
Direct Endoplasmic Reticulum Targeting by the Selective Alkylphospholipid Analog and Antitumor Ether Lipid Edelfosine as a Therapeutic Approach in Pancreatic Cancer


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. The Pancreas
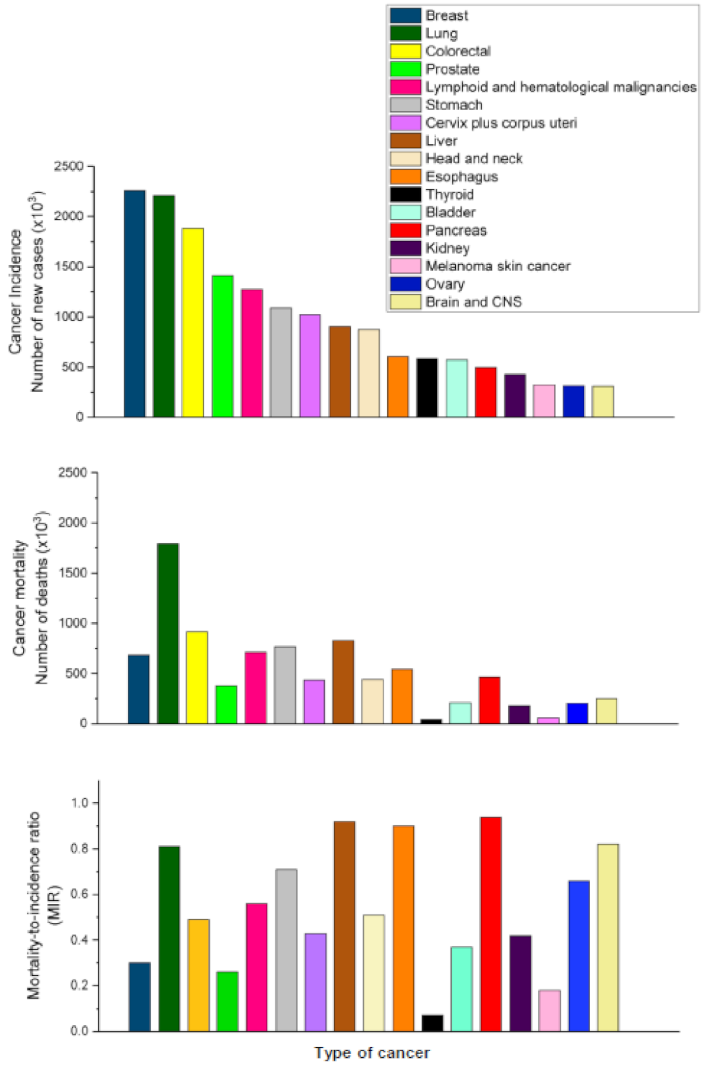
2. Pancreatic Cancer
3. Differences in Mutated Genes between PDAC and PanNET
4. Pancreatic Cancer Stages
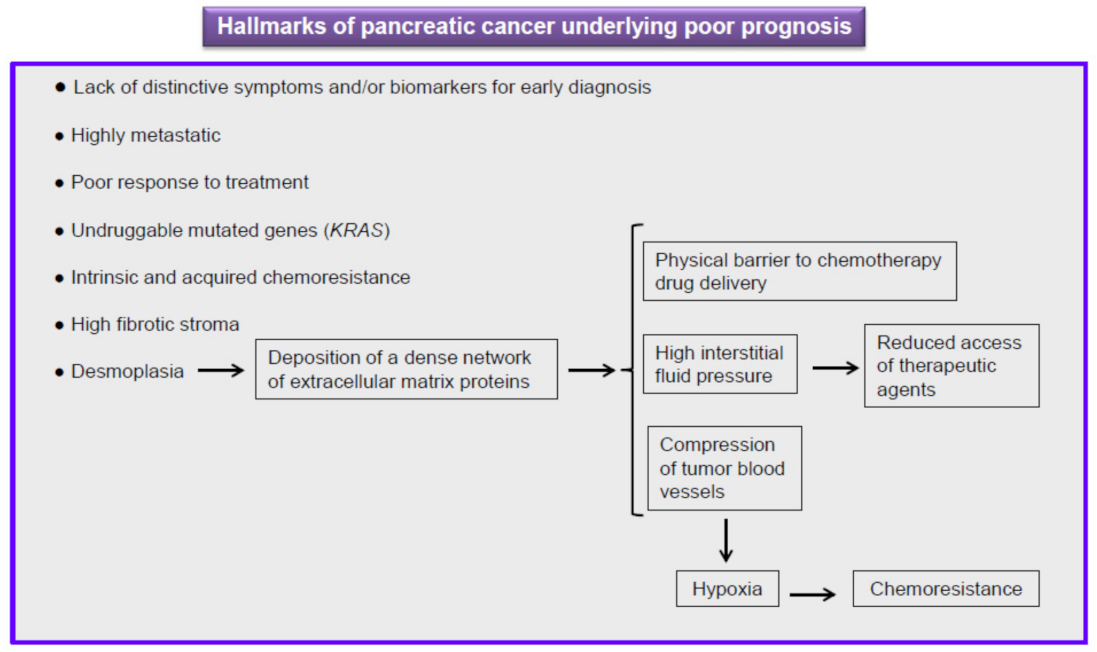
5. Current Therapy against Pancreatic Cancer Is Ineffective
6. Aging and Pancreas
7. Endoplasmic Reticulum and Pancreas
8. ER Stress and Cancer Resistance
9. ER Stress and Antitumor Drugs
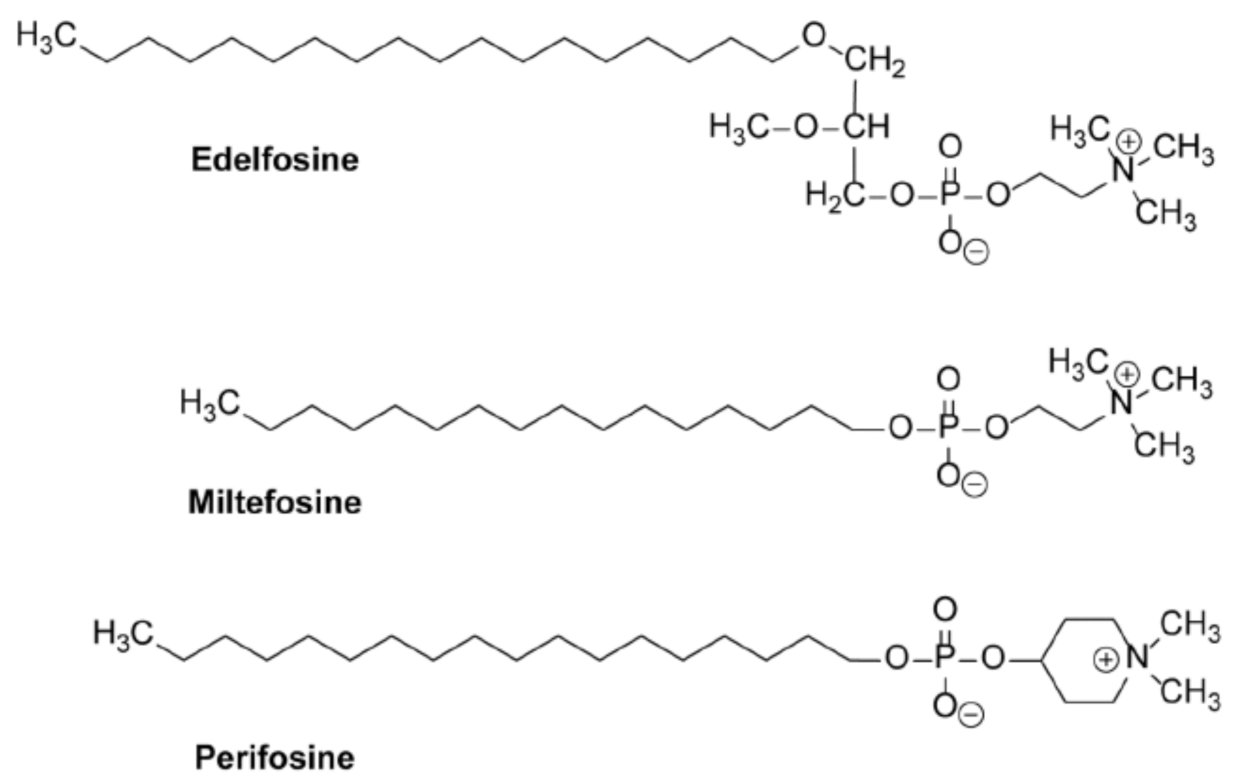
10. Alkylphospholipid Analogs as Selective Antitumor Drugs against Cancer Cells by Targeting Subcellular Structures
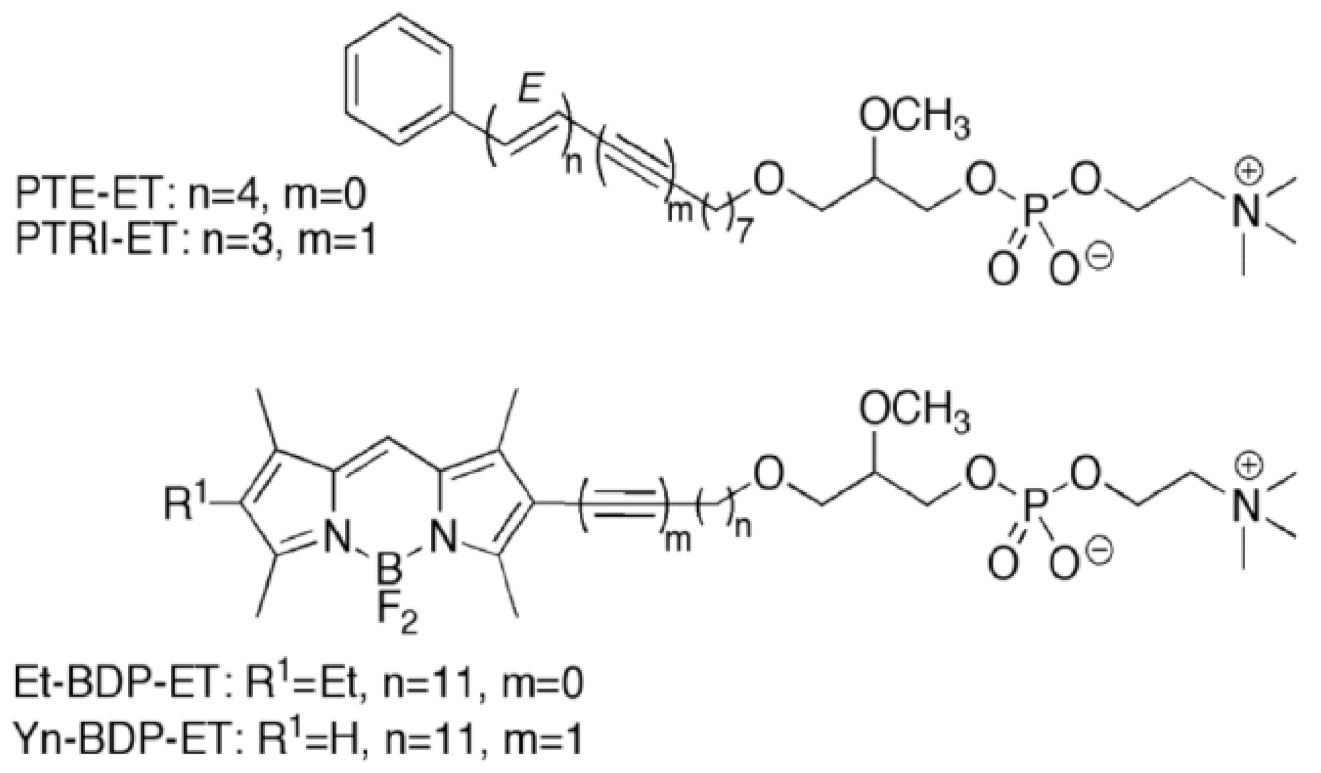
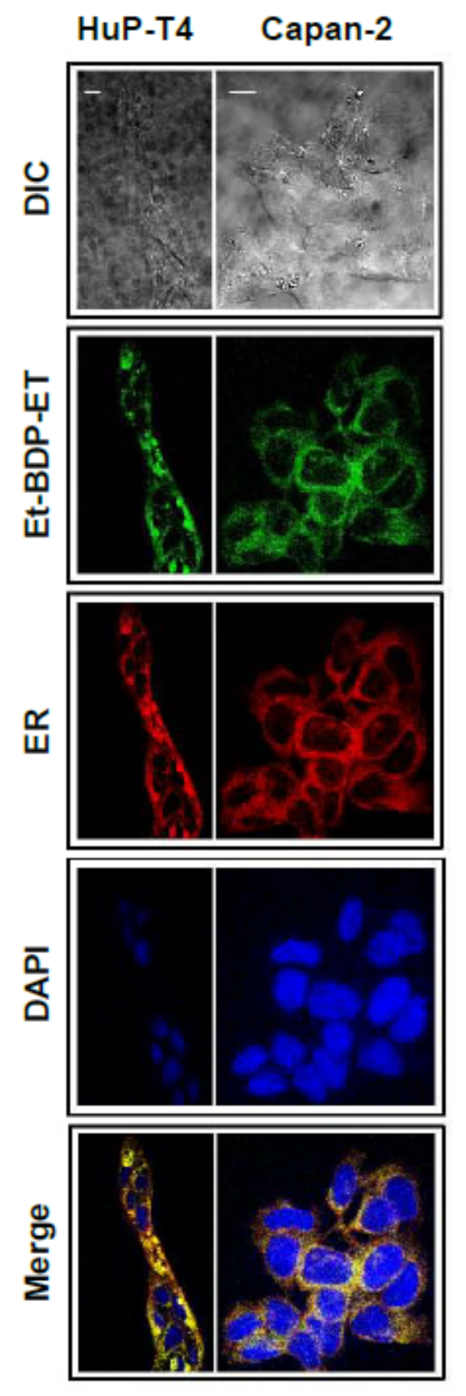
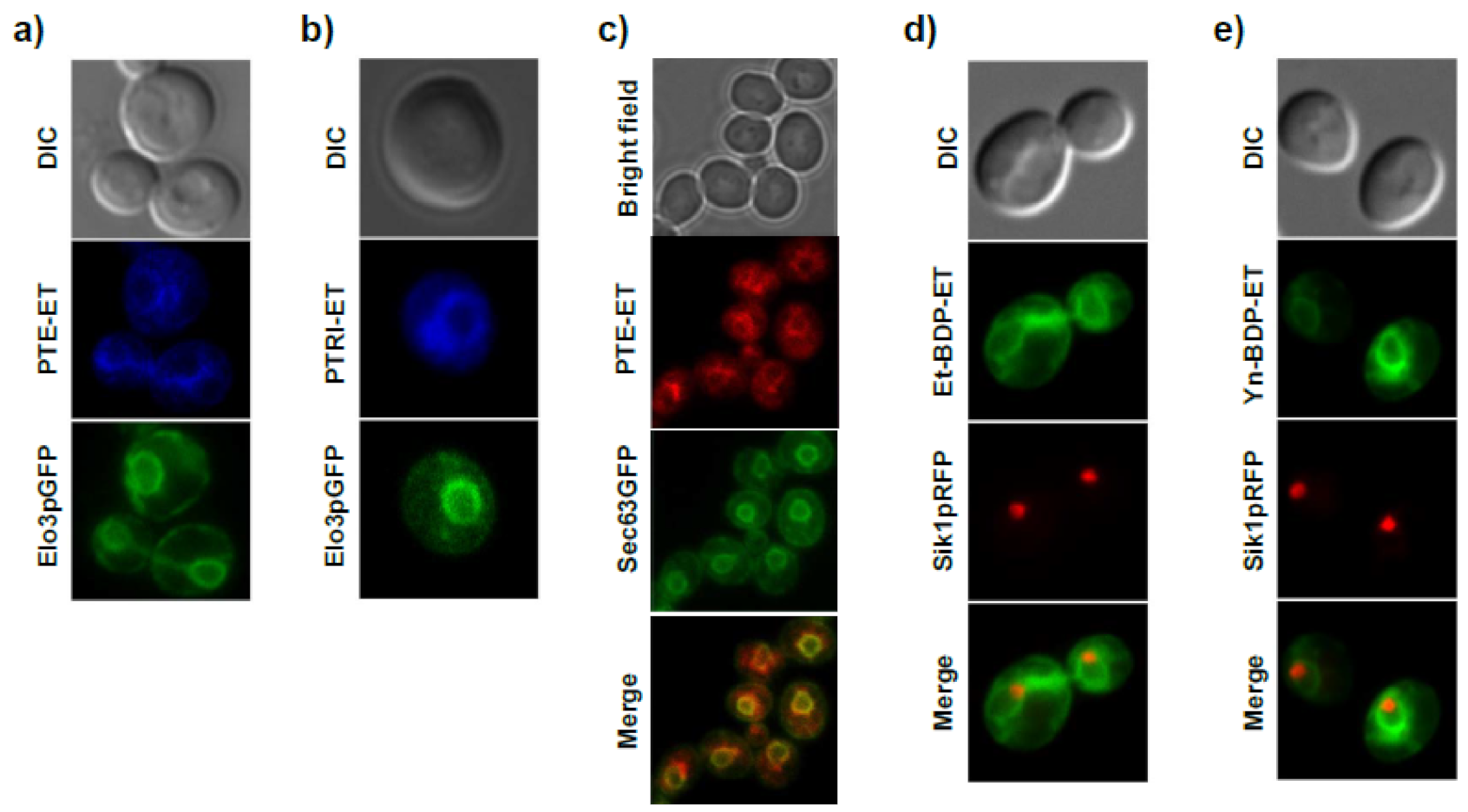
11. Accumulation of the Ether Lipid Edelfosine in the ER of Pancreatic Cancer Cells
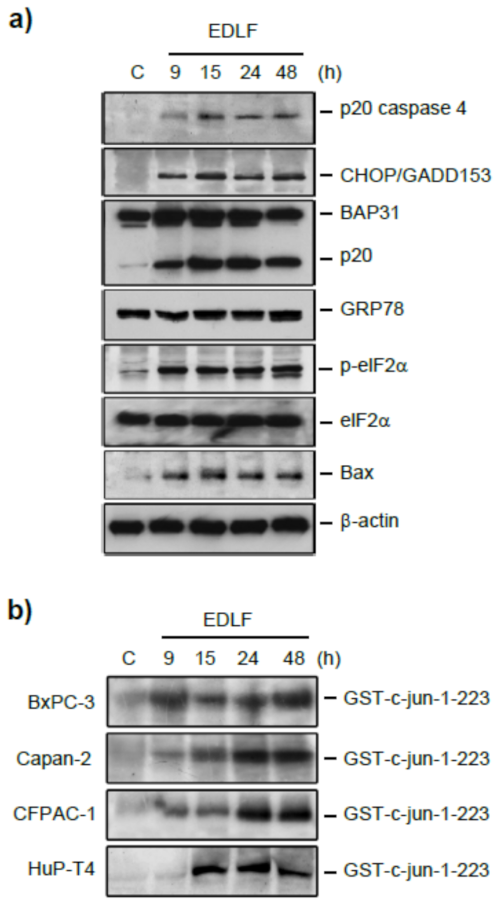
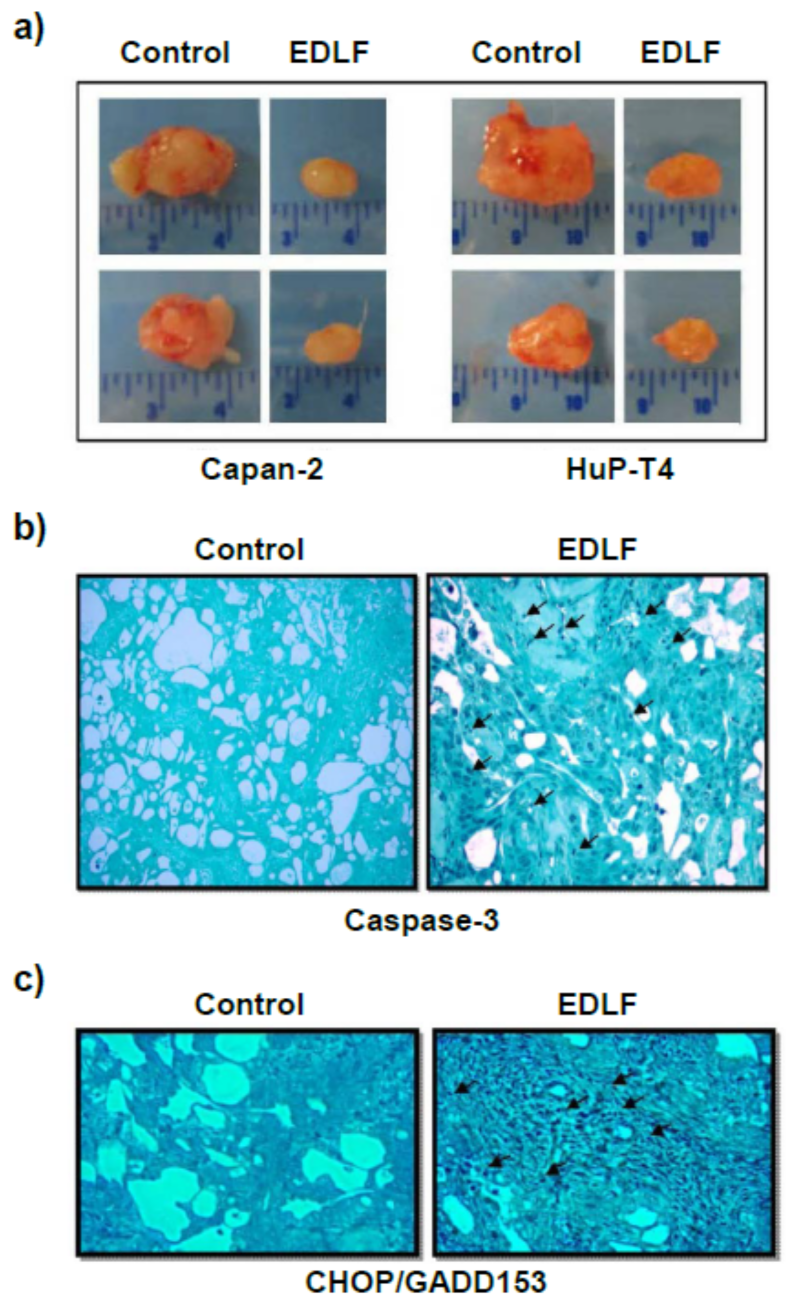
12. Induction of ER Stress-Mediated Apoptosis following Treatment with the Ether Lipid Edelfosine in Pancreatic Cancer Cells
13. ER Stress and Mitochondrial Connection in the Induction of Apoptosis in Pancreatic Cancer Cells by the Ether Lipid Edelfosine
14. Novel Approaches for the Potentiation of ER-Mediated Apoptosis in Pancreatic Cancer Cells Induced by Edelfosine
15. Outlook
16. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AELs | Antitumor ether lipids |
| ALPs | Alkyl-lysophospholipid analogs |
