The 3.0 Cell Communication: New Insights in the Usefulness of Tunneling Nanotubes for Glioblastoma Treatment

 ,
,  , , , ,
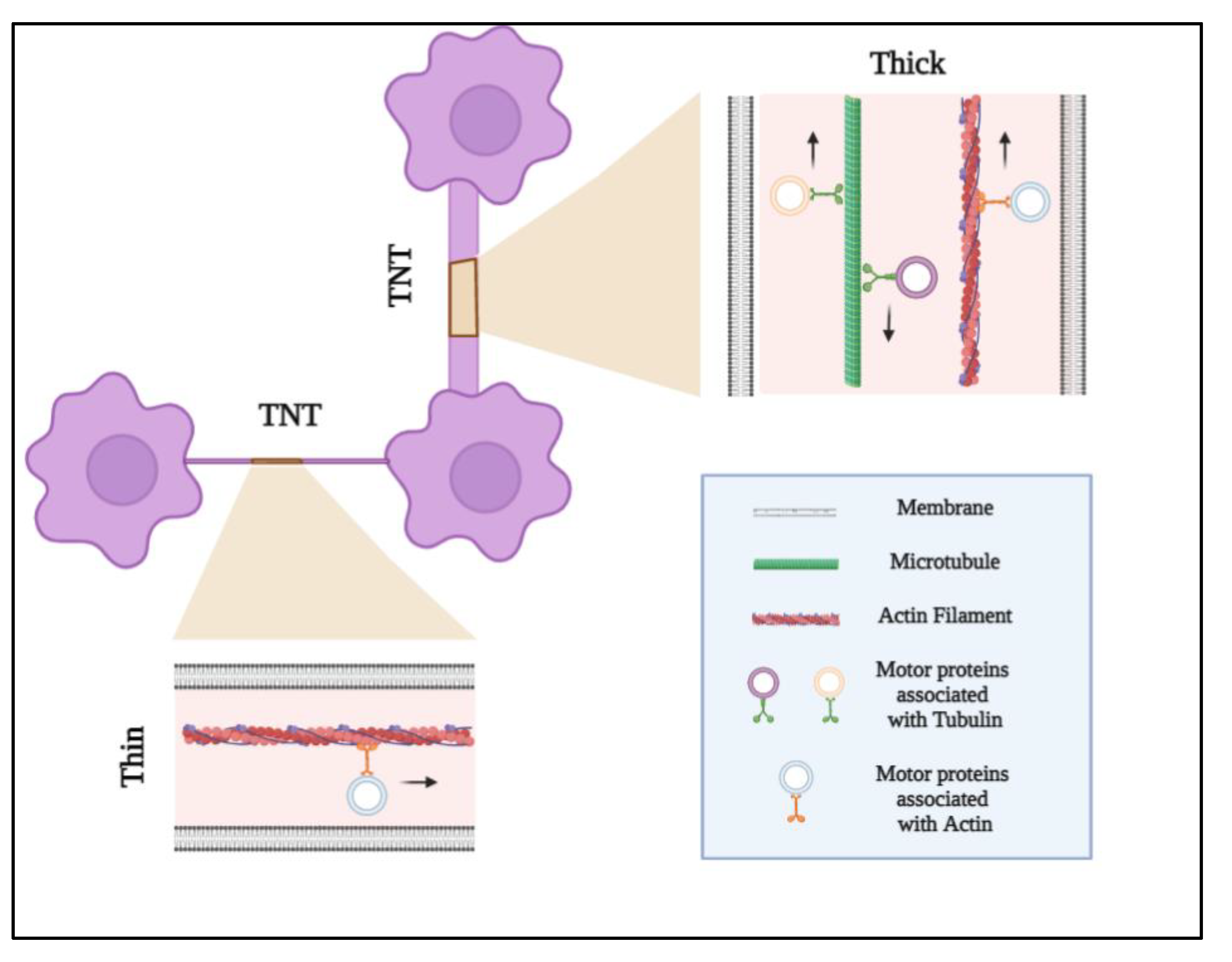
, , , , 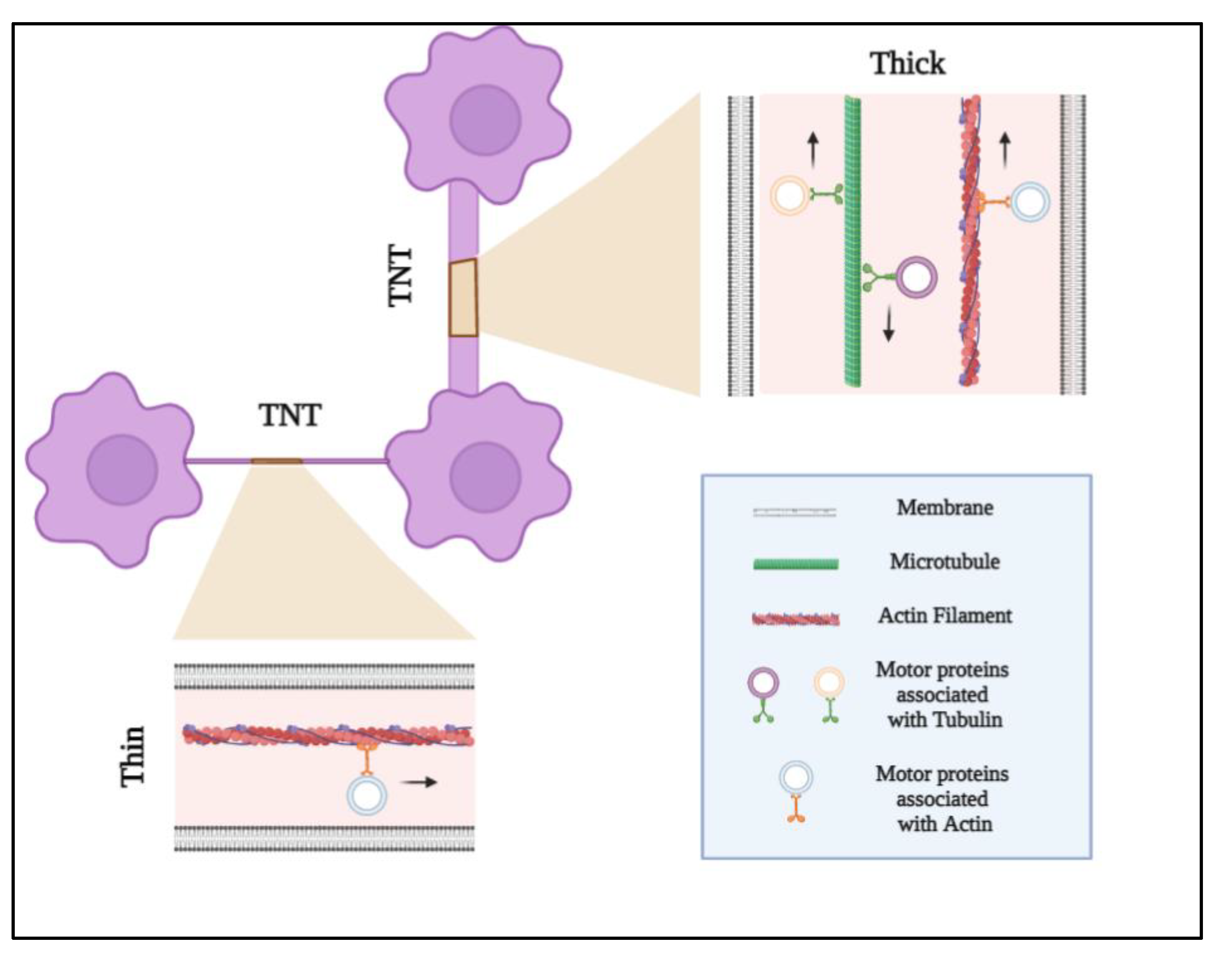{kind=link}
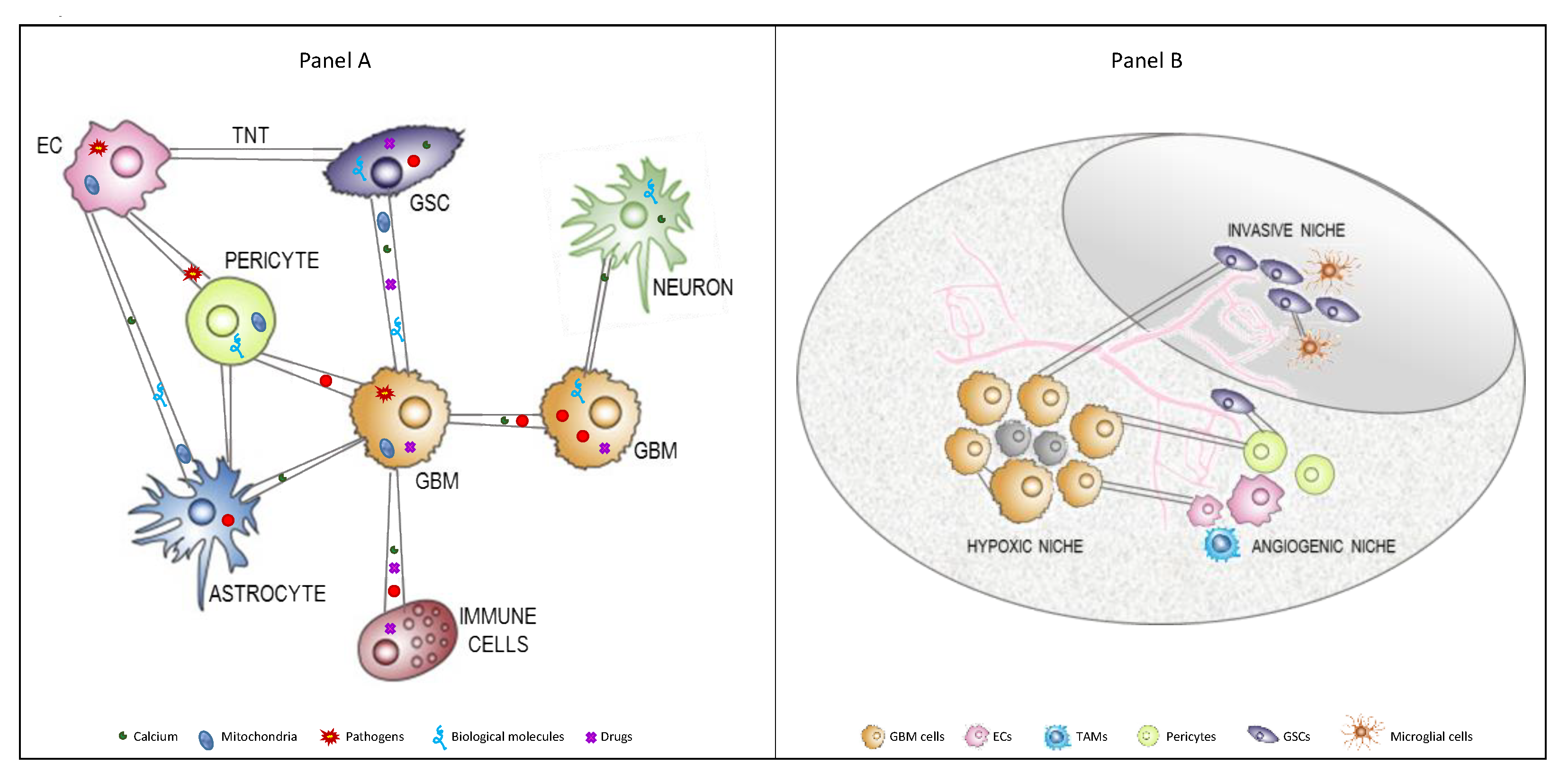{kind=link}
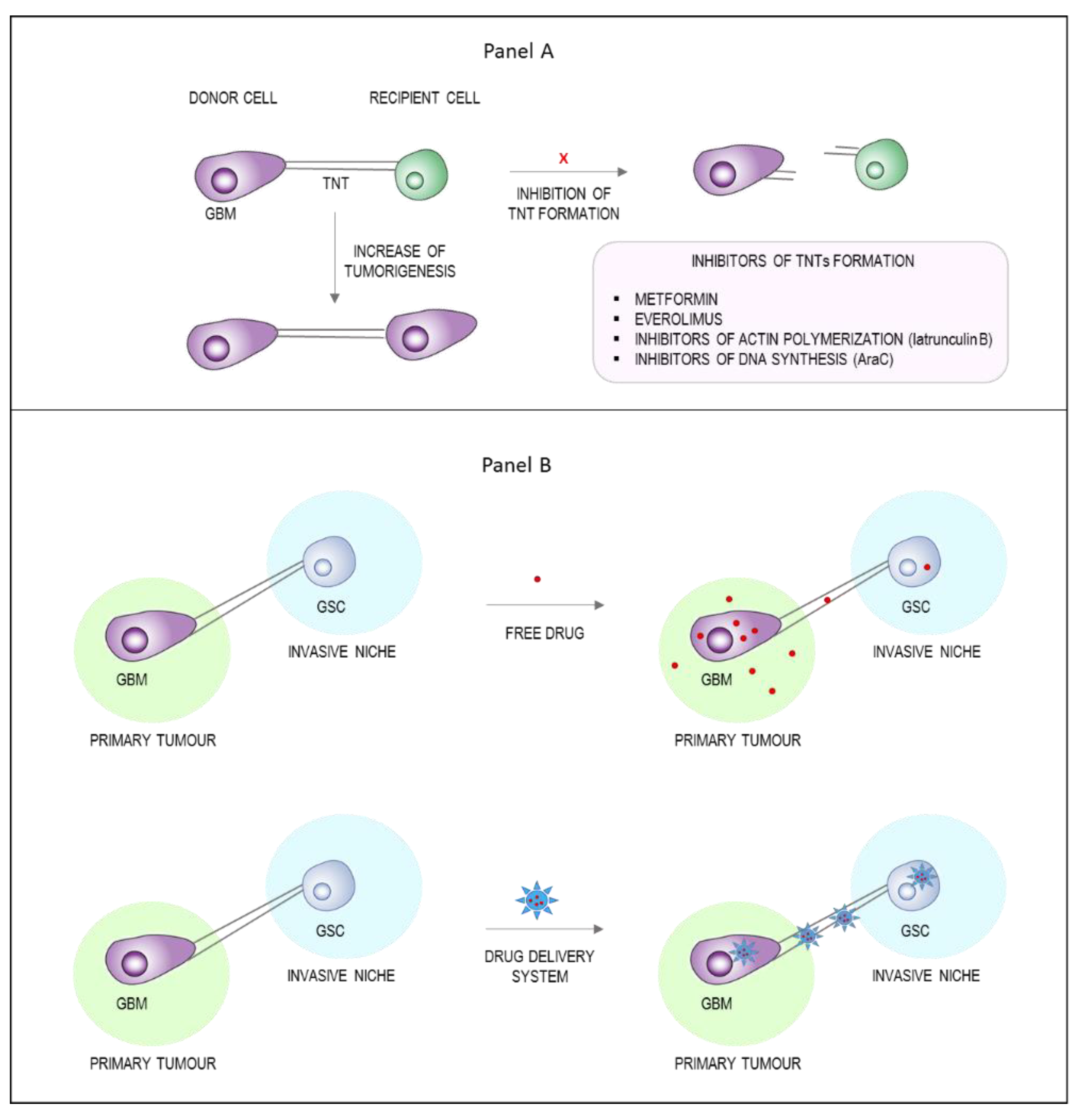{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
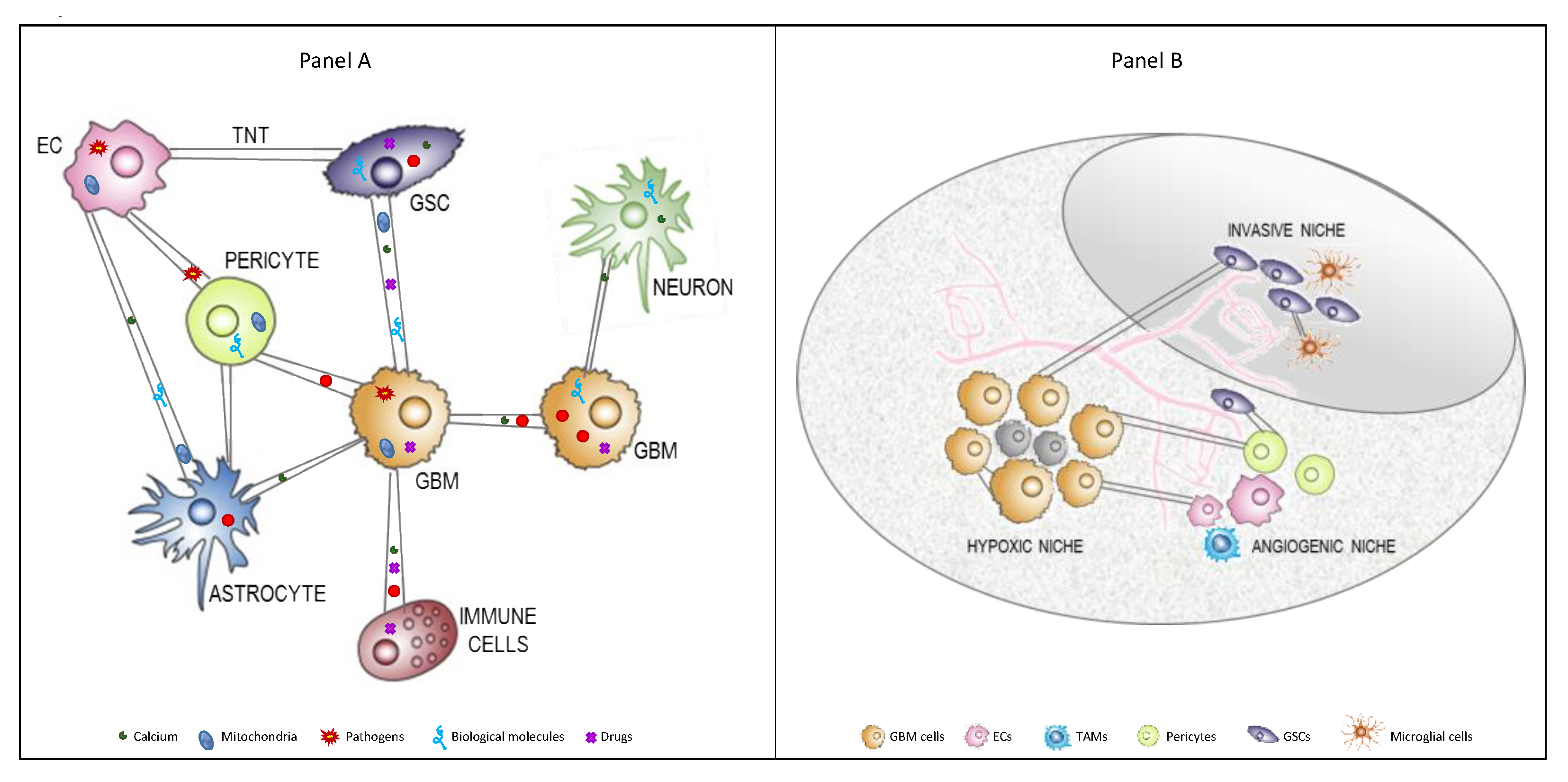
2. Main Cell Types Interacting in Glioblastoma Microenvironment
2.1. Tumor Immune Cells
2.2. Glioblastoma Stem Cells
2.3. Other Brain Cells
3. Cell Communication Modalities
4. Tunneling Nanotubes
4.1. TNT Discovery, Characteristics, and Classification
4.2. TNTs Formation
4.3. TNT Function, Biological Effects, and Mechanisms of Transport
5. Tunneling Nanotubes in Glioblastoma
5.1. Formation of TNTs in GBM
5.2. Role of TNTs in GBM
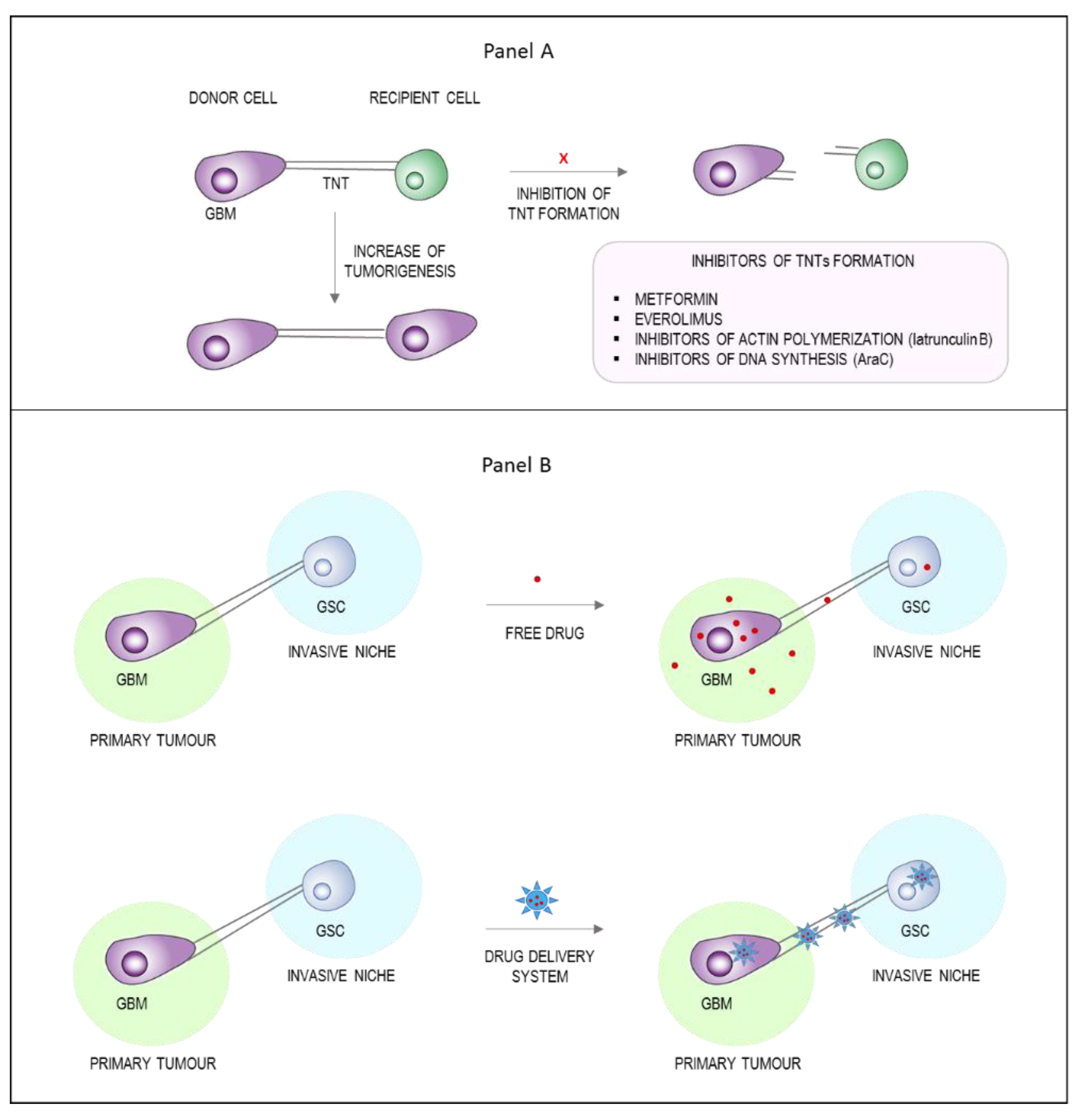
6. TNTs as a Novel Strategy to Enhance Tumor Drug Delivery
6.1. Inhibition of TNTs Formation
6.2. Exploiting TNTs as Drug Delivery Channels
6.3. Insights and TNTs Research Outlook
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gimple, R.C.; Bhargava, S.; Dixit, D.; Rich, J.N. Glioblastoma stem cells: Lessons from the tumor hierarchy in a lethal cancer. Genes Dev. 2019, 33, 591–609. [Google Scholar] [CrossRef]
- Hanif, F.; Muzaffar, K.; Perveen, K.; Malhi, S.; Simjee, S. Glioblastoma Multiforme: A Review of its Epidemiology and Pathogenesis through Clinical Presentation and Treatment. Asian Pac. J. Cancer Prev. 2017, 18, 3–9. [Google Scholar] [CrossRef]
- Fabian, D.; Eibl, M.D.P.G.P.; Alnahhas, I.; Sebastian, N.; Giglio, P.; Puduvalli, V.; Gonzalez, J.; Palmer, J.D. Treatment of Glioblastoma (GBM) with the Addition of Tumor-Treating Fields (TTF): A Review. Cancers 2019, 11, 174. [Google Scholar] [CrossRef] [Green Version]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weller, M.; Bent, M.V.D.; Preusser, M.; Le Rhun, E.; Tonn, J.C.; Minniti, G.; Bendszus, M.; Balana, C.; Chinot, O.; Dirven, L.; et al. EANO guidelines on the diagnosis and treatment of diffuse gliomas of adulthood. Nat. Rev. Clin. Oncol. 2021, 18, 170–186. [Google Scholar] [CrossRef] [PubMed]
- Westphal, M.; Lamszus, K. The neurobiology of gliomas: From cell biology to the development of therapeutic approaches. Nat. Rev. Neurosci. 2011, 12, 495–508. [Google Scholar] [CrossRef]
- Komotar, R.J.; Otten, M.L.; Moise, G.; Connolly, J.E.S. Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma—A Critical Review. Clin. Med. Oncol. 2008, 2, 421–422. [Google Scholar] [CrossRef] [PubMed]
