
Tumor-Associated Microglia and Macrophages in the Glioblastoma Microenvironment and Their Implications for Therapy

Abstract
:Simple Summary
Abstract
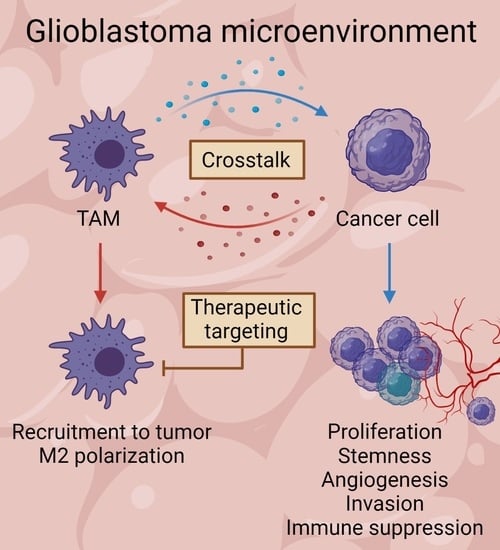
1. Introduction
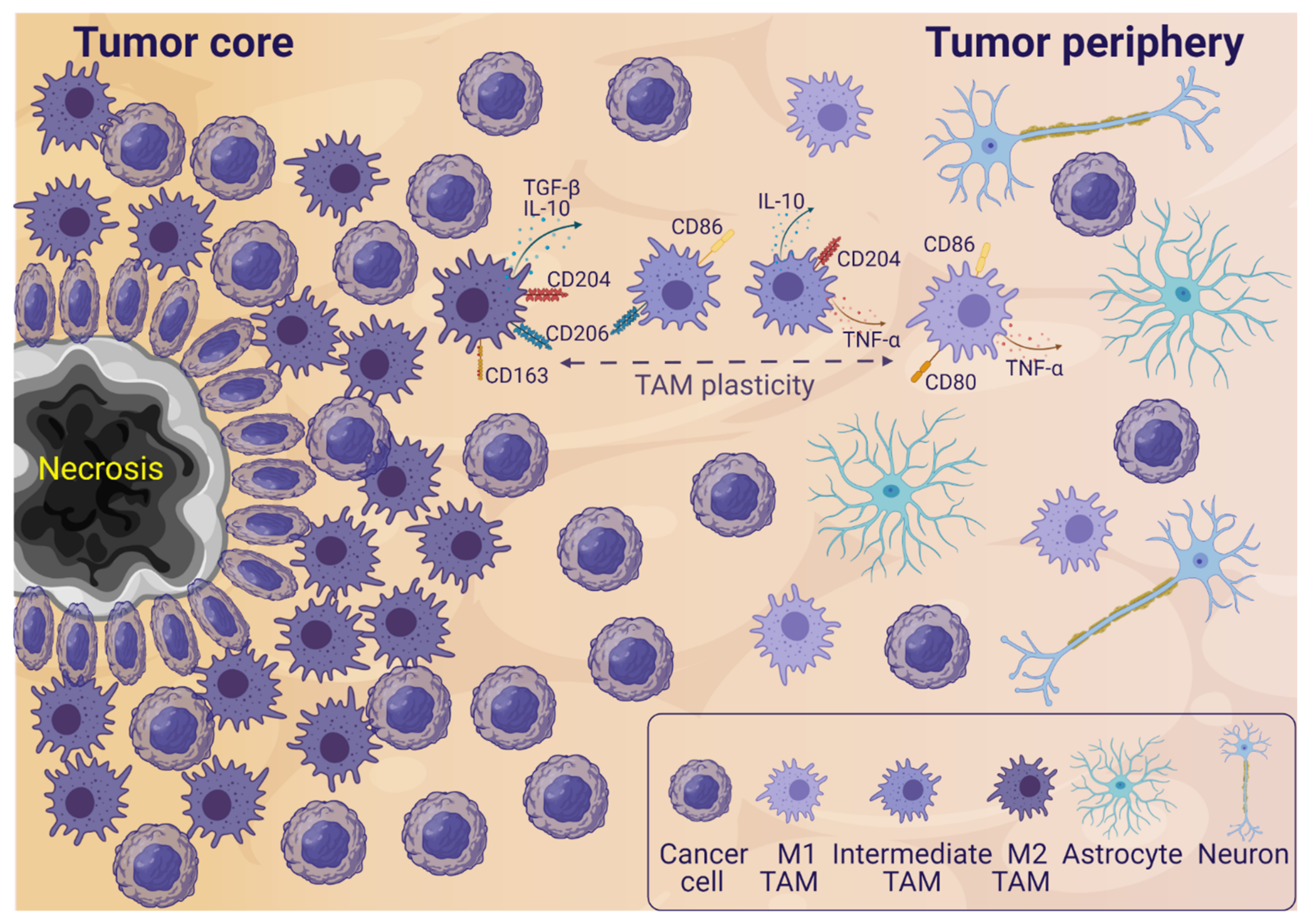
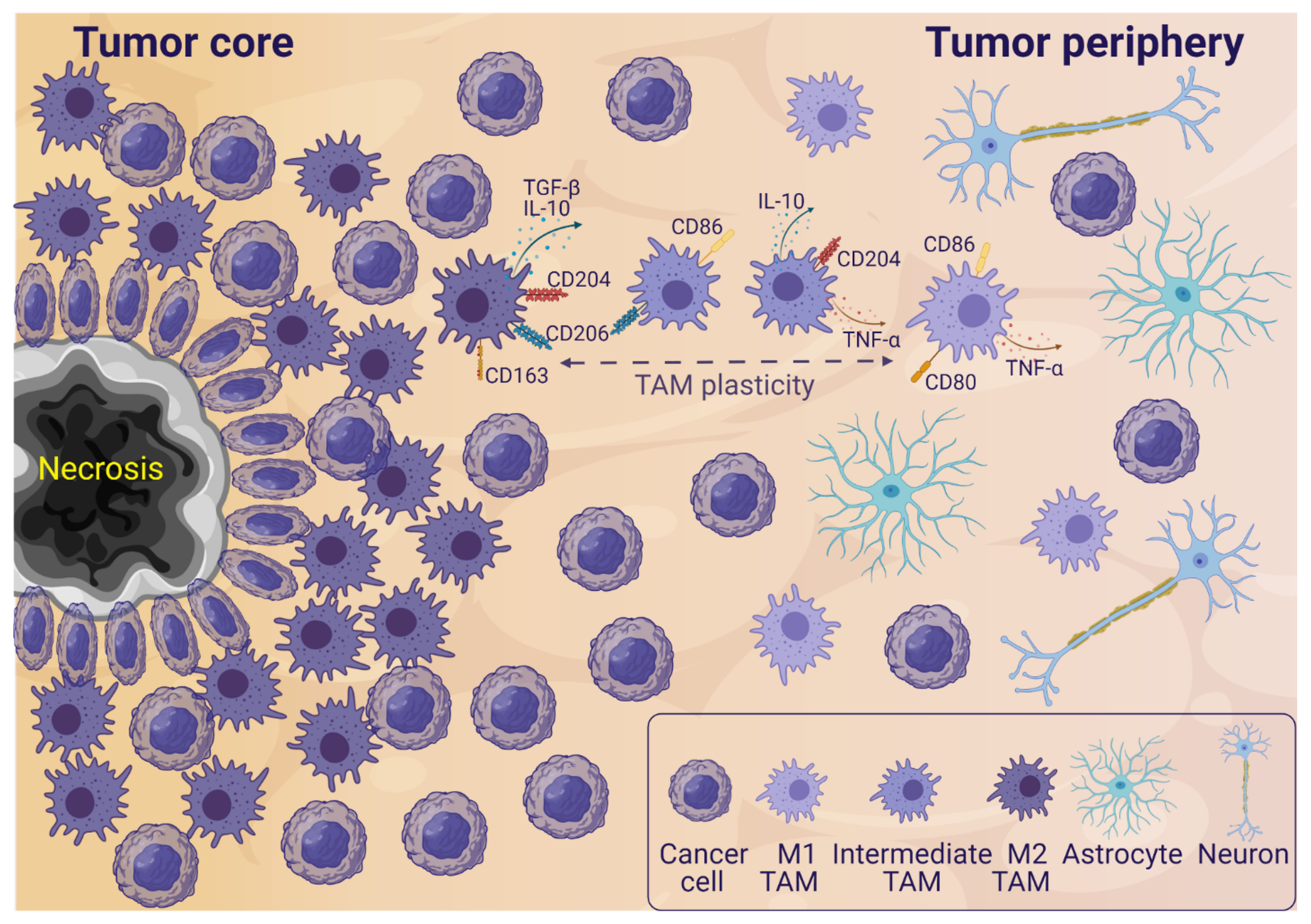
2. TAMs in the Glioblastoma Microenvironment
3. Glioblastoma and TAM Crosstalk
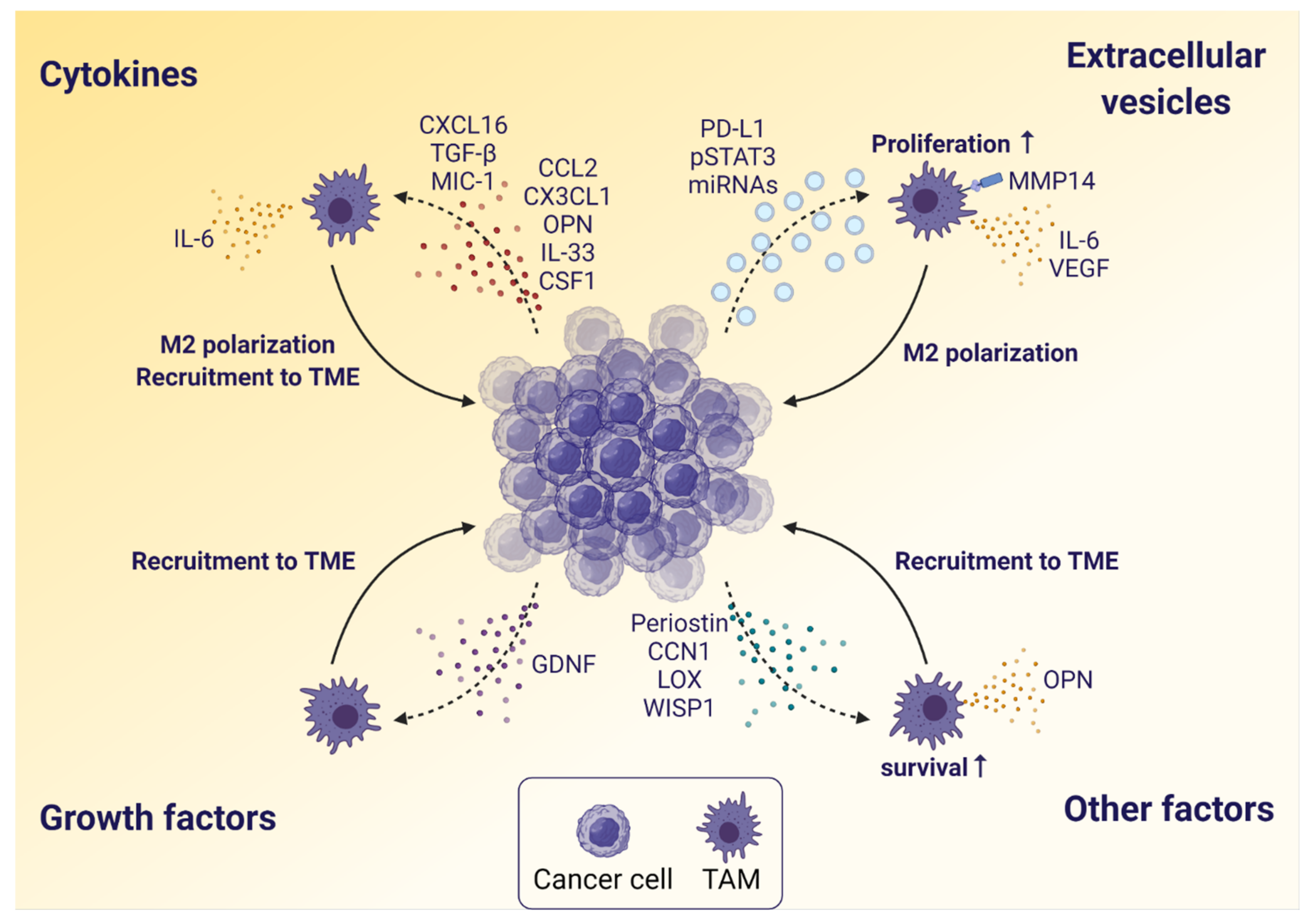
3.1. The Influence of Glioblastoma Cells on TAMs
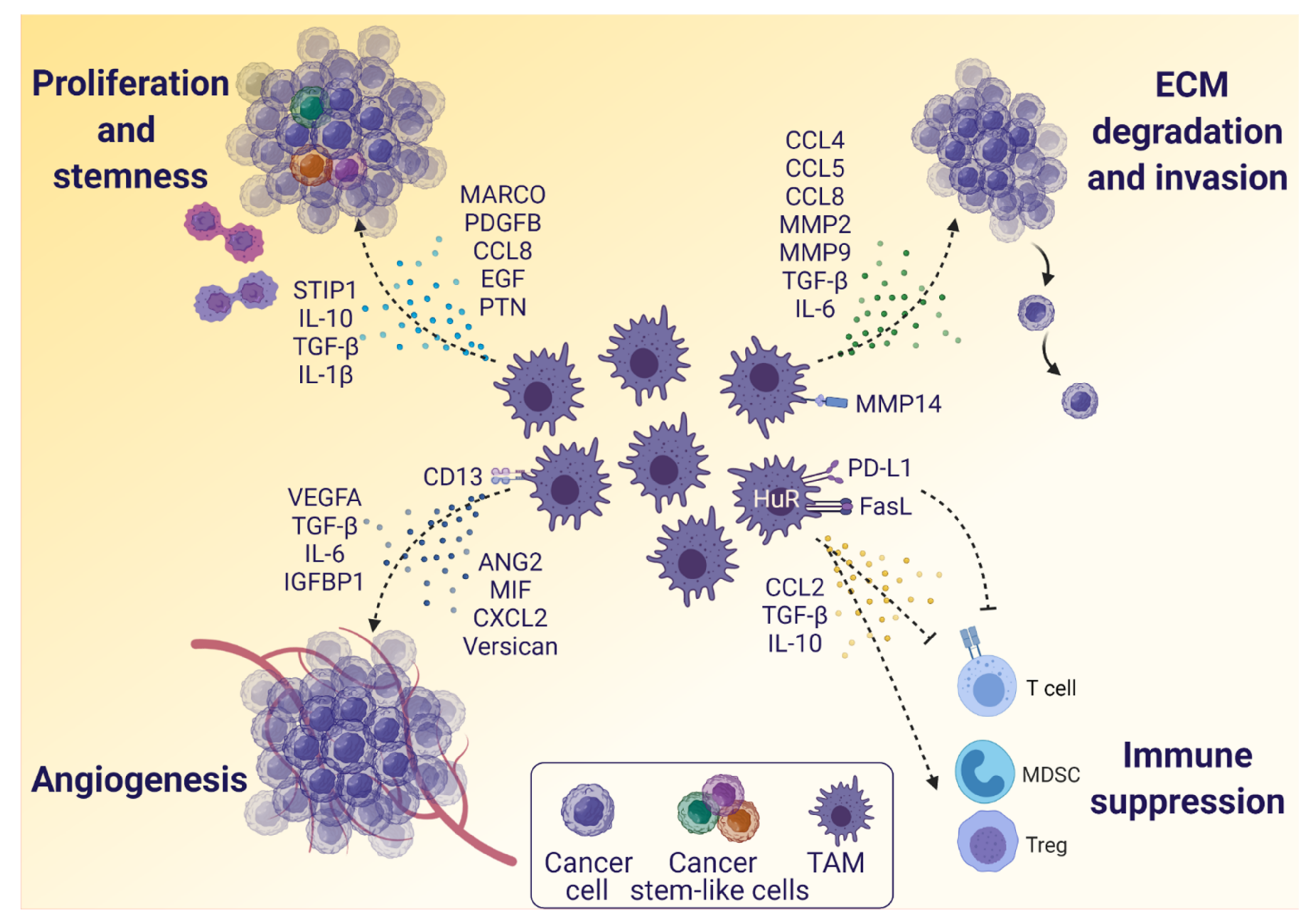
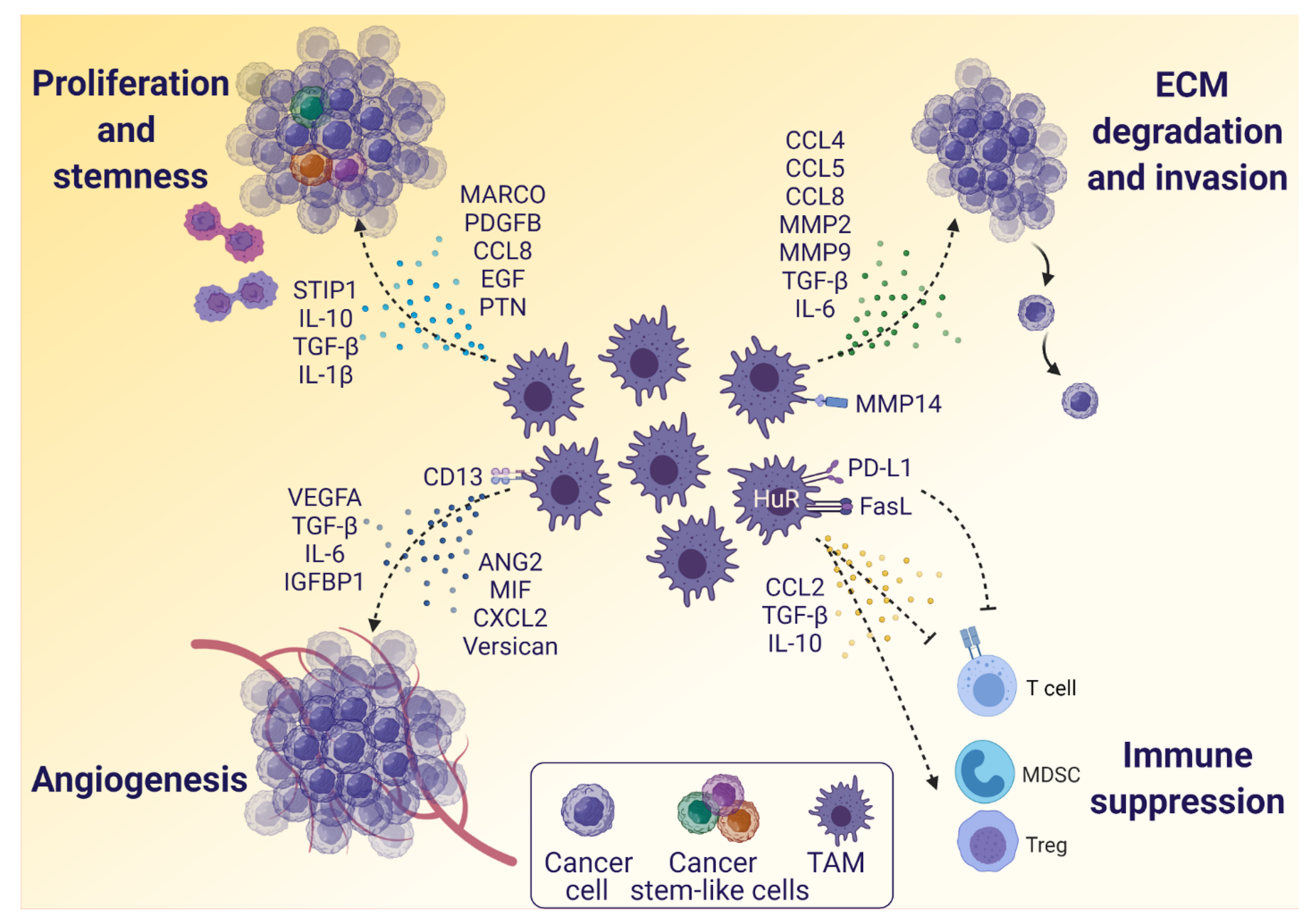
3.2. The Influence of TAMs on Glioblastoma Cells
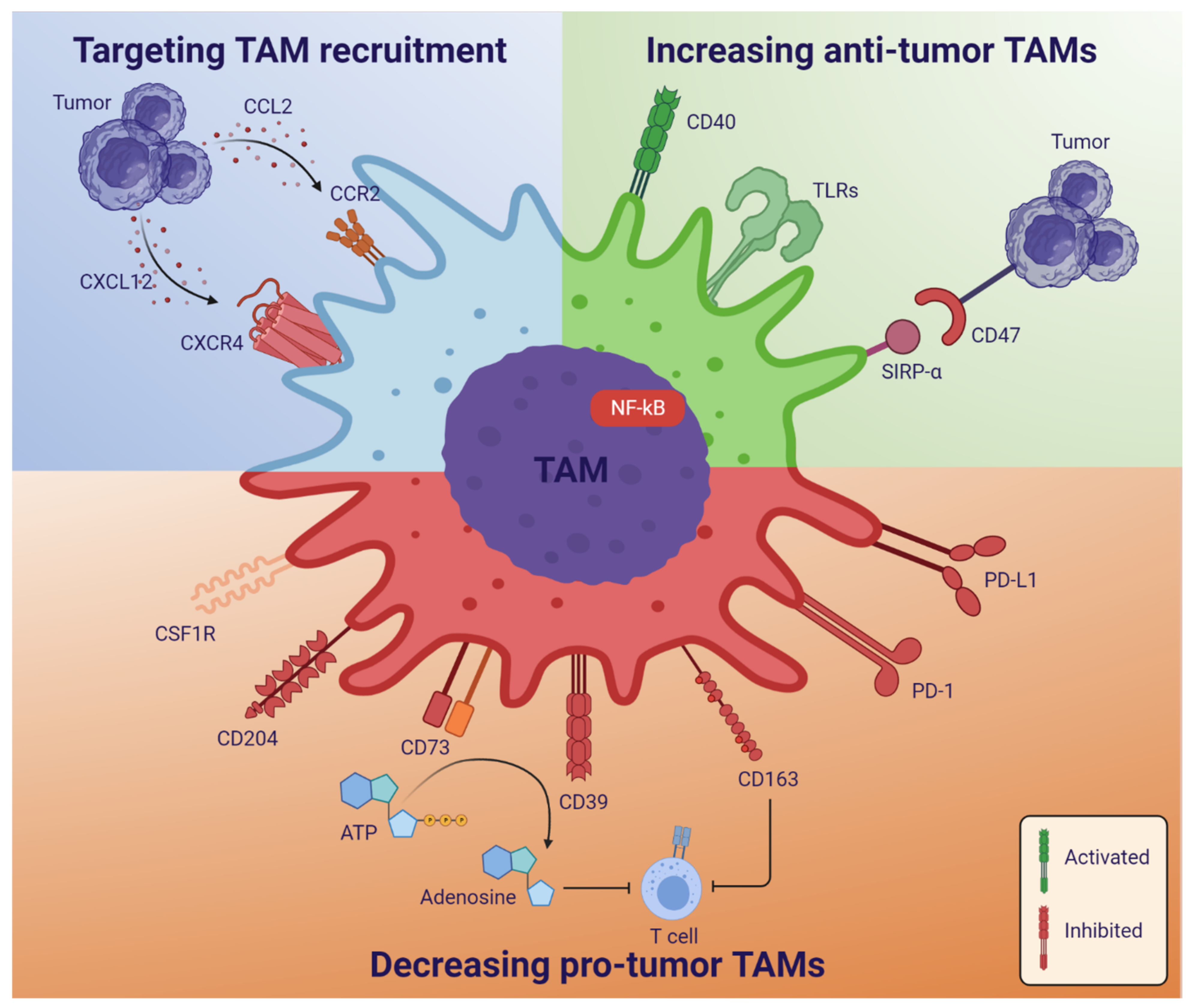
4. Glioblastoma Treatment—Therapies Targeting TAMs and Challenges
4.1. Targeting TAM Recruitment
4.2. Reprogramming TAM Polarization and Increasing Anti-Tumor TAMs
4.3. Decreasing Tumor-Promoting TAMs
4.4. Combination Therapies and Translational Challenges
5. Single-Cell Omics in Characterization of the Glioblastoma Microenvironment
6. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tykocki, T.; Eltayeb, M. Ten-year survival in glioblastoma. A systematic review. J. Clin. Neurosci. 2018, 54, 7–13. [Google Scholar] [CrossRef]
- Weenink, B.; French, P.J.; Smitt, P.A.E.S.; Debets, R.; Geurts, M. Immunotherapy in glioblastoma: Current shortcomings and future perspectives. Cancers 2020, 12, 751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Badie, B.; Schartner, J.M. Flow Cytometric Characterization of Tumor-associated Macrophages in Experimental Gliomas. Neurosurgery 2000, 46, 957–962. [Google Scholar] [CrossRef] [PubMed]
- Charles, N.A.; Holland, E.C.; Gilbertson, R.; Glass, R.; Kettenmann, H. The brain tumor microenvironment. Glia 2011, 59, 1169–1180. [Google Scholar] [CrossRef]
- Sørensen, M.D.; Dahlrot, R.H.; Boldt, H.B.; Hansen, S.; Kristensen, B.W. Tumour-associated microglia/macrophages predict poor prognosis in high-grade gliomas and correlate with an aggressive tumour subtype. Neuropathol. Appl. Neurobiol. 2018, 44, 185–206. [Google Scholar] [CrossRef] [Green Version]
- Caponegro, M.D.; Moffitt, R.A.; Tsirka, S.E. Expression of neuropilin-1 is linked to glioma associated microglia and macrophages and correlates with unfavorable prognosis in high grade gliomas. Oncotarget 2018, 9, 35655–35665. [Google Scholar] [CrossRef] [Green Version]
- Hambardzumyan, D.; Gutmann, D.H.; Kettenmann, H.; Roesch, S.; Rapp, C.; Dettling, S.; Herold-Mende, C. The role of microglia and macrophages in glioma maintenance and progression. Nat. Neurosci. 2015, 19, 20–27. [Google Scholar] [CrossRef] [Green Version]
- Han, S.; Ma, E.; Wang, X.; Yu, C.; Dong, T.; Zhan, W.; Wei, X.; Liang, G.; Feng, S. Rescuing defective tumor-infiltrating T-cell proliferation in glioblastoma patients. Oncol. Lett. 2016, 12, 2924–2929. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Lage, M.; Lynch, T.M.; Bi, Y.; Cocito, C.; Way, G.P.; Pal, S.; Haller, J.; Yan, R.E.; Ziober, A.; Nguyen, A.; et al. Immune landscapes associated with different glioblastoma molecular subtypes. Acta Neuropathol. Commun. 2019, 7, 203. [Google Scholar] [CrossRef] [Green Version]
- Woroniecka, K.; Chongsathidkiet, P.; Rhodin, K.; Kemeny, H.; Dechant, C.; Harrison Farber, S.; Elsamadicy, A.A.; Cui, X.; Koyama, S.; Jackson, C.; et al. T-cell exhaustion signatures vary with tumor type and are severe in glioblastoma. Clin. Cancer Res. 2018, 24, 4175–4186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woroniecka, K.I.; Rhodin, K.E.; Chongsathidkiet, P.; Keith, K.A.; Fecci, P.E. T-Cell dysfunction in glioblastoma: Applying a new framework. Clin. Cancer Res. 2018, 24, 3792–3802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schiffer, D.; Annovazzi, L.; Casalone, C.; Corona, C.; Mellai, M. Glioblastoma: Microenvironment and niche concept. Cancers 2019, 11, 5. [Google Scholar] [CrossRef] [Green Version]
- Szulzewsky, F.; Arora, S.; de Witte, L.; Ulas, T.; Markovic, D.; Schultze, J.L.; Holland, E.C.; Synowitz, M.; Wolf, S.A.; Kettenmann, H. Human glioblastoma-associated microglia/monocytes express a distinct RNA profile compared to human control and murine samples. Glia 2016, 64, 1416–1436. [Google Scholar] [CrossRef]
- Wang, Q.; Hu, B.; Hu, X.; Kim, H.; Squatrito, M.; Scarpace, L.; deCarvalho, A.C.; Lyu, S.; Li, P.; Li, Y.; et al. Tumor Evolution of Glioma-Intrinsic Gene Expression Subtypes Associates with Immunological Changes in the Microenvironment. Cancer Cell 2017, 32, 42–56.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, J.; Chen, P.; Gupta, P.; Ott, M.; Zamler, D.; Kassab, C.; Bhat, K.P.; Curran, M.A.; De Groot, J.F.; Heimberger, A.B. Immune biology of glioma-associated macrophages and microglia: Functional and therapeutic implications. Neuro-Oncology 2020, 22, 5. [Google Scholar] [CrossRef]
- Bowman, R.L.; Klemm, F.; Akkari, L.; Pyonteck, S.M.; Sevenich, L.; Quail, D.F.; Dhara, S.; Simpson, K.; Gardner, E.E.; Iacobuzio-Donahue, C.A.; et al. Macrophage Ontogeny Underlies Differences in Tumor-Specific Education in Brain Malignancies. Cell Rep. 2016, 17, 2445–2459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, K.; Noubade, R.; Manzanillo, P.; Ota, N.; Foreman, O.; Hackney, J.A.; Friedman, B.A.; Pappu, R.; Scearce-Levie, K.; Ouyang, W. Mice deficient in NRROS show abnormal microglial development and neurological disorders. Nat. Immunol. 2017, 18, 633–641. [Google Scholar] [CrossRef]
- Kronenberg, G.; Uhlemann, R.; Richter, N.; Klempin, F.; Wegner, S.; Staerck, L.; Wolf, S.; Uckert, W.; Kettenmann, H.; Endres, M.; et al. Distinguishing features of microglia- and monocyte-derived macrophages after stroke. Acta Neuropathol. 2018, 135, 551–568. [Google Scholar] [CrossRef]
- Murata, Y.; Sugimoto, K.; Yang, C.; Harada, K.; Gono, R.; Harada, T.; Miyashita, Y.; Higashisaka, K.; Katada, R.; Tanaka, J.; et al. Activated microglia-derived macrophage-like cells exacerbate brain edema after ischemic stroke correlate with astrocytic expression of aquaporin-4 and interleukin-1 alpha release. Neurochem. Int. 2020, 140, 104848. [Google Scholar] [CrossRef]
- Müller, A.; Brandenburg, S.; Turkowski, K.; Müller, S.; Vajkoczy, P. Resident microglia, and not peripheral macrophages, are the main source of brain tumor mononuclear cells. Int. J. Cancer 2015, 137, 278–288. [Google Scholar] [CrossRef] [PubMed]
- Bennett, M.L.; Bennett, F.C.; Liddelow, S.A.; Ajami, B.; Zamanian, J.L.; Fernhoff, N.B.; Mulinyawe, S.B.; Bohlen, C.J.; Adil, A.; Tucker, A.; et al. New tools for studying microglia in the mouse and human CNS. Proc. Natl. Acad. Sci. USA 2016, 113, E1738–E1746. [Google Scholar] [CrossRef] [Green Version]
- Mildner, A.; Huang, H.; Radke, J.; Stenzel, W.; Priller, J. P2Y12 receptor is expressed on human microglia under physiological conditions throughout development and is sensitive to neuroinflammatory diseases. Glia 2017, 65, 375–387. [Google Scholar] [CrossRef] [PubMed]
- Müller, S.; Kohanbash, G.; Liu, S.J.; Alvarado, B.; Carrera, D.; Bhaduri, A.; Watchmaker, P.B.; Yagnik, G.; Di Lullo, E.; Malatesta, M.; et al. Single-cell profiling of human gliomas reveals macrophage ontogeny as a basis for regional differences in macrophage activation in the tumor microenvironment. Genome Biol. 2017, 18, 234. [Google Scholar] [CrossRef] [PubMed]
