J. Mol. Sci. 2012, 13, 15588–15600. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, N.; Riley, C.; Oliva, K.; Rice, G.; Quinn, M. Ascites induces modulation of α6β1 integrin and urokinase plasminogen activator receptor expression and associated functions in ovarian carcinoma. Br. J. Cancer 2005, 92, 1475–1485. [Google Scholar] [CrossRef] [PubMed]
- Gillan, L.; Matei, D.; Fishman, D.A.; Gerbin, C.S.; Karlan, B.Y.; Chang, D.D. Periostin secreted by epithelial ovarian carcinoma is a ligand for αVβ3 and αVβ5 integrins and promotes cell motility. Cancer Res. 2002, 62, 5358–5364. [Google Scholar]
- Lane, D.; Goncharenko-Khaider, N.; Rancourt, C.; Piché, A. Ovarian cancer ascites protects from TRAIL-induced cell death through alphavbeta5 integrin-mediated focal adhesion kinase and Akt activation. Oncogene 2010, 29, 3519–3531. [Google Scholar] [CrossRef] [PubMed]
- Carduner, L.; Leroy-Dudal, J.; Picot, C.R.; Gallet, O.; Carreiras, F.; Kellouche, S. Ascites-induced shift along epithelial-mesenchymal spectrum in ovarian cancer cells: Enhancement of their invasive behavior partly dependant on αv integrins. Clin. Exp. Metastasis 2014, 31, 675–688. [Google Scholar] [CrossRef]
- Scalici, J.M.; Harrer, C.; Allen, A.; Jazaeri, A.; Atkins, K.A.; McLachlan, K.R.; Slack-Davis, J.K. Inhibition of α4β1 integrin increases ovarian cancer response to carboplatin. Gynecol. Oncol. 2014, 132, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Casey, R.C.; Burleson, K.M.; Skubitz, K.M.; Pambuccian, S.E.; Oegema, T.R.; Ruff, L.E.; Skubitz, A.P.N. Β1-Integrins Regulate the Formation and Adhesion of Ovarian Carcinoma Multicellular Spheroids. Am. J. Pathol. 2001, 159, 2071–2080. [Google Scholar] [CrossRef]
- Yokoyama, Y.; Sedgewick, G.; Ramakrishnan, S. Endostatin binding to ovarian cancer cells inhibits peritoneal attachment and dissemination. Cancer Res. 2007, 67, 10813–10822. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, A.S.; Hagan, S.; Rath, O.; Kolch, W. MAP kinase signalling pathways in cancer. Oncogene 2007, 26, 3279–3290. [Google Scholar] [CrossRef]
- Mills, G.B.; May, C.; McGill, M.; Roifman, C.M.; Mellors, A. A putative new growth factor in ascitic fluid from ovarian cancer patients: Identification, characterization, and mechanism of action. Cancer Res. 1988, 48, 1066–1071. [Google Scholar]
- Freedman, R.S.; Deavers, M.; Liu, J.; Wang, E. Peritoneal inflammation—A microenvironment for Epithelial Ovarian Cancer (EOC). J. Transl Med. 2004, 2, 23. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Kong, X.; Dou, Q.; Ye, J.; Xu, D.; Shang, H.; Xu, K.; Song, Y. Evaluation of tumor markers for the differential diagnosis of benign and malignant ascites. Ann. Hepatol. 2014, 13, 357–363. [Google Scholar] [CrossRef]
- Lane, D.; Matte, I.; Garde-Granger, P.; Laplante, C.; Carignan, A.; Rancourt, C.; Piché, A. Inflammation-regulating factors in ascites as predictive biomarkers of drug resistance and progression-free survival in serous epithelial ovarian cancers. BMC Cancer 2015, 15, 492. [Google Scholar] [CrossRef]
- Matte, I.; Garde-Granger, P.; Bessette, P.; Piché, A. Ascites from ovarian cancer patients stimulates MUC16 mucin expression and secretion in human peritoneal mesothelial cells through an Akt-dependent pathway. BMC Cancer 2019, 19, 406. [Google Scholar] [CrossRef] [PubMed]
- Jia, D.; Nagaoka, Y.; Katsumata, M.; Orsulic, S. Inflammation is a key contributor to ovarian cancer cell seeding. Sci. Rep. 2018, 8, 12394. [Google Scholar] [CrossRef] [PubMed]
- Browning, L.; Patel, M.R.; Horvath, E.B.; Tawara, K.; Jorcyk, C.L. IL-6 and ovarian cancer: Inflammatory cytokines in promotion of metastasis. Cancer Manag. Res. 2018, 10, 6685–6693. [Google Scholar] [CrossRef]
- Giuntoli, R.L.; Webb, T.J.; Zoso, A.; Rogers, O.; Diaz-Montes, T.P.; Bristow, R.E.; Oelke, M. Ovarian cancer-associated ascites demonstrates altered immune environment: Implications for antitumor immunity. Anticancer Res. 2009, 29, 2875–2884. [Google Scholar]
- Choy, E.; Rose-John, S. Interleukin-6 as a Multifunctional Regulator: Inflammation, Immune Response, and Fibrosis. J. Scleroderma Relat. Disord. 2017, 2, S1–S5. [Google Scholar] [CrossRef]
- Yin, X.; Wu, L.; Yang, H.; Yang, H. Prognostic significance of neutrophil–lymphocyte ratio (NLR) in patients with ovarian cancer. Medicine 2019, 98, e17475. [Google Scholar] [CrossRef]
- Reinartz, S.; Finkernagel, F.; Adhikary, T.; Rohnalter, V.; Schumann, T.; Schober, Y.; Nockher, W.A.; Nist, A.; Stiewe, T.; Jansen, J.M.; et al. A transcriptome-based global map of signaling pathways in the ovarian cancer microenvironment associated with clinical outcome. Genome Biol. 2016, 17, 108. [Google Scholar] [CrossRef] [PubMed]
- Hart, K.M.; Byrne, K.T.; Molloy, M.J.; Usherwood, E.M.; Berwin, B. IL-10 immunomodulation of myeloid cells regulates a murine model of ovarian cancer. Front. Immunol 2011, 2, 29. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Chen, F.; Xu, Q.; Han, L.; Xu, J.; Gao, L.; Sun, X.; Li, Y.; Li, Y.; Qian, M.; et al. Ovarian Cancer Microenvironment: An Overlooked or Overestimated Dark Side? J. Clin. Trials Oncol. 2017, 1, 19. [Google Scholar]
- Alldredge, J.; Flies, D.; Higuchi, T.; Ma, T.; Adams, S.F. Impaired interleukin-10 signaling and ovarian cancer growth in the peritoneal cavity. J. Clin. Oncol. 2014, 32, e22094. [Google Scholar] [CrossRef]
- Onallah, H.; Davidson, B.; Reich, R. Diverse Effects of Lysophosphatidic Acid Receptors on Ovarian Cancer Signaling Pathways. J. Oncol. 2019, 2019, 7547469. [Google Scholar] [CrossRef]
- Yung, Y.C.; Stoddard, N.C.; Chun, J. LPA receptor signaling: Pharmacology, physiology, and pathophysiology. J. Lipid Res. 2014, 55, 1192–1214. [Google Scholar] [CrossRef] [PubMed]
- De La Franier, B.; Thompson, M. Detection of the Ovarian Cancer Biomarker Lysophosphatidic Acid in Serum. Biosensors 2020, 10, 13. [Google Scholar] [CrossRef] [PubMed]
- Sedláková, I.; Vávrová, J.; Tošner, J.; Hanousek, L. Lysophosphatidic acid (LPA)—A perspective marker in ovarian cancer. Tumour Biol. J. Int. Soc. Oncodev. Biol. Med. 2011, 32, 311–316. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Shen, Z.; Wiper, D.W.; Wu, M.; Morton, R.E.; Elson, P.; Kennedy, A.W.; Belinson, J.; Markman, M.; Casey, G. Lysophosphatidic Acid as a Potential Biomarker for Ovarian and Other Gynecologic Cancers. JAMA 1998, 280, 719–723. [Google Scholar] [CrossRef]
- Said, N.A.; Elmarakby, A.A.; Imig, J.D.; Fulton, D.J.; Motamed, K. SPARC ameliorates ovarian cancer-associated inflammation. Neoplasia 2008, 10, 1092–1104. [Google Scholar] [CrossRef]
- Cai, Q.; Zhao, Z.; Antalis, C.; Yan, L.; Del Priore, G.; Hamed, A.H.; Stehman, F.B.; Schilder, J.M.; Xu, Y. Elevated and secreted phospholipase A2 activities as new potential therapeutic targets in human epithelial ovarian cancer. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2012, 26, 3306–3320. [Google Scholar] [CrossRef] [PubMed]
- Schulte, R.R.; Linkous, A.G.; Hallahan, D.E.; Yazlovitskaya, E.M. Cytosolic phospholipase A2 as a molecular target for the radiosensitization of ovarian cancer. Cancer Lett. 2011, 304, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Cui, L.; Pan, Y.; Shao, D.; Zheng, X.; Zhang, F.; Zhang, H.; He, K.; Chen, L. Berberine inhibits the chemotherapy-induced repopulation by suppressing the arachidonic acid metabolic pathway and phosphorylation of FAK in ovarian cancer. Cell Prolif. 2017, 50, e12393. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Zaman, M.M.; Vlasakov, I.; Roy, R.; Huang, L.; Martin, C.R.; Freedman, S.D.; Serhan, C.N.; Moses, M.A. Adipocytes promote ovarian cancer chemoresistance. Sci. Rep. 2019, 9, 13316. [Google Scholar] [CrossRef]
- Punnonen, R.; Seppälä, E.; Punnonen, K.; Heinonen, P.K. Fatty acid composition and arachidonic acid metabolites in ascitic fluid of patients with ovarian cancer. Prostaglandins Leukot. Med. 1986, 22, 153–158. [Google Scholar] [CrossRef]
- Wang, D.; DuBois, R.N. Eicosanoids and cancer. Nat. Rev. Cancer 2010, 10, 181–193. [Google Scholar] [CrossRef] [PubMed]
- Wilson, A.J.; Fadare, O.; Beeghly-Fadiel, A.; Son, D.-S.; Liu, Q.; Zhao, S.; Saskowski, J.; Uddin, M.J.; Daniel, C.; Crews, B.; et al. Aberrant over-expression of COX-1 intersects multiple pro-tumorigenic pathways in high-grade serous ovarian cancer. Oncotarget 2015, 6, 21353–21368. [Google Scholar] [CrossRef]
- Gupta, R.A.; Tejada, L.V.; Tong, B.J.; Das, S.K.; Morrow, J.D.; Dey, S.K.; DuBois, R.N. Cyclooxygenase-1 is overexpressed and promotes angiogenic growth factor production in ovarian cancer. Cancer Res. 2003, 63, 906–911. [Google Scholar]