| APLs | Alkylphospholipid analogs |
| ATLs | Antitumor lipids |
| ATF6 | Activating transcription factor 6 |
| BAP31 | B-cell receptor–associated protein 31 |
| BiP | Binding immunoglobulin protein |
| BODIPY | 4,4-Difluoro-4-bora-3a,4a-diaza-s-indacene; boron-dipyrromethene |
| CHOP | CCAAT/enhancer-binding protein homologous protein |
| ER | Endoplasmic reticulum |
| ERK | Extracellular signal-regulated kinase |
| GADD153 | Growth-arrest and DNA-damage-inducible gene 153 |
| IRE1 | Inositol-requiring enzyme 1 (IRE1) |
| FOLFIRINOX | Folinic acid, 5-fluorouracil, irinotecan, and oxaliplatin |
| GRP78 | 78-kDa glucose-regulated protein |
| HDI | Human Development Index |
| JNK | c-Jun NH2-terminal kinase |
| MAMs | Mitochondria-associated membranes |
| MAPK | Mitogen-activated protein kinase |
| MEK | MAPK/ERK kinase |
| MIR | Mortality-to-incidence ratio |
| MM | Multiple myeloma |
| mTOR | Mammalian target of rapamycin |
| Nab-paclitaxel | Nanoparticle albumin-bound paclitaxel (Abrazane) |
| PanNET | Pancreatic neuroendocrine tumor |
| PDAC | Pancreatic ductal adenocarcinoma |
| PERK | Protein kinase RNA (PKR)-like ER kinase |
| PI3K | Phosphoinositide-3-kinase |
| RAF | Rapidly accelerated fibrosarcoma |
| RFP | Red fluorescent protein |
| SCID | Severe combined immunodeficiency |
| UPR | Unfolded protein response |
References
- Talathi, S.S.; Zimmerman, R.; Young, M. Anatomy, Abdomen and Pelvis, Pancreas. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2021. Available online: https://www.ncbi.nlm.nih.gov/books/NBK532912/ (accessed on 15 June 2021).