- Weller, M.; Cloughesy, T.; Perry, J.R.; Wick, W. Standards of care for treatment of recurrent glioblastoma—Are we there yet? Neuro-Oncology 2013, 15, 4–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stupp, R.; Hegi, M.; Mason, W.P.; Bent, M.V.D.; Taphoorn, M.J.; Janzer, R.C.; Ludwin, S.K.; Allgeier, A.; Fisher, B.; Belanger, K.; et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009, 10, 459–466. [Google Scholar] [CrossRef]
- Ohka, F.; Natsume, A.; Wakabayashi, T. Current Trends in Targeted Therapies for Glioblastoma Multiforme. Neurol. Res. Int. 2012, 2012, 1–13. [Google Scholar] [CrossRef]
- Thakkar, J.P.; Dolecek, T.A.; Horbinski, C.; Ostrom, Q.; Lightner, D.D.; Barnholtz-Sloan, J.; Villano, J.L. Epidemiologic and Molecular Prognostic Review of Glioblastoma. Cancer Epidemiol. Biomark. Prev. 2014, 23, 1985–1996. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Lee, I.-H.; Cho, H.J.; Park, C.-K.; Jung, Y.-S.; Kim, Y.; Nam, S.H.; Kim, B.S.; Johnson, M.D.; Kong, D.-S.; et al. Spatiotemporal Evolution of the Primary Glioblastoma Genome. Cancer Cell 2015, 28, 318–328. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.; Zheng, S.; Amini, S.S.; Virk, S.M.; Mikkelsen, T.; Brat, D.J.; Grimsby, J.; Sougnez, C.; Muller, F.; Hu, J.; et al. Whole-genome and multisector exome sequencing of primary and post-treatment glioblastoma reveals patterns of tumor evolution. Genome Res. 2015, 25, 316–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perrin, S.L.; Samuel, M.S.; Koszyca, B.; Brown, M.P.; Ebert, L.M.; Oksdath, M.; Gomez, G.A. Glioblastoma heterogeneity and the tumour microenvironment: Implications for preclinical research and development of new treatments. Biochem. Soc. Trans. 2019, 47, 625–638. [Google Scholar] [CrossRef] [PubMed]
- Venkatesh, V.S.; Lou, E. Tunneling nanotubes: A bridge for heterogeneity in glioblastoma and a new therapeutic target? Cancer Rep. 2019, 2, e1185. [Google Scholar] [CrossRef] [Green Version]
- Sahebjam, S.; Sharabi, A.; Lim, M.; Kesarwani, P.; Chinnaiyan, P. Immunotherapy and radiation in glioblastoma. J. Neurooncol. 2017, 134, 531–539. [Google Scholar] [CrossRef] [PubMed]
- De Leo, A.; Ugolini, A.; Veglia, F. Myeloid Cells in Glioblastoma Microenvironment. Cells 2020, 10, 18. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Feng, X.; Herting, C.J.; Garcia, V.A.; Nie, K.; Pong, W.W.; Rasmussen, R.; Dwivedi, B.; Seby, S.; Wolf, S.A.; et al. Cellular and Molecular Identity of Tumor-Associated Macrophages in Glioblastoma. Cancer Res. 2017, 77, 2266–2278. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Guo, G.; Guan, H.; Yu, Y.; Lu, J.; Yu, J. Challenges and potential of PD-1/PD-L1 checkpoint blockade immunotherapy for glioblastoma. J. Exp. Clin. Cancer Res. 2019, 38, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Goods, B.A.; Hernandez, A.L.; Lowther, D.E.; Lucca, L.E.; Lerner, B.A.; Gunel, M.; Raddassi, K.; Coric, V.; Hafler, D.A.; Love, J. Functional differences between PD-1+ and PD-1- CD4+ effector T cells in healthy donors and patients with glioblastoma multiforme. PLoS ONE 2017, 12, e0181538. [Google Scholar] [CrossRef] [Green Version]