- Munkarah, A.R.; Morris, R.; Baumann, P.; Deppe, G.; Malone, J.; Diamond, M.P.; Saed, G.M. Effects of Prostaglandin E2 on Proliferation and Apoptosis of Epithelial Ovarian Cancer Cells. J. Soc. Gynecol. Investig. 2002, 9, 168–173. [Google Scholar] [CrossRef] [PubMed]
- Qiu, X.; Cheng, J.C.; Chang, H.M.; Leung, P.C. COX2 and PGE2 mediate EGF-induced E-cadherin-independent human ovarian cancer cell invasion. Endocr.-Relat. Cancer 2014, 21, 533–543. [Google Scholar] [CrossRef][Green Version]
- Spinella, F.; Rosanò, L.; Di Castro, V.; Natali, P.G.; Bagnato, A. Endothelin-1-induced prostaglandin E2-EP2, EP4 signaling regulates vascular endothelial growth factor production and ovarian carcinoma cell invasion. J. Biol. Chem. 2004, 279, 46700–46705. [Google Scholar] [CrossRef]
- Spinella, F.; Rosanò, L.; Elia, G.; Di Castro, V.; Natali, P.G.; Bagnato, A. Endothelin-1 stimulates cyclooxygenase-2 expression in ovarian cancer cells through multiple signaling pathways: Evidence for involvement of transactivation of the epidermal growth factor receptor. J. Cardiovasc. Pharmacol. 2004, 44 (Suppl. 1), S140–S143. [Google Scholar] [CrossRef]
- Zhang, X.; Yan, K.; Deng, L.; Liang, J.; Liang, H.; Feng, D.; Ling, B. Cyclooxygenase 2 Promotes Proliferation and Invasion in Ovarian Cancer Cells via the PGE2/NF-κB Pathway. Cell Transplant. 2019, 28, 1S–13S. [Google Scholar] [CrossRef]
- Reader, J.C.; Staats, P.; Goloubeva, O.; Jian, N.; Roque, D.M.; Matheny, M.E.; Fulton, A.M.; Rao, G.G. Functional studies of EP4 in ovarian cancer. Cancer Res. 2016, 76, 5169. [Google Scholar]
- Serhan, K.; Gartung, A.; Panigrahy, D. Drawing a link between the thromboxane A2 pathway and the role of platelets and tumor cells in ovarian cancer. Prostaglandins Other Lipid Mediat. 2018, 137, 40–45. [Google Scholar] [CrossRef] [PubMed]
- Guo, A.M.; Liu, X.; Al-Wahab, Z.; Maddippati, K.R.; Ali-Fehmi, R.; Scicli, A.G.; Munkarah, A.R. Role of 12-lipoxygenase in regulation of ovarian cancer cell proliferation and survival. Cancer Chemother. Pharmacol. 2011, 68, 1273–1283. [Google Scholar] [CrossRef]
- Liu, Q.; Tan, W.; Che, J.; Yuan, D.; Zhang, L.; Sun, Y.; Yue, X.; Xiao, L.; Jin, Y. 12-HETE facilitates cell survival by activating the integrin-linked kinase/NF-κB pathway in ovarian cancer. Cancer Manag. Res. 2018, 10, 5825–5838. [Google Scholar] [CrossRef] [PubMed]
- Freedman, R.S.; Wang, E.; Voiculescu, S.; Patenia, R.; Bassett, R.L.; Deavers, M.; Marincola, F.M.; Yang, P.; Newman, R.A. Comparative analysis of peritoneum and tumor eicosanoids and pathways in advanced ovarian cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2007, 13, 5736–5744. [Google Scholar] [CrossRef]
- Rocconi, R.P.; Kirby, T.O.; Seitz, R.S.; Beck, R.; Straughn, J.M.; Alvarez, R.D.; Huh, W.K. Lipoxygenase pathway receptor expression in ovarian cancer. Reprod. Sci. 2008, 15, 321–326. [Google Scholar] [CrossRef] [PubMed]
- Wen, Z.; Liu, H.; Li, M.; Li, B.; Gao, W.; Shao, Q.; Fan, B.; Zhao, F.; Wang, Q.; Xie, Q.; et al. Increased metabolites of 5-lipoxygenase from hypoxic ovarian cancer cells promote tumor-associated macrophage infiltration. Oncogene 2015, 34, 1241–1252. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Park, S.Y.; Kim, J.-H. Leukotriene B4 receptor-2 contributes to chemoresistance of SK-OV-3 ovarian cancer cells through activation of signal transducer and activator of transcription-3-linked cascade. Biochim. Biophys. Acta 2016, 1863, 236–243. [Google Scholar] [CrossRef] [PubMed]
- Seo, J.-M.; Park, S.; Kim, J.-H. Leukotriene B4 receptor-2 promotes invasiveness and metastasis of ovarian cancer cells through signal transducer and activator of transcription 3 (STAT3)-dependent up-regulation of matrix metalloproteinase 2. J. Biol. Chem. 2012, 287, 13840–13849. [Google Scholar] [CrossRef]
- Hong, G.; Baudhuin, L.M.; Xu, Y. Sphingosine-1-phosphate modulates growth and adhesion of ovarian cancer cells. FEBS Lett 1999, 460, 513–518. [Google Scholar] [CrossRef]
- Schwartz, B.M.; Hong, G.; Morrison, B.H.; Wu, W.; Baudhuin, L.M.; Xiao, Y.J.; Mok, S.C.; Xu, Y. Lysophospholipids increase interleukin-8 expression in ovarian cancer cells. Gynecol. Oncol. 2001, 81, 291–300. [Google Scholar] [CrossRef] [PubMed]
- Dai, L.; Liu, Y.; Xie, L.; Wu, X.; Qiu, L.; Di, W. Sphingosine kinase 1/sphingosine-1-phosphate (S1P)/S1P receptor axis is involved in ovarian cancer angiogenesis. Oncotarget 2017, 8, 74947–74961. [Google Scholar] [CrossRef]
- Zhang, H.; Wang, Q.; Zhao, Q.; Di, W. MiR-124 inhibits the migration and invasion of ovarian cancer cells by targeting SphK1. J. Ovarian Res. 2013, 6, 84. [Google Scholar] [CrossRef] [PubMed]
- White, M.D.; Chan, L.; Antoon, J.W.; Beckman, B.S. Targeting ovarian cancer and chemoresistance through selective inhibition of sphingosine kinase-2 with ABC294640. Anticancer Res. 2013, 33, 3573–3579. [Google Scholar]
- Lee, K.-B.; Byun, H.-J.; Park, S.H.; Park, C.-Y.; Lee, S.-H.; Rho, S.B. CYR61 controls p53 and NF-κB expression through PI3K/Akt/mTOR pathways in carboplatin-induced ovarian cancer cells. Cancer Lett. 2012, 315, 86–95. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Cai, M.; Zhao, S.; Wang, H.; Li, M.; Yao, S.; Jiang, N. CYR61 overexpression associated with the development and poor prognosis of ovarian carcinoma. Med. Oncol. 2014, 31, 117. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Huo, R.; Li, N.; Li, H.H.; Zhai, T.; Li, H.H.; Shen, B.; Ye, J.; Fu, R.; Di, W. CYR61, a potential biomarker of tumor inflammatory response in epithelial ovarian cancer microenvironment of tumor progress. BMC Cancer 2019, 19, 1140. [Google Scholar] [CrossRef] [PubMed]
- Chu, C.S.; Kim, S.H.; June, C.H.; Coukos, G. Immunotherapy opportunities in ovarian cancer. Expert Rev. Anticancer Ther. 2008, 8, 243–257. [Google Scholar] [CrossRef]
- Chaplin, D.D. Overview of the immune response. J. Allergy Clin. Immunol. 2010, 125, S3–S23. [Google Scholar] [CrossRef]
- Bamias, A.; Tsiatas, M.L.; Kafantari, E.; Liakou, C.; Rodolakis, A.; Voulgaris, Z.; Vlahos, G.; Papageorgiou, T.; Tsitsilonis, O.; Bamia, C.; et al. Significant differences of lymphocytes isolated from ascites of patients with ovarian cancer compared to blood and tumor lymphocytes. Association of CD3+CD56+ cells with platinum resistance. Gynecol. Oncol. 2007, 106, 75–81. [Google Scholar] [CrossRef]
- Rådestad, E.; Klynning, C.; Stikvoort, A.; Mogensen, O.; Nava, S.; Magalhaes, I.; Uhlin, M. Immune profiling and identification of prognostic immune-related risk factors in human ovarian cancer. Oncoimmunology 2019, 8, e1535730. [Google Scholar] [CrossRef]
- Vazquez, J.; Chavarria, M.; Lopez, G.E.; Felder, M.A.; Kapur, A.; Romo Chavez, A.; Karst, N.; Barroilhet, L.; Patankar, M.S.; Stanic, A.K. Identification of unique clusters of T, dendritic, and innate lymphoid cells in the peritoneal fluid of ovarian cancer patients. Am. J. Reprod. Immunol. 2020, 84, e13284. [Google Scholar] [CrossRef]
- Correa, R.J.M.; Peart, T.; Valdes, Y.R.; DiMattia, G.E.; Shepherd, T.G. Modulation of AKT activity is associated with reversible dormancy in ascites-derived epithelial ovarian cancer spheroids. Carcinogenesis 2012, 33, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Orecchioni, M.; Ghosheh, Y.; Pramod, A.B.; Ley, K. Macrophage Polarization: Different Gene Signatures in M1(LPS+) vs. Classically and M2(LPS-) vs. Alternatively Activated Macrophages. Front. Immunol. 2019, 10, 1084. [Google Scholar] [CrossRef]
- Gartung, A.; Yang, J.; Sukhatme, V.P.; Bielenberg, D.R.; Fernandes, D.; Chang, J.; Schmidt, B.A.; Hwang, S.H.; Zurakowski, D.; Huang, S.; et al. Suppression of chemotherapy-induced cytokine/lipid mediator surge and ovarian cancer by a dual COX-2/sEH inhibitor. Proc. Natl. Acad. Sci. USA 2019, 116, 1698–1703. [Google Scholar] [CrossRef] [PubMed]
- Reader, J.; Harper, A.K.; Legesse, T.; Staats, P.N.; Goloubeva, O.; Rao, G.G.; Fulton, A.; Roque, D.M. EP4 and Class III β-Tubulin Expression in Uterine Smooth Muscle Tumors: Implications for Prognosis and Treatment. Cancers 2019, 11, 1590. [Google Scholar] [CrossRef]
- Sweat, R.S.; Stapor, P.C.; Murfee, W.L. Relationships Between Lymphangiogenesis and Angiogenesis During Inflammation in Rat Mesentery Microvascular Networks. Lymphat. Res. Biol. 2012, 10, 198–207. [Google Scholar] [CrossRef] [PubMed]