- Venteicher, A.S.; Tirosh, I.; Hebert, C.; Yizhak, K.; Neftel, C.; Filbin, M.G.; Hovestadt, V.; Escalante, L.E.; Shaw, M.L.; Rodman, C.; et al. Decoupling genetics, lineages, and microenvironment in IDH-mutant gliomas by single-cell RNA-seq. Science 2017, 355, eaai8478. [Google Scholar] [CrossRef] [Green Version]
- Roesch, S.; Rapp, C.; Dettling, S.; Herold-Mende, C. When immune cells turn bad—tumor-associated microglia/macrophages in glioma. Int. J. Mol. Sci. 2018, 19, 436. [Google Scholar] [CrossRef] [Green Version]
- Mosser, D.M.; Edwards, J.P. Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 2008, 8, 958–969. [Google Scholar] [CrossRef]
- Gabrusiewicz, K.; Rodriguez, B.; Wei, J.; Hashimoto, Y.; Healy, L.M.; Maiti, S.N.; Thomas, G.; Zhou, S.; Wang, Q.; Elakkad, A.; et al. Glioblastoma-infiltrated innate immune cells resemble M0 macrophage phenotype. JCI Insight 2016, 1, e85841. [Google Scholar] [CrossRef]
- Chen, Z.; Feng, X.; Herting, C.J.; Garcia, V.A.; Nie, K.; Pong, W.W.; Rasmussen, R.; Dwivedi, B.; Seby, S.; Wolf, S.A.; et al. Cellular and Molecular Identity of Tumor-Associated Macrophages in Glioblastoma. Cancer Res. 2017, 77, 2266–2278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sousa, C.; Golebiewska, A.; Poovathingal, S.K.; Kaoma, T.; Pires-Afonso, Y.; Martina, S.; Coowar, D.; Azuaje, F.; Skupin, A.; Balling, R.; et al. Single-cell transcriptomics reveals distinct inflammation-induced microglia signatures. EMBO Rep. 2018, 19, e46171. [Google Scholar] [CrossRef]
- Ochocka, N.; Segit, P.; Walentynowicz, K.A.; Wojnicki, K.; Cyranowski, S.; Swatler, J.; Mieczkowski, J.; Kaminska, B. Single-cell RNA sequencing reveals functional heterogeneity of glioma-associated brain macrophages. Nat. Commun. 2021, 12, 1151. [Google Scholar] [CrossRef]
- Ravi, V.; Neidert, N.; Will, P.; Joseph, K.; Maier, J.; Kückelhaus, J.; Vollmer, L.; Goeldner, J.; Behringer, S.; Scherer, F.; et al. Lineage and Spatial Mapping of Glioblastoma-associated Immunity. bioRxiv. 2021. [Google Scholar] [CrossRef]
- Pombo Antunes, A.R.; Scheyltjens, I.; Lodi, F.; Messiaen, J.; Antoranz, A.; Duerinck, J.; Kancheva, D.; Martens, L.; De Vlaminck, K.; Van Hove, H.; et al. Single-cell profiling of myeloid cells in glioblastoma across species and disease stage reveals macrophage competition and specialization. Nat. Neurosci. 2021, 24, 595–610. [Google Scholar] [CrossRef]
- Puchalski, R.B.; Shah, N.; Miller, J.; Dalley, R.; Nomura, S.R.; Yoon, J.-G.; Smith, K.A.; Lankerovich, M.; Bertagnolli, D.; Bickley, K.; et al. An anatomic transcriptional atlas of human glioblastoma. Science 2018, 360, 660–663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamura, R.; Ohara, K.; Sasaki, H.; Morimoto, Y.; Kosugi, K.; Yoshida, K.; Toda, M. Difference in Immunosuppressive Cells Between Peritumoral Area and Tumor Core in Glioblastoma. World Neurosurg. 2018, 120, e601–e610. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Xue, H.; Shao, Q.; Wang, J.; Guo, X.; Chen, X.; Zhang, J.; Xu, S.; Li, T.; Zhang, P.; et al. Hypoxia promotes glioma-associated macrophage infiltration via periostin and subsequent M2 polarization by upregulating TGF-beta and M-CSFR. Oncotarget 2016, 7, 80521–80542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darmanis, S.; Sloan, S.A.; Croote, D.; Mignardi, M.; Chernikova, S.; Samghababi, P.; Zhang, Y.; Neff, N.; Kowarsky, M.; Caneda, C.; et al. Single-Cell RNA-Seq Analysis of Infiltrating Neoplastic Cells at the Migrating Front of Human Glioblastoma. Cell Rep. 2017, 21, 1399–1410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komohara, Y.; Horlad, H.; Ohnishi, K.; Fujiwara, Y.; Bai, B.; Nakagawa, T.; Suzu, S.; Nakamura, H.; Kuratsu, J.; Takeya, M. Importance of direct macrophage-tumor cell interaction on progression of human glioma. Cancer Sci. 2012, 103, 2165–2172. [Google Scholar] [CrossRef] [PubMed]
- Prosniak, M.; Harshyne, L.A.; Andrews, D.W.; Kenyon, L.C.; Bedelbaeva, K.; Apanasovich, T.V.; Heber-Katz, E.; Curtis, M.T.; Cotzia, P.; Hooper, D.C. Glioma grade is associated with the accumulation and activity of cells bearing M2 monocyte markers. Clin. Cancer Res. 2013, 19, 3776–3786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verhaak, R.G.W.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [Green Version]
- Patel, A.P.; Tirosh, I.; Trombetta, J.J.; Shalek, A.K.; Gillespie, S.M.; Wakimoto, H.; Cahill, D.P.; Nahed, B.V.; Curry, W.T.; Martuza, R.L.; et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 2014, 344, 1396–1401. [Google Scholar] [CrossRef] [Green Version]
- Sottoriva, A.; Spiteri, I.; Piccirillo, S.G.M.; Touloumis, A.; Collins, V.P.; Marioni, J.C.; Curtis, C.; Watts, C.; Tavaré, S. Intratumor heterogeneity in human glioblastoma reflects cancer evolutionary dynamics. Proc. Natl. Acad. Sci. USA 2013, 110, 4009–4014. [Google Scholar] [CrossRef] [Green Version]
- Kaffes, I.; Szulzewsky, F.; Chen, Z.; Herting, C.J.; Gabanic, B.; Velázquez Vega, J.E.; Shelton, J.; Switchenko, J.M.; Ross, J.L.; McSwain, L.F.; et al. Human Mesenchymal glioblastomas are characterized by an increased immune cell presence compared to Proneural and Classical tumors. Oncoimmunology 2019, 8, e1655360. [Google Scholar] [CrossRef] [PubMed]
- Wu, A.; Wei, J.; Kong, L.Y.; Wang, Y.; Priebe, W.; Qiao, W.; Sawaya, R.; Heimberger, A.B. Glioma cancer stem cells induce immunosuppressive macrophages/microglia. Neuro-Oncology 2010, 12, 1113–1125. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Pan, Y.; Gutmann, D.H. Genetic and genomic alterations differentially dictate low-grade glioma growth through cancer stem cell-specific chemokine recruitment of T cells and microglia. Neuro-Oncology 2019, 21, 1250–1262. [Google Scholar] [CrossRef] [PubMed]
- Lepore, F.; D’Alessandro, G.; Antonangeli, F.; Santoro, A.; Esposito, V.; Limatola, C.; Trettel, F. CXCL16/CXCR6 axis drives microglia/macrophages phenotype in physiological conditions and plays a crucial role in glioma. Front. Immunol. 2018, 9, 2750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Held-Feindt, J.; Hattermann, K.; Müerköster, S.S.; Wedderkopp, H.; Knerlich-Lukoschus, F.; Ungefroren, H.; Mehdorn, H.M.; Mentlein, R. CX3CR1 promotes recruitment of human glioma-infiltrating microglia/macrophages (GIMs). Exp. Cell Res. 2010, 316, 1553–1566. [Google Scholar] [CrossRef] [PubMed]
- De Boeck, A.; Ahn, B.Y.; D’Mello, C.; Lun, X.; Menon, S.V.; Alshehri, M.M.; Szulzewsky, F.; Shen, Y.; Khan, L.; Dang, N.H.; et al. Glioma-derived IL-33 orchestrates an inflammatory brain tumor microenvironment that accelerates glioma progression. Nat. Commun. 2020, 11, 4997. [Google Scholar] [CrossRef]
- Wei, J.; Marisetty, A.; Schrand, B.; Gabrusiewicz, K.; Hashimoto, Y.; Ott, M.; Grami, Z.; Kong, L.Y.; Ling, X.; Caruso, H.; et al. Osteopontin mediates glioblastoma-associated macrophage infiltration and is a potential therapeutic target. J. Clin. Invest. 2019, 129, 137–149. [Google Scholar] [CrossRef]
- Chen, P.; Zhao, D.; Li, J.; Liang, X.; Li, J.; Chang, A.; Henry, V.K.; Lan, Z.; Spring, D.J.; Rao, G.; et al. Symbiotic Macrophage-Glioma Cell Interactions Reveal Synthetic Lethality in PTEN-Null Glioma. Cancer Cell 2019, 35, 868–884.e6. [Google Scholar] [CrossRef] [PubMed]