- Bardhan, K.; Anagnostou, T.; Boussiotis, V.A. The PD1:PD-L1/2 Pathway from Discovery to Clinical Implementation. Front. Immunol. 2016, 7, 550. [Google Scholar] [CrossRef] [Green Version]
- Litak, J.; Mazurek, M.; Grochowski, C.; Kamieniak, P.; Roliński, J. PD-L1/PD-1 Axis in Glioblastoma Multiforme. Int. J. Mol. Sci. 2019, 20, 5347. [Google Scholar] [CrossRef] [Green Version]
- Taiarol, L.; Formicola, B.; Magro, R.D.; Sesana, S.; Re, F. An update of nanoparticle-based approaches for glioblastoma multiforme immunotherapy. Nanomed. 2020, 15, 1861–1871. [Google Scholar] [CrossRef] [PubMed]
- Grauer, O.M.; Nierkens, S.; Bennink, E.; Toonen, L.W.; Boon, L.; Wesseling, P.; Sutmuller, R.P.; Adema, G.J. CD4+FoxP3+ regulatory T cells gradually accumulate in gliomas during tumor growth and efficiently suppress antiglioma immune responsesin vivo. Int. J. Cancer 2007, 121, 95–105. [Google Scholar] [CrossRef] [PubMed]
- Jain, P.; Coisne, C.; Enzmann, G.; Rottapel, R.; Engelhardt, B. α4β1Integrin Mediates the Recruitment of Immature Dendritic Cells across the Blood-Brain Barrier during Experimental Autoimmune Encephalomyelitis. J. Immunol. 2010, 184, 7196–7206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, S.D.; McMahon, E.J.; Schreiner, B.; Bailey, S.L. Antigen Presentation in the CNS by Myeloid Dendritic Cells Drives Progression of Relapsing Experimental Autoimmune Encephalomyelitis. Ann. N. Y. Acad. Sci. 2007, 1103, 179–191. [Google Scholar] [CrossRef] [PubMed]
- D’Agostino, P.M.; Gottfried-Blackmore, A.; Anandasabapathy, N.; Bulloch, K. Brain dendritic cells: Biology and pathology. Acta Neuropathol. 2012, 124, 599–614. [Google Scholar] [CrossRef]
- Klemm, F.; Maas, R.R.; Bowman, R.L.; Kornete, M.; Soukup, K.; Nassiri, S.; Brouland, J.-P.; Iacobuzio-Donahue, C.A.; Brennan, C.; Tabar, V.; et al. Interrogation of the Microenvironmental Landscape in Brain Tumors Reveals Disease-Specific Alterations of Immune Cells. Cell 2020, 181, 1643–1660.e17. [Google Scholar] [CrossRef]
- Friebel, E.; Kapolou, K.; Unger, S.; Núñez, N.G.; Utz, S.; Rushing, E.J.; Regli, L.; Weller, M.; Greter, M.; Tugues, S.; et al. Single-Cell Mapping of Human Brain Cancer Reveals Tumor-Specific Instruction of Tissue-Invading Leukocytes. Cell 2020, 181, 1626–1642.e20. [Google Scholar] [CrossRef]
- Pappolla, M.; Sambamurti, K.; Vidal, R.; Pacheco-Quinto, J.; Poeggeler, B.; Matsubara, E. Evidence for lymphatic Aβ clearance in Alzheimer’s transgenic mice. Neurobiol. Dis. 2014, 71, 215–219. [Google Scholar] [CrossRef] [Green Version]