- Roque, D.M.; Bellone, S.; Buza, N.; Romani, C.; Cocco, E.; Bignotti, E.; Ravaggi, A.; Rutherford, T.J.; Schwartz, P.E.; Pecorelli, S.; et al. Class III β-tubulin overexpression in ovarian clear cell and serous carcinoma as a maker for poor overall survival after platinum/taxane chemotherapy and sensitivity to patupilone. Am. J. Obstet. Gynecol. 2013, 209, 62–e1. [Google Scholar] [CrossRef]
- Roque, D.M.; Buza, N.; Glasgow, M.; Bellone, S.; Bortolomai, I.; Gasparrini, S.; Cocco, E.; Ratner, E.; Silasi, D.-A.; Azodi, M.; et al. Class III β-tubulin overexpression within the tumor microenvironment is a prognostic biomarker for poor overall survival in ovarian cancer patients treated with neoadjuvant carboplatin/paclitaxel. Clin. Exp. Metastasis 2014, 31, 101–110. [Google Scholar] [CrossRef]
- Diakos, C.I.; Charles, K.A.; McMillan, D.C.; Clarke, S.J. Cancer-related inflammation and treatment effectiveness. Lancet Oncol. 2014, 15, e493–e503. [Google Scholar] [CrossRef]
- Reinartz, S.; Schumann, T.; Finkernagel, F.; Wortmann, A.; Jansen, J.M.; Meissner, W.; Krause, M.; Schwörer, A.-M.; Wagner, U.; Müller-Brüsselbach, S.; et al. Mixed-polarization phenotype of ascites-associated macrophages in human ovarian carcinoma: Correlation of CD163 expression, cytokine levels and early relapse. Int. J. Cancer 2014, 134, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Adhikary, T.; Wortmann, A.; Finkernagel, F.; Lieber, S.; Nist, A.; Stiewe, T.; Wagner, U.; Müller-Brüsselbach, S.; Reinartz, S.; Müller, R. Interferon signaling in ascites-associated macrophages is linked to a favorable clinical outcome in a subgroup of ovarian carcinoma patients. BMC Genom. 2017, 18, 243. [Google Scholar] [CrossRef] [PubMed]
- Nowak, M.; Klink, M.A.-O. The Role of Tumor-Associated Macrophages in the Progression and Chemoresistance of Ovarian Cancer. Cell 2020, 9, 1299. [Google Scholar] [CrossRef]
- Cheng, H.; Wang, Z.; Fu, L.; Xu, T. Macrophage Polarization in the Development and Progression of Ovarian Cancers: An Overview. Front. Oncol. 2019, 9, 421. [Google Scholar] [CrossRef]
- Hagemann, T.; Wilson, J.; Kulbe, H.; Li, N.F.; Leinster, D.A.; Charles, K.; Klemm, F.; Pukrop, T.; Binder, C.; Balkwill, F.R. Macrophages induce invasiveness of epithelial cancer cells via NF-kappa B and JNK. J. Immunol. 2005, 175, 1197–1205. [Google Scholar] [CrossRef]
- Dijkgraaf, E.M.; Heusinkveld, M.; Tummers, B.; Vogelpoel, L.T.C.; Goedemans, R.; Jha, V.; Nortier, J.W.R.; Welters, M.J.P.; Kroep, J.R.; van der Burg, S.H. Chemotherapy Alters Monocyte Differentiation to Favor Generation of Cancer-Supporting M2 Macrophages in the Tumor Microenvironment. Cancer Res. 2013, 73, 2480. [Google Scholar] [CrossRef] [PubMed]
- Colvin, E.K. Tumor-Associated Macrophages Contribute to Tumor Progression in Ovarian Cancer. Front. Oncol. 2014, 4, 137. [Google Scholar] [CrossRef] [PubMed]
- Yin, M.; Li, X.; Tan, S.; Zhou, H.J.; Ji, W.; Bellone, S.; Xu, X.; Zhang, H.; Santin, A.D.; Lou, G.; et al. Tumor-associated macrophages drive spheroid formation during early transcoelomic metastasis of ovarian cancer. J. Clin. Investig. 2016, 126, 4157–4173. [Google Scholar] [CrossRef]
- Lai, P.; Rabinowich, H.; Crowley-Nowick, P.A.; Bell, M.C.; Mantovani, G.; Whiteside, T.L. Alterations in expression and function of signal-transducing proteins in tumor-associated T and natural killer cells in patients with ovarian carcinoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 1996, 2, 161–173. [Google Scholar]
- Castriconi, R.; Cantoni, C.; Della Chiesa, M.; Vitale, M.; Marcenaro, E.; Conte, R.; Biassoni, R.; Bottino, C.; Moretta, L.; Moretta, A. Transforming growth factor beta 1 inhibits expression of NKp30 and NKG2D receptors: Consequences for the NK-mediated killing of dendritic cells. Proc. Natl. Acad. Sci. USA 2003, 100, 4120–4125. [Google Scholar] [CrossRef]
- Greppi, M.; Tabellini, G.; Patrizi, O.; Candiani, S.; Decensi, A.; Parolini, S.; Sivori, S.; Pesce, S.; Paleari, L.; Marcenaro, E. Strengthening the AntiTumor NK Cell Function for the Treatment of Ovarian Cancer. Int. J. Mol. Sci. 2019, 20, 890. [Google Scholar] [CrossRef]
- Nham, T.; Poznanski, S.M.; Fan, I.Y.; Shenouda, M.M.; Chew, M.V.; Lee, A.J.; Vahedi, F.; Karimi, Y.; Butcher, M.; Lee, D.A.; et al. Ex vivo-expanded NK cells from blood and ascites of ovarian cancer patients are cytotoxic against autologous primary ovarian cancer cells. Cancer Immunol. Immunother. CII 2018, 67, 575–587. [Google Scholar] [CrossRef]
- Roque, D.M.; Santin, A.D. Antigen-specific immunotherapy for ovarian cancer. Future Med. 2012, 136–154. [Google Scholar] [CrossRef]
- Curiel, T.J.; Cheng, P.; Mottram, P.; Alvarez, X.; Moons, L.; Evdemon-Hogan, M.; Wei, S.; Zou, L.; Kryczek, I.; Hoyle, G.; et al. Dendritic cell subsets differentially regulate angiogenesis in human ovarian cancer. Cancer Res. 2004, 64, 5535–5538. [Google Scholar] [CrossRef]
- Wefers, C.; Duiveman-de Boer, T.; Yigit, R.; Zusterzeel, P.L.M.; van Altena, A.M.; Massuger, L.F.A.G.; De Vries, I.J.M. Survival of Ovarian Cancer Patients Is Independent of the Presence of DC and T Cell Subsets in Ascites. Front. Immunol. 2018, 9, 3156. [Google Scholar] [CrossRef] [PubMed]
- Brencicova, E.; Jagger, A.L.; Evans, H.G.; Georgouli, M.; Laios, A.; Attard Montalto, S.; Mehra, G.; Spencer, J.; Ahmed, A.A.; Raju-Kankipati, S.; et al. Interleukin-10 and prostaglandin E2 have complementary but distinct suppressive effects on Toll-like receptor-mediated dendritic cell activation in ovarian carcinoma. PLoS ONE 2017, 12, e0175712. [Google Scholar] [CrossRef]
- Gabrilovich, D.I. Myeloid-Derived Suppressor Cells. Cancer Immunol. Res. 2017, 5, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Baert, T.; Vankerckhoven, A.; Riva, M.; Van Hoylandt, A.; Thirion, G.; Holger, G.; Mathivet, T.; Vergote, I.; Coosemans, A. Myeloid Derived Suppressor Cells: Key Drivers of Immunosuppression in Ovarian Cancer. Front. Immunol. 2019, 10, 1273. [Google Scholar] [CrossRef]
- Mosmann, T.R.; Cherwinski, H.; Bond, M.W.; Giedlin, M.A.; Coffman, R.L. Two types of murine helper T cell clone. I. Definition according to profiles of lymphokine activities and secreted proteins. J. Immunol. 1986, 136, 2348–2357. [Google Scholar]
- Berger, A. Th1 and Th2 responses: What are they? BMJ (Clin. Res. Ed.) 2000, 321, 424. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhang, X.L.; Zhang, G.H.; Gao, Y.F. Artesunate promotes Th1 differentiation from CD4+ T cells to enhance cell apoptosis in ovarian cancer via miR-142. Braz J. Med. Biol Res. 2019, 52, e7992. [Google Scholar] [CrossRef]
- Hao, C.J.; Li, J.; Liu, P.; Li, X.L.; Hu, Y.Q.; Sun, J.C.; Wei, Y. Effects of the balance between type 1 and type 2 T helper cells on ovarian cancer. Genet. Mol. Res. GMR 2016, 15, 15027936. [Google Scholar] [CrossRef]
- Abiko, K.; Mandai, M.; Hamanishi, J.; Yoshioka, Y.; Matsumura, N.; Baba, T.; Yamaguchi, K.; Murakami, R.; Yamamoto, A.; Kharma, B.; et al. PD-L1 on tumor cells is induced in ascites and promotes peritoneal dissemination of ovarian cancer through CTL dysfunction. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2013, 19, 1363–1374. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Li, Y.; Zhu, B. T-cell exhaustion in the tumor microenvironment. Cell Death Dis. 2015, 6, e1792. [Google Scholar] [CrossRef] [PubMed]
- Fan, C.A.; Reader, J.; Roque, D.M. Review of Immune Therapies Targeting Ovarian Cancer. Curr. Treat. Options Oncol. 2018, 19, 74. [Google Scholar] [CrossRef]
- Song, M.; Sandoval, T.A.; Chae, C.-S.; Chopra, S.; Tan, C.; Rutkowski, M.R.; Raundhal, M.; Chaurio, R.A.; Payne, K.K.; Konrad, C.; et al. IRE1α–XBP1 controls T cell function in ovarian cancer by regulating mitochondrial activity. Nature 2018, 562, 423–428. [Google Scholar] [CrossRef]
- Kryczek, I.; Banerjee, M.; Cheng, P.; Vatan, L.; Szeliga, W.; Wei, S.; Huang, E.; Finlayson, E.; Simeone, D.; Welling, T.H.; et al. Phenotype, distribution, generation, and functional and clinical relevance of Th17 cells in the human tumor environments. Blood 2009, 114, 1141–1149. [Google Scholar] [CrossRef]
- Ou, Y.; Cannon, M.J.; Nakagawa, M. Regulatory T Cells in Gynecologic Cancer. MOJ Immunol. 2018, 6, 34–42. [Google Scholar] [CrossRef]
- Landskron, J.; Helland, Ø.; Torgersen, K.M.; Aandahl, E.M.; Gjertsen, B.T.; Bjørge, L.; Taskén, K. Activated regulatory and memory T-cells accumulate in malignant ascites from ovarian carcinoma patients. Cancer Immunol. Immunother. CII 2015, 64, 337–347. [Google Scholar] [CrossRef]
- Chen, Y.-L.; Chang, M.-C.; Chen, C.-A.; Lin, H.-W.; Cheng, W.-F.; Chien, C.-L. Depletion of Regulatory T Lymphocytes Reverses the Imbalance between Pro- and Anti-Tumor Immunities via Enhancing Antigen-Specific T Cell Immune Responses. PLoS ONE 2012, 7, e47190. [Google Scholar] [CrossRef] [PubMed]