- Sohn, T.A.; Yeo, C.J.; Cameron, J.L.; Koniaris, L.; Kaushal, S.; Abrams, R.A.; Sauter, P.K.; Coleman, J.; Hruban, R.H.; Lillemoe, K.D. Resected adenocarcinoma of the pancreas-616 patients: Results, outcomes, and prognostic indicators. J. Gastrointest. Surg. 2000, 4, 567–579. [Google Scholar] [CrossRef]
- Artinyan, A.; Soriano, P.A.; Prendergast, C.; Low, T.; Ellenhorn, J.D.; Kim, J. The anatomic location of pancreatic cancer is a prognostic factor for survival. HPB (Oxford) 2008, 10, 371–376. [Google Scholar] [CrossRef] [PubMed]
- Das, S.L.; Kennedy, J.I.; Murphy, R.; Phillips, A.R.; Windsor, J.A.; Petrov, M.S. Relationship between the exocrine and endocrine pancreas after acute pancreatitis. World J. Gastroenterol. 2014, 20, 17196–17205. [Google Scholar] [CrossRef]
- Weiss, F.U.; Halangk, W.; Lerch, M.M. New advances in pancreatic cell physiology and pathophysiology. Best Pract. Res. Clin. Gastroenterol. 2008, 22, 3–15. [Google Scholar] [CrossRef]
- Reichert, M.; Rustgi, A.K. Pancreatic ductal cells in development, regeneration, and neoplasia. J. Clin. Investig. 2011, 121, 4572–4578. [Google Scholar] [CrossRef] [PubMed]
- Pelosi, E.; Castelli, G.; Testa, U. Pancreatic Cancer: Molecular Characterization, Clonal Evolution and Cancer Stem Cells. Biomedicines 2017, 5, 65. [Google Scholar] [CrossRef]
- Storz, P. Acinar cell plasticity and development of pancreatic ductal adenocarcinoma. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 296–304. [Google Scholar] [CrossRef]
- Kelgiorgi, D.; Dervenis, C. Pancreatic neuroendocrine tumors: The basics, the gray zone, and the target. F1000Res 2017, 6, 663. [Google Scholar] [CrossRef] [PubMed]
- Klimstra, D.S.; Modlin, I.R.; Coppola, D.; Lloyd, R.V.; Suster, S. The pathologic classification of neuroendocrine tumors: A review of nomenclature, grading, and staging systems. Pancreas 2010, 39, 707–712. [Google Scholar] [CrossRef] [PubMed]
- Burns, W.R.; Edil, B.H. Neuroendocrine pancreatic tumors: Guidelines for management and update. Curr. Treat. Options Oncol. 2012, 13, 24–34. [Google Scholar] [CrossRef]
- Khanna, L.; Prasad, S.R.; Sunnapwar, A.; Kondapaneni, S.; Dasyam, A.; Tammisetti, V.S.; Salman, U.; Nazarullah, A.; Katabathina, V.S. Pancreatic Neuroendocrine Neoplasms: 2020 Update on Pathologic and Imaging Findings and Classification. Radiographics 2020, 40, 1240–1262. [Google Scholar] [CrossRef]
- Brooks, J.C.; Shavelle, R.M.; Vavra-Musser, K.N. Life expectancy in pancreatic neuroendocrine cancer. Clin. Res. Hepatol. Gastroenterol. 2019, 43, 88–97. [Google Scholar] [CrossRef]
- Ahrendt, S.A.; Pitt, H.A. Surgical management of pancreatic cancer. Oncology (Williston Park) 2002, 16, 725–734. [Google Scholar] [PubMed]
- Vincent, A.; Herman, J.; Schulick, R.; Hruban, R.H.; Goggins, M. Pancreatic cancer. Lancet 2011, 378, 607–620. [Google Scholar] [CrossRef]
- Bond-Smith, G.; Banga, N.; Hammond, T.M.; Imber, C.J. Pancreatic adenocarcinoma. BMJ 2012, 344, e2476. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer Statistics. CA Cancer J. Clin. 2017, 67, 7–30. [Google Scholar] [CrossRef]
- McGuigan, A.; Kelly, P.; Turkington, R.C.; Jones, C.; Coleman, H.G.; McCain, R.S. Pancreatic cancer: A review of clinical diagnosis, epidemiology, treatment and outcomes. World J. Gastroenterol. 2018, 24, 4846–4861. [Google Scholar] [CrossRef] [PubMed]
- Rawla, P.; Sunkara, T.; Gaduputi, V. Epidemiology of Pancreatic Cancer: Global Trends, Etiology and Risk Factors. World J. Oncol. 2019, 10, 10–27. [Google Scholar] [CrossRef]
- International Agency for Research on Cancer, WHO. Very high HDI. The Global Cancer Observatory. Globocan 2020. WHO. 2021. Available online: https://gco.iarc.fr/today/data/factsheets/populations/981-very-high-hdi-fact-sheets.pdf (accessed on 15 June 2021).
- Fidler, M.M.; Soerjomataram, I.; Bray, F. A global view on cancer incidence and national levels of the human development index. Int. J. Cancer 2016, 139, 2436–2446. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- International Agency for Research on Cancer, WHO. Low HDI. The Global Cancer Observatory. Globocan 2020. WHO. 2021. Available online: https://gco.iarc.fr/today/data/factsheets/populations/984-low-hdi-fact-sheets.pdf (accessed on 15 June 2021).
- Goodarzi, E.; Dehkordi, A.H.; Beiranvand, R.; Naemi, H.; Khazaei, Z. Epidemiology of the Incidence and Mortality of Pancreas Cancer and its Relationship with the Human Development Index (HDI) in the World: An Ecological Study in 2018. Curr. Pharm. Des. 2020, 26, 5163–5173. [Google Scholar] [CrossRef]
- Veisani, Y.; Jenabi, E.; Khazaei, S.; Nematollahi, S. Global incidence and mortality rates in pancreatic cancer and the association with the Human Development Index: Decomposition approach. Public Health 2018, 156, 87–91. [Google Scholar] [CrossRef] [PubMed]
- Wong, M.C.S.; Jiang, J.Y.; Liang, M.; Fang, Y.; Yeung, M.S.; Sung, J.J.Y. Global temporal patterns of pancreatic cancer and association with socioeconomic development. Sci. Rep. 2017, 7, 3165. [Google Scholar] [CrossRef] [PubMed]
- Ataey, A.; Jafarvand, E.; Adham, D.; Moradi-Asl, E. The Relationship Between Obesity, Overweight, and the Human Development Index in World Health Organization Eastern Mediterranean Region Countries. J. Prev. Med. Public Health 2020, 53, 98–105. [Google Scholar] [CrossRef]
- Arnold, M.; Pandeya, N.; Byrnes, G.; Renehan, P.A.G.; Stevens, G.A.; Ezzati, P.M.; Ferlay, J.; Miranda, J.J.; Romieu, I.; Dikshit, R.; et al. Global burden of cancer attributable to high body-mass index in 2012: A population-based study. Lancet Oncol. 2015, 16, 36–46. [Google Scholar] [CrossRef]
- Paternoster, S.; Falasca, M. The intricate relationship between diabetes, obesity and pancreatic cancer. Biochim. Biophys. Acta. Rev. Cancer 2020, 1873, 188326. [Google Scholar] [CrossRef] [PubMed]
- International Agency for Research on Cancer, WHO. World. The Global Cancer Observatory. Globocan 2020. WHO. 2021. Available online: https://gco.iarc.fr/today/data/factsheets/populations/900-world-fact-sheets.pdf (accessed on 15 June 2021).
- Ferlay, J.; Partensky, C.; Bray, F. More deaths from pancreatic cancer than breast cancer in the EU by 2017. Acta Oncol. 2016, 55, 1158–1160. [Google Scholar] [CrossRef] [PubMed]
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting cancer incidence and deaths to 2030: The unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014, 74, 2913–2921. [Google Scholar] [CrossRef]
- Lennerz, J.K.; Stenzinger, A. Allelic ratio of KRAS mutations in pancreatic cancer. Oncologist 2015, 20, e8–e9. [Google Scholar] [CrossRef]
- Kleeff, J.; Korc, M.; Apte, M.; La Vecchia, C.; Johnson, C.D.; Biankin, A.V.; Neale, R.E.; Tempero, M.; Tuveson, D.A.; Hruban, R.H.; et al. Pancreatic cancer. Nat. Rev. Dis. Primers 2016, 2, 16022. [Google Scholar] [CrossRef]
- Jiao, Y.; Shi, C.; Edil, B.H.; de Wilde, R.F.; Klimstra, D.S.; Maitra, A.; Schulick, R.D.; Tang, L.H.; Wolfgang, C.L.; Choti, M.A.; et al. DAXX/ATRX, MEN1, and mTOR pathway genes are frequently altered in pancreatic neuroendocrine tumors. Science 2011, 331, 1199–1203. [Google Scholar] [CrossRef]
- Chan, C.S.; Laddha, S.V.; Lewis, P.W.; Koletsky, M.S.; Robzyk, K.; Da Silva, E.; Torres, P.J.; Untch, B.R.; Li, J.; Bose, P.; et al. ATRX, DAXX or MEN1 mutant pancreatic neuroendocrine tumors are a distinct alpha-cell signature subgroup. Nat. Commun. 2018, 9, 4158. [Google Scholar] [CrossRef] [PubMed]
- Lopez, N.E.; Prendergast, C.; Lowy, A.M. Borderline resectable pancreatic cancer: Definitions and management. World J. Gastroenterol. 2014, 20, 10740–10751. [Google Scholar] [CrossRef] [PubMed]
- Tamburrino, D.; Partelli, S.; Crippa, S.; Manzoni, A.; Maurizi, A.; Falconi, M. Selection criteria in resectable pancreatic cancer: A biological and morphological approach. World J. Gastroenterol. 2014, 20, 11210–11215. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.H.; Jang, J.Y.; Kang, J.S.; Kim, J.R.; Han, Y.; Kim, E.; Kwon, W.; Kim, S.W. Recent treatment patterns and survival outcomes in pancreatic cancer according to clinical stage based on single-center large-cohort data. Ann. Hepatobiliary Pancreat. Surg. 2018, 22, 386–396. [Google Scholar] [CrossRef]
- Barugola, G.; Partelli, S.; Marcucci, S.; Sartori, N.; Capelli, P.; Bassi, C.; Pederzoli, P.; Falconi, M. Resectable pancreatic cancer: Who really benefits from resection? Ann. Surg. Oncol. 2009, 16, 3316–3322. [Google Scholar] [CrossRef] [PubMed]
- Evans, D.B. What Makes a Pancreatic Cancer Resectable? Am. Soc. Clin. Oncol. Educ. Book 2018, 38, 300–305. [Google Scholar] [CrossRef] [PubMed]
- Lambert, A.; Schwarz, L.; Borbath, I.; Henry, A.; Van Laethem, J.L.; Malka, D.; Ducreux, M.; Conroy, T. An update on treatment options for pancreatic adenocarcinoma. Ther. Adv. Med. Oncol. 2019, 11, 1758835919875568. [Google Scholar] [CrossRef]
- Christians, K.K.; Heimler, J.W.; George, B.; Ritch, P.S.; Erickson, B.A.; Johnston, F.; Tolat, P.P.; Foley, W.D.; Evans, D.B.; Tsai, S. Survival of patients with resectable pancreatic cancer who received neoadjuvant therapy. Surgery 2016, 159, 893–900. [Google Scholar] [CrossRef] [PubMed]
- Cucchetti, A.; Ercolani, G.; Taffurelli, G.; Serenari, M.; Maroni, L.; Pezzilli, R.; Del Gaudio, M.; Ravaioli, M.; Cescon, M.; Pinna, A.D. A comprehensive analysis on expected years of life lost due to pancreatic cancer. Pancreatology 2016, 16, 449–453. [Google Scholar] [CrossRef]
- Huang, L.; Jansen, L.; Balavarca, Y.; Babaei, M.; van der Geest, L.; Lemmens, V.; Van Eycken, L.; De Schutter, H.; Johannesen, T.B.; Primic-Zakelj, M.; et al. Stratified survival of resected and overall pancreatic cancer patients in Europe and the USA in the early twenty-first century: A large, international population-based study. BMC Med. 2018, 16, 125. [Google Scholar] [CrossRef] [PubMed]
- Bosetti, C.; Bertuccio, P.; Negri, E.; La Vecchia, C.; Zeegers, M.P.; Boffetta, P. Pancreatic cancer: Overview of descriptive epidemiology. Mol. Carcinog. 2012, 51, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Oberstein, P.E.; Olive, K.P. Pancreatic cancer: Why is it so hard to treat? Therap. Adv. Gastroenterol. 2013, 6, 321–337. [Google Scholar] [CrossRef] [PubMed]
- Wong, H.H.; Lemoine, N.R. Biological approaches to therapy of pancreatic cancer. Pancreatology 2008, 8, 431–461. [Google Scholar] [CrossRef] [PubMed]