- Cserr, H.F.; Harling-Berg, C.J.; Knopf, P.M. Drainage of Brain Extracellular Fluid into Blood and Deep Cervical Lymph and its Immunological Significance. Brain Pathol. 1992, 2, 269–276. [Google Scholar] [CrossRef]
- Louveau, A.; Harris, T.; Kipnis, J. Revisiting the Mechanisms of CNS Immune Privilege. Trends Immunol. 2015, 36, 569–577. [Google Scholar] [CrossRef] [Green Version]
- Iliff, J.J.; Wang, M.; Liao, Y.; Plogg, B.A.; Peng, W.; Gundersen, G.A.; Benveniste, H.; Vates, G.E.; Deane, R.; Goldman, S.; et al. A Paravascular Pathway Facilitates CSF Flow Through the Brain Parenchyma and the Clearance of Interstitial Solutes, Including Amyloid. Sci. Transl. Med. 2012, 4, 147ra111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Louveau, A.; Smirnov, I.; Keyes, T.; Eccles, J.; Rouhani, S.; Peske, J.D.; Derecki, N.C.; Castle, D.; Mandell, J.W.; Lee, K.S.; et al. Structural and functional features of central nervous system lymphatic vessels. Nat. Cell Biol. 2015, 523, 337–341. [Google Scholar] [CrossRef]
- Louveau, A.; Plog, B.A.; Antila, S.; Alitalo, K.; Nedergaard, M.; Kipnis, J. Understanding the functions and relationships of the glymphatic system and meningeal lymphatics. J. Clin. Investig. 2017, 127, 3210–3219. [Google Scholar] [CrossRef] [Green Version]
- Broz, M.L.; Binnewies, M.; Boldajipour, B.; Nelson, A.E.; Pollack, J.L.; Erle, D.; Barczak, A.; Rosenblum, M.D.; Daud, A.; Barber, D.L.; et al. Dissecting the Tumor Myeloid Compartment Reveals Rare Activating Antigen-Presenting Cells Critical for T Cell Immunity. Cancer Cell 2014, 26, 638–652. [Google Scholar] [CrossRef] [Green Version]
- Salmon, H.; Idoyaga, J.; Rahman, A.; Leboeuf, M.; Remark, R.; Jordan, S.; Casanova-Acebes, M.; Khudoynazarova, M.; Agudo, J.; Tung, N.; et al. Expansion and Activation of CD103 + Dendritic Cell Progenitors at the Tumor Site Enhances Tumor Responses to Therapeutic PD-L1 and BRAF Inhibition. Immunity 2016, 44, 924–938. [Google Scholar] [CrossRef] [Green Version]
- Sánchez-Paulete, A.R.; Cueto, F.J.; Lopez, M.M.; Labiano, S.; Morales-Kastresana, A.; E Rodriguez-Ruiz, M.; Jure-Kunkel, M.; Azpilikueta, A.; Aznar, M.A.; Quetglas, J.I.; et al. Cancer Immunotherapy with Immunomodulatory Anti-CD137 and Anti–PD-1 Monoclonal Antibodies Requires BATF3-Dependent Dendritic Cells. Cancer Discov. 2016, 6, 71–79. [Google Scholar] [CrossRef] [Green Version]