- Cheng, W.-F.; Hung, C.-F.; Chai, C.-Y.; Chen, C.-A.; Lee, C.-N.; Su, Y.-N.; Tseng, W.-Y.I.; Hsieh, C.-Y.; Shih, I.-M.; Wang, T.-L.; et al. Generation and Characterization of an Ascitogenic Mesothelin-Expressing Tumor Model. Cancer 2007, 110, 420–431. [Google Scholar] [CrossRef]
- Govindaraj, C.; Scalzo-Inguanti, K.; Madondo, M.; Hallo, J.; Flanagan, K.; Quinn, M.; Plebanski, M. Impaired Th1 immunity in ovarian cancer patients is mediated by TNFR2+ Tregs within the tumor microenvironment. Clin. Immunol. 2013, 149, 97–110. [Google Scholar] [CrossRef]
- Li, J.; Feng, G.; Liu, J.; Rong, R.; Luo, F.; Guo, L.; Zhu, T.; Wang, G.; Chu, Y. Renal cell carcinoma may evade the immune system by converting CD4+Foxp3- T cells into CD4+CD25+Foxp3+ regulatory T cells: Role of tumor COX-2-derived PGE2. Mol. Med. Rep. 2010, 3, 959–963. [Google Scholar] [CrossRef]
- Facciabene, A.; Peng, X.; Hagemann, I.S.; Balint, K.; Barchetti, A.; Wang, L.P.; Gimotty, P.A.; Gilks, C.B.; Lal, P.; Zhang, L.; et al. Tumour hypoxia promotes tolerance and angiogenesis via CCL28 and T(reg) cells. Nature 2011, 475, 226–230. [Google Scholar] [CrossRef] [PubMed]
- Kampan, N.C.; Madondo, M.T.; McNally, O.M.; Stephens, A.N.; Quinn, M.A.; Plebanski, M. Interleukin 6 Present in Inflammatory Ascites from Advanced Epithelial Ovarian Cancer Patients Promotes Tumor Necrosis Factor Receptor 2-Expressing Regulatory T Cells. Front. Immunol. 2017, 8, 1482. [Google Scholar] [CrossRef]
- Ballabio, A.; Bonifacino, J.S. Lysosomes as dynamic regulators of cell and organismal homeostasis. Nat. Rev. Mol. Cell Biol. 2020, 21, 101–118. [Google Scholar] [CrossRef] [PubMed]
- Sancak, Y.; Bar-Peled, L.; Zoncu, R.; Markhard, A.L.; Nada, S.; Sabatini, D.M. Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell 2010, 141, 290–303. [Google Scholar] [CrossRef] [PubMed]
- Kimmelman, A.C.; White, E. Autophagy and Tumor Metabolism. Cell Metab. 2017, 25, 1037–1043. [Google Scholar] [CrossRef]
- Beratis, N.G.; Kaperonis, A.; Eliopoulou, M.I.; Kourounis, G.; Tzingounis, V.A. Increased activity of lysosomal enzymes in the peritoneal fluid of patients with gynecologic cancers and pelvic inflammatory disease. J. Cancer Res. Clin. Oncol. 2005, 131, 371–376. [Google Scholar] [CrossRef]
- Xu, Y.; Cao, X.; Zhang, S.; Zhang, Y.; Shen, Z. High expression of LAMP1 as a prognostic marker in patients with epithelial ovarian cancer. Int. J. Clin. Exp. Pathol. 2017, 10, 9104–9111. [Google Scholar] [PubMed]
- Meunier, L.; Puiffe, M.-L.; Le Page, C.; Filali-Mouhim, A.; Chevrette, M.; Tonin, P.N.; Provencher, D.M.; Mes-Masson, A.-M. Effect of Ovarian Cancer Ascites on Cell Migration and Gene Expression in an Epithelial Ovarian Cancer In Vitro Model. Transl. Oncol. 2010, 3, 230–238. [Google Scholar] [CrossRef]
- Fang, C.H.; Lin, Y.T.; Liang, C.M.; Liang, S.M. A novel c-Kit/phospho-prohibitin axis enhances ovarian cancer stemness and chemoresistance via Notch3-PBX1 and β-catenin-ABCG2 signaling. J. Biomed. Sci 2020, 27, 42. [Google Scholar] [CrossRef]
- Ostrowski, M.; Carmo, N.B.; Krumeich, S.; Fanget, I.; Raposo, G.; Savina, A.; Moita, C.F.; Schauer, K.; Hume, A.N.; Freitas, R.P.; et al. Rab27a and Rab27b control different steps of the exosome secretion pathway. Nat. Cell Biol. 2010, 12, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Dorayappan, K.D.P.; Wanner, R.; Wallbillich, J.J.; Saini, U.; Zingarelli, R.; Suarez, A.A.; Cohn, D.E.; Selvendiran, K. Hypoxia-induced exosomes contribute to a more aggressive and chemoresistant ovarian cancer phenotype: A novel mechanism linking STAT3/Rab proteins. Oncogene 2018, 37, 3806–3821. [Google Scholar] [CrossRef]
- Safaei, R.; Larson, B.J.; Cheng, T.C.; Gibson, M.A.; Otani, S.; Naerdemann, W.; Howell, S.B. Abnormal lysosomal trafficking and enhanced exosomal export of cisplatin in drug-resistant human ovarian carcinoma cells. Mol. Cancer 2005, 4, 1595–1604. [Google Scholar] [CrossRef]
- Alharbi, M.; Lai, A.; Sharma, S.; Kalita-de Croft, P.; Godbole, N.; Campos, A.; Guanzon, D.; Salas-Burgos, A.; Carrion, F.; Zuñiga, F.A.; et al. Extracellular Vesicle Transmission of Chemoresistance to Ovarian Cancer Cells Is Associated with Hypoxia-Induced Expression of Glycolytic Pathway Proteins, and Prediction of Epithelial Ovarian Cancer Disease Recurrence. Cancers 2021, 13, 3388. [Google Scholar] [CrossRef] [PubMed]
- Doherty, J.R.; Cleveland, J.L. Targeting lactate metabolism for cancer therapeutics. J. Clin. Investig. 2013, 123, 3685–3692. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.-L.; Xia, H.H.-X.; Zhu, S.-L. Ascitic Fluid Analysis in the Differential Diagnosis of Ascites: Focus on Cirrhotic Ascites. J. Clin. Transl. Hepatol. 2014, 2, 58–64. [Google Scholar] [CrossRef]
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef]
- Sorrin, A.J.; Kemal Ruhi, M.; Ferlic, N.A.; Karimnia, V.; Polacheck, W.J.; Celli, J.P.; Huang, H.-C.; Rizvi, I. Photodynamic Therapy and the Biophysics of the Tumor Microenvironment. Photochem. Photobiol. 2020, 96, 232–259. [Google Scholar] [CrossRef]
- Nakamura, K.; Sawada, K.; Kobayashi, M.; Miyamoto, M.; Shimizu, A.; Yamamoto, M.; Kinose, Y.; Kimura, T. Role of the Exosome in Ovarian Cancer Progression and Its Potential as a Therapeutic Target. Cancers 2019, 11, 1147. [Google Scholar] [CrossRef]
- Feng, W.; Dean, D.C.; Hornicek, F.J.; Shi, H.; Duan, Z. Exosomes promote pre-metastatic niche formation in ovarian cancer. Mol. Cancer 2019, 18, 124. [Google Scholar] [CrossRef]
- Taylor, D.D.; Gerçel-Taylor, C. Tumour-derived exosomes and their role in cancer-associated T-cell signalling defects. Br. J. Cancer 2005, 92, 305–311. [Google Scholar] [CrossRef]
- Wei, M.; Yang, T.; Chen, X.; Wu, Y.; Deng, X.; He, W.; Yang, J.; Wang, Z. Malignant ascites-derived exosomes promote proliferation and induce carcinoma-associated fibroblasts transition in peritoneal mesothelial cells. Oncotarget 2017, 8, 42262–42271. [Google Scholar] [CrossRef] [PubMed]
- Liang, R.; Chen, X.; Chen, L.; Wan, F.; Chen, K.; Sun, Y.; Zhu, X. STAT3 signaling in ovarian cancer: A potential therapeutic target. J. Cancer 2020, 11, 837–848. [Google Scholar] [CrossRef] [PubMed]
- Comisso, E.; Scarola, M.; Rosso, M.; Piazza, S.; Marzinotto, S.; Ciani, Y.; Orsaria, M.; Mariuzzi, L.; Schneider, C.; Schoeftner, S.; et al. OCT4 controls mitotic stability and inactivates the RB tumor suppressor pathway to enhance ovarian cancer aggressiveness. Oncogene 2017, 36, 4253–4266. [Google Scholar] [CrossRef]
- Nichols, J.; Zevnik, B.; Anastassiadis, K.; Niwa, H.; Klewe-Nebenius, D.; Chambers, I.; Schöler, H.; Smith, A. Formation of Pluripotent Stem Cells in the Mammalian Embryo Depends on the POU Transcription Factor Oct4. Cell 1998, 95, 379–391. [Google Scholar] [CrossRef]
- Wang, Y.-J.; Herlyn, M. The emerging roles of Oct4 in tumor-initiating cells. Am. J. Physiology.-Cell Physiol. 2015, 309, C709–C718. [Google Scholar] [CrossRef] [PubMed]
- Ruan, Z.; Yang, X.; Cheng, W. OCT4 accelerates tumorigenesis through activating JAK/STAT signaling in ovarian cancer side population cells. Cancer Manag. Res. 2018, 11, 389–399. [Google Scholar] [CrossRef] [PubMed]
- Gupta, V.; Yull, F.; Khabele, D. Bipolar Tumor-Associated Macrophages in Ovarian Cancer as Targets for Therapy. Cancers 2018, 10, 366. [Google Scholar] [CrossRef]
- Condeelis, J.; Pollard, J.W. Macrophages: Obligate Partners for Tumor Cell Migration, Invasion, and Metastasis. Cell 2006, 124, 263–266. [Google Scholar] [CrossRef] [PubMed]
- Sica, A.; Larghi, P.; Mancino, A.; Rubino, L.; Porta, C.; Totaro, M.G.; Rimoldi, M.; Biswas, S.K.; Allavena, P.; Mantovani, A. Macrophage polarization in tumour progression. Semin. Cancer Biol. 2008, 18, 349–355. [Google Scholar] [CrossRef] [PubMed]
- Takaishi, K.; Komohara, Y.; Tashiro, H.; Ohtake, H.; Nakagawa, T.; Katabuchi, H.; Takeya, M. Involvement of M2-polarized macrophages in the ascites from advanced epithelial ovarian carcinoma in tumor progression via Stat3 activation. Cancer Sci. 2010, 101, 2128–2136. [Google Scholar] [CrossRef]
- Hagemann, T.; Wilson, J.; Burke, F.; Kulbe, H.; Li, N.F.; Plüddemann, A.; Charles, K.; Gordon, S.; Balkwill, F.R. Ovarian Cancer Cells Polarize Macrophages Toward A Tumor-Associated Phenotype. J. Immunol. 2006, 176, 5023–5032. [Google Scholar] [CrossRef]
- Kalluri, R. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 2016, 16, 582–598. [Google Scholar] [CrossRef]