- Katz, M.H.; Hu, C.Y.; Fleming, J.B.; Pisters, P.W.; Lee, J.E.; Chang, G.J. Clinical calculator of conditional survival estimates for resected and unresected survivors of pancreatic cancer. Arch. Surg. 2012, 147, 513–519. [Google Scholar] [CrossRef]
- Maitra, A.; Hruban, R.H. Pancreatic cancer. Annu. Rev. Pathol. 2008, 3, 157–188. [Google Scholar] [CrossRef] [PubMed]
- Adamska, A.; Domenichini, A.; Falasca, M. Pancreatic Ductal Adenocarcinoma: Current and Evolving Therapies. Int. J. Mol. Sci. 2017, 18, 1338. [Google Scholar] [CrossRef]
- Papke, B.; Der, C.J. Drugging RAS: Know the enemy. Science 2017, 355, 1158–1163. [Google Scholar] [CrossRef]
- Hajatdoost, L.; Sedaghat, K.; Walker, E.J.; Thomas, J.; Kosari, S. Chemotherapy in Pancreatic Cancer: A Systematic Review. Medicina (Kaunas) 2018, 54, 48. [Google Scholar] [CrossRef]
- Stathis, A.; Moore, M.J. Advanced pancreatic carcinoma: Current treatment and future challenges. Nat. Rev. Clin. Oncol. 2010, 7, 163–172. [Google Scholar] [CrossRef]
- Faluyi, O.O.; Connor, J.L.; Chatterjee, M.; Ikin, C.; Wong, H.; Palmer, D.H. Advanced pancreatic adenocarcinoma outcomes with transition from devolved to centralised care in a regional Cancer Centre. Br. J. Cancer 2017, 116, 424–431. [Google Scholar] [CrossRef][Green Version]
- Garrido-Laguna, I.; Hidalgo, M. Pancreatic cancer: From state-of-the-art treatments to promising novel therapies. Nat. Rev. Clin. Oncol. 2015, 12, 319–334. [Google Scholar] [CrossRef]
- Abbassi, R.; Algul, H. Palliative chemotherapy in pancreatic cancer-treatment sequences. Transl. Gastroenterol. Hepatol. 2019, 4, 56. [Google Scholar] [CrossRef] [PubMed]
- Jiang, B.; Zhou, L.; Lu, J.; Wang, Y.; Liu, C.; You, L.; Guo, J. Stroma-Targeting Therapy in Pancreatic Cancer: One Coin with Two Sides? Front. Oncol. 2020, 10, 576399. [Google Scholar] [CrossRef]
- Heldin, C.H.; Rubin, K.; Pietras, K.; Ostman, A. High interstitial fluid pressure–an obstacle in cancer therapy. Nat. Rev. Cancer 2004, 4, 806–813. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.; Saiyin, H.; Fu, D.; Li, J. Stroma–A Double-Edged Sword in Pancreatic Cancer: A Lesson from Targeting Stroma in Pancreatic Cancer with Hedgehog Signaling Inhibitors. Pancreas 2018, 47, 382–389. [Google Scholar] [CrossRef] [PubMed]
- Kota, J.; Hancock, J.; Kwon, J.; Korc, M. Pancreatic cancer: Stroma and its current and emerging targeted therapies. Cancer Lett. 2017, 391, 38–49. [Google Scholar] [CrossRef] [PubMed]
- Suker, M.; Beumer, B.R.; Sadot, E.; Marthey, L.; Faris, J.E.; Mellon, E.A.; El-Rayes, B.F.; Wang-Gillam, A.; Lacy, J.; Hosein, P.J.; et al. FOLFIRINOX for locally advanced pancreatic cancer: A systematic review and patient-level meta-analysis. Lancet Oncol. 2016, 17, 801–810. [Google Scholar] [CrossRef]
- Thota, R.; Maitra, A.; Berlin, J.D. Preclinical Rationale for the Phase III Trials in Metastatic Pancreatic Cancer: Is Wishful Thinking Clouding Successful Drug Development for Pancreatic Cancer? Pancreas 2017, 46, 143–150. [Google Scholar] [CrossRef]
- Burris, H.A., 3rd; Moore, M.J.; Andersen, J.; Green, M.R.; Rothenberg, M.L.; Modiano, M.R.; Cripps, M.C.; Portenoy, R.K.; Storniolo, A.M.; Tarassoff, P.; et al. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: A randomized trial. J. Clin. Oncol. 1997, 15, 2403–2413. [Google Scholar] [CrossRef]
- Von Hoff, D.D.; Ervin, T.; Arena, F.P.; Chiorean, E.G.; Infante, J.; Moore, M.; Seay, T.; Tjulandin, S.A.; Ma, W.W.; Saleh, M.N.; et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N. Engl. J. Med. 2013, 369, 1691–1703. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, D.; El-Maraghi, R.H.; Hammel, P.; Heinemann, V.; Kunzmann, V.; Sastre, J.; Scheithauer, W.; Siena, S.; Tabernero, J.; Teixeira, L.; et al. nab-Paclitaxel plus gemcitabine for metastatic pancreatic cancer: Long-term survival from a phase III trial. J. Natl. Cancer Inst. 2015, 107, dju413. [Google Scholar] [CrossRef] [PubMed]
- Thota, R.; Pauff, J.M.; Berlin, J.D. Treatment of metastatic pancreatic adenocarcinoma: A review. Oncology (Williston Park) 2014, 28, 70–74. [Google Scholar] [PubMed]
- Roviello, G.; Ramello, M.; Catalano, M.; D’Angelo, A.; Conca, R.; Gasperoni, S.; Dreoni, L.; Petrioli, R.; Ianza, A.; Nobili, S.; et al. Association between neutropenia and survival to nab-paclitaxel and gemcitabine in patients with metastatic pancreatic cancer. Sci. Rep. 2020, 10, 19281. [Google Scholar] [CrossRef] [PubMed]
- Conroy, T.; Desseigne, F.; Ychou, M.; Bouche, O.; Guimbaud, R.; Becouarn, Y.; Adenis, A.; Raoul, J.L.; Gourgou-Bourgade, S.; de la Fouchardiere, C.; et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N. Engl. J. Med. 2011, 364, 1817–1825. [Google Scholar] [CrossRef]
- Gourgou-Bourgade, S.; Bascoul-Mollevi, C.; Desseigne, F.; Ychou, M.; Bouche, O.; Guimbaud, R.; Becouarn, Y.; Adenis, A.; Raoul, J.L.; Boige, V.; et al. Impact of FOLFIRINOX compared with gemcitabine on quality of life in patients with metastatic pancreatic cancer: Results from the PRODIGE 4/ACCORD 11 randomized trial. J. Clin. Oncol. 2012, 31, 23–29. [Google Scholar] [CrossRef]
- Lambert, A.; Gavoille, C.; Conroy, T. Current status on the place of FOLFIRINOX in metastatic pancreatic cancer and future directions. Ther. Adv. Gastroenterol. 2017, 10, 631–645. [Google Scholar] [CrossRef]
- Sawada, M.; Kasuga, A.; Mie, T.; Furukawa, T.; Taniguchi, T.; Fukuda, K.; Yamada, Y.; Takeda, T.; Kanata, R.; Matsuyama, M.; et al. Modified FOLFIRINOX as a second-line therapy following gemcitabine plus nab-paclitaxel therapy in metastatic pancreatic cancer. BMC Cancer 2020, 20, 449. [Google Scholar] [CrossRef]
- Cavanna, L.; Stroppa, E.M.; Citterio, C.; Mordenti, P.; Di Nunzio, C.; Peveri, S.; Orlandi, E.; Vecchia, S. Modified FOLFIRINOX for unresectable locally advanced/metastatic pancreatic cancer. A real-world comparison of an attenuated with a full dose in a single center experience. Onco Targets Ther. 2019, 12, 3077–3085. [Google Scholar] [CrossRef]
- Tong, H.; Fan, Z.; Liu, B.; Lu, T. The benefits of modified FOLFIRINOX for advanced pancreatic cancer and its induced adverse events: A systematic review and meta-analysis. Sci. Rep. 2018, 8, 8666. [Google Scholar] [CrossRef]
- Karandish, F.; Mallik, S. Biomarkers and Targeted Therapy in Pancreatic Cancer. Biomark. Cancer 2016, 8, 27–35. [Google Scholar] [CrossRef]
- Tanaka, S. Molecular Pathogenesis and Targeted Therapy of Pancreatic Cancer. Ann. Surg. Oncol. 2016, 23 (Suppl. 2), S197–S205. [Google Scholar] [CrossRef] [PubMed]
- Amanam, I.; Chung, V. Targeted Therapies for Pancreatic Cancer. Cancers (Basel) 2018, 10, 36. [Google Scholar] [CrossRef]
- Qian, Y.; Gong, Y.; Fan, Z.; Luo, G.; Huang, Q.; Deng, S.; Cheng, H.; Jin, K.; Ni, Q.; Yu, X.; et al. Molecular alterations and targeted therapy in pancreatic ductal adenocarcinoma. J. Hematol. Oncol. 2020, 13, 130. [Google Scholar] [CrossRef]
- Schneider, G.; Schmid, R.M. Genetic alterations in pancreatic carcinoma. Mol. Cancer 2003, 2, 15. [Google Scholar] [CrossRef] [PubMed]
- Makohon-Moore, A.; Brosnan, J.A.; Iacobuzio-Donahue, C.A. Pancreatic cancer genomics: Insights and opportunities for clinical translation. Genome Med. 2013, 5, 26. [Google Scholar] [CrossRef]
- Nelson, S.R.; Walsh, N. Genetic Alterations Featuring Biological Models to Tailor Clinical Management of Pancreatic Cancer Patients. Cancers (Basel) 2020, 12, 1233. [Google Scholar] [CrossRef] [PubMed]
- Waddell, N.; Pajic, M.; Patch, A.M.; Chang, D.K.; Kassahn, K.S.; Bailey, P.; Johns, A.L.; Miller, D.; Nones, K.; Quek, K.; et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature 2015, 518, 495–501. [Google Scholar] [CrossRef]
- Witkiewicz, A.K.; McMillan, E.A.; Balaji, U.; Baek, G.; Lin, W.C.; Mansour, J.; Mollaee, M.; Wagner, K.U.; Koduru, P.; Yopp, A.; et al. Whole-exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets. Nat. Commun. 2015, 6, 6744. [Google Scholar] [CrossRef]
- Bailey, P.; Chang, D.K.; Nones, K.; Johns, A.L.; Patch, A.M.; Gingras, M.C.; Miller, D.K.; Christ, A.N.; Bruxner, T.J.; Quinn, M.C.; et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016, 531, 47–52. [Google Scholar] [CrossRef]
- Mollinedo, F.; Gajate, C. Novel therapeutic approaches for pancreatic cancer by combined targeting of RAF-->MEK-->ERK signaling and autophagy survival response. Ann. Transl. Med. 2019, 7, S153. [Google Scholar] [CrossRef] [PubMed]
- Infante, J.R.; Somer, B.G.; Park, J.O.; Li, C.P.; Scheulen, M.E.; Kasubhai, S.M.; Oh, D.Y.; Liu, Y.; Redhu, S.; Steplewski, K.; et al. A randomised, double-blind, placebo-controlled trial of trametinib, an oral MEK inhibitor, in combination with gemcitabine for patients with untreated metastatic adenocarcinoma of the pancreas. Eur. J. Cancer 2014, 50, 2072–2081. [Google Scholar] [CrossRef] [PubMed]
- Van Cutsem, E.; Hidalgo, M.; Canon, J.L.; Macarulla, T.; Bazin, I.; Poddubskaya, E.; Manojlovic, N.; Radenkovic, D.; Verslype, C.; Raymond, E.; et al. Phase I/II trial of pimasertib plus gemcitabine in patients with metastatic pancreatic cancer. Int. J. Cancer 2018, 143, 2053–2064. [Google Scholar] [CrossRef]
- Kinsey, C.G.; Camolotto, S.A.; Boespflug, A.M.; Guillen, K.P.; Foth, M.; Truong, A.; Schuman, S.S.; Shea, J.E.; Seipp, M.T.; Yap, J.T.; et al. Protective autophagy elicited by RAF-->MEK-->ERK inhibition suggests a treatment strategy for RAS-driven cancers. Nat. Med. 2019, 25, 620–627. [Google Scholar] [CrossRef]
- Bryant, K.L.; Stalnecker, C.A.; Zeitouni, D.; Klomp, J.E.; Peng, S.; Tikunov, A.P.; Gunda, V.; Pierobon, M.; Waters, A.M.; George, S.D.; et al. Combination of ERK and autophagy inhibition as a treatment approach for pancreatic cancer. Nat. Med. 2019, 25, 628–640. [Google Scholar] [CrossRef]
- Chantarojanasiri, T.; Hirooka, Y.; Ratanachu-Ek, T.; Kawashima, H.; Ohno, E.; Goto, H. Evolution of pancreas in aging: Degenerative variation or early changes of disease? J. Med. Ultrason. 2015, 42, 177–183. [Google Scholar] [CrossRef]
- Wang, L.; Zheng, S. Pancreatic senescence and its clinical manifestations. Aging Med. 2020, 3, 48–52. [Google Scholar] [CrossRef]
- Caglar, V.; Songur, A.; Yagmurca, M.; Acar, M.; Toktas, M.; Gonul, Y. Age-related volumetric changes in pancreas: A stereological study on computed tomography. Surg. Radiol. Anat. 2012, 34, 935–941. [Google Scholar] [CrossRef] [PubMed]
- Janssen, J.; Papavassiliou, I. Effect of aging and diffuse chronic pancreatitis on pancreas elasticity evaluated using semiquantitative EUS elastography. Ultraschall Med. 2014, 35, 253–258. [Google Scholar] [CrossRef]