- Verhaak, R.G.; Hoadley, K.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated Genomic Analysis Identifies Clinically Relevant Subtypes of Glioblastoma Characterized by Abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galli, R.; Binda, E.; Orfanelli, U.; Cipelletti, B.; Gritti, A.; De Vitis, S.; Fiocco, R.; Foroni, C.; DiMeco, F.; Vescovi, A. Isolation and Characterization of Tumorigenic, Stem-like Neural Precursors from Human Glioblastoma. Cancer Res. 2004, 64, 7011–7021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spinelli, C.; Montermini, L.; Meehan, B.; Brisson, A.R.; Tan, S.; Choi, D.; Nakano, I.; Rak, J. Molecular subtypes and differentiation programmes of glioma stem cells as determinants of extracellular vesicle profiles and endothelial cell-stimulating activities. J. Extracell. Vesicles 2018, 7, 1490144. [Google Scholar] [CrossRef]
- Bao, S.; Wu, Q.; Sathornsumetee, S.; Hao, Y.; Li, Z.; Hjelmeland, A.B.; Shideng, B.; McLendon, R.E.; Bigner, D.D.; Rich, J.N. Stem Cell–like Glioma Cells Promote Tumor Angiogenesis through Vascular Endothelial Growth Factor. Cancer Res. 2006, 66, 7843–7848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Folkins, C.; Shaked, Y.; Man, S.; Tang, T.; Lee, C.R.; Zhu, Z.; Hoffman, R.M.; Kerbel, R.S. Glioma Tumor Stem-Like Cells Promote Tumor Angiogenesis and Vasculogenesis via Vascular Endothelial Growth Factor and Stromal-Derived Factor 1. Cancer Res. 2009, 69, 7243–7251. [Google Scholar] [CrossRef] [Green Version]
- Virolle, T. Cellules souches cancéreuses dans les glioblastomes. Bull Cancer 2017, 104, 1075–1079. [Google Scholar] [CrossRef] [PubMed]
- Peleli, M.; Moustakas, A.; Papapetropoulos, A. Endothelial-Tumor Cell Interaction in Brain and CNS Malignancies. Int. J. Mol. Sci. 2020, 21, 7371. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Cortés, M.; Bellido, D.D.; Oliver, F.J. Vasculogenic Mimicry: Become an Endothelial Cell “But Not So Much”. Front. Oncol. 2019, 9, 803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vitiani, L.R.; Pallini, R.; Biffoni, M.; Todaro, M.; Invernici, G.; Cenci, T.; Maira, G.; Parati, E.A.; Stassi, G.; Larocca, L.M.; et al. Tumour vascularization via endothelial differentiation of glioblastoma stem-like cells. Nat. Cell Biol. 2010, 468, 824–830. [Google Scholar] [CrossRef]
- Badr, C.E.; Silver, D.J.; Siebzehnrubl, F.A.; Deleyrolle, L.P. Metabolic heterogeneity and adaptability in brain tumors. Cell. Mol. Life Sci. 2020, 77, 5101–5119. [Google Scholar] [CrossRef] [PubMed]
- Di Tomaso, T.; Mazzoleni, S.; Wang, E.; Sovena, G.; Clavenna, D.; Franzin, A.; Mortini, P.; Ferrone, S.; Doglioni, C.; Marincola, F.M.; et al. Immunobiological Characterization of Cancer Stem Cells Isolated from Glioblastoma Patients. Clin. Cancer Res. 2010, 16, 800–813. [Google Scholar] [CrossRef] [Green Version]
- Gilbertson, R.J.; Rich, J.N. Making a tumour’s bed: Glioblastoma stem cells and the vascular niche. Nat. Rev. Cancer 2007, 7, 733–736. [Google Scholar] [CrossRef]