- Platten, M.; Kretz, A.; Naumann, U.; Aulwurm, S.; Egashira, K.; Isenmann, S.; Weller, M. Monocyte chemoattractant protein-1 increases microglial infiltration and aggressiveness of gliomas. Ann. Neurol. 2003, 54, 388–392. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Sarkar, S.; Cua, R.; Zhou, Y.; Hader, W.; Wee Yong, V. A dialog between glioma and microglia that promotes tumor invasiveness through the CCL2/CCR2/interleukin-6 axis. Carcinogenesis 2012, 33, 312–319. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Alvarez-Breckenridge, C.A.; Wang, Q.E.; Yu, J. TGF-β signaling and its targeting for glioma treatment. Am. J. Cancer Res. 2015, 5, 945–955. [Google Scholar]
- Ku, M.C.; Wolf, S.A.; Respondek, D.; Matyash, V.; Pohlmann, A.; Waiczies, S.; Waiczies, H.; Niendorf, T.; Synowitz, M.; Glass, R.; et al. GDNF mediates glioblastoma-induced microglia attraction but not astrogliosis. Acta Neuropathol. 2013, 125, 609–620. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Huo, R.; Li, N.; Li, H.; Zhai, T.; Li, H.; Shen, B.; Ye, J.; Fu, R.; Di, W. CYR61, a potential biomarker of tumor inflammatory response in epithelial ovarian cancer microenvironment of tumor progress. BMC Cancer 2019, 19, 1140. [Google Scholar] [CrossRef]
- Yan, S.; Liu, H.; Liu, Z.; Peng, F.; Jiang, F.; Li, L.; Fu, R. CCN1 stimulated the osteoblasts via PTEN/AKT/GSK3β/cyclinD1 signal pathway in Myeloma Bone Disease. Cancer Med. 2020, 9, 737–744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uneda, A.; Kurozumi, K.; Fujimura, A.; Fujii, K.; Ishida, J.; Shimazu, Y.; Otani, Y.; Tomita, Y.; Hattori, Y.; Matsumoto, Y.; et al. Differentiated glioblastoma cells accelerate tumor progression by shaping the tumor microenvironment via CCN1-mediated macrophage infiltration. Acta Neuropathol. Commun. 2021, 9, 29. [Google Scholar] [CrossRef]
- Zhou, W.; Ke, S.Q.; Huang, Z.; Flavahan, W.; Fang, X.; Paul, J.; Wu, L.; Sloan, A.E.; McLendon, R.E.; Li, X.; et al. Periostin secreted by glioblastoma stem cells recruits M2 tumour-associated macrophages and promotes malignant growth. Nat. Cell Biol. 2015, 2, 170–182. [Google Scholar] [CrossRef] [Green Version]
- Tao, W.; Chu, C.; Zhou, W.; Huang, Z.; Zhai, K.; Fang, X.; Huang, Q.; Zhang, A.; Wang, X.; Yu, X.; et al. Dual Role of WISP1 in maintaining glioma stem cells and tumor-supportive macrophages in glioblastoma. Nat. Commun. 2020, 11, 3015. [Google Scholar] [CrossRef]
- Yekula, A.A.; Yekula, A.A.; Muralidharan, K.; Kang, K.; Carter, B.S.; Balaj, L. Extracellular Vesicles in Glioblastoma Tumor Microenvironment. Front. Immunol. 2020, 10, 3137. [Google Scholar] [CrossRef]
- Gabrusiewicz, K.; Li, X.; Wei, J.; Hashimoto, Y.; Marisetty, A.L.; Ott, M.; Wang, F.; Hawke, D.; Yu, J.; Healy, L.M.; et al. Glioblastoma stem cell-derived exosomes induce M2 macrophages and PD-L1 expression on human monocytes. Oncoimmunology 2018, 7, e1412909. [Google Scholar] [CrossRef] [PubMed]
- De Vrij, J.; Niek Maas, S.L.; Kwappenberg, K.M.C.; Schnoor, R.; Kleijn, A.; Dekker, L.; Luider, T.M.; De Witte, L.D.; Litjens, M.; Van Strien, M.E.; et al. Glioblastoma-derived extracellular vesicles modify the phenotype of monocytic cells. Int. J. Cancer 2015, 137, 1630–1642. [Google Scholar] [CrossRef] [Green Version]
- Qian, M.; Wang, S.; Guo, X.; Wang, J.; Zhang, Z.; Qiu, W.; Gao, X.; Chen, Z.; Xu, J.; Zhao, R.; et al. Hypoxic glioma-derived exosomes deliver microRNA-1246 to induce M2 macrophage polarization by targeting TERF2IP via the STAT3 and NF-κB pathways. Oncogene 2020, 39, 428–442. [Google Scholar] [CrossRef]
- Abels, E.R.; Maas, S.L.N.; Nieland, L.; Wei, Z.; Cheah, P.S.; Tai, E.; Kolsteeg, C.J.; Dusoswa, S.A.; Ting, D.T.; Hickman, S.; et al. Glioblastoma-Associated Microglia Reprogramming Is Mediated by Functional Transfer of Extracellular miR-21. Cell Rep. 2019, 28, 3105–3119.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; Zhang, C.; Maimela, N.R.; Yang, L.; Zhang, Z.; Ping, Y.; Huang, L.; Zhang, Y. Molecular and clinical characterization of CD163 expression via large-scale analysis in glioma. Oncoimmunology 2019, 8, 1601478. [Google Scholar] [CrossRef]
- Gürsel, D.B.; Shin, B.J.; Burkhardt, J.K.; Kesavabhotla, K.; Schlaff, C.D.; Boockvar, J.A. Glioblastoma stem-like cells-biology and therapeutic implications. Cancers 2011, 3, 2655–2666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nørøxe, D.S.; Poulsen, H.S.; Lassen, U. Hallmarks of glioblastoma: A systematic review. ESMO Open 2016, 1, e000144. [Google Scholar] [CrossRef]
- Chen, J.; Li, Y.; Yu, T.S.; McKay, R.M.; Burns, D.K.; Kernie, S.G.; Parada, L.F. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature 2012, 488, 522–526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, R.; Chadalavada, K.; Wilshire, J.; Kowalik, U.; Hovinga, K.E.; Geber, A.; Fligelman, B.; Leversha, M.; Brennan, C.; Tabar, V. Glioblastoma stem-like cells give rise to tumour endothelium. Nature 2010, 468, 829–833. [Google Scholar] [CrossRef]
- Zhang, S.; Zhao, B.S.; Zhou, A.; Lin, K.; Zheng, S.; Lu, Z.; Chen, Y.; Sulman, E.P.; Xie, K.; Bögler, O.; et al. m6A Demethylase ALKBH5 Maintains Tumorigenicity of Glioblastoma Stem-like Cells by Sustaining FOXM1 Expression and Cell Proliferation Program. Cancer Cell 2017, 31, 591–606.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guerra-Rebollo, M.; Garrido, C.; Sánchez-Cid, L.; Soler-Botija, C.; Meca-Cortés, O.; Rubio, N.; Blanco, J. Targeting of replicating CD133 and OCT4/SOX2 expressing glioma stem cells selects a cell population that reinitiates tumors upon release of therapeutic pressure. Sci. Rep. 2019, 9, 9549. [Google Scholar] [CrossRef] [PubMed]
- Saadeh, F.S.; Mahfouz, R.; Assi, H.I. Egfr as a clinical marker in glioblastomas and other gliomas. Int. J. Biol. Markers 2018, 33, 22–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coniglio, S.J.; Eugenin, E.; Dobrenis, K.; Stanley, E.R.; West, B.L.; Symons, M.H.; Segall, J.E. Microglial stimulation of glioblastoma invasion involves epidermal growth factor receptor (EGFR) and colony stimulating factor 1 receptor (CSF-1R) signaling. Mol. Med. 2012, 18, 519–527. [Google Scholar] [CrossRef]
- Zhou, R.; Qian, D.; Hua, X.; Hu, M.; Chen, H.; Wang, B. Epidermal Growth Factor (EGF) Promotes Human Malignant Glioma Invasion by Mediating Secretion of Human Cytomegalovirus Infected Monocyte-Derived Macrophages. Precis. Med. 2018, 3, 1097. [Google Scholar] [CrossRef]
- Erlich, R.B.; Kahn, S.A.; Lima, F.R.S.; Muras, A.G.; Martins, R.A.P.; Linden, R.; Chiarini, L.B.; Martins, V.R.; Neto, V.M. STI1 promotes glioma proliferation through MAPK and PI3K pathways. Glia 2007, 55, 1690–1698. [Google Scholar] [CrossRef]
- Da Fonseca, A.C.C.; Romão, L.; Amaral, R.F.; Assad Kahn, S.; Lobo, D.; Martins, S.; Marcondes de Souza, J.; Moura-Neto, V.; Lima, F.R.S. Microglial stress inducible protein 1 promotes proliferation and migration in human glioblastoma cells. Neuroscience 2012, 200, 130–141. [Google Scholar] [CrossRef]
- Qi, L.; Yu, H.; Zhang, Y.; Zhao, D.; Lv, P.; Zhong, Y.; Xu, Y. IL-10 secreted by M2 macrophage promoted tumorigenesis through interaction with JAK2 in glioma. Oncotarget 2016, 7, 71673–71685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, J.; Xu, Z.; Duan, H.; Ji, H.; Zhen, Z.; Li, B.; Wang, H.; Tang, H.; Zhou, J.; Guo, T.; et al. Tumor-associated macrophage interleukin-β promotes glycerol-3-phosphate dehydrogenase activation, glycolysis and tumorigenesis in glioma cells. Cancer Sci. 2020, 111, 1979–1990. [Google Scholar] [CrossRef]