- Sato, T.; Ito, K.; Tamada, T.; Sone, T.; Noda, Y.; Higaki, A.; Kanki, A.; Tanimoto, D.; Higashi, H. Age-related changes in normal adult pancreas: MR imaging evaluation. Eur. J. Radiol. 2012, 81, 2093–2098. [Google Scholar] [CrossRef] [PubMed]
- Detlefsen, S.; Sipos, B.; Feyerabend, B.; Kloppel, G. Pancreatic fibrosis associated with age and ductal papillary hyperplasia. Virchows Arch. 2005, 447, 800–805. [Google Scholar] [CrossRef]
- Riccillo, F.L.; Bracamonte, M.I.; Console, G.M.; Gomez Dumm, C.L. Histomorphological and quantitative immunohistochemical changes in the rat pancreas during aging. Biocell 2004, 28, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Lohr, J.M.; Panic, N.; Vujasinovic, M.; Verbeke, C.S. The ageing pancreas: A systematic review of the evidence and analysis of the consequences. J. Intern. Med. 2018, 283, 446–460. [Google Scholar] [CrossRef] [PubMed]
- Ilic, M.; Ilic, I. Epidemiology of pancreatic cancer. World J. Gastroenterol. 2016, 22, 9694–9705. [Google Scholar] [CrossRef]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Liu, J.; Xia, G.; Lei, S.; Huang, X. Survival of pancreatic cancer patients is negatively correlated with age at diagnosis: A population-based retrospective study. Sci. Rep. 2020, 10, 7048. [Google Scholar] [CrossRef]
- Leung, P.S.; Ip, S.P. Pancreatic acinar cell: Its role in acute pancreatitis. Int. J. Biochem. Cell Biol. 2006, 38, 1024–1030. [Google Scholar] [CrossRef]
- Case, R.M. Synthesis, intracellular transport and discharge of exportable proteins in the pancreatic acinar cell and other cells. Biol. Rev. Camb. Philos. Soc. 1978, 53, 211–354. [Google Scholar] [CrossRef]
- Cabrera, O.; Berman, D.M.; Kenyon, N.S.; Ricordi, C.; Berggren, P.O.; Caicedo, A. The unique cytoarchitecture of human pancreatic islets has implications for islet cell function. Proc. Natl. Acad. Sci. USA 2006, 103, 2334–2339. [Google Scholar] [CrossRef]
- Da Silva Xavier, G. The Cells of the Islets of Langerhans. J. Clin. Med. 2018, 7, 54. [Google Scholar] [CrossRef]
- Oyadomari, S.; Araki, E.; Mori, M. Endoplasmic reticulum stress-mediated apoptosis in pancreatic beta-cells. Apoptosis 2002, 7, 335–345. [Google Scholar] [CrossRef]
- Corazzari, M.; Gagliardi, M.; Fimia, G.M.; Piacentini, M. Endoplasmic Reticulum Stress, Unfolded Protein Response, and Cancer Cell Fate. Front. Oncol. 2017, 7, 78. [Google Scholar] [CrossRef]
- Robinson, C.M.; Talty, A.; Logue, S.E.; Mnich, K.; Gorman, A.M.; Samali, A. An Emerging Role for the Unfolded Protein Response in Pancreatic Cancer. Cancers (Basel) 2021, 13, 261. [Google Scholar] [CrossRef]
- Wang, X.Z.; Harding, H.P.; Zhang, Y.; Jolicoeur, E.M.; Kuroda, M.; Ron, D. Cloning of mammalian Ire1 reveals diversity in the ER stress responses. EMBO J. 1998, 17, 5708–5717. [Google Scholar] [CrossRef]
- Huang, G.; Yao, J.; Zeng, W.; Mizuno, Y.; Kamm, K.E.; Stull, J.T.; Harding, H.P.; Ron, D.; Muallem, S. ER stress disrupts Ca2+-signaling complexes and Ca2+ regulation in secretory and muscle cells from PERK-knockout mice. J. Cell Sci. 2006, 119, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Haze, K.; Yoshida, H.; Yanagi, H.; Yura, T.; Mori, K. Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol. Biol. Cell 1999, 10, 3787–3799. [Google Scholar] [CrossRef] [PubMed]
- Pommier, A.; Anaparthy, N.; Memos, N.; Kelley, Z.L.; Gouronnec, A.; Yan, R.; Auffray, C.; Albrengues, J.; Egeblad, M.; Iacobuzio-Donahue, C.A.; et al. Unresolved endoplasmic reticulum stress engenders immune-resistant, latent pancreatic cancer metastases. Science 2018, 360, eaao4908. [Google Scholar] [CrossRef] [PubMed]
- Sah, R.P.; Garg, S.K.; Dixit, A.K.; Dudeja, V.; Dawra, R.K.; Saluja, A.K. Endoplasmic reticulum stress is chronically activated in chronic pancreatitis. J. Biol. Chem. 2014, 289, 27551–27561. [Google Scholar] [CrossRef] [PubMed]
- New, M.; Van Acker, T.; Long, J.S.; Sakamaki, J.I.; Ryan, K.M.; Tooze, S.A. Molecular Pathways Controlling Autophagy in Pancreatic Cancer. Front. Oncol. 2017, 7, 28. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Navas, R.; Munder, M.; Mollinedo, F. Depletion of L-arginine induces autophagy as a cytoprotective response to endoplasmic reticulum stress in human T lymphocytes. Autophagy 2012, 8, 1557–1576. [Google Scholar] [CrossRef]
- Chen, X.; Cubillos-Ruiz, J.R. Endoplasmic reticulum stress signals in the tumour and its microenvironment. Nat. Rev. Cancer 2021, 21, 71–88. [Google Scholar] [CrossRef]
- Garcia-Carbonero, N.; Li, W.; Cabeza-Morales, M.; Martinez-Useros, J.; Garcia-Foncillas, J. New Hope for Pancreatic Ductal Adenocarcinoma Treatment Targeting Endoplasmic Reticulum Stress Response: A Systematic Review. Int. J. Mol. Sci. 2018, 19, 2468. [Google Scholar] [CrossRef] [PubMed]
- Nawrocki, S.T.; Carew, J.S.; Dunner, K., Jr.; Boise, L.H.; Chiao, P.J.; Huang, P.; Abbruzzese, J.L.; McConkey, D.J. Bortezomib inhibits PKR-like endoplasmic reticulum (ER) kinase and induces apoptosis via ER stress in human pancreatic cancer cells. Cancer Res. 2005, 65, 11510–11519. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Liang, S.Q.; Yang, H.; Luthi, U.; Riether, C.; Berezowska, S.; Marti, T.M.; Hall, S.R.R.; Bruggmann, R.; Kocher, G.J.; et al. Increased sensitivity to apoptosis upon endoplasmic reticulum stress-induced activation of the unfolded protein response in chemotherapy-resistant malignant pleural mesothelioma. Br. J. Cancer 2018, 119, 65–75. [Google Scholar] [CrossRef] [PubMed]
- Mandic, A.; Hansson, J.; Linder, S.; Shoshan, M.C. Cisplatin induces endoplasmic reticulum stress and nucleus-independent apoptotic signaling. J. Biol. Chem. 2003, 278, 9100–9106. [Google Scholar] [CrossRef]
- Carracedo, A.; Gironella, M.; Lorente, M.; Garcia, S.; Guzman, M.; Velasco, G.; Iovanna, J.L. Cannabinoids induce apoptosis of pancreatic tumor cells via endoplasmic reticulum stress-related genes. Cancer Res. 2006, 66, 6748–6755. [Google Scholar] [CrossRef]
- Limonta, P.; Moretti, R.M.; Marzagalli, M.; Fontana, F.; Raimondi, M.; Montagnani Marelli, M. Role of Endoplasmic Reticulum Stress in the Anticancer Activity of Natural Compounds. Int. J. Mol. Sci. 2019, 20, 961. [Google Scholar] [CrossRef] [PubMed]
- Nawrocki, S.T.; Carew, J.S.; Pino, M.S.; Highshaw, R.A.; Dunner, K., Jr.; Huang, P.; Abbruzzese, J.L.; McConkey, D.J. Bortezomib sensitizes pancreatic cancer cells to endoplasmic reticulum stress-mediated apoptosis. Cancer Res. 2005, 65, 11658–11666. [Google Scholar] [CrossRef]
- Avril, T.; Vauleon, E.; Chevet, E. Endoplasmic reticulum stress signaling and chemotherapy resistance in solid cancers. Oncogenesis 2017, 6, e373. [Google Scholar] [CrossRef]
- Nikesitch, N.; Lee, J.M.; Ling, S.; Roberts, T.L. Endoplasmic reticulum stress in the development of multiple myeloma and drug resistance. Clin. Transl. Immunol. 2018, 7, e1007. [Google Scholar] [CrossRef]
- Sitia, R.; Braakman, I. Quality control in the endoplasmic reticulum protein factory. Nature 2003, 426, 891–894. [Google Scholar] [CrossRef] [PubMed]
- Ellgaard, L.; Helenius, A. Quality control in the endoplasmic reticulum. Nat. Rev. Mol. Cell Biol. 2003, 4, 181–191. [Google Scholar] [CrossRef]
- Walter, P.; Ron, D. The unfolded protein response: From stress pathway to homeostatic regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef]
- Iurlaro, R.; Munoz-Pinedo, C. Cell death induced by endoplasmic reticulum stress. FEBS J. 2016, 283, 2640–2652. [Google Scholar] [CrossRef]
- Breckenridge, D.G.; Germain, M.; Mathai, J.P.; Nguyen, M.; Shore, G.C. Regulation of apoptosis by endoplasmic reticulum pathways. Oncogene 2003, 22, 8608–8618. [Google Scholar] [CrossRef]
- Hitomi, J.; Katayama, T.; Eguchi, Y.; Kudo, T.; Taniguchi, M.; Koyama, Y.; Manabe, T.; Yamagishi, S.; Bando, Y.; Imaizumi, K.; et al. Involvement of caspase-4 in endoplasmic reticulum stress-induced apoptosis and Abeta-induced cell death. J. Cell Biol. 2004, 165, 347–356. [Google Scholar] [CrossRef]
- Merksamer, P.I.; Papa, F.R. The UPR and cell fate at a glance. J. Cell Sci. 2010, 123, 1003–1006. [Google Scholar] [CrossRef]
- Gajate, C.; Matos-da-Silva, M.; Dakir, E.L.; Fonteriz, R.I.; Alvarez, J.; Mollinedo, F. Antitumor alkyl-lysophospholipid analog edelfosine induces apoptosis in pancreatic cancer by targeting endoplasmic reticulum. Oncogene 2012, 31, 2627–2639. [Google Scholar] [CrossRef]
- Hetz, C. The unfolded protein response: Controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol. 2012, 13, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Sano, R.; Reed, J.C. ER stress-induced cell death mechanisms. Biochim. Biophys. Acta 2013, 1833, 3460–3470. [Google Scholar] [CrossRef] [PubMed]
- Bonilla, X.; Dakir, E.H.; Mollinedo, F.; Gajate, C. Endoplasmic reticulum targeting in Ewing’s sarcoma by the alkylphospholipid analog edelfosine. Oncotarget 2015, 6, 14596–14613. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C.; Papa, F.R. The Unfolded Protein Response and Cell Fate Control. Mol. Cell 2018, 69, 169–181. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.S. The glucose-regulated proteins: Stress induction and clinical applications. Trends Biochem. Sci. 2001, 26, 504–510. [Google Scholar] [CrossRef]
- Park, K.W.; Eun Kim, G.; Morales, R.; Moda, F.; Moreno-Gonzalez, I.; Concha-Marambio, L.; Lee, A.S.; Hetz, C.; Soto, C. The Endoplasmic Reticulum Chaperone GRP78/BiP Modulates Prion Propagation in vitro and in vivo. Sci. Rep. 2017, 7, 44723. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Lv, Y.; Zhao, N.; Guan, G.; Wang, J. Protein kinase R-like ER kinase and its role in endoplasmic reticulum stress-decided cell fate. Cell Death Dis. 2015, 6, e1822. [Google Scholar] [CrossRef] [PubMed]
- Breckenridge, D.G.; Stojanovic, M.; Marcellus, R.C.; Shore, G.C. Caspase cleavage product of BAP31 induces mitochondrial fission through endoplasmic reticulum calcium signals, enhancing cytochrome c release to the cytosol. J. Cell Biol. 2003, 160, 1115–1127. [Google Scholar] [CrossRef]
- Iwasawa, R.; Mahul-Mellier, A.L.; Datler, C.; Pazarentzos, E.; Grimm, S. Fis1 and Bap31 bridge the mitochondria-ER interface to establish a platform for apoptosis induction. EMBO J. 2011, 30, 556–568. [Google Scholar] [CrossRef]
- Bian, Z.M.; Elner, S.G.; Elner, V.M. Dual involvement of caspase-4 in inflammatory and ER stress-induced apoptotic responses in human retinal pigment epithelial cells. Investig. Ophthalmol. Vis. Sci. 2009, 50, 6006–6014. [Google Scholar] [CrossRef]