- Norden, A.D.; Drappatz, J.; Wen, P.Y. Novel anti-angiogenic therapies for malignant gliomas. Lancet Neurol. 2008, 7, 1152–1160. [Google Scholar] [CrossRef]
- Grabner, G.; Nöbauer, I.; Elandt, K.; Kronnerwetter, C.; Woehrer, A.; Marosi, C.; Prayer, D.; Trattnig, S.; Preusser, M. Longitudinal brain imaging of five malignant glioma patients treated with bevacizumab using susceptibility-weighted magnetic resonance imaging at 7 T. Magn. Reson. Imaging 2012, 30, 139–147. [Google Scholar] [CrossRef]
- Chamberlain, M.C.; Neuwelt, E.A.; Thompson, E.M.; Frenkel, E.P. The paradoxical effect of bevacizumab in the therapy of malignant gliomas. Neurology 2011, 77, 803–804. [Google Scholar] [CrossRef]
- Zhang, H.; Zhou, Y.; Cui, B.; Liu, Z.; Shen, H. Novel insights into astrocyte-mediated signaling of proliferation, invasion and tumor immune microenvironment in glioblastoma. Biomed. Pharmacother. 2020, 126, 110086. [Google Scholar] [CrossRef] [PubMed]
- Biasoli, D.; Sobrinho, M.F.; Da Fonseca, A.C.C.; De Matos, D.G.; Romao, L.; Maciel, R.D.M.; Rehen, S.; Moura-Neto, V.; Borges, H.L.; Lima, F.R.S. Glioblastoma cells inhibit astrocytic p53-expression favoring cancer malignancy. Oncogenesis 2014, 3, e123. [Google Scholar] [CrossRef] [Green Version]
- Broekman, M.L.; Maas, S.L.N.; Abels, E.R.; Mempel, T.R.; Krichevsky, A.M.; Breakefield, X.O. Multidimensional communication in the microenvirons of glioblastoma. Nat. Rev. Neurol. 2018, 14, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Lawn, S.; Krishna, N.; Pisklakova, A.; Qu, X.; Fenstermacher, D.A.; Fournier, M.; Vrionis, F.D.; Tran, N.; Chan, J.A.; Kenchappa, R.S.; et al. Neurotrophin Signaling via TrkB and TrkC Receptors Promotes the Growth of Brain Tumor-initiating Cells. J. Biol. Chem. 2015, 290, 3814–3824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venkatesh, H.S.; Johung, T.B.; Caretti, V.; Noll, A.; Tang, Y.; Nagaraja, S.; Gibson, E.M.; Mount, C.; Polepalli, J.; Mitra, S.S.; et al. Neuronal Activity Promotes Glioma Growth through Neuroligin-3 Secretion. Cell 2015, 161, 803–816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dolma, S.; Selvadurai, H.J.; Lan, X.; Lee, L.; Kushida, M.; Voisin, V.; Whetstone, H.; So, M.; Aviv, T.; Park, N.; et al. Inhibition of Dopamine Receptor D4 Impedes Autophagic Flux, Proliferation, and Survival of Glioblastoma Stem Cells. Cancer Cell 2016, 29, 859–873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gillespie, S.; Monje, M. An active role for neurons in glioma progression: Making sense of Scherer’s structures. Neuro-Oncology 2018, 20, 1292–1299. [Google Scholar] [CrossRef] [PubMed]