- Bronzert, D.A.; Pantazis, P.; Antoniades, H.N.; Kasid, A.; Davidson, N.; Dickson, R.B.; Lippman, M.E. Synthesis and secretion of platelet-derived growth factor by human breast cancer cell lines. Proc. Natl. Acad. Sci. USA 1987, 84, 5763–5767. [Google Scholar] [CrossRef]
- Cai, J.; Tang, H.; Xu, L.; Wang, X.; Yang, C.; Ruan, S.; Guo, J.; Hu, S.; Wang, Z. Fibroblasts in omentum activated by tumor cells promote ovarian cancer growth, adhesion and invasiveness. Carcinogenesis 2011, 33, 20–29. [Google Scholar] [CrossRef]
- Dasari, S.; Fang, Y.; Mitra, A.K. Cancer Associated Fibroblasts: Naughty Neighbors That Drive Ovarian Cancer Progression. Cancers 2018, 10, 406. [Google Scholar] [CrossRef]
- Yeung, T.-L.; Leung, C.S.; Wong, K.-K.; Samimi, G.; Thompson, M.S.; Liu, J.; Zaid, T.M.; Ghosh, S.; Birrer, M.J.; Mok, S.C. TGF-β modulates ovarian cancer invasion by upregulating CAF-derived versican in the tumor microenvironment. Cancer Res. 2013, 73, 5016–5028. [Google Scholar] [CrossRef]
- Mitra, A.K.; Zillhardt, M.; Hua, Y.; Tiwari, P.; Murmann, A.E.; Peter, M.E.; Lengyel, E. MicroRNAs reprogram normal fibroblasts into cancer-associated fibroblasts in ovarian cancer. Cancer Discov. 2012, 2, 1100–1108. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Tan, Y.; Yu, W.; Zheng, S.; Zhang, S.; Sun, L.; Ding, K. Small role with big impact: miRNAs as communicators in the cross-talk between cancer-associated fibroblasts and cancer cells. Int. J. Biol. Sci. 2017, 13, 339–348. [Google Scholar] [CrossRef] [PubMed]
- Falcone, G.; Felsani, A.; D’Agnano, I. Signaling by exosomal microRNAs in cancer. J. Exp. Clin. Cancer Res. CR 2015, 34, 32. [Google Scholar] [CrossRef] [PubMed]
- Gao, Q.; Yang, Z.; Xu, S.; Li, X.; Yang, X.; Jin, P.; Liu, Y.; Zhou, X.; Zhang, T.; Gong, C.; et al. Heterotypic CAF-tumor spheroids promote early peritoneal metastatis of ovarian cancer. J. Exp. Med. 2019, 216, 688–703. [Google Scholar] [CrossRef] [PubMed]
- Andrisic, L.; Dudzik, D.; Barbas, C.; Milkovic, L.; Grune, T.; Zarkovic, N. Short overview on metabolomics approach to study pathophysiology of oxidative stress in cancer. Redox Biol. 2018, 14, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Hasin, Y.; Seldin, M.; Lusis, A. Multi-omics approaches to disease. Genome Biol. 2017, 18, 83. [Google Scholar] [CrossRef]
- Shruthi, B.S.; Vinodhkumar, S.P. Proteomics: A new perspective for cancer. Adv. Biomed. Res. 2016, 5, 67. [Google Scholar] [CrossRef] [PubMed]
- Elzek, M.A.; Rodland, K.D. Proteomics of ovarian cancer: Functional insights and clinical applications. Cancer Metastasis Rev. 2015, 34, 83–96. [Google Scholar] [CrossRef]
- Tuck, M.; Turgeon, D.K.; Brenner, D.E. Serum and plasma collection: Preanalytical variables and standard operating procedures in biomarker research. In Proteomic and Metabolomic Approaches to Biomarker Discovery; Issaq, H.J., Veenstra, T.D.B.T.P., Eds.; Academic Press: Boston, MA, USA, 2013. [Google Scholar] [CrossRef]
- Cruz, I.N.; Coley, H.M.; Kramer, H.B.; Madhuri, T.K.; Safuwan, N.A.M.M.; Angelino, A.R.; Yang, M. Proteomics analysis of ovarian cancer cell lines and tissues reveals drug resistance-associated proteins. Cancer Genom. Proteom. 2017, 14, 35–52. [Google Scholar] [CrossRef]
- Swiatly, A.; Horala, A.; Matysiak, J.; Hajduk, J.; Nowak-Markwitz, E.; Kokot, Z.J. Understanding ovarian cancer: iTRAQ-based proteomics for biomarker discovery. Int. J. Mol. Sci. 2018, 19, 2240. [Google Scholar] [CrossRef]
- Zhang, W.; Ou, X.; Wu, X. Proteomics profiling of plasma exosomes in epithelial ovarian cancer: A potential role in the coagulation cascade, diagnosis and prognosis. Int. J. Oncol. 2019, 54, 1719–1733. [Google Scholar] [CrossRef]
- Zadeh Fakhar, H.B.; Zali, H.; Rezaie-Tavirani, M.; Darkhaneh, R.F.; Babaabasi, B. Proteome profiling of low grade serous ovarian cancer. J. Ovarian Res. 2019, 12, 1–14. [Google Scholar] [CrossRef]
- Ahmed, N.; Greening, D.; Samardzija, C.; Escalona, R.M.; Chen, M.; Findlay, J.K.; Kannourakis, G. Unique proteome signature of post-chemotherapy ovarian cancer ascites-derived tumor cells. Sci. Rep. 2016, 6, 30061. [Google Scholar] [CrossRef]
- Kuk, C.; Kulasingam, V.; Gunawardana, C.G.; Smith, C.R.; Batruch, I.; Diamandis, E.P. Mining the ovarian cancer ascites proteome for potential ovarian cancer biomarkers. Mol. Cell. Proteom. MCP 2009, 8, 661–669. [Google Scholar] [CrossRef]
- Shender, V.O.; Pavlyukov, M.S.; Ziganshin, R.H.; Arapidi, G.P.; Kovalchuk, S.I.; Anikanov, N.A.; Altukhov, I.A.; Alexeev, D.G.; Butenko, I.O.; Shavarda, A.L.; et al. Proteome-metabolome profiling of ovarian cancer ascites reveals novel components involved in intercellular communication. Mol. Cell. Proteom. MCP 2014, 13, 3558–3571. [Google Scholar] [CrossRef] [PubMed]
- Suidan, R.S.; Sun, C.C.; Westin, S.N.; Coleman, R.L.; Mills, G.B.; Meyer, L.A. The management of malignant ascites and impact on quality of life outcomes in women with ovarian cancer. Expert Rev. Qual. Life Cancer Care 2016, 1, 231–238. [Google Scholar] [CrossRef] [PubMed]
- Avraham-Chakim, L.; Elad, D.; Zaretsky, U.; Kloog, Y.; Jaffa, A.; Grisaru, D. Fluid-flow induced wall shear stress and epithelial ovarian cancer peritoneal spreading. PLoS ONE 2013, 8, e60965. [Google Scholar] [CrossRef]
- Hyler, A.R.; Baudoin, N.C.; Brown, M.S.; Stremler, M.A.; Cimini, D.; Davalos, R.V.; Schmelz, E.M. Fluid shear stress impacts ovarian cancer cell viability, subcellular organization, and promotes genomic instability. PLoS ONE 2018, 13, e0194170. [Google Scholar] [CrossRef]
- Zhang, W.C.; Shyh-Chang, N.; Yang, H.; Rai, A.; Umashankar, S.; Ma, S.; Soh, B.S.; Sun, L.L.; Tai, B.C.; Nga, M.E.; et al. Glycine decarboxylase activity drives non-small cell lung cancer tumor-initiating cells and tumorigenesis. Cell 2012, 148, 259–272. [Google Scholar] [CrossRef] [PubMed]
- Baenke, F.; Peck, B.; Miess, H.; Schulze, A. Hooked on fat: The role of lipid synthesis in cancer metabolism and tumour development. Dis. Models Mech. 2013, 6, 1353–1363. [Google Scholar] [CrossRef]
- Kuo, C.-Y.; Ann, D.K. When fats commit crimes: Fatty acid metabolism, cancer stemness and therapeutic resistance. Cancer Commun. 2018, 38, 47. [Google Scholar] [CrossRef]
- Li, J.; Condello, S.; Thomes-Pepin, J.; Ma, X.; Xia, Y.; Hurley, T.D.; Matei, D.; Cheng, J.X. Lipid Desaturation Is a Metabolic Marker and Therapeutic Target of Ovarian Cancer Stem Cells. Cell Stem Cell 2017, 20, 303–314. [Google Scholar] [CrossRef] [PubMed]
- Ehmsen, S.; Pedersen, M.H.; Wang, G.; Terp, M.G.; Arslanagic, A.; Hood, B.L.; Conrads, T.P.; Leth-Larsen, R.; Ditzel, H.J. Increased Cholesterol Biosynthesis Is a Key Characteristic of Breast Cancer Stem Cells Influencing Patient Outcome. Cell Rep. 2019, 27, 3927–3938. [Google Scholar] [CrossRef] [PubMed]
- Teeuwssen, M.; Fodde, R. Wnt Signaling in Ovarian Cancer Stemness, EMT, and Therapy Resistance. J. Clin. Med. 2019, 8, 1658. [Google Scholar] [CrossRef] [PubMed]
- Roane, B.M.; Arend, R.C.; Birrer, M.J. Review: Targeting the Transforming Growth Factor-Beta Pathway in Ovarian Cancer. Cancers 2019, 11, 668. [Google Scholar] [CrossRef]
- Cole, A.J.; Dwight, T.; Gill, A.J.; Dickson, K.-A.; Zhu, Y.; Clarkson, A.; Gard, G.B.; Maidens, J.; Valmadre, S.; Clifton-Bligh, R.; et al. Assessing mutant p53 in primary high-grade serous ovarian cancer using immunohistochemistry and massively parallel sequencing. Sci. Rep. 2016, 6, 26191. [Google Scholar] [CrossRef]
- McGivern, N.; El-Helali, A.; Mullan, P.; McNeish, I.A.; Paul Harkin, D.; Kennedy, R.D.; McCabe, N. Activation of MAPK signalling results in resistance to saracatinib (AZD0530) in ovarian cancer. Oncotarget 2017, 9, 4722–4736. [Google Scholar] [CrossRef]
- Elschenbroich, S.; Ignatchenko, V.; Clarke, B.; Kalloger, S.E.; Boutros, P.C.; Gramolini, A.O.; Shaw, P.; Jurisica, I.; Kislinger, T. In-Depth Proteomics of Ovarian Cancer Ascites: Combining Shotgun Proteomics and Selected Reaction Monitoring Mass Spectrometry. J. Proteome Res. 2011, 10, 2286–2299. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.-L.; Lu, Z.; Bast, R.C., Jr. The role of biomarkers in the management of epithelial ovarian cancer. Expert Rev. Mol. Diagn. 2017, 17, 577–591. [Google Scholar] [CrossRef]
- Cantón-Romero, J.C.; Miranda-Díaz, A.G.; Bañuelos-Ramírez, J.L.; Carrillo-Ibarra, S.; Sifuentes-Franco, S.; Castellanos-González, J.A.; Rodríguez-Carrizalez, A.D. Markers of Oxidative Stress and Inflammation in Ascites and Plasma in Patients with Platinum-Sensitive, Platinum-Resistant, and Platinum-Refractory Epithelial Ovarian Cancer. Oxidative Med. Cell. Longev. 2017, 2017, 2873030. [Google Scholar] [CrossRef]