- Reiling, J.H.; Sabatini, D.M. Increased mTORC1 signaling UPRegulates stress. Mol. Cell 2008, 29, 533–535. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, S.G.; Gromada, J.; Urano, F. Endoplasmic reticulum stress and pancreatic beta-cell death. Trends Endocrinol. Metab. 2011, 22, 266–274. [Google Scholar]
- Ghosh, R.; Colon-Negron, K.; Papa, F.R. Endoplasmic reticulum stress, degeneration of pancreatic islet beta-cells, and therapeutic modulation of the unfolded protein response in diabetes. Mol. Metab. 2019, 27S, S60–S68. [Google Scholar] [CrossRef] [PubMed]
- Gajate, C.; Mollinedo, F. Biological activities, mechanisms of action and biomedical prospect of the antitumor ether phospholipid ET-18-OCH3 (Edelfosine), a proapoptotic agent in tumor cells. Curr. Drug Metab. 2002, 3, 491–525. [Google Scholar] [CrossRef]
- Mollinedo, F.; Gajate, C.; Martin-Santamaria, S.; Gago, F. ET-18-OCH3 (edelfosine): A selective antitumour lipid targeting apoptosis through intracellular activation of Fas/CD95 death receptor. Curr. Med. Chem. 2004, 11, 3163–3184. [Google Scholar] [CrossRef] [PubMed]
- Mollinedo, F. Antitumor ether lipids: Proapoptotic agents with multiple therapeutic indications. Expert Opin. Ther. Pat. 2007, 17, 385–405. [Google Scholar] [CrossRef]
- Mollinedo, F. Antitumor alkylphospholipid analogs: A promising and growing family of synthetic cell membrane-targeting molecules for cancer treatment. Anticancer Agents Med. Chem. 2014, 14, 495–498. [Google Scholar] [CrossRef][Green Version]
- Gajate, C.; Mollinedo, F. Lipid rafts, endoplasmic reticulum and mitochondria in the antitumor action of the alkylphospholipid analog edelfosine. Anticancer Agents Med. Chem. 2014, 14, 509–527. [Google Scholar] [CrossRef]
- Sundar, S.; Chatterjee, M. Visceral leishmaniasis–current therapeutic modalities. Indian J. Med. Res. 2006, 123, 345–352. [Google Scholar]
- Sundar, S.; Jha, T.K.; Thakur, C.P.; Engel, J.; Sindermann, H.; Fischer, C.; Junge, K.; Bryceson, A.; Berman, J. Oral miltefosine for Indian visceral leishmaniasis. N. Engl. J. Med. 2002, 347, 1739–1746. [Google Scholar] [CrossRef]
- Sundar, S.; Rai, M. Treatment of visceral leishmaniasis. Expert Opin. Pharm. 2005, 6, 2821–2829. [Google Scholar] [CrossRef] [PubMed]
- Sundar, S.; Singh, A.; Rai, M.; Prajapati, V.K.; Singh, A.K.; Ostyn, B.; Boelaert, M.; Dujardin, J.C.; Chakravarty, J. Efficacy of miltefosine in the treatment of visceral leishmaniasis in India after a decade of use. Clin. Infect. Dis. 2012, 55, 543–550. [Google Scholar] [CrossRef]
- Ghobrial, I.M.; Roccaro, A.; Hong, F.; Weller, E.; Rubin, N.; Leduc, R.; Rourke, M.; Chuma, S.; Sacco, A.; Jia, X.; et al. Clinical and translational studies of a phase II trial of the novel oral Akt inhibitor perifosine in relapsed or relapsed/refractory Waldenstrom’s macroglobulinemia. Clin. Cancer Res. 2010, 16, 1033–1041. [Google Scholar] [CrossRef]
- Richardson, P.G.; Wolf, J.; Jakubowiak, A.; Zonder, J.; Lonial, S.; Irwin, D.; Densmore, J.; Krishnan, A.; Raje, N.; Bar, M.; et al. Perifosine plus bortezomib and dexamethasone in patients with relapsed/refractory multiple myeloma previously treated with bortezomib: Results of a multicenter phase I/II trial. J. Clin. Oncol. 2011, 29, 4243–4249. [Google Scholar] [CrossRef]
- Jakubowiak, A.J.; Richardson, P.G.; Zimmerman, T.; Alsina, M.; Kaufman, J.L.; Kandarpa, M.; Kraftson, S.; Ross, C.W.; Harvey, C.; Hideshima, T.; et al. Perifosine plus lenalidomide and dexamethasone in relapsed and relapsed/refractory multiple myeloma: A Phase I Multiple Myeloma Research Consortium study. Br. J. Haematol. 2012, 158, 472–480. [Google Scholar] [CrossRef] [PubMed]
- Richardson, P.G.; Eng, C.; Kolesar, J.; Hideshima, T.; Anderson, K.C. Perifosine, an oral, anti-cancer agent and inhibitor of the Akt pathway: Mechanistic actions, pharmacodynamics, pharmacokinetics, and clinical activity. Expert Opin. Drug Metab. Toxicol. 2012, 8, 623–633. [Google Scholar] [CrossRef]
- Richardson, P.G.; Nagler, A.; Ben-Yehuda, D.; Badros, A.; Hari, P.; Hajek, R.; Spicka, I.; Kaya, H.; Le Blanc, R.; Yoon, S.-S.; et al. Randomized Placebo-Controlled Phase III Study of Perifosine Combined with Bortezomib and Dexamethasone in Relapsed, Refractory Multiple Myeloma Patients Previously Treated with Bortezomib. Blood 2013, 122, 3189. [Google Scholar] [CrossRef]
- Friedman, D.R.; Lanasa, M.C.; Davis, P.H.; Allgood, S.D.; Matta, K.M.; Brander, D.M.; Chen, Y.; Davis, E.D.; Volkheimer, A.D.; Moore, J.O.; et al. Perifosine treatment in chronic lymphocytic leukemia: Results of a phase II clinical trial and in vitro studies. Leuk. Lymphoma 2014, 55, 1067–1075. [Google Scholar] [CrossRef] [PubMed]
- Bendell, J.C.; Nemunaitis, J.; Vukelja, S.J.; Hagenstad, C.; Campos, L.T.; Hermann, R.C.; Sportelli, P.; Gardner, L.; Richards, D.A. Randomized placebo-controlled phase II trial of perifosine plus capecitabine as second- or third-line therapy in patients with metastatic colorectal cancer. J. Clin. Oncol. 2011, 29, 4394–4400. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Becher, O.J.; Millard, N.E.; Modak, S.; Kushner, B.H.; Haque, S.; Spasojevic, I.; Trippett, T.M.; Gilheeney, S.W.; Khakoo, Y.; Lyden, D.C.; et al. A phase I study of single-agent perifosine for recurrent or refractory pediatric CNS and solid tumors. PLoS ONE 2017, 12, e0178593. [Google Scholar] [CrossRef] [PubMed]
- Becher, O.J.; Gilheeney, S.W.; Khakoo, Y.; Lyden, D.C.; Haque, S.; De Braganca, K.C.; Kolesar, J.M.; Huse, J.T.; Modak, S.; Wexler, L.H.; et al. A phase I study of perifosine with temsirolimus for recurrent pediatric solid tumors. Pediatr. Blood Cancer 2017, 64, e26409. [Google Scholar] [CrossRef] [PubMed]
- Kushner, B.H.; Cheung, N.V.; Modak, S.; Becher, O.J.; Basu, E.M.; Roberts, S.S.; Kramer, K.; Dunkel, I.J. A phase I/Ib trial targeting the PI3K/Akt pathway using perifosine: Long-term progression-free survival of patients with resistant neuroblastoma. Int. J. Cancer 2017, 140, 480–484. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, K.; Kagabu, M.; Mizuno, M.; Oda, K.; Aoki, D.; Mabuchi, S.; Kamiura, S.; Yamaguchi, S.; Aoki, Y.; Saito, T.; et al. Phase II basket trial of perifosine monotherapy for recurrent gynecologic cancer with or without PIK3CA mutations. Invest. New Drugs 2017, 35, 800–812. [Google Scholar] [CrossRef]
- Kaley, T.J.; Panageas, K.S.; Mellinghoff, I.K.; Nolan, C.; Gavrilovic, I.T.; DeAngelis, L.M.; Abrey, L.E.; Holland, E.C.; Lassman, A.B. Phase II trial of an AKT inhibitor (perifosine) for recurrent glioblastoma. J. Neurooncol. 2019, 144, 403–407. [Google Scholar] [CrossRef]
- Kaley, T.J.; Panageas, K.S.; Pentsova, E.I.; Mellinghoff, I.K.; Nolan, C.; Gavrilovic, I.; DeAngelis, L.M.; Abrey, L.E.; Holland, E.C.; Omuro, A.; et al. Phase I clinical trial of temsirolimus and perifosine for recurrent glioblastoma. Ann. Clin. Transl. Neurol. 2020, 7, 429–436. [Google Scholar] [CrossRef]
- Dorlo, T.P.; Balasegaram, M.; Beijnen, J.H.; de Vries, P.J. Miltefosine: A review of its pharmacology and therapeutic efficacy in the treatment of leishmaniasis. J. Antimicrob. Chemother. 2012, 67, 2576–2597. [Google Scholar] [CrossRef]
- Leonard, R.; Hardy, J.; van Tienhoven, G.; Houston, S.; Simmonds, P.; David, M.; Mansi, J. Randomized, double-blind, placebo-controlled, multicenter trial of 6% miltefosine solution, a topical chemotherapy in cutaneous metastases from breast cancer. J. Clin. Oncol. 2001, 19, 4150–4159. [Google Scholar] [CrossRef] [PubMed]
- Gajate, C.; Mollinedo, F. Edelfosine and perifosine induce selective apoptosis in multiple myeloma by recruitment of death receptors and downstream signaling molecules into lipid rafts. Blood 2007, 109, 711–719. [Google Scholar] [CrossRef] [PubMed]
- Kny, G. Über Lysolecithin-Analoga–Synthese und biologische Eigenschaften. Diploma Thesis, University of Freiburg, Freiburg, Germany, 1969. [Google Scholar]
- Mollinedo, F.; Fernandez-Luna, J.L.; Gajate, C.; Martin-Martin, B.; Benito, A.; Martinez-Dalmau, R.; Modolell, M. Selective induction of apoptosis in cancer cells by the ether lipid ET-18-OCH3 (Edelfosine): Molecular structure requirements, cellular uptake, and protection by Bcl-2 and Bcl-XL. Cancer Res. 1997, 57, 1320–1328. [Google Scholar] [PubMed]
- Hoffman, D.R.; Hoffman, L.H.; Snyder, F. Cytotoxicity and metabolism of alkyl phospholipid analogues in neoplastic cells. Cancer Res. 1986, 46, 5803–5809. [Google Scholar]
- Magistrelli, A.; Villa, P.; Benfenati, E.; Modest, E.J.; Salmona, M.; Tacconi, M.T. Fate of 1-O-octadecyl-2-O-methyl-rac-glycero-3-phosphocholine (ET18-OME) in malignant cells, normal cells, and isolated and perfused rat liver. Drug Metab. Dispos. 1995, 23, 113–118. [Google Scholar]
- Varela-M, R.E.; Villa-Pulgarin, J.A.; Yepes, E.; Muller, I.; Modolell, M.; Munoz, D.L.; Robledo, S.M.; Muskus, C.E.; Lopez-Aban, J.; Muro, A.; et al. In Vitro and In Vivo Efficacy of Ether Lipid Edelfosine against Leishmania spp. and SbV-Resistant Parasites. PLoS Negl. Trop. Dis. 2012, 6, e1612. [Google Scholar] [CrossRef][Green Version]
- Villa-Pulgarin, J.A.; Gajate, C.; Botet, J.; Jimenez, A.; Justies, N.; Varela-M, R.E.; Cuesta-Marban, A.; Muller, I.; Modolell, M.; Revuelta, J.L.; et al. Mitochondria and lipid raft-located FOF1-ATP synthase as major therapeutic targets in the antileishmanial and anticancer activities of ether lipid edelfosine. PLoS Negl. Trop. Dis. 2017, 11, e0005805. [Google Scholar] [CrossRef]
- Yepes, E.; Varela-M, R.E.; Lopez-Aban, J.; Dakir, E.H.; Mollinedo, F.; Muro, A. In vitro and in vivo anti-schistosomal activity of the alkylphospholipid analog edelfosine. PLoS ONE 2014, 9, e109431. [Google Scholar] [CrossRef]
- Yepes, E.; Varela-M, R.E.; Lopez-Aban, J.; Rojas-Caraballo, J.; Muro, A.; Mollinedo, F. Inhibition of Granulomatous Inflammation and Prophylactic Treatment of Schistosomiasis with a Combination of Edelfosine and Praziquantel. PLoS Negl. Trop. Dis. 2015, 9, e0003893. [Google Scholar] [CrossRef]
- Legarda-Ceballos, A.L.; Rojas-Caraballo, J.; Lopez-Aban, J.; Ruano, A.L.; Yepes, E.; Gajate, C.; Mollinedo, F.; Muro, A. The alkylphospholipid edelfosine shows activity against Strongyloides venezuelensis and induces apoptosis-like cell death. Acta. Trop. 2016, 162, 180–187. [Google Scholar] [CrossRef] [PubMed]