- Hong, X.; Sin, W.C.; Harris, A.; Naus, C.C. Gap junctions modulate glioma invasion by direct transfer of microRNA. Oncotarget 2015, 6, 15566–15577. [Google Scholar] [CrossRef] [Green Version]
- Balça-Silva, J.; Matias, D.; Dubois, L.G.; Carneiro, B.; Carmo, A.D.; Girão, H.; Ferreira, F.; Ferrer, V.P.; Chimelli, L.; Filho, P.N.; et al. The Expression of Connexins and SOX2 Reflects the Plasticity of Glioma Stem-Like Cells. Transl. Oncol. 2017, 10, 555–569. [Google Scholar] [CrossRef] [PubMed]
- Sinyuk, M.; Mulkearns-Hubert, E.; Reizes, O.; Lathia, J. Cancer Connectors: Connexins, Gap Junctions, and Communication. Front. Oncol. 2018, 8, 646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Q.; Boire, A.; Jin, X.; Valiente, M.; Er, E.E.; Lopez-Soto, A.; Jacob, L.S.; Patwa, R.; Shah, H.; Xu, K.; et al. Carcinoma–astrocyte gap junctions promote brain metastasis by cGAMP transfer. Nat. Cell Biol. 2016, 533, 493–498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Artesi, M.; Kroonen, J.; Bredel, M.; Nguyen-Khac, M.; Deprez, M.; Schoysman, L.; Poulet, C.; Chakravarti, A.; Kim, H.; Scholtens, D.; et al. Connexin 30 expression inhibits growth of human malignant gliomas but protects them against radiation therapy. Neuro-Oncology 2015, 17, 392–406. [Google Scholar] [CrossRef] [PubMed]
- Aasen, T.; Leithe, E.; Graham, S.V.; Kameritsch, P.; Mayan, M.D.; Mesnil, M.; Pogoda, K.; Tabernero, A. Connexins in cancer: Bridging the gap to the clinic. Oncogene 2019, 38, 4429–4451. [Google Scholar] [CrossRef] [Green Version]
- Valdebenito, S.; Audia, A.; Bhat, K.P.; Okafo, G.; Eugenin, E.A. Tunneling Nanotubes Mediate Adaptation of Glioblastoma Cells to Temozolomide and Ionizing Radiation Treatment. iScience 2020, 23, 101450. [Google Scholar] [CrossRef] [PubMed]
- Yekula, A.; Yekula, A.; Muralidharan, K.; Kang, K.; Carter, B.S.; Balaj, L. Extracellular Vesicles in Glioblastoma Tumor Microenvironment. Front. Immunol. 2020, 10, 3137. [Google Scholar] [CrossRef]
- Zaborowski, M.; Balaj, L.; Breakefield, X.O.; Lai, C.P.-K. Extracellular Vesicles: Composition, Biological Relevance, and Methods of Study. Bioscience 2015, 65, 783–797. [Google Scholar] [CrossRef] [Green Version]
- Matarredona, E.R.; Pastor, A.M. Extracellular Vesicle-Mediated Communication between the Glioblastoma and Its Microenvironment. Cells 2019, 9, 96. [Google Scholar] [CrossRef] [Green Version]
- Fonseca, P.; Vardaki, I.; Occhionero, A.; Panaretakis, T. Metabolic and Signaling Functions of Cancer Cell-Derived Extracellular Vesicles. Int. Rev. Cell Mol. Biol. 2016, 326, 175–199. [Google Scholar]
- D’Asti, E.; Chennakrishnaiah, S.; Lee, T.H.; Rak, J. Extracellular Vesicles in Brain Tumor Progression. Cell. Mol. Neurobiol. 2016, 36, 383–407. [Google Scholar] [CrossRef]