- Bruna, A.; Darken, R.S.; Rojo, F.; Ocaña, A.; Peñuelas, S.; Arias, A.; Paris, R.; Tortosa, A.; Mora, J.; Baselga, J.; et al. High TGFβ-Smad Activity Confers Poor Prognosis in Glioma Patients and Promotes Cell Proliferation Depending on the Methylation of the PDGF-B Gene. Cancer Cell 2007, 11, 147–160. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Kuang, W.; Zhou, Q.; Zhang, Y. TGF-β1 secreted by M2 phenotype macrophages enhances the stemness and migration of glioma cells via the SMAD2/3 signalling pathway. Int. J. Mol. Med. 2018, 42, 3395–3403. [Google Scholar] [CrossRef] [Green Version]
- Sa, J.K.; Chang, N.; Lee, H.W.; Cho, H.J.; Ceccarelli, M.; Cerulo, L.; Yin, J.; Kim, S.S.; Caruso, F.P.; Lee, M.; et al. Transcriptional regulatory networks of tumor-associated macrophages that drive malignancy in mesenchymal glioblastoma. Genome Biol. 2020, 21, 216. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Chen, L.; Dang, W.-Q.; Cao, M.-F.; Xiao, J.-F.; Lv, S.-Q.; Jiang, W.-J.; Yao, X.-H.; Lu, H.-M.; Miao, J.-Y.; et al. CCL8 secreted by tumor-associated macrophages promotes invasion and stemness of glioblastoma cells via ERK1/2 signaling. Lab. Investig. 2020, 100, 619–629. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, T.; Yang, N.; Xu, S.; Li, X.; Wang, D. Hypoxia and macrophages promote glioblastoma invasion by the CCL4-CCR5 axis. Oncol. Rep. 2016, 36, 3522–3528. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Ping, Y.F.; Zhou, W.; He, Z.C.; Chen, C.; Bian, B.S.J.; Zhang, L.; Chen, L.; Lan, X.; Zhang, X.C.; et al. Tumour-associated macrophages secrete pleiotrophin to promote PTPRZ1 signalling in glioblastoma stem cells for tumour growth. Nat. Commun. 2017, 8, 15080. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Ye, J.; Shen, F.; Zhu, Y.; Yeghiazarians, Y.; Zhu, W.; Chen, Y.; Lawton, M.T.; Young, W.L.; Yang, G.Y. Interleukin-6 stimulates circulating blood-derived endothelial progenitor cell angiogenesis in vitro. J. Cereb. Blood Flow Metab. 2008, 28, 90–98. [Google Scholar] [CrossRef] [Green Version]
- Zhu, C.; Kros, J.M.; Cheng, C.; Mustafa, D. The contribution of tumor-Associated macrophages in glioma neo-Angiogenesis and implications for anti-Angiogenic strategies. Neuro-Oncology 2017, 19, 1435–1446. [Google Scholar] [CrossRef] [Green Version]
- Ahir, B.K.; Engelhard, H.H.; Lakka, S.S. Tumor Development and Angiogenesis in Adult Brain Tumor: Glioblastoma. Mol. Neurobiol. 2020, 57, 2461–2478. [Google Scholar] [CrossRef] [Green Version]
- Nijaguna, M.B.; Patil, V.; Urbach, S.; Shwetha, S.D.; Sravani, K.; Hegde, A.S.; Chandramouli, B.A.; Arivazhagan, A.; Marin, P.; Santosh, V.; et al. Glioblastoma-derived macrophage colony-stimulating factor (MCSF) induces microglial release of insulin-like growth factor-binding protein 1 (IGFBP1) to promote angiogenesis. J. Biol. Chem. 2015, 290, 23401–23415. [Google Scholar] [CrossRef] [Green Version]
- Osterberg, N.; Ferrara, N.; Vacher, J.; Gaedicke, S.; Niedermann, G.; Weyerbrock, A.; Doostkam, S.; Schaefer, H.E.; Plate, K.H.; Machein, M.R. Decrease of VEGF-A in myeloid cells attenuates glioma progression and prolongs survival in an experimental glioma model. Neuro. Oncol. 2016, 18, 939–949. [Google Scholar] [CrossRef] [Green Version]
- Gilbert, M.R.; Dignam, J.J.; Armstrong, T.S.; Wefel, J.S.; Blumenthal, D.T.; Vogelbaum, M.A.; Colman, H.; Chakravarti, A.; Pugh, S.; Won, M.; et al. A randomized trial of bevacizumab for newly diagnosed glioblastoma. N. Engl. J. Med. 2014, 370, 699–708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wick, W.; Gorlia, T.; Bendszus, M.; Taphoorn, M.; Sahm, F.; Harting, I.; Brandes, A.A.; Taal, W.; Domont, J.; Idbaih, A.; et al. Lomustine and Bevacizumab in Progressive Glioblastoma. N. Engl. J. Med. 2017, 377, 1954–1963. [Google Scholar] [CrossRef]
- Goumans, M.J.; Valdimarsdottir, G.; Itoh, S.; Rosendahl, A.; Sideras, P.; Ten Dijke, P. Balancing the activation state of the endothelium via two distinct TGF-β type I receptors. EMBO J. 2002, 21, 1743–1753. [Google Scholar] [CrossRef]
- Wang, X.B.; Tian, X.Y.; Li, Y.; Li, B.; Li, Z. Elevated expression of macrophage migration inhibitory factor correlates with tumor recurrence and poor prognosis of patients with gliomas. J. Neurooncol. 2012, 106, 43–45. [Google Scholar] [CrossRef]
- Ha, W.; Sevim-Nalkiran, H.; Zaman, A.M.; Matsuda, K.; Khasraw, M.; Nowak, A.K.; Chung, L.; Baxter, R.C.; McDonald, K.L. Ibudilast sensitizes glioblastoma to temozolomide by targeting Macrophage Migration Inhibitory Factor (MIF). Sci. Rep. 2019, 9, 2905. [Google Scholar] [CrossRef] [Green Version]
- Castro, B.A.; Flanigan, P.; Jahangiri, A.; Hoffman, D.; Chen, W.; Kuang, R.; De Lay, M.; Yagnik, G.; Wagner, J.R.; Mascharak, S.; et al. Macrophage migration inhibitory factor downregulation: A novel mechanism of resistance to anti-angiogenic therapy. Oncogene 2017, 36, 3749–3759. [Google Scholar] [CrossRef] [Green Version]
- Mangano, K.; Mazzon, E.; Basile, M.S.; Di Marco, R.; Bramanti, P.; Mammana, S.; Petralia, M.C.; Fagone, P.; Nicoletti, F. Pathogenic role for macrophage migration inhibitory factor in glioblastoma and its targeting with specific inhibitors as novel tailored therapeutic approach. Oncotarget 2018, 9, 17951–17970. [Google Scholar] [CrossRef] [Green Version]
- Guo, X.; Xu, S.; Gao, X.; Wang, J.; Xue, H.; Chen, Z.; Zhang, J.; Guo, X.; Qian, M.; Qiu, W.; et al. Macrophage migration inhibitory factor promotes vasculogenic mimicry formation induced by hypoxia via CXCR4/AKT/EMT pathway in human glioblastoma cells. Oncotarget 2017, 8, 80358–80372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munaut, C.; Boniver, J.; Foidart, J.M.; Deprez, M. Macrophage migration inhibitory factor (MIF) expression in human glioblastomas correlates with vascular endothelial growth factor (VEGF) expression. Neuropathol. Appl. Neurobiol. 2002, 28, 452–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, P.S.; Wen, J.; Ang, L.C.; Sheng, W.; Viloria-Petit, A.; Wang, Y.; Wu, Y.; Kerbel, R.S.; Yang, B.B. Versican/PG-M G3 domain promotes tumor growth and angiogenesis. FASEB J. 2004, 18, 754–756. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Yee, A.J. Versican V2 isoform enhances angiogenesis by regulating endothelial cell activities and fibronectin expression. FEBS Lett. 2013, 587, 185–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brandenburg, S.; Müller, A.; Turkowski, K.; Radev, Y.T.; Rot, S.; Schmidt, C.; Bungert, A.D.; Acker, G.; Schorr, A.; Hippe, A.; et al. Resident microglia rather than peripheral macrophages promote vascularization in brain tumors and are source of alternative pro-angiogenic factors. Acta Neuropathol. 2016, 131, 365–378. [Google Scholar] [CrossRef]
- Blank, A.; Kremenetskaia, I.; Urbantat, R.M.; Acker, G.; Turkowski, K.; Radke, J.; Schneider, U.C.; Vajkoczy, P.; Brandenburg, S. Microglia/macrophages express alternative proangiogenic factors depending on granulocyte content in human glioblastoma. J. Pathol. 2021, 253, 160–173. [Google Scholar] [CrossRef]
- Koh, I.; Cha, J.; Park, J.; Choi, J.; Kang, S.G.; Kim, P. The mode and dynamics of glioblastoma cell invasion into a decellularized tissue-derived extracellular matrix-based three-dimensional tumor model. Sci. Rep. 2018, 8, 4608. [Google Scholar] [CrossRef]
- Wu, C.Y.-J.; Chen, C.H.; Lin, C.Y.; Feng, L.Y.; Lin, Y.C.; Wei, K.C.; Huang, C.Y.; Fang, J.Y.; Chen, P.Y. CCL5 of glioma-associated microglia/macrophages regulates glioma migration and invasion via calcium-dependent matrix metalloproteinase 2. Neuro-Oncology 2020, 22, 253–266. [Google Scholar] [CrossRef]