- Mollinedo, F.; de la Iglesia-Vicente, J.; Gajate, C.; Estella-Hermoso de Mendoza, A.; Villa-Pulgarin, J.A.; de Frias, M.; Roue, G.; Gil, J.; Colomer, D.; Campanero, M.A.; et al. In vitro and In vivo selective antitumor activity of Edelfosine against mantle cell lymphoma and chronic lymphocytic leukemia involving lipid rafts. Clin. Cancer Res. 2010, 16, 2046–2054. [Google Scholar] [CrossRef]
- Mollinedo, F.; de la Iglesia-Vicente, J.; Gajate, C.; Estella-Hermoso de Mendoza, A.; Villa-Pulgarin, J.A.; Campanero, M.A.; Blanco-Prieto, M.J. Lipid raft-targeted therapy in multiple myeloma. Oncogene 2010, 29, 3748–3757. [Google Scholar] [CrossRef] [PubMed]
- Reis-Sobreiro, M.; Gajate, C.; Mollinedo, F. Involvement of mitochondria and recruitment of Fas/CD95 signaling in lipid rafts in resveratrol-mediated antimyeloma and antileukemia actions. Oncogene 2009, 28, 3221–3234. [Google Scholar] [CrossRef]
- Mollinedo, F.; Gajate, C.; Morales, A.I.; del Canto-Janez, E.; Justies, N.; Collia, F.; Rivas, J.V.; Modolell, M.; Iglesias, A. Novel anti-inflammatory action of edelfosine lacking toxicity with protective effect in experimental colitis. J. Pharmacol. Exp. Ther. 2009, 329, 439–449. [Google Scholar] [CrossRef] [PubMed]
- Gajate, C.; Del Canto-Janez, E.; Acuña, A.U.; Amat-Guerri, F.; Geijo, E.; Santos-Beneit, A.M.; Veldman, R.J.; Mollinedo, F. Intracellular triggering of Fas aggregation and recruitment of apoptotic molecules into Fas-enriched rafts in selective tumor cell apoptosis. J. Exp. Med. 2004, 200, 353–365. [Google Scholar] [CrossRef]
- Gajate, C.; Fonteriz, R.I.; Cabaner, C.; Alvarez-Noves, G.; Alvarez-Rodriguez, Y.; Modolell, M.; Mollinedo, F. Intracellular triggering of Fas, independently of FasL, as a new mechanism of antitumor ether lipid-induced apoptosis. Int. J. Cancer 2000, 85, 674–682. [Google Scholar] [CrossRef]
- Gajate, C.; Mollinedo, F. The antitumor ether lipid ET-18-OCH3 induces apoptosis through translocation and capping of Fas/CD95 into membrane rafts in human leukemic cells. Blood 2001, 98, 3860–3863. [Google Scholar] [CrossRef] [PubMed]
- Mollinedo, F.; Gajate, C. Fas/CD95 death receptor and lipid rafts: New targets for apoptosis-directed cancer therapy. Drug Resist. Updat. 2006, 9, 51–73. [Google Scholar] [CrossRef]
- Gajate, C.; Gonzalez-Camacho, F.; Mollinedo, F. Involvement of raft aggregates enriched in Fas/CD95 death-inducing signaling complex in the antileukemic action of edelfosine in Jurkat cells. PLoS ONE 2009, 4, e5044. [Google Scholar] [CrossRef] [PubMed]
- Mollinedo, F.; Gajate, C. Lipid rafts as major platforms for signaling regulation in cancer. Adv. Biol. Regul. 2015, 57, 130–146. [Google Scholar] [CrossRef] [PubMed]
- Gajate, C.; Mollinedo, F. Lipid rafts and raft-mediated supramolecular entities in the regulation of CD95 death receptor apoptotic signaling. Apoptosis 2015, 20, 584–606. [Google Scholar] [CrossRef]
- Gajate, C.; Mollinedo, F. Lipid raft-mediated Fas/CD95 apoptotic signaling in leukemic cells and normal leukocytes and therapeutic implications. J. Leukoc. Biol. 2015, 98, 739–759. [Google Scholar] [CrossRef] [PubMed]
- Gajate, C.; Mollinedo, F. Fas/CD95, lipid rafts and cancer. In TRAIL, Fas Ligand, TNF and TLR3 in Cancer; Micheau, O., Ed.; Springer International Publishing AG: Cham, Switzerland, 2017; pp. 187–228. [Google Scholar]
- Mollinedo, F.; Gajate, C. Lipid rafts as signaling hubs in cancer cell survival/death and invasion: Implications in tumor progression and therapy. J. Lipid Res. 2020, 61, 611–635. [Google Scholar] [CrossRef]
- Gajate, C.; Gonzalez-Camacho, F.; Mollinedo, F. Lipid raft connection between extrinsic and intrinsic apoptotic pathways. Biochem. Biophys. Res. Commun. 2009, 380, 780–784. [Google Scholar] [CrossRef]
- Mollinedo, F.; Gajate, C. Lipid rafts and clusters of apoptotic signaling molecule-enriched rafts in cancer therapy. Future Oncol. 2010, 6, 811–821. [Google Scholar] [CrossRef]
- Mollinedo, F.; Gajate, C. Lipid rafts, death receptors and CASMERs: New insights for cancer therapy. Future Oncol. 2010, 6, 491–494. [Google Scholar] [CrossRef]
- Zaremberg, V.; Gajate, C.; Cacharro, L.M.; Mollinedo, F.; McMaster, C.R. Cytotoxicity of an anti-cancer lysophospholipid through selective modification of lipid raft composition. J. Biol. Chem. 2005, 280, 38047–38058. [Google Scholar] [CrossRef]
- Reis-Sobreiro, M.; Roue, G.; Moros, A.; Gajate, C.; de la Iglesia-Vicente, J.; Colomer, D.; Mollinedo, F. Lipid raft-mediated Akt signaling as a therapeutic target in mantle cell lymphoma. Blood Cancer J. 2013, 3, e118. [Google Scholar] [CrossRef]
- Czyz, O.; Bitew, T.; Cuesta-Marban, A.; McMaster, C.R.; Mollinedo, F.; Zaremberg, V. Alteration of plasma membrane organization by an anticancer lysophosphatidylcholine analogue induces intracellular acidification and internalization of plasma membrane transporters in yeast. J. Biol. Chem. 2013, 288, 8419–8432. [Google Scholar] [CrossRef]
- Cuesta-Marban, A.; Botet, J.; Czyz, O.; Cacharro, L.M.; Gajate, C.; Hornillos, V.; Delgado, J.; Zhang, H.; Amat-Guerri, F.; Acuña, A.U.; et al. Drug uptake, lipid rafts, and vesicle trafficking modulate resistance to an anticancer lysophosphatidylcholine analogue in yeast. J. Biol. Chem. 2013, 288, 8405–8418. [Google Scholar] [CrossRef]
- Nieto-Miguel, T.; Gajate, C.; Mollinedo, F. Differential Targets and Subcellular Localization of Antitumor Alkyl-lysophospholipid in Leukemic Versus Solid Tumor Cells. J. Biol. Chem. 2006, 281, 14833–14840. [Google Scholar] [CrossRef]
- Mollinedo, F. Death receptors in multiple myeloma and therapeutic opportunities. In Myeloma Therapy. Pursuing the Plasma Cell; Lonial, S., Ed.; Humana Press: Totowa, NJ, USA, 2008; pp. 393–419. [Google Scholar]
- White-Gilbertson, S.; Hua, Y.; Liu, B. The role of endoplasmic reticulum stress in maintaining and targeting multiple myeloma: A double-edged sword of adaptation and apoptosis. Front. Genet. 2013, 4, 109. [Google Scholar] [CrossRef] [PubMed]
- Karaskov, E.; Scott, C.; Zhang, L.; Teodoro, T.; Ravazzola, M.; Volchuk, A. Chronic palmitate but not oleate exposure induces endoplasmic reticulum stress, which may contribute to INS-1 pancreatic beta-cell apoptosis. Endocrinology 2006, 147, 3398–3407. [Google Scholar] [CrossRef] [PubMed]
- Cnop, M.; Ladriere, L.; Hekerman, P.; Ortis, F.; Cardozo, A.K.; Dogusan, Z.; Flamez, D.; Boyce, M.; Yuan, J.; Eizirik, D.L. Selective inhibition of eukaryotic translation initiation factor 2 alpha dephosphorylation potentiates fatty acid-induced endoplasmic reticulum stress and causes pancreatic beta-cell dysfunction and apoptosis. J. Biol. Chem. 2007, 282, 3989–3997. [Google Scholar] [CrossRef] [PubMed]
- Cunha, D.A.; Hekerman, P.; Ladriere, L.; Bazarra-Castro, A.; Ortis, F.; Wakeham, M.C.; Moore, F.; Rasschaert, J.; Cardozo, A.K.; Bellomo, E.; et al. Initiation and execution of lipotoxic ER stress in pancreatic beta-cells. J. Cell Sci. 2008, 121, 2308–2318. [Google Scholar] [CrossRef] [PubMed]
- Quesada, E.; Delgado, J.; Gajate, C.; Mollinedo, F.; Acuña, A.U.; Amat-Guerri, F. Fluorescent phenylpolyene analogues of the ether phospholipid edelfosine for the selective labeling of cancer cells. J. Med. Chem. 2004, 47, 5333–5335. [Google Scholar] [CrossRef]
- Kowada, T.; Maeda, H.; Kikuchi, K. BODIPY-based probes for the fluorescence imaging of biomolecules in living cells. Chem. Soc. Rev. 2015, 44, 4953–4972. [Google Scholar] [CrossRef]
- Lu, H.; Shen, Z. Editorial: BODIPYs and Their Derivatives: The Past, Present and Future. Front. Chem. 2020, 8, 290. [Google Scholar] [CrossRef]
- Mollinedo, F.; Fernandez, M.; Hornillos, V.; Delgado, J.; Amat-Guerri, F.; Acuña, A.U.; Nieto-Miguel, T.; Villa-Pulgarin, J.A.; Gonzalez-Garcia, C.; Cena, V.; et al. Involvement of lipid rafts in the localization and dysfunction effect of the antitumor ether phospholipid edelfosine in mitochondria. Cell Death Dis. 2011, 2, e158. [Google Scholar] [CrossRef]
- Klee, M.; Pimentel-Muinos, F.X. Bcl-XL specifically activates Bak to induce swelling and restructuring of the endoplasmic reticulum. J. Cell Biol. 2005, 168, 723–734. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Gajate, C.; Yu, L.P.; Fang, Y.X.; Mollinedo, F. Mitochondrial-derived ROS in edelfosine-induced apoptosis in yeasts and tumor cells. Acta Pharmacol. Sin. 2007, 28, 888–894. [Google Scholar] [CrossRef] [PubMed]
- Mollinedo, F. Lipid raft involvement in yeast cell growth and death. Front. Oncol. 2012, 2, 140. [Google Scholar] [CrossRef] [PubMed]
- Casas, C. GRP78 at the Centre of the Stage in Cancer and Neuroprotection. Front. Neurosci. 2017, 11, 177. [Google Scholar] [CrossRef] [PubMed]
- Bi, X.; Zhang, G.; Wang, X.; Nguyen, C.; May, H.I.; Li, X.; Al-Hashimi, A.A.; Austin, R.C.; Gillette, T.G.; Fu, G.; et al. Endoplasmic Reticulum Chaperone GRP78 Protects Heart from Ischemia/Reperfusion Injury through Akt Activation. Circ. Res. 2018, 122, 1545–1554. [Google Scholar] [CrossRef] [PubMed]
- Nieto-Miguel, T.; Fonteriz, R.I.; Vay, L.; Gajate, C.; Lopez-Hernandez, S.; Mollinedo, F. Endoplasmic Reticulum Stress in the Proapoptotic Action of Edelfosine in Solid Tumor Cells. Cancer Res. 2007, 67, 10368–10378. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Wey, S.; Zhang, Y.; Ye, R.; Lee, A.S. Role of the unfolded protein response regulator GRP78/BiP in development, cancer, and neurological disorders. Antioxid. Redox Signal. 2009, 11, 2307–2316. [Google Scholar] [CrossRef]
- Gajate, C.; Santos-Beneit, A.; Modolell, M.; Mollinedo, F. Involvement of c-Jun NH2-terminal kinase activation and c-Jun in the induction of apoptosis by the ether phospholipid 1-O-octadecyl-2-O-methyl-rac-glycero-3-phosphocholine. Mol. Pharmacol. 1998, 53, 602–612. [Google Scholar] [CrossRef]
- Bennett, B.L.; Sasaki, D.T.; Murray, B.W.; O’Leary, E.C.; Sakata, S.T.; Xu, W.; Leisten, J.C.; Motiwala, A.; Pierce, S.; Satoh, Y.; et al. SP600125, an anthrapyrazolone inhibitor of Jun N-terminal kinase. Proc. Natl. Acad. Sci. USA 2001, 98, 13681–13686. [Google Scholar] [CrossRef]
- Nishitoh, H.; Matsuzawa, A.; Tobiume, K.; Saegusa, K.; Takeda, K.; Inoue, K.; Hori, S.; Kakizuka, A.; Ichijo, H. ASK1 is essential for endoplasmic reticulum stress-induced neuronal cell death triggered by expanded polyglutamine repeats. Genes Dev. 2002, 16, 1345–1355. [Google Scholar] [CrossRef]