- Kampan, N.C.; Madondo, M.T.; Reynolds, J.; Hallo, J.; McNally, O.M.; Jobling, T.W.; Stephens, A.N.; Quinn, M.A.; Plebanski, M. Pre-operative sera interleukin-6 in the diagnosis of high-grade serous ovarian cancer. Sci. Rep. 2020, 10, 2213. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Liu, W.; Wang, X.; Wang, X.; Sun, H. Prognostic value of serum IL-8 and IL-10 in patients with ovarian cancer undergoing chemotherapy. Oncol. Lett. 2019, 17, 2365–2369. [Google Scholar] [CrossRef]
- Zhang, W.; Ling, D.; Tan, J.; Zhang, J.; Li, L. Expression of urokinase plasminogen activator and plasminogen activator inhibitor type-1 in ovarian cancer and its clinical significance. Oncol. Rep. 2013, 29, 637–645. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Xiao, M.; Zhang, Z.; Cui, R.; Jiang, X.; Wang, S.; Bai, H.; Liu, C.; Zhang, Z. Potential interaction between lysophosphatidic acid and tumor-associated macrophages in ovarian carcinoma. J. Inflamm. 2020, 17, 23. [Google Scholar] [CrossRef] [PubMed]
- Okła, K.; Surówka, J.; Frąszczak, K.; Czerwonka, A.; Kaławaj, K.; Wawruszak, A.; Kotarski, J.; Wertel, I. Assessment of the clinicopathological relevance of mesothelin level in plasma, peritoneal fluid, and tumor tissue of epithelial ovarian cancer patients. Tumor Biol. 2018, 40, 1010428318804937. [Google Scholar] [CrossRef]
- Meyers, M.A. Distribution of intra-abdominal malignant seeding: Dependency on dynamics of flow of ascitic fluid. Am. J. Roentgenol. Radium Ther. Nucl. Med. 1973, 119, 198–206. [Google Scholar] [CrossRef]
- Mitchell, G.A.G. The spread of acute intraperitoneal effusions. BJS 1940, 28, 291–313. [Google Scholar] [CrossRef]
- Carmignani, C.P.; Sugarbaker, T.A.; Bromley, C.M.; Sugarbaker, P.H. Intraperitoneal cancer dissemination: Mechanisms of the patterns of spread. Cancer Metastasis Rev. 2003, 22, 465–472. [Google Scholar] [CrossRef]
- Feki, A.; Berardi, P.; Bellingan, G.; Major, A.; Krause, K.-H.; Petignat, P.; Zehra, R.; Pervaiz, S.; Irminger-Finger, I. Dissemination of intraperitoneal ovarian cancer: Discussion of mechanisms and demonstration of lymphatic spreading in ovarian cancer model. Crit. Rev. Oncol. /Hematol. 2009, 72, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Sugarbaker, P.H. Observations concerning cancer spread within the peritoneal cavity and concepts supporting an ordered pathophysiology. Cancer Treat. Res. 1996, 82, 79–100. [Google Scholar] [CrossRef]
- Bricou, A.; Batt, R.E.; Chapron, C. Peritoneal fluid flow influences anatomical distribution of endometriotic lesions: Why Sampson seems to be right. Eur. J. Obstet. Gynecol. Reprod. Biol. 2008, 138, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Drye, J.C. Intraperitoneal pressure in the human. Surg. Gynecol. Obstet. 1948, 87, 472–475. [Google Scholar]
- Novak, C.; Horst, E.; Mehta, G. Review: Mechanotransduction in ovarian cancer: Shearing into the unknown. APL Bioeng. 2018, 2, 31701. [Google Scholar] [CrossRef] [PubMed]
- Nathanson, S.D.; Nelson, L. Interstitial fluid pressure in breast cancer, benign breast conditions, and breast parenchyma. Ann. Surg. Oncol. 1994, 1, 333–338. [Google Scholar] [CrossRef] [PubMed]
- Polacheck, W.J.; Charest, J.L.; Kamm, R.D. Interstitial flow influences direction of tumor cell migration through competing mechanisms. Proc. Natl. Acad. Sci. USA 2011, 108, 11115–11120. [Google Scholar] [CrossRef]
- Rofstad, E.K.; Galappathi, K.; Mathiesen, B.S. Tumor Interstitial Fluid Pressure-A Link between Tumor Hypoxia, Microvascular Density, and Lymph Node Metastasis. Neoplasia 2014, 16, 586–594. [Google Scholar] [CrossRef] [PubMed]
- Milosevic, M.; Lunt, S.J.; Fyles, A.; Hill, R.P. Interstitial fluid pressure in tumors: Therapeutic barrier and biomarker of angiogenesis. Future Oncol. 2008, 4, 793–802. [Google Scholar] [CrossRef]
- Rutkowski, J.M.; Swartz, M.A. A driving force for change: Interstitial flow as a morphoregulator. Trends Cell Biol. 2007, 17, 44–50. [Google Scholar] [CrossRef]
- Swartz, M.A.; Fleury, M.E. Interstitial Flow and Its Effects in Soft Tissues. Annu. Rev. Biomed. Eng. 2007, 9, 229–256. [Google Scholar] [CrossRef] [PubMed]
- Heldin, C.H.; Rubin, K.; Pietras, K.; Östman, A. High interstitial fluid pressure—An obstacle in cancer therapy. Nat. Rev. Cancer 2004, 4, 806–813. [Google Scholar] [CrossRef]
- Munson, J.M.; Shieh, A.C. Interstitial fluid flow in cancer: Implications for disease progression and treatment. Cancer Manag. Res. 2014, 6, 317–328. [Google Scholar] [CrossRef]
- Shieh, A.C.; Rozansky, H.A.; Hinz, B.; Swartz, M.A. Tumor cell invasion is promoted by interstitial flow-induced matrix priming by stromal fibroblasts. Cancer Res. 2011, 71, 790–800. [Google Scholar] [CrossRef] [PubMed]
- Flessner, M.F.; Choi, J.; He, Z.; Credit, K. Physiological characterization of human ovarian cancer cells in a rat model of intraperitoneal antineoplastic therapy. J. Appl. Physiol. 2004, 97, 1518–1526. [Google Scholar] [CrossRef] [PubMed]
- Flessner, M.F.; Choi, J.; Credit, K.; Deverkadra, R.; Henderson, K. Resistance of tumor interstitial pressure to the penetration of intraperitoneally delivered antibodies into metastatic ovarian tumors. Clin. Cancer Res. 2005, 11, 3117–3125. [Google Scholar] [CrossRef]
- Klymenko, Y.; Wates, R.B.; Weiss-Bilka, H.; Lombard, R.; Liu, Y.; Campbell, L.; Kim, O.; Wagner, D.; Ravosa, M.J.; Stack, M.S. Modeling the effect of ascites-induced compression on ovarian cancer multicellular aggregates. Dis. Models Mech. 2018, 11, dmm034199. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Credit, K.; Henderson, K.; Deverkadra, R.; He, Z.; Wiig, H.; Vanpelt, H.; Flessner, M.F. Intraperitoneal immunotherapy for metastatic ovarian carcinoma: Resistance of intratumoral collagen to antibody penetration. Clin. Cancer Res. 2006, 12, 1906–1912. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.K.; Tong, R.T.; Munn, L.L. Effect of vascular normalization by antiangiogenic therapy on interstitial hypertension, peritumor edema, and lymphatic metastasis: Insights from a mathematical model. Cancer Res. 2007, 67, 2729–2735. [Google Scholar] [CrossRef] [PubMed]
- Steuperaert, M.; Debbaut, C.; Carlier, C.; De Wever, O.; Descamps, B.; Vanhove, C.; Ceelen, W.; Segers, P. A 3D CFD model of the interstitial fluid pressure and drug distribution in heterogeneous tumor nodules during intraperitoneal chemotherapy. Drug Deliv. 2019, 26, 404–415. [Google Scholar] [CrossRef]
- Ip, C.K.M.; Li, S.-S.; Tang, M.Y.H.; Sy, S.K.H.; Ren, Y.; Shum, H.C.; Wong, A.S.T. Stemness and chemoresistance in epithelial ovarian carcinoma cells under shear stress. Sci. Rep. 2016, 6, 26788. [Google Scholar] [CrossRef] [PubMed]
- Li, S.-S.; Ip, C.K.M.; Tang, M.Y.H.; Tang, M.K.S.; Tong, Y.; Zhang, J.; Hassan, A.A.; Mak, A.S.C.; Yung, S.; Chan, T.-M.; et al. Sialyl Lewisx-P-selectin cascade mediates tumor–mesothelial adhesion in ascitic fluid shear flow. Nat. Commun. 2019, 10, 2406. [Google Scholar] [CrossRef]
- Sun, L.; Wen, J.; Wang, L.; Wen, Q.; Wu, J.; Bie, M. Fluid shear stress-induced IL-8/CXCR signaling in human ovarian cancer cells. Transl. Cancer Res. 2019, 8. [Google Scholar] [CrossRef]
- Davies, P.F. Flow-mediated endothelial mechanotransduction. Physiol. Rev. 1995, 75, 519–560. [Google Scholar] [CrossRef]
- Jeffrey, B.; Udaykumar, H.S.; Schulze, K.S.; Carmignani, C.P.; Sugarbaker, T.A.; Bromley, C.M.; Sugarbaker, P.H.; Niedbala, M.J.; Crickard, K.; Bernacki, R.J. Flow fields generated by peristaltic reflex in isolated guinea pig ileum: Impact of contraction depth and shoulders. Am. J. Physiol.-Gastrointest. Liver Physiol. 2003, 285, 465–472. [Google Scholar] [CrossRef] [PubMed]
- Gupta, G.P.; Massagué, J. Cancer metastasis: Building a framework. Cell 2006, 127, 679–695. [Google Scholar] [CrossRef]
- Chambers, A.F.; Groom, A.C.; MacDonald, I.C. Dissemination and growth of cancer cells in metastatic sites. Nat. Rev. Cancer 2002, 2, 563–572. [Google Scholar] [CrossRef] [PubMed]
- Valastyan, S.; Weinberg, R.A. Tumor metastasis: Molecular insights and evolving paradigms. Cell 2011, 147, 275–292. [Google Scholar] [CrossRef]
- Ahmed, K.N.; Abubaker, M.; Quinn, J.F.; Ahmed, N.; Abubaker, K.; Findlay, J.; Quinn, M.; Ahmed, K.N.; Abubaker, M.; Quinn, J.F. Epithelial Mesenchymal Transition and Cancer Stem Cell-Like Phenotypes Facilitate Chemoresistance in Recurrent Ovarian Cancer. Curr. Cancer Drug Targets 2010, 10, 268–278. [Google Scholar] [CrossRef]