- Markovic, D.S.; Glass, R.; Synowitz, M.; Van Rooijen, N.; Kettenmann, H. Microglia stimulate the invasiveness of glioma cells by increasing the activity of metalloprotease-2. J. Neuropathol. Exp. Neurol. 2005, 64, 754–762. [Google Scholar] [CrossRef] [Green Version]
- Gjorgjevski, M.; Hannen, R.; Carl, B.; Li, Y.; Landmann, E.; Buchholz, M.; Bartsch, J.W.; Nimsky, C. Molecular profiling of the tumor microenvironment in glioblastoma patients: Correlation of microglia/macrophage polarization state with metalloprotease expression profiles and survival. Biosci. Rep. 2019, 39, BSR20182361. [Google Scholar] [CrossRef] [Green Version]
- Yan, W.; Zhang, W.; Sun, L.; Liu, Y.; You, G.; Wang, Y.; Kang, C.; You, Y.; Jiang, T. Identification of MMP-9 specific microRNA expression profile as potential targets of anti-invasion therapy in glioblastoma multiforme. Brain Res. 2011, 1411, 108–115. [Google Scholar] [CrossRef] [PubMed]
- Lakka, S.S.; Jasti, S.L.; Gondi, C.; Boyd, D.; Chandrasekar, N.; Dinh, D.H.; Olivero, W.C.; Gujrati, M.; Rao, J.S. Downregulation of MMP-9 in ERK-mutated stable transfectants inhibits glioma invasion in vitro. Oncogene 2002, 21, 5601–5608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solga, R.; Behrens, J.; Ziemann, A.; Riou, A.; Berwanger, C.; Becker, L.; Garrett, L.; de Angelis, M.H.; Fischer, L.; Coras, R.; et al. CRN2 binds to TIMP4 and MMP14 and promotes perivascular invasion of glioblastoma cells. Eur. J. Cell Biol. 2019, 98, 151046. [Google Scholar] [CrossRef] [PubMed]
- Wesolowska, A.; Kwiatkowska, A.; Slomnicki, L.; Dembinski, M.; Master, A.; Sliwa, M.; Franciszkiewicz, K.; Chouaib, S.; Kaminska, B. Microglia-derived TGF-β as an important regulator of glioblastoma invasion—An inhibition of TGF-β-dependent effects by shRNA against human TGF-β type II receptor. Oncogene 2008, 27, 918–930. [Google Scholar] [CrossRef] [Green Version]
- Wick, W.; Platten, M.; Weller, M. Glioma cell invasion: Regulation of metalloproteinase activity by TGF-β. J. Neurooncol. 2001, 53, 177–185. [Google Scholar] [CrossRef]
- Li, R.; Li, G.; Deng, L.; Liu, Q.; Dai, J.; Shen, J.; Zhang, J. IL-6 augments the invasiveness of U87MG human glioblastoma multiforme cells via up-regulation of MMP-2 and fascin-1. Oncol. Rep. 2010, 23, 1553–1559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, Z.F.; Liu, H.; Gao, R.; Zhou, M.; Ma, J.; Zhang, Y.; Zhao, J.; Chen, Y.; Zhang, T.; Huang, F.; et al. Tumor cell-released autophagosomes (TRAPs) promote immunosuppression through induction of M2-like macrophages with increased expression of PD-L1. J. Immunother. Cancer 2018, 6, 151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyazaki, T.; Ishikawa, E.; Matsuda, M.; Sugii, N.; Kohzuki, H.; Akutsu, H.; Sakamoto, N.; Takano, S.; Matsumura, A. Infiltration of CD163-positive macrophages in glioma tissues after treatment with anti-PD-L1 antibody and role of PI3Kγ inhibitor as a combination therapy with anti-PD-L1 antibody in in vivo model using temozolomide-resistant murine glioma-initiating cell. Brain Tumor Pathol. 2020, 37, 41–49. [Google Scholar] [CrossRef]
- Bloch, O.; Crane, C.A.; Kaur, R.; Safaee, M.; Rutkowski, M.J.; Parsa, A.T. Gliomas promote immunosuppression through induction of B7-H1 expression in tumor-associated macrophages. Clin. Cancer Res. 2013, 19, 3165–3175. [Google Scholar] [CrossRef] [Green Version]
- Ene, C.I.; Ene, C.I.; Kreuser, S.A.; Jung, M.; Jung, M.; Zhang, H.; Arora, S.; White Moyes, K.; Szulzewsky, F.; Barber, J.; et al. Anti-PD-L1 antibody direct activation of macrophages contributes to a radiation-induced abscopal response in glioblastoma. Neuro-Oncology 2020, 22, 639–651. [Google Scholar] [CrossRef]
- Gratas, C.; Tohma, Y.; Van Meir, E.G.; Klein, M.; Tenan, M.; Ishii, N.; Tachibana, O.; Kleihues, P.; Ohgaki, H. Fas ligond expression in glioblastoma cell lines and primary astrocytic brain tumors. Brain Pathol. 1997, 7, 863–869. [Google Scholar] [CrossRef]
- Badie, B.; Schartner, J.; Prabakaran, S.; Paul, J.; Vorpahl, J. Expression of Fas ligand by microglia: Possible role in glioma immune evasion. J. Neuroimmunol. 2001, 120, 19–24. [Google Scholar] [CrossRef]
- Chang, A.L.; Miska, J.; Wainwright, D.A.; Dey, M.; Rivetta, C.V.; Yu, D.; Kanojia, D.; Pituch, K.C.; Qiao, J.; Pytel, P.; et al. CCL2 produced by the glioma microenvironment is essential for the recruitment of regulatory t cells and myeloid-derived suppressor cells. Cancer Res. 2016, 76, 5671–5682. [Google Scholar] [CrossRef] [Green Version]
- Wagner, S.; Czub, S.; Greif, M.; Vince, G.H.; Süss, N.; Kerkau, S.; Rieckmann, P.; Roggendorf, W.; Roosen, K.; Tonn, J.C. Microglial/macrophage expression of interleukin 10 in human glioblastomas. Int. J. Cancer 1999, 82, 12–16. [Google Scholar] [CrossRef]
- Hattermann, K.; Sebens, S.; Helm, O.; Schmitt, A.D.; Mentlein, R.; Mehdorn, H.M.; Held-Feindt, J. Chemokine expression profile of freshly isolated human glioblastoma- associated macrophages/microglia. Oncol. Rep. 2014, 32, 270–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, D.A.; Massagué, J. TGF-β directly targets cytotoxic T cell functions during tumor evasion of immune surveillance. Cancer Cell 2005, 8, 369–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, T.; Terai, M.; Tamura, Y.; Alexeev, V.; Mastrangelo, M.J.; Selvan, S.R. Interleukin 10 in the tumor microenvironment: A target for anticancer immunotherapy. Immunol. Res. 2011, 51, 170–182. [Google Scholar] [CrossRef]
- Holmes, B.; Benavides-Serrato, A.; Freeman, R.S.; Landon, K.A.; Bashir, T.; Nishimura, R.N.; Gera, J. MTORC2/AKT/HSF1/HuR constitute a feed-forward loop regulating Rictor expression and tumor growth in glioblastoma. Oncogene 2018, 37, 732–743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Leavenworth, J.W.; Hjelmeland, A.B.; Smith, R.; Patel, N.; Borg, B.; Si, Y.; King, P.H. Deletion of the RNA regulator HuR in tumor-associated microglia and macrophages stimulates anti-tumor immunity and attenuates glioma growth. Glia 2019, 67, 2424–2439. [Google Scholar] [CrossRef] [PubMed]
- Kioi, M.; Vogel, H.; Schultz, G.; Hoffman, R.M.; Harsh, G.R.; Brown, J.M. Inhibition of vasculogenesis, but not angiogenesis, prevents the recurrence of glioblastoma after irradiation in mice. J. Clin. Invest. 2010, 120, 694–705. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.M.; Thomas, R.; Nagpal, S.; Recht, L. Macrophage exclusion after radiation therapy (MERT): A new and effective way to increase the therapeutic ratio of radiotherapy. Radiother. Oncol. 2020, 144, 159–164. [Google Scholar] [CrossRef]
- Akkari, L.; Bowman, R.L.; Tessier, J.; Klemm, F.; Handgraaf, S.M.; de Groot, M.; Quail, D.F.; Tillard, L.; Gadiot, J.; Huse, J.T.; et al. Dynamic changes in glioma macrophage populations after radiotherapy reveal CSF-1R inhibition as a strategy to overcome resistance. Sci. Transl. Med. 2020, 12, eaaw7843. [Google Scholar] [CrossRef]
- Leblond, M.M.; Pérès, E.A.; Helaine, C.; Gérault, A.N.; Moulin, D.; Anfray, C.; Divoux, D.; Petit, E.; Bernaudin, M.; Valable, S. M2 macrophages are more resistant than M1 macrophages following radiation therapy in the context of glioblastoma. Oncotarget 2017, 8, 72597–72612. [Google Scholar] [CrossRef] [Green Version]