- Pace, K.R.; Dutt, R.; Galileo, D.S. Exosomal L1CAM Stimulates Glioblastoma Cell Motility, Proliferation, and Invasiveness. Int. J. Mol. Sci. 2019, 20, 3982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lane, R.; Simon, T.; Vintu, M.; Solkin, B.; Koch, B.; Stewart, N.; Benstead-Hume, G.; Pearl, F.M.G.; Critchley, G.; Stebbing, J.; et al. Cell-derived extracellular vesicles can be used as a biomarker reservoir for glioblastoma tumor subtyping. Commun. Biol. 2019, 2, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Himes, B.T.; E Peterson, T.; De Mooij, T.; Garcia, M.L.M.C.; Jung, M.-Y.; Uhm, S.; Yan, D.; Tyson, J.; Jin-Lee, H.J.; Parney, D.; et al. The role of extracellular vesicles and PD-L1 in glioblastoma-mediated immunosuppressive monocyte induction. Neuro-Oncology 2020, 22, 967–978. [Google Scholar] [CrossRef]
- Rustom, A.; Saffrich, R.; Markovic, I.; Walther, P.; Gerdes, H.-H. Nanotubular Highways for Intercellular Organelle Transport. Science 2004, 303, 1007–1010. [Google Scholar] [CrossRef] [Green Version]
- Gurke, S.; Barroso, J.F.V.; Gerdes, H.-H. The art of cellular communication: Tunneling nanotubes bridge the divide. Histochem. Cell Biol. 2008, 129, 539–550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veranič, P.; Lokar, M.; Schütz, G.J.; Weghuber, J.; Wieser, S.; Hägerstrand, H.; Kralj-Iglič, V.; Iglič, A. Different Types of Cell-to-Cell Connections Mediated by Nanotubular Structures. Biophys. J. 2008, 95, 4416–4425. [Google Scholar] [CrossRef] [Green Version]
- Errede, M.; Mangieri, D.; Longo, G.; Girolamo, F.; De Trizio, I.; Vimercati, A.; Serio, G.; Frei, K.; Perris, R.; Virgintino, D. Tunneling nanotubes evoke pericyte/endothelial communication during normal and tumoral angiogenesis. Fluids Barriers CNS 2018, 15, 28. [Google Scholar] [CrossRef] [Green Version]
- Drab, M.; Stopar, D.; Kralj-Iglič, V.; Iglič, A. Inception Mechanisms of Tunneling Nanotubes. Cells 2019, 8, 626. [Google Scholar] [CrossRef] [Green Version]
- Önfelt, B.; Nedvetzki, S.; Benninger, R.; Purbhoo, M.A.; Sowinski, S.; Hume, A.; Seabra, M.; Neil, M.; French, P.; Davis, D. Structurally Distinct Membrane Nanotubes between Human Macrophages Support Long-Distance Vesicular Traffic or Surfing of Bacteria. J. Immunol. 2006, 177, 8476–8483. [Google Scholar] [CrossRef] [Green Version]
- Vidulescu, C.; Clejan, S.; O’Connor, K.C. Vesicle traffic through intercellular bridges in DU 145 human prostate cancer cells. J. Cell. Mol. Med. 2004, 8, 388–396. [Google Scholar] [CrossRef]
- Önfelt, B.; Nedvetzki, S.; Yanagi, K.; Davis, D. Cutting Edge: Membrane Nanotubes Connect Immune Cells. J. Immunol. 2004, 173, 1511–1513. [Google Scholar] [CrossRef] [Green Version]
- Quah, B.J.C.; Barlow