- Iwatsuki, M.; Mimori, K.; Yokobori, T.; Ishi, H.; Beppu, T.; Nakamori, S.; Baba, H.; Mori, M. Epithelial-mesenchymal transition in cancer development and its clinical significance. Cancer Sci. 2010, 101, 293–299. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.C.; Mallidi, S.; Liu, J.; Chiang, C.T.; Mai, Z.; Goldschmidt, R.; Ebrahim-Zadeh, N.; Rizvi, I.; Hasan, T. Photodynamic Therapy Synergizes with Irinotecan to Overcome Compensatory Mechanisms and Improve Treatment Outcomes in Pancreatic Cancer. Cancer Res. 2016, 76, 1066–1077. [Google Scholar] [CrossRef]
- Huang, H.C.; Rizvi, I.; Liu, J.; Anbil, S.; Kalra, A.; Lee, H.; Baglo, Y.; Paz, N.; Hayden, D.; Pereira, S.; et al. Photodynamic Priming Mitigates Chemotherapeutic Selection Pressures and Improves Drug Delivery. Cancer Res. 2018, 78, 558–571. [Google Scholar] [CrossRef]
- Anbil, S.; Pigula, M.; Huang, H.C.; Mallidi, S.; Broekgaarden, M.; Baglo, Y.; De Silva, P.; Simeone, D.M.; Mino-Kenudson, M.; Maytin, E.V.; et al. Vitamin D Receptor Activation and Photodynamic Priming Enables Durable Low-dose Chemotherapy. Mol. Cancer 2020, 19, 1308–1319. [Google Scholar] [CrossRef]
- Celli, J.P.; Rizvi, I.; Evans, C.L.; Abu-Yousif, A.O.; Hasan, T. Quantitative imaging reveals heterogeneous growth dynamics and treatment-dependent residual tumor distributions in a three-dimensional ovarian cancer model. J. Biomed. Opt. 2010, 15, 051603. [Google Scholar] [CrossRef][Green Version]
- Celli, J.P.; Spring, B.Q.; Rizvi, I.; Evans, C.L.; Samkoe, K.S.; Verma, S.; Pogue, B.W.; Hasan, T. Imaging and photodynamic therapy: Mechanisms, monitoring, and optimization. Chem. Rev. 2010, 110, 2795–2838. [Google Scholar] [CrossRef]
- Rizvi, I.; Celli, J.P.; Evans, C.L.; Abu-Yousif, A.O.; Muzikansky, A.; Pogue, B.W.; Finkelstein, D.; Hasan, T. Synergistic Enhancement of Carboplatin Efficacy with Photodynamic Therapy in a Three-Dimensional Model for Micrometastatic Ovarian Cancer. Cancer Res. 2010, 70, 9319–9328. [Google Scholar] [CrossRef] [PubMed]
- Rizvi, I.; Dinh, T.A.; Yu, W.; Chang, Y.; Sherwood, M.E.; Hasan, T. Photoimmunotherapy and irradiance modulation reduce chemotherapy cycles and toxicity in a murine model for ovarian carcinomatosis: Perspective and results. Isr. J. Chem. 2012, 52, 776–787. [Google Scholar] [CrossRef]
- Spring, B.Q.; Abu-Yousif, A.O.; Palanisami, A.; Rizvi, I.; Zheng, X.; Mai, Z.; Anbil, S.; Sears, R.B.; Mensah, L.B.; Goldschmidt, R.; et al. Selective treatment and monitoring of disseminated cancer micrometastases in vivo using dual-function, activatable immunoconjugates. Proc. Natl. Acad. Sci. USA 2014, 111, E933–E942. [Google Scholar] [CrossRef]
- Spring, B.Q.; Rizvi, I.; Xu, N.; Hasan, T. The role of photodynamic therapy in overcoming cancer drug resistance. Photochem. Photobiol. Sci. 2015, 14, 1476–1491. [Google Scholar] [CrossRef] [PubMed]
- Duska, L.R.; Hamblin, M.R.; Miller, J.L.; Hasan, T. Combination photoimmunotherapy and cisplatin: Effects on human ovarian cancer ex vivo. J. Natl. Cancer Inst. 1999, 91, 1557–1563. [Google Scholar] [CrossRef]
- Zuluaga, M.F.; Lange, N. Combination of photodynamic therapy with anti-cancer agents. Curr. Med. Chem. 2008, 15, 1655–1673. [Google Scholar] [CrossRef] [PubMed]
- Paz, H.; Pathak, N.; Yang, J. Invading one step at a time: The role of invadopodia in tumor metastasis. Oncogene 2014, 33, 4193–4202. [Google Scholar] [CrossRef]
- Domcke, S.; Sinha, R.; Levine, D.A.; Sander, C.; Schultz, N. Evaluating cell lines as tumour models by comparison of genomic profiles. Nat. Commun. 2013, 4, 2126. [Google Scholar] [CrossRef]
- Hernandez, L.; Kim, M.K.; Lyle, L.T.; Bunch, K.P.; House, C.D.; Ning, F.; Noonan, A.M.; Annunziata, C.M. Characterization of ovarian cancer cell lines as in vivo models for preclinical studies. Gynecol. Oncol. 2016, 142, 332–340. [Google Scholar] [CrossRef]
- Mitra, A.K.; Davis, D.A.; Tomar, S.; Roy, L.; Gurler, H.; Xie, J.; Lantvit, D.D.; Cardenas, H.; Fang, F.; Liu, Y.; et al. In vivo tumor growth of high-grade serous ovarian cancer cell lines. Gynecol. Oncol. 2015, 138, 372–377. [Google Scholar] [CrossRef] [PubMed]
- Bobbs, A.S.; Cole, J.M.; Cowden Dahl, K.D. Emerging and Evolving Ovarian Cancer Animal Models. Cancer Growth Metastasis 2015, 8, 29–36. [Google Scholar] [CrossRef]
- De Haven Brandon, A.; Box, G.; Hallsworth, A.; Court, W.; Matthews, N.; Herodek, B.; Arteagabeitia, A.B.; Valenti, M.; Kirkin, V. Identification of ovarian high-grade serous carcinoma cell lines that show estrogen-sensitive growth as xenografts in immunocompromised mice. Sci. Rep. 2020, 10, 10799. [Google Scholar] [CrossRef] [PubMed]
- Molpus, K.L.; Koelliker, D.; Atkins, L.; Kato, D.T.; Buczek-Thomas, J.; Fuller, A.F., Jr.; Hasan, T. Characterization of a xenograft model of human ovarian carcinoma which produces intraperitoneal carcinomatosis and metastases in mice. Int. J. Cancer 1996, 68, 588–595. [Google Scholar] [CrossRef]
- Hamilton, T.C.; Young, R.C.; Louie, K.G.; Behrens, B.C.; McKoy, W.M.; Grotzinger, K.R.; Ozols, R.F. Characterization of a xenograft model of human ovarian carcinoma which produces ascites and intraabdominal carcinomatosis in mice. Cancer Res. 1984, 44, 5286–5290. [Google Scholar]
- Asem, M.; Young, A.; Oyama, C.; ClaureDeLaZerda, A.; Liu, Y.; Ravosa, M.J.; Gupta, V.; Jewell, A.; Khabele, D.; Stack, M.S. Ascites-induced compression alters the peritoneal microenvironment and promotes metastatic success in ovarian cancer. Sci. Rep. 2020, 10, 11913. [Google Scholar] [CrossRef]
- Gotlieb, W.H.; Feldman, B.; Feldman-Moran, O.; Zmira, N.; Kreizer, D.; Segal, Y.; Elran, E.; Ben-Baruch, G. Intraperitoneal pressures and clinical parameters of total paracentesis for palliation of symptomatic ascites in ovarian cancer. Gynecol. Oncol. 1998, 71, 381–385. [Google Scholar] [CrossRef]
- Matsuzaki, S.; Vernis, L.; Bonnin, M.; Houlle, C.; Fournet-Fayard, A.; Rosano, G.; Lafaye, A.L.; Chartier, C.; Barriere, A.; Storme, B.; et al. Effects of low intraperitoneal pressure and a warmed, humidified carbon dioxide gas in laparoscopic surgery: A randomized clinical trial. Sci. Rep. 2017, 7, 11287. [Google Scholar] [CrossRef] [PubMed]
- Austefjord, M.W.; Gerdes, H.H.; Wang, X. Tunneling nanotubes: Diversity in morphology and structure. Commun. Integr. Biol. 2014, 7, e27934. [Google Scholar] [CrossRef] [PubMed]
- Lou, E.; Fujisawa, S.; Barlas, A.; Romin, Y.; Manova-Todorova, K.; Moore, M.A.; Subramanian, S. Tunneling Nanotubes: A new paradigm for studying intercellular communication and therapeutics in cancer. Commun. Integr. Biol. 2012, 5, 399–403. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.; Jensen, T.P.; Reierson, K.; Mathews, B.K.; Bhagra, A.; Franco-Sadud, R.; Grikis, L.; Mader, M.; Dancel, R.; Lucas, B.P.; et al. Recommendations on the Use of Ultrasound Guidance for Adult Abdominal Paracentesis: A Position Statement of the Society of Hospital Medicine. J. Hosp. Med. 2019, 14, E7–E15. [Google Scholar] [CrossRef]
- Runyon, B.A. Management of adult patients with ascites due to cirrhosis: An update. Hepatology 2009, 49, 2087–2107. [Google Scholar] [CrossRef] [PubMed]
- Aghajanian, C.; Blank, S.V.; Goff, B.A.; Judson, P.L.; Teneriello, M.G.; Husain, A.; Sovak, M.A.; Yi, J.; Nycum, L.R. OCEANS: A randomized, double-blind, placebo-controlled phase III trial of chemotherapy with or without bevacizumab in patients with platinum-sensitive recurrent epithelial ovarian, primary peritoneal, or fallopian tube cancer. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2012, 30, 2039–2045. [Google Scholar] [CrossRef] [PubMed]
- Pujade-Lauraine, E.; Hilpert, F.; Weber, B.; Reuss, A.; Poveda, A.; Kristensen, G.; Sorio, R.; Vergote, I.B.; Witteveen, P.; Bamias, A.; et al. AURELIA: A randomized phase III trial evaluating bevacizumab (BEV) plus chemotherapy (CT) for platinum (PT)-resistant recurrent ovarian cancer (OC). J. Clin. Oncol. 2012, 30, LBA5002. [Google Scholar] [CrossRef]
- Numnum, T.M.; Rocconi, R.P.; Whitworth, J.; Barnes, M.N. The use of bevacizumab to palliate symptomatic ascites in patients with refractory ovarian carcinoma. Gynecol. Oncol. 2006, 102, 425–428. [Google Scholar] [CrossRef] [PubMed]
- Holash, J.; Davis, S.; Papadopoulos, N.; Croll, S.D.; Ho, L.; Russell, M.; Boland, P.; Leidich, R.; Hylton, D.; Burova, E.; et al. VEGF-Trap: A VEGF blocker with potent antitumor effects. Proc. Natl. Acad. Sci. USA 2002, 99, 11393–11398. [Google Scholar] [CrossRef]
- Byrne, A.T.; Ross, L.; Holash, J.; Nakanishi, M.; Hu, L.; Hofmann, J.I.; Yancopoulos, G.D.; Jaffe, R.B. Vascular endothelial growth factor-trap decreases tumor burden, inhibits ascites, and causes dramatic vascular remodeling in an ovarian cancer model. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2003, 9, 5721–5728. [Google Scholar]