- Bhat, K.P.L.; Balasubramaniyan, V.; Vaillant, B.; Ezhilarasan, R.; Hummelink, K.; Hollingsworth, F.; Wani, K.; Heathcock, L.; James, J.D.; Goodman, L.D.; et al. Mesenchymal differentiation mediated by NF-κB promotes radiation resistance in glioblastoma. Cancer Cell 2013, 24, 331–346. [Google Scholar] [CrossRef] [Green Version]
- Pienta, K.J.; Machiels, J.-P.; Schrijvers, D.; Alekseev, B.; Shkolnik, M.; Crabb, S.J.; Li, S.; Seetharam, S.; Puchalski, T.A.; Takimoto, C.; et al. Phase 2 study of carlumab (CNTO 888), a human monoclonal antibody against CC-chemokine ligand 2 (CCL2), in metastatic castration-resistant prostate cancer. Investig. New Drugs 2013, 31, 760–768. [Google Scholar] [CrossRef]
- Brana, I.; Calles, A.; LoRusso, P.M.; Yee, L.K.; Puchalski, T.A.; Seetharam, S.; Zhong, B.; de Boer, C.J.; Tabernero, J.; Calvo, E. Carlumab, an anti-C-C chemokine ligand 2 monoclonal antibody, in combination with four chemotherapy regimens for the treatment of patients with solid tumors: An open-label, multicenter phase 1b study. Target. Oncol. 2015, 10, 111–123. [Google Scholar] [CrossRef] [PubMed]
- Nywening, T.M.; Wang-Gillam, A.; Sanford, D.E.; Belt, B.A.; Panni, R.Z.; Cusworth, B.M.; Toriola, A.T.; Nieman, R.K.; Worley, L.A.; Yano, M.; et al. Targeting tumour-associated macrophages with CCR2 inhibition in combination with FOLFIRINOX in patients with borderline resectable and locally advanced pancreatic cancer: A single-centre, open-label, dose-finding, non-randomised, phase 1b trial. Lancet Oncol. 2016, 17, 651–662. [Google Scholar] [CrossRef] [Green Version]
- Thomas, R.P.; Nagpal, S.; Iv, M.; Soltys, S.G.; Bertrand, S.; Pelpola, J.S.; Ball, R.; Yang, J.; Sundaram, V.; Lavezo, J.; et al. Macrophage exclusion after radiation therapy (MERT): A First in Human Phase I/II Trial using a CXCR4 Inhibitor in Glioblastoma. Clin. Cancer Res. 2019, 25, 6948–6957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, E.Q.; Duda, D.G.; Muzikansky, A.; Gerstner, E.R.; Kuhn, J.G.; Reardon, D.A.; Nayak, L.; Norden, A.D.; Doherty, L.; LaFrankie, D.; et al. Phase I and biomarker study of plerixafor and bevacizumab in recurrent high-grade glioma. Clin. Cancer Res. 2018, 24, 4643–4649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sikic, B.I.; Lakhani, N.; Patnaik, A.; Shah, S.A.; Chandana, S.R.; Rasco, D.; Colevas, A.D.; O’Rourke, T.; Narayanan, S.; Papadopoulos, K.; et al. First-in-human, first-in-class phase i trial of the anti-CD47 antibody Hu5F9-G4 in patients with advanced cancers. J. Clin. Oncol. 2019, 37, 946–953. [Google Scholar] [CrossRef]
- Grégoire, H.; Roncali, L.; Rousseau, A.; Chérel, M.; Delneste, Y.; Jeannin, P.; Hindré, F.; Garcion, E. Targeting Tumor Associated Macrophages to Overcome Conventional Treatment Resistance in Glioblastoma. Front. Pharmacol. 2020, 11, 368. [Google Scholar] [CrossRef] [Green Version]
- Adams, S.; Kozhaya, L.; Martiniuk, F.; Meng, T.C.; Chiriboga, L.; Liebes, L.; Hochman, T.; Shuman, N.; Axelrod, D.; Speyer, J.; et al. Topical TLR7 agonist imiquimod can induce immune-mediated rejection of skin metastases in patients with breast cancer. Clin. Cancer Res. 2012, 18, 6748–6757. [Google Scholar] [CrossRef] [Green Version]
- Achyut, B.R.; Angara, K.; Jain, M.; Borin, T.F.; Rashid, M.H.; Iskander, A.S.M.; Ara, R.; Kolhe, R.; Howard, S.; Venugopal, N.; et al. Canonical NFκB signaling in myeloid cells is required for the glioblastoma growth. Sci. Rep. 2017, 7, 13754. [Google Scholar] [CrossRef] [Green Version]
- Barberi, T.; Martin, A.; Suresh, R.; Barakat, D.J.; Harris-Bookman, S.; Drake, C.G.; Lim, M.; Friedman, A.D. Absence of host NF-κB p50 induces murine glioblastoma tumor regression, increases survival, and decreases T-cell induction of tumor-associated macrophage M2 polarization. Cancer Immunol. Immunother. 2018, 67, 1491–1503. [Google Scholar] [CrossRef] [PubMed]
- Suresh, R.; Barakat, D.J.; Barberi, T.; Zheng, L.; Jaffee, E.; Pienta, K.J.; Friedman, A.D. NF-κB p50-deficient immature myeloid cell (p50-IMC) adoptive transfer slows the growth of murine prostate and pancreatic ductal carcinoma. J. Immunother. Cancer 2020, 8, e000244. [Google Scholar] [CrossRef] [Green Version]
- Tap, W.D.; Wainberg, Z.A.; Anthony, S.P.; Ibrahim, P.N.; Zhang, C.; Healey, J.H.; Chmielowski, B.; Staddon, A.P.; Cohn, A.L.; Shapiro, G.I.; et al. Structure-Guided Blockade of CSF1R Kinase in Tenosynovial Giant-Cell Tumor. N. Engl. J. Med. 2015, 373, 428–437. [Google Scholar] [CrossRef] [Green Version]
- Butowski, N.; Colman, H.; De Groot, J.F.; Omuro, A.M.; Nayak, L.; Wen, P.Y.; Cloughesy, T.F.; Marimuthu, A.; Haidar, S.; Perry, A.; et al. Orally administered colony stimulating factor 1 receptor inhibitor PLX3397 in recurrent glioblastoma: An Ivy Foundation Early Phase Clinical Trials Consortium phase II study. Neuro. Oncol. 2016, 18, 557–564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pyonteck, S.M.; Akkari, L.; Schuhmacher, A.J.; Bowman, R.L.; Sevenich, L.; Quail, D.F.; Olson, O.C.; Quick, M.L.; Huse, J.T.; Teijeiro, V.; et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat. Med. 2013, 19, 1264–1272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almahariq, M.F.; Quinn, T.J.; Kesarwani, P.; Kant, S.; Miller, C.R.; Chinnaiyan, P. Inhibition of colony-stimulating factor-1 receptor enhances the efficacy of radiotherapy and reduces immune suppression in glioblastoma. In Vivo 2021, 35, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Cloughesy, T.F.; Mochizuki, A.Y.; Orpilla, J.R.; Hugo, W.; Lee, A.H.; Davidson, T.B.; Wang, A.C.; Ellingson, B.M.; Rytlewski, J.A.; Sanders, C.M.; et al. Neoadjuvant anti-PD-1 immunotherapy promotes a survival benefit with intratumoral and systemic immune responses in recurrent glioblastoma. Nat. Med. 2019, 25, 477–486. [Google Scholar] [CrossRef]
- Reardon, D.A.; Brandes, A.A.; Omuro, A.; Mulholland, P.; Lim, M.; Wick, A.; Baehring, J.; Ahluwalia, M.S.; Roth, P.; Bähr, O.; et al. Effect of Nivolumab vs Bevacizumab in Patients with Recurrent Glioblastoma: The CheckMate 143 Phase 3 Randomized Clinical Trial. JAMA Oncol. 2020, 6, 1003–1010. [Google Scholar] [CrossRef] [PubMed]
- Khasraw, M.; Reardon, D.A.; Weller, M.; Sampson, J.H. PD-1 Inhibitors: Do they have a Future in the Treatment of Glioblastoma? Clin. Cancer Res. 2020, 26, 5287–5296. [Google Scholar] [CrossRef]
- Goswami, S.; Walle, T.; Cornish, A.E.; Basu, S.; Anandhan, S.; Fernandez, I.; Vence, L.; Blando, J.; Zhao, H.; Yadav, S.S.; et al. Immune profiling of human tumors identifies CD73 as a combinatorial target in glioblastoma. Nat. Med. 2020, 26, 39–46. [Google Scholar] [CrossRef]
- Ghalamfarsa, G.; Kazemi, M.H.; Raoofi Mohseni, S.; Masjedi, A.; Hojjat-Farsangi, M.; Azizi, G.; Yousefi, M.; Jadidi-Niaragh, F. CD73 as a potential opportunity for cancer immunotherapy. Expert Opin. Ther. Targets 2019, 23, 127–142. [Google Scholar] [CrossRef] [PubMed]
- Takenaka, M.C.; Gabriely, G.; Rothhammer, V.; Mascanfroni, I.D.; Wheeler, M.A.; Chao, C.C.; Gutiérrez-Vázquez, C.; Kenison, J.; Tjon, E.C.; Barroso, A.; et al. Control of tumor-associated macrophages and T cells in glioblastoma via AHR and CD39. Nat. Neurosci. 2019, 22, 729–740. [Google Scholar] [CrossRef]
- Etzerodt, A.; Tsalkitzi, K.; Maniecki, M.; Damsky, W.; Delfini, M.; Baudoin, E.; Moulin, M.; Bosenberg, M.; Graversen, J.H.; Auphan-Anezin, N.; et al. Specific targeting of CD163+ TAMs mobilizes inflammatory monocytes and promotes T cell-mediated tumor regression. J. Exp. Med. 2019, 216, 2394–2411. [Google Scholar] [CrossRef]