- Nieto-Miguel, T.; Gajate, C.; Gonzalez-Camacho, F.; Mollinedo, F. Proapoptotic role of Hsp90 by its interaction with c-Jun N-terminal kinase in lipid rafts in edelfosine-mediated antileukemic therapy. Oncogene 2008, 27, 1779–1787. [Google Scholar] [CrossRef]
- Bello, C.; Bai, J.; Zambron, B.K.; Elias-Rodriguez, P.; Gajate, C.; Robina, I.; Caffa, I.; Cea, M.; Montecucco, F.; Nencioni, A.; et al. Induction of cell killing and autophagy by amphiphilic pyrrolidine derivatives on human pancreatic cancer cells. Eur. J. Med. Chem. 2018, 150, 457–478. [Google Scholar] [CrossRef]
- Nguyen, M.; Breckenridge, D.G.; Ducret, A.; Shore, G.C. Caspase-resistant BAP31 inhibits fas-mediated apoptotic membrane fragmentation and release of cytochrome c from mitochondria. Mol. Cell Biol. 2000, 20, 6731–6740. [Google Scholar] [CrossRef]
- Vance, J.E. Phospholipid synthesis in a membrane fraction associated with mitochondria. J. Biol. Chem. 1990, 265, 7248–7256. [Google Scholar] [CrossRef]
- Giorgi, C.; Missiroli, S.; Patergnani, S.; Duszynski, J.; Wieckowski, M.R.; Pinton, P. Mitochondria-associated membranes: Composition, molecular mechanisms, and physiopathological implications. Antioxid. Redox Signal. 2015, 22, 995–1019. [Google Scholar] [CrossRef] [PubMed]
- Missiroli, S.; Patergnani, S.; Caroccia, N.; Pedriali, G.; Perrone, M.; Previati, M.; Wieckowski, M.R.; Giorgi, C. Mitochondria-associated membranes (MAMs) and inflammation. Cell Death Dis. 2018, 9, 329. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, M.; Hayashi, T.; Su, T.P. The role of cholesterol in the association of endoplasmic reticulum membranes with mitochondria. Biochem. Biophys. Res. Commun. 2012, 417, 635–639. [Google Scholar] [CrossRef] [PubMed]
- Mollinedo, F.; Gajate, C. Mitochondrial Targeting Involving Cholesterol-Rich Lipid Rafts in the Mechanism of Action of the Antitumor Ether Lipid and Alkylphospholipid Analog Edelfosine. Pharmaceutics 2021, 13, 763. [Google Scholar] [CrossRef]
- Flis, V.V.; Daum, G. Lipid transport between the endoplasmic reticulum and mitochondria. Cold Spring Harb. Perspect. Biol. 2013, 5, a013235. [Google Scholar] [CrossRef]
- Missiroli, S.; Danese, A.; Iannitti, T.; Patergnani, S.; Perrone, M.; Previati, M.; Giorgi, C.; Pinton, P. Endoplasmic reticulum-mitochondria Ca2+ crosstalk in the control of the tumor cell fate. Biochim. Biophys. Acta. Mol. Cell Res. 2017, 1864, 858–864. [Google Scholar] [CrossRef]
- Busto, J.V.; del Canto-Jañez, E.; Goñi, F.M.; Mollinedo, F.; Alonso, A. Combination of the anti-tumour cell ether lipid edelfosine with sterols abolishes haemolytic side effects of the drug. J. Chem. Biol. 2008, 1, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Ausili, A.; Martinez-Valera, P.; Torrecillas, A.; Gomez-Murcia, V.; de Godos, A.M.; Corbalan-Garcia, S.; Teruel, J.A.; Gomez Fernandez, J.C. Anticancer Agent Edelfosine Exhibits a High Affinity for Cholesterol and Disorganizes Liquid-Ordered Membrane Structures. Langmuir 2018, 34, 8333–8346. [Google Scholar] [CrossRef] [PubMed]
- Ausili, A.; Torrecillas, A.; Aranda, F.J.; Mollinedo, F.; Gajate, C.; Corbalan-Garcia, S.; de Godos, A.; Gomez-Fernandez, J.C. Edelfosine Is Incorporated into Rafts and Alters Their Organization. J. Phys. Chem. B 2008, 112, 11643–11654. [Google Scholar] [CrossRef] [PubMed]
- Munder, M.; Mollinedo, F.; Calafat, J.; Canchado, J.; Gil-Lamaignere, C.; Fuentes, J.M.; Luckner, C.; Doschko, G.; Soler, G.; Eichmann, K.; et al. Arginase I is constitutively expressed in human granulocytes and participates in fungicidal activity. Blood 2005, 105, 2549–2556. [Google Scholar] [CrossRef]
- Munder, M.; Schneider, H.; Luckner, C.; Giese, T.; Langhans, C.D.; Fuentes, J.M.; Kropf, P.; Mueller, I.; Kolb, A.; Modolell, M.; et al. Suppression of T-cell functions by human granulocyte arginase. Blood 2006, 108, 1627–1634. [Google Scholar] [CrossRef] [PubMed]
- Mollinedo, F. Neutrophil Degranulation, Plasticity, and Cancer Metastasis. Trends Immunol. 2019, 40, 228–242. [Google Scholar] [CrossRef]
- Wheatley, D.N. Arginine deprivation and metabolomics: Important aspects of intermediary metabolism in relation to the differential sensitivity of normal and tumour cells. Semin. Cancer Biol. 2005, 15, 247–253. [Google Scholar] [CrossRef]
- Wheatley, D.N.; Philip, R.; Campbell, E. Arginine deprivation and tumour cell death: Arginase and its inhibition. Mol. Cell Biochem. 2003, 244, 177–185. [Google Scholar] [CrossRef]
- García-Navas, R.; Gajate, C.; Mollinedo, F. Neutrophils drive endoplasmic reticulum stress-mediated apoptosis in cancer cells through arginase-1 release. Sci. Rep. 2021, 11, 12574. [Google Scholar] [CrossRef] [PubMed]
- Lecot, P.; Sarabi, M.; Pereira Abrantes, M.; Mussard, J.; Koenderman, L.; Caux, C.; Bendriss-Vermare, N.; Michallet, M.C. Neutrophil Heterogeneity in Cancer: From Biology to Therapies. Front. Immunol. 2019, 10, 2155. [Google Scholar] [CrossRef]
- Szczerba, B.M.; Castro-Giner, F.; Vetter, M.; Krol, I.; Gkountela, S.; Landin, J.; Scheidmann, M.C.; Donato, C.; Scherrer, R.; Singer, J.; et al. Neutrophils escort circulating tumour cells to enable cell cycle progression. Nature 2019, 566, 553–557. [Google Scholar] [CrossRef] [PubMed]
- Ogata, M.; Hino, S.; Saito, A.; Morikawa, K.; Kondo, S.; Kanemoto, S.; Murakami, T.; Taniguchi, M.; Tanii, I.; Yoshinaga, K.; et al. Autophagy is activated for cell survival after endoplasmic reticulum stress. Mol. Cell Biol. 2006, 26, 9220–9231. [Google Scholar] [CrossRef]
- Hoyer-Hansen, M.; Jaattela, M. Connecting endoplasmic reticulum stress to autophagy by unfolded protein response and calcium. Cell Death Differ. 2007, 14, 1576–1582. [Google Scholar] [CrossRef]
- Mizushima, N.; Yoshimori, T. How to interpret LC3 immunoblotting. Autophagy 2007, 3, 542–545. [Google Scholar] [CrossRef] [PubMed]
- Reid, M.D.; Basturk, O.; Thirabanjasak, D.; Hruban, R.H.; Klimstra, D.S.; Bagci, P.; Altinel, D.; Adsay, V. Tumor-infiltrating neutrophils in pancreatic neoplasia. Mod. Pathol. 2011, 24, 1612–1619. [Google Scholar] [CrossRef] [PubMed]
- Chao, T.; Furth, E.E.; Vonderheide, R.H. CXCR2-Dependent Accumulation of Tumor-Associated Neutrophils Regulates T-cell Immunity in Pancreatic Ductal Adenocarcinoma. Cancer Immunol. Res. 2016, 4, 968–982. [Google Scholar] [CrossRef]
- Wu, L.; Saxena, S.; Awaji, M.; Singh, R.K. Tumor-Associated Neutrophils in Cancer: Going Pro. Cancers (Basel) 2019, 11, 564. [Google Scholar] [CrossRef] [PubMed]
- Shaul, M.E.; Fridlender, Z.G. Tumour-associated neutrophils in patients with cancer. Nat. Rev. Clin. Oncol. 2019, 16, 601–620. [Google Scholar] [CrossRef] [PubMed]
- Huber, M.; Brehm, C.U.; Gress, T.M.; Buchholz, M.; Alashkar Alhamwe, B.; von Strandmann, E.P.; Slater, E.P.; Bartsch, J.W.; Bauer, C.; Lauth, M. The Immune Microenvironment in Pancreatic Cancer. Int. J. Mol. Sci. 2020, 21, 7307. [Google Scholar] [CrossRef]
- Zhang, Y.; Chandra, V.; Riquelme Sanchez, E.; Dutta, P.; Quesada, P.R.; Rakoski, A.; Zoltan, M.; Arora, N.; Baydogan, S.; Horne, W.; et al. Interleukin-17-induced neutrophil extracellular traps mediate resistance to checkpoint blockade in pancreatic cancer. J. Exp. Med. 2020, 217, e20190354. [Google Scholar] [CrossRef] [PubMed]
- Naso, J.R.; Topham, J.T.; Karasinska, J.M.; Lee, M.K.C.; Kalloger, S.E.; Wong, H.L.; Nelson, J.; Moore, R.A.; Mungall, A.J.; Jones, S.J.M.; et al. Tumor infiltrating neutrophils and gland formation predict overall survival and molecular subgroups in pancreatic ductal adenocarcinoma. Cancer Med. 2021, 10, 1155–1165. [Google Scholar] [CrossRef] [PubMed]
- Charles Jacob, H.K.; Charles Richard, J.L.; Signorelli, R.; Kashuv, T.; Lavania, S.; Vaish, U.; Boopathy, R.; Middleton, A.; Boone, M.M.; Sundaram, R.; et al. Modulation of Early Neutrophil Granulation: The Circulating Tumor Cell-Extravesicular Connection in Pancreatic Ductal Adenocarcinoma. Cancers (Basel) 2021, 13, 2727. [Google Scholar] [CrossRef]
- Carbone, D.; Parrino, B.; Cascioferro, S.; Pecoraro, C.; Giovannetti, E.; Di Sarno, V.; Musella, S.; Auriemma, G.; Cirrincione, G.; Diana, P. 1,2,4-Oxadiazole topsentin analogs with antiproliferative activity against pancreatic cancer cells, targeting GSK3β kinase. ChemMedChem 2021, 16, 537–554. [Google Scholar] [CrossRef]
- Cascioferro, S.; Li Petri, G.; Parrino, B.; Carbone, D.; Funel, N.; Bergonzini, C.; Mantini, G.; Dekker, H.; Geerke, D.; Peters, G.J.; et al. Imidazo [2,1-b][1,3,4]thiadiazoles with antiproliferative activity against primary and gemcitabine-resistant pancreatic cancer cells. Eur. J. Med. Chem. 2020, 189, 112088. [Google Scholar] [CrossRef]
- Li Petri, G.; Cascioferro, S.; El Hassouni, B.; Carbone, D.; Parrino, B.; Cirrincione, G.; Peters, G.J.; Diana, P.; Giovannetti, E. Biological evaluation of the antiproliferative and anti-migratory activity of a series of 3-(6-phenylimidazo[2,1-b][1,3,4]thiadiazol-2-yl)-1H-indole derivatives against pancreatic cancer cells. Anticancer Res. 2019, 39, 3615–3620. [Google Scholar] [CrossRef]
- Cascioferro, S.; Li Petri, G.; Parrino, B.; El Hassouni, B.; Carbone, D.; Arizza, V.; Perricone, U.; Padova, A.; Funel, N.; Peters, G.J.; et al. 3-(6-Phenylimidazo [2,1-b][1,3,4]thiadiazol-2-yl)-1H-indole derivatives as new anticancer agents in the treatment of pancreatic ductal adenocarcinoma. Molecules 2020, 25, 329. [Google Scholar] [CrossRef]











Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mollinedo, F.; Gajate, C. Direct Endoplasmic Reticulum Targeting by the Selective Alkylphospholipid Analog and Antitumor Ether Lipid Edelfosine as a Therapeutic Approach in Pancreatic Cancer. Cancers 2021, 13, 4173. https://doi.org/10.3390/cancers13164173
Mollinedo F, Gajate C. Direct Endoplasmic Reticulum Targeting by the Selective Alkylphospholipid Analog and Antitumor Ether Lipid Edelfosine as a Therapeutic Approach in Pancreatic Cancer. Cancers. 2021; 13(16):4173. https://doi.org/10.3390/cancers13164173
Chicago/Turabian StyleMollinedo, Faustino, and Consuelo Gajate. 2021. "Direct Endoplasmic Reticulum Targeting by the Selective Alkylphospholipid Analog and Antitumor Ether Lipid Edelfosine as a Therapeutic Approach in Pancreatic Cancer" Cancers 13, no. 16: 4173. https://doi.org/10.3390/cancers13164173
APA StyleMollinedo, F., & Gajate, C. (2021). Direct Endoplasmic Reticulum Targeting by the Selective Alkylphospholipid Analog and Antitumor Ether Lipid Edelfosine as a Therapeutic Approach in Pancreatic Cancer. Cancers, 13(16), 4173. https://doi.org/10.3390/cancers13164173