- Gotlieb, W.H.; Amant, F.; Advani, S.; Goswami, C.; Hirte, H.; Provencher, D.; Somani, N.; Yamada, S.D.; Tamby, J.-F.; Vergote, I. Intravenous aflibercept for treatment of recurrent symptomatic malignant ascites in patients with advanced ovarian cancer: A phase 2, randomised, double-blind, placebo-controlled study. Lancet. Oncol. 2012, 13, 154–162. [Google Scholar] [CrossRef]
- Jatoi, A.; Nieva, J.J.; Qin, R.; Loprinzi, C.L.; Wos, E.J.; Novotny, P.J.; Moore, D.F., Jr.; Mowat, R.B.; Bechar, N.; Pajon, E.R., Jr.; et al. A pilot study of long-acting octreotide for symptomatic malignant ascites. Oncology 2012, 82, 315–320. [Google Scholar] [CrossRef]
- Chirivi, R.G.; Garofalo, A.; Crimmin, M.J.; Bawden, L.J.; Stoppacciaro, A.; Brown, P.D.; Giavazzi, R. Inhibition of the metastatic spread and growth of B16-BL6 murine melanoma by a synthetic matrix metalloproteinase inhibitor. Int. J. Cancer 1994, 58, 460–464. [Google Scholar] [CrossRef]
- Davies, B.; Brown, P.D.; East, N.; Crimmin, M.J.; Balkwill, F.R. A synthetic matrix metalloproteinase inhibitor decreases tumor burden and prolongs survival of mice bearing human ovarian carcinoma xenografts. Cancer Res. 1993, 53, 2087–2091. [Google Scholar] [PubMed]
- Wang, X.; Fu, X.; Brown, P.D.; Crimmin, M.J.; Hoffman, R.M. Matrix metalloproteinase inhibitor BB-94 (batimastat) inhibits human colon tumor growth and spread in a patient-like orthotopic model in nude mice. Cancer Res. 1994, 54, 4726–4728. [Google Scholar] [PubMed]
- Watson, S.A.; Morris, T.M.; Parsons, S.L.; Steele, R.J.; Brown, P.D. Therapeutic effect of the matrix metalloproteinase inhibitor, batimastat, in a human colorectal cancer ascites model. Br. J. Cancer 1996, 74, 1354–1358. [Google Scholar] [CrossRef] [PubMed]
- Watson, S.A.; Morris, T.M.; Robinson, G.; Crimmin, M.J.; Brown, P.D.; Hardcastle, J.D. Inhibition of organ invasion by the matrix metalloproteinase inhibitor batimastat (BB-94) in two human colon carcinoma metastasis models. Cancer Res. 1995, 55, 3629–3633. [Google Scholar]
- Parsons, S.L.; Watson, S.A.; Steele, R.J. Phase I/II trial of batimastat, a matrix metalloproteinase inhibitor, in patients with malignant ascites. Eur. J. Surg. Oncol. J. Eur. Soc. Surg. Oncol. Br. Assoc. Surg. Oncol. 1997, 23, 526–531. [Google Scholar] [CrossRef]
- Beattie, G.J.; Smyth, J.F. Phase I study of intraperitoneal metalloproteinase inhibitor BB94 in patients with malignant ascites. Clin. Cancer Res. 1998, 4, 1899–1902. [Google Scholar]
- Mackey, J.R.; Wood, L.; Nabholtz, J.; Jensen, J.; Venner, P. A phase II trial of triamcinolone hexacetanide for symptomatic recurrent malignant ascites. J. Pain Symptom Manag. 2000, 19, 193–199. [Google Scholar] [CrossRef]
- Stuart, G.C.; Nation, J.G.; Snider, D.D.; Thunberg, P. Intraperitoneal interferon in the management of malignant ascites. Cancer 1993, 71, 2027–2030. [Google Scholar] [CrossRef]
- Räth, U.; Kaufmann, M.; Schmid, H.; Hofmann, J.; Wiedenmann, B.; Kist, A.; Kempeni, J.; Schlick, E.; Bastert, G.; Kommerell, B. Effect of intraperitoneal recombinant human tumour necrosis factor alpha on malignant ascites. Eur. J. Cancer 1991, 27, 121–125. [Google Scholar] [CrossRef]
- Katano, M.; Torisu, M. New approach to management of malignant ascites with a streptococcal preparation, OK-432. II. Intraperitoneal inflammatory cell-mediated tumor cell destruction. Surgery 1983, 93, 365–373. [Google Scholar]
- Torisu, M.; Katano, M.; Kimura, Y.; Itoh, H.; Takesue, M. New approach to management of malignant ascites with a streptococcal preparation, OK-432. I. Improvement of host immunity and prolongation of survival. Surgery 1983, 93, 357–364. [Google Scholar]
- Mahler, F.; Rapin, C.H.; Macgee, W. Corynebacterium parvum as palliative treatment in malignant ascites. J. Palliat. Care 1988, 4, 58–62. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.-M.; Chan, L.-Y.; Wang, S.-M.; Wu, M.-F.; Chern, J.-W.; Roffler, S.R. Cure of malignant ascites and generation of protective immunity by monoclonal antibody–targeted activation of a glucuronide prodrug in rats. Int. J. Cancer 1997, 73, 392–402. [Google Scholar] [CrossRef]
- Hird, V.; Thomas, H.; Stewart, J.S.; Epenetos, A.A. Malignant ascites: Review of the literature, and an update on monoclonal antibody-targeted therapy. Eur. J. Obstet. Gynecol. Reprod. Biol. 1989, 32, 37–45. [Google Scholar] [CrossRef]
- Heiss, M.M.; Murawa, P.; Koralewski, P.; Kutarska, E.; Kolesnik, O.O.; Ivanchenko, V.V.; Dudnichenko, A.S.; Aleknaviciene, B.; Razbadauskas, A.; Gore, M.; et al. The trifunctional antibody catumaxomab for the treatment of malignant ascites due to epithelial cancer: Results of a prospective randomized phase II/III trial. Int. J. Cancer 2010, 127, 2209–2221. [Google Scholar] [CrossRef]
- Tochner, Z.; Mitchell, J.B.; Harrington, F.S.; Smith, P.; Russo, D.T.; Russo, A. Treatment of murine intraperitoneal ovarian ascitic tumor with hematoporphyrin derivative and laser light. Cancer Res. 1985, 45, 2983–2987. [Google Scholar] [PubMed]
- Tochner, Z.; Mitchell, J.B.; Smith, P.; Harrington, F.; Glatstein, E.; Russo, D.; Russo, A. Photodynamic therapy of ascites tumours within the peritoneal cavity. Br. J. Cancer 1986, 53, 733–736. [Google Scholar] [CrossRef]
- Fotopoulou, C.; Berg, T.; Hausen, A.; Hennig, R.; Jalan, R.; Malagó, M.; Capel, J.; De Gottardi, A.; Stirnimann, G. Continuous low flow ascites drainage through the urinary bladder via the Alfapump system in palliative patients with malignant ascites. BMC Palliat. Care 2019, 18, 109. [Google Scholar] [CrossRef] [PubMed]
- Bellot, P.; Welker, M.-W.; Soriano, G.; von Schaewen, M.; Appenrodt, B.; Wiest, R.; Whittaker, S.; Tzonev, R.; Handshiev, S.; Verslype, C.; et al. Automated low flow pump system for the treatment of refractory ascites: A multi-center safety and efficacy study. J. Hepatol. 2013, 58, 922–927. [Google Scholar] [CrossRef] [PubMed]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- Valle, S.J.; Alzahrani, N.A.; Alzahrani, S.E.; Liauw, W.; Morris, D.L. Laparoscopic hyperthermic intraperitoneal chemotherapy (HIPEC) for refractory malignant ascites in patients unsuitable for cytoreductive surgery. Int. J. Surg. 2015, 23, 176–180. [Google Scholar] [CrossRef] [PubMed]


| Cytokine/Growth Factor | Role |
|---|---|
| Cancer Antigen 125 (CA-125) | |
| Epidermal Growth Factor (EGF) |
|
| Interleukin-6 (IL-6) |
|
| Interleukin-8 (IL-8) | |
| Interleukin-10 (IL-10) | |
| Lysophosphatidic Acid (LPA) |
|
| Urokinase Plasminogen Activator (uPA) | |
| Vascular Endothelial Growth Factor (VEGF) |
| Reference | Major Findings |
|---|---|
| Avraham-Chakim et al., 2013. [287] |
|
| Rizvi et al., 2013. [9] |
|
| Ip et al., 2016. [329] |
|
| Hyler et al., 2018. [288] |
|
| Li et al., 2019. [330] |
|
| Sun et al., 2019. [331] |
|
| Nath et al., 2020. [10] |
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rickard, B.P.; Conrad, C.; Sorrin, A.J.; Ruhi, M.K.; Reader, J.C.; Huang, S.A.; Franco, W.; Scarcelli, G.; Polacheck, W.J.; Roque, D.M.; et al. Malignant Ascites in Ovarian Cancer: Cellular, Acellular, and Biophysical Determinants of Molecular Characteristics and Therapy Response. Cancers 2021, 13, 4318. https://doi.org/10.3390/cancers13174318
Rickard BP, Conrad C, Sorrin AJ, Ruhi MK, Reader JC, Huang SA, Franco W, Scarcelli G, Polacheck WJ, Roque DM, et al. Malignant Ascites in Ovarian Cancer: Cellular, Acellular, and Biophysical Determinants of Molecular Characteristics and Therapy Response. Cancers. 2021; 13(17):4318. https://doi.org/10.3390/cancers13174318
Chicago/Turabian StyleRickard, Brittany P., Christina Conrad, Aaron J. Sorrin, Mustafa Kemal Ruhi, Jocelyn C. Reader, Stephanie A. Huang, Walfre Franco, Giuliano Scarcelli, William J. Polacheck, Dana M. Roque, and et al. 2021. "Malignant Ascites in Ovarian Cancer: Cellular, Acellular, and Biophysical Determinants of Molecular Characteristics and Therapy Response" Cancers 13, no. 17: 4318. https://doi.org/10.3390/cancers13174318
APA StyleRickard, B. P., Conrad, C., Sorrin, A. J., Ruhi, M. K., Reader, J. C., Huang, S. A., Franco, W., Scarcelli, G., Polacheck, W. J., Roque, D. M., del Carmen, M. G., Huang, H.-C., Demirci, U., & Rizvi, I. (2021). Malignant Ascites in Ovarian Cancer: Cellular, Acellular, and Biophysical Determinants of Molecular Characteristics and Therapy Response. Cancers, 13(17), 4318. https://doi.org/10.3390/cancers13174318