- Yuan, Y.; Zhao, Q.; Zhao, S.; Zhang, P.; Zhao, H.; Li, Z.; Du, Y.; Tian, X.; Lu, J. Characterization of transcriptome profile and clinical features of a novel immunotherapy target CD204 in diffuse glioma. Cancer Med. 2019, 8, 3811–3821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giordano, F.A.; Link, B.; Glas, M.; Herrlinger, U.; Wenz, F.; Umansky, V.; Brown, J.M.; Herskind, C. Targeting the Post-Irradiation Tumor Microenvironment in Glioblastoma via Inhibition of CXCL12. Cancers 2019, 11, 272. [Google Scholar] [CrossRef] [Green Version]
- Stupp, R.; Hegi, M.E.; Mason, W.P.; van den Bent, M.J.; Taphoorn, M.J.; Janzer, R.C.; Ludwin, S.K.; Allgeier, A.; Fisher, B.; Belanger, K.; et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009, 10, 459–466. [Google Scholar] [CrossRef]
- Willingham, S.B.; Volkmer, J.-P.; Gentles, A.J.; Sahoo, D.; Dalerba, P.; Mitra, S.S.; Wang, J.; Contreras-Trujillo, H.; Martin, R.; Cohen, J.D.; et al. The CD47-signal regulatory protein alpha (SIRPa) interaction is a therapeutic target for human solid tumors. Proc. Natl. Acad. Sci. USA 2012, 109, 6662–6667. [Google Scholar] [CrossRef] [Green Version]
- Zhu, H.; Leiss, L.; Yang, N.; Rygh, C.B.; Mitra, S.S.; Cheshier, S.H.; Weissman, I.L.; Huang, B.; Miletic, H.; Bjerkvig, R.; et al. Surgical debulking promotes recruitment of macrophages and triggers glioblastoma phagocytosis in combination with CD47 blocking immunotherapy. Oncotarget 2017, 8, 12145–12157. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Hutter, G.; Kahn, S.A.; Azad, T.D.; Gholamin, S.; Xu, C.Y.; Liu, J.; Achrol, A.S.; Richard, C.; Sommerkamp, P.; et al. Anti-CD47 Treatment Stimulates Phagocytosis of Glioblastoma by M1 and M2 Polarized Macrophages and Promotes M1 Polarized Macrophages In Vivo. PLoS ONE 2016, 11, e0153550. [Google Scholar] [CrossRef]
- Ma, D.; Liu, S.; Lal, B.; Wei, S.; Wang, S.; Zhan, D.; Zhang, H.; Lee, R.S.; Gao, P.; Lopez-Bertoni, H.; et al. Extracellular Matrix Protein Tenascin C Increases Phagocytosis Mediated by CD47 Loss of Function in Glioblastoma. Cancer Res. 2019, 79, 2697–2708. [Google Scholar] [CrossRef] [Green Version]
- Hutter, G.; Theruvath, J.; Graef, C.M.; Zhang, M.; Schoen, M.K.; Manz, E.M.; Bennett, M.L.; Olson, A.; Azad, T.D.; Sinha, R.; et al. Microglia are effector cells of CD47-SIRPα antiphagocytic axis disruption against glioblastoma. Proc. Natl. Acad. Sci. USA 2019, 116, 997–1006. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Chen, W.; Fan, J.; Wang, S.; Xian, Z.; Luan, J.; Li, Y.; Wang, Y.; Nan, Y.; Luo, M.; et al. Disrupting CD47-SIRPα axis alone or combined with autophagy depletion for the therapy of glioblastoma. Carcinogenesis 2018, 39, 689–699. [Google Scholar] [CrossRef] [Green Version]
- Hsu, S.P.C.; Chen, Y.C.; Chiang, H.C.; Huang, Y.C.; Huang, C.C.; Wang, H.E.; Wang, Y.S.; Chi, K.H. Rapamycin and hydroxychloroquine combination alters macrophage polarization and sensitizes glioblastoma to immune checkpoint inhibitors. J. Neurooncol. 2020, 146, 417–426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, Y.; Mu, R.; Wang, Z.; Xing, P.; Zhang, J.; Dong, L.; Wang, C. A toll-like receptor agonist mimicking microbial signal to generate tumor-suppressive macrophages. Nat. Commun. 2019, 10, 2272. [Google Scholar] [CrossRef] [Green Version]
- Werner, J.-M.; Kuhl, S.; Ulrich, K.; Krischek, B.; Stavrinou, P.; Goldbrunner, R.; Timmer, M. Expression of CD40 Correlates Negatively with Overall and Progression-Free Survival of Low- and High-Grade Gliomas. World Neurosurg. 2019, 130, e17–e25. [Google Scholar] [CrossRef] [PubMed]
- Hoves, S.; Ooi, C.-H.; Wolter, C.; Sade, H.; Bissinger, S.; Schmittnaegel, M.; Ast, O.; Giusti, A.M.; Wartha, K.; Runza, V.; et al. Rapid activation of tumor-associated macrophages boosts preexisting tumor immunity. J. Exp. Med. 2018, 215, 859–876. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, T.; Natoli, G. Transcriptional regulation of macrophage polarization: Enabling diversity with identity. Nat. Rev. Immunol. 2011, 11, 750–761. [Google Scholar] [CrossRef]
- Saccani, A.; Schioppa, T.; Porta, C.; Biswas, S.K.; Nebuloni, M.; Vago, L.; Bottazzi, B.; Colombo, M.P.; Mantovani, A.; Sica, A. p50 nuclear factor-κB overexpression in tumor-associated macrophages inhibits M1 inflammatory responses and antitumor resistance. Cancer Res. 2006, 66, 11432–11440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porta, C.; Rimoldi, M.; Raes, G.; Brys, L.; Ghezzi, P.; Di Liberto, D.; Dieli, F.; Ghisletti, S.; Natoli, G.; De Baetselier, P.; et al. Tolerance and M2 (alternative) macrophage polarization are related processes orchestrated by p50 nuclear factor κB. Proc. Natl. Acad. Sci. USA 2009, 106, 14978–14983. [Google Scholar] [CrossRef] [Green Version]
- Cho, H.R.; Jeon, H.; Park, C.K.; Park, S.H.; Choi, S.H. Radiogenomics Profiling for Glioblastoma-related Immune Cells Reveals CD49d Expression Correlation with MRI parameters and Prognosis. Sci. Rep. 2018, 8, 16022. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Ramesh, A.; Kulkarni, A. Targeting macrophages: A novel avenue for cancer drug discovery. Expert Opin. Drug Discov. 2020, 15, 561–574. [Google Scholar] [CrossRef] [PubMed]
- Yan, D.; Kowal, J.; Akkari, L.; Schuhmacher, A.J.; Huse, J.T.; West, B.L.; Joyce, J.A. Inhibition of colony stimulating factor-1 receptor abrogates microenvironment-mediated therapeutic resistance in gliomas. Oncogene 2017, 36, 6049–6058. [Google Scholar] [CrossRef] [PubMed]
- Gordon, S.R.; Maute, R.L.; Dulken, B.W.; Hutter, G.; George, B.M.; McCracken, M.N.; Gupta, R.; Tsai, J.M.; Sinha, R.; Corey, D.; et al. PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature 2017, 545, 495–499. [Google Scholar] [CrossRef] [PubMed]
- Rao, G.; Latha, K.; Ott, M.; Sabbagh, A.; Marisetty, A.; Ling, X.; Zamler, D.; Doucette, T.A.; Yang, Y.; Kong, L.Y.; et al. Anti-PD-1 induces M1 polarization in the glioma microenvironment and exerts therapeutic efficacy in the absence of CD8 cytotoxic T cells. Clin. Cancer Res. 2020, 26, 4699–4712. [Google Scholar] [CrossRef]
- Moesta, A.K.; Li, X.Y.; Smyth, M.J. Targeting CD39 in cancer. Nat. Rev. Immunol. 2020, 20, 739–755. [Google Scholar] [CrossRef]
- Cosset, É.; Weis, S.M.; Cheresh, D.A. Re-thinking the preclinical development of GBM therapeutics. Oncoscience 2018, 5, 11–12. [Google Scholar] [CrossRef] [PubMed]
- Tritz, Z.P.; Ayasoufi, K.; Johnson, A.J. Anti-PD-1 checkpoint blockade monotherapy in the orthotopic GL261 glioma model: The devil is in the detail. Neuro-Oncol. Adv. 2021, 3, vdab066. [Google Scholar] [CrossRef] [PubMed]
- De La Rochere, P.; Guil-Luna, S.; Decaudin, D.; Azar, G.; Sidhu, S.S.; Piaggio, E. Humanized Mice for the Study of Immuno-Oncology. Trends Immunol. 2018, 39, 748–763. [Google Scholar] [CrossRef]
- Allen, T.M.; Brehm, M.A.; Bridges, S.; Ferguson, S.; Kumar, P.; Mirochnitchenko, O.; Palucka, K.; Pelanda, R.; Sanders-Beer, B.; Shultz, L.D.; et al. Humanized immune system mouse models: Progress, challenges and opportunities. Nat. Immunol. 2019, 20, 770–774. [Google Scholar] [CrossRef]
- Sarkaria, J.N.; Hu, L.S.; Parney, I.F.; Pafundi, D.H.; Brinkmann, D.H.; Laack, N.N.; Giannini, C.; Burns, T.C.; Kizilbash, S.H.; Laramy, J.K.; et al. Is the blood-brain barrier really disrupted in all glioblastomas? A critical assessment of existing clinical data. Neuro-Oncology 2018, 20, 184–191. [Google Scholar] [CrossRef]
- Wang, L.B.; Karpova, A.; Gritsenko, M.A.; Kyle, J.E.; Cao, S.; Li, Y.; Rykunov, D.; Colaprico, A.; Rothstein, J.H.; Hong, R.; et al. Proteogenomic and metabolomic characterization of human glioblastoma. Cancer Cell 2021, 39, 509–528.e20. [Google Scholar] [CrossRef] [PubMed]
- Stuart, T.; Butler, A.; Hoffman, P.; Hafemeister, C.; Papalexi, E.; Mauck, W.M.; Hao, Y.; Stoeckius, M.; Smibert, P.; Satija, R. Comprehensive Integration of Single-Cell Data. Cell 2019, 177, 1888–1902.e21. [Google Scholar] [CrossRef] [PubMed]
- Yasen, A.; Aini, A.; Wang, H.; Li, W.; Zhang, C.; Ran, B.; Tuxun, T.; Maimaitinijiati, Y.; Shao, Y.; Aji, T.; et al. Progress and applications of single-cell sequencing techniques. Infect. Genet. Evol. 2020, 80, 104198. [Google Scholar] [CrossRef] [PubMed]
- Neftel, C.; Laffy, J.; Filbin, M.G.; Hara, T.; Shore, M.E.; Rahme, G.J.; Richman, A.R.; Silverbush, D.; Shaw, M.L.; Hebert, C.M.; et al. An Integrative Model of Cellular States, Plasticity, and Genetics for Glioblastoma. Cell 2019, 178, 835–849.e21. [Google Scholar] [CrossRef] [PubMed]
- Yuan, D.; Tao, Y.; Chen, G.; Shi, T. Systematic expression analysis of ligand-receptor pairs reveals important cell-to-cell interactions inside glioma. Cell Commun. Signal. 2019, 17, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalita-de Croft, P.; Sadeghi Rad, H.; Gasper, H.; O’Byrne, K.; Lakhani, S.R.; Kulasinghe, A. Spatial profiling technologies and applications for brain cancers. Expert Rev. Mol. Diagn. 2021, 21, 323–332. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Targeting Strategy | Target | Clinical/Experimental | Result | Reference |
|---|---|---|---|---|
| TAM recruitment | CCL2-CCR2 axis | Clinical phase 1/2 trials with anti-CCL2 mAb or CCR2 small molecule inhibitor in different solid tumors | Carlumab, anti-CCL2 mAb: well-tolerated, but no durable anti-tumor effect PF-04136309, CCR2 small molecule inhibitor: well tolerated, some anti-tumor effect | [132,133] [134] |
| CXCL12-CXCR4 axis | Clinical phase 1/2 trials with CXCR4 antagonist in glioblastoma and high grade gliomas | Plerixafor, CXCR4 antagonist: - Glioblastoma patients treated with concurrent chemoradiation. Treatment well-tolerated and improved median survival (21.3 months) - Follow-up trial of NCT01977677 with whole brain radiation. Results are pending - High grade glioma (mostly glioblastoma) patients treated with Plerixafor in combination with Bevacizumab. Treatment well-tolerated, but no effect on survival | NCT01977677 [135] NCT03746080 [136] | |
| Reprogramming TAM polarization and increasing anti-tumor TAMs | SIRPα-CD47 pathway | Clinical phase 1 trial with anti-CD47 mAb in solid tumors | Hu5F9-G4 mAb: well tolerated, but final results are pending | [137] |
| TLRs | Clinical phase 1/2 trials with TLR agonist small molecule inhibitors in solid cancers and lymphomas | Several TLR agonists are tested: there is limited effects in general, except the exception below Imiquimod: well-tolerated, and partial responses observed in skin/chest wall metastases in breast cancer patients In glioblastoma, TLR agonists are tested as immune adjuvants for immunotherapy: results are pending | [138] [139] NCT03392545 NCT01204684 | |
| CD40 | Clinical phase 1 trials in pediatric CNS tumors, solid tumors and gliomas with anti-CD40 antibodies | Anti-CD40 mAb in pediatric CNS tumors: results are pending Anti-CD40 mAb in combination with anti-PD-L1 and D2C7-IT in recurrent glioma: results are pending Anti-CD40 mAb in combination with CSF1R inhibitor: results are pending | NCT03389802 NCT04547777 NCT02760797 | |
| NF-kB | Experimental mouse models of glioblastoma, prostate and pancreatic cancer | NF-kB inhibition in TAMs leads to M1 polarization | [140,141,142] | |
| Decreasing tumor-promoting TAMs | CSF1R | Numerous clinical trials in solid and hematological tumors testing small molecule inhibitors and monoclonal antibodies Experimental mouse models of glioblastoma | PLX3397 small molecule inhibitor: - FDA approval of for tynosynovial giant cell tumor - Phase 2 trial: well tolerated, but no anti-tumor effect in glioblastoma - Phase 1/2 trial in glioblastoma, in combination with radio- and chemotherapy: results are pending BLZ945 small molecule inhibitor: - Phase 1/2 trial in advanced solid tumors in combination with anti-PD-1: results are pending - M2 polarization in glioblastoma mouse models - Improves the effect of radiation therapy by delaying or preventing recurrence | [143] [144] NCT01790503 NCT02829723 [145] [129, 146] |
| PD-1/PD-L1 | Clinical trial with anti-PD-1 in recurrent glioblastoma Clinical phase 3 trial with anti-PD-1 in recurrent glioblastoma Clinical phase 3 trials with anti-PD-1 in newly diagnosed glioblastoma | Improved survival with neoadjuvant pembrolizumab treatment compared to adjuvant Comparison of Nivolumab with Bevacizumab. No difference in overall survival, but more durable responses with PD-1 blockade Nivolumab compared to TMZ/TMZ+ radiation in patients with unmethylated/methylated MGMT promoter. Results for all patients show no effect on overall survival. However, MGMT promoter methylated patients had increased survival | [147] [148] NCT02617589 NCT02667587 [149] | |
| CD73 | Experimental glioblastoma model with CD73-/- mice Clinical phase 1 and 2 trials in solid tumors | Significant improved survival of CD73-/- mice after anti-PD-1 and anti-CTLA-4 treatment compared to untreated mice as well as treated wildtype mice CD73 inhibition is tested alone or in combination with immune checkpoint inhibitors: results are pending | [150] [151] | |
| CD39 | Experimental glioma mouse model and TCGA glioblastoma patient data Clinical phase 1 trial in advanced solid tumors | CD39 expression in TAMs associates with glioma grade and contributes to T cell dysfunction Anti-CD39 antibody (SRF617): results are pending | [152] NCT04336098 | |
| CD163 | Experimental melanoma mouse model Experimental in vitro set-up with human glioma-derived cells | Depletion of CD163+ (M2-like) TAM population leads to tumor regression Human-derived CD163+ TAMs inhibit effector T cells | [153] [66] | |
| CD204 | TCGA, CGGA, and glioma patient cohorts | CD204 expression is associated with survival and correlates with malignancy grade in gliomas | [7,154] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Andersen, R.S.; Anand, A.; Harwood, D.S.L.; Kristensen, B.W. Tumor-Associated Microglia and Macrophages in the Glioblastoma Microenvironment and Their Implications for Therapy. Cancers 2021, 13, 4255. https://doi.org/10.3390/cancers13174255
Andersen RS, Anand A, Harwood DSL, Kristensen BW. Tumor-Associated Microglia and Macrophages in the Glioblastoma Microenvironment and Their Implications for Therapy. Cancers. 2021; 13(17):4255. https://doi.org/10.3390/cancers13174255
Chicago/Turabian StyleAndersen, Rikke Sick, Atul Anand, Dylan Scott Lykke Harwood, and Bjarne Winther Kristensen. 2021. "Tumor-Associated Microglia and Macrophages in the Glioblastoma Microenvironment and Their Implications for Therapy" Cancers 13, no. 17: 4255. https://doi.org/10.3390/cancers13174255
APA StyleAndersen, R. S., Anand, A., Harwood, D. S. L., & Kristensen, B. W. (2021). Tumor-Associated Microglia and Macrophages in the Glioblastoma Microenvironment and Their Implications for Therapy. Cancers, 13(17), 4255. https://doi.org/10.3390/cancers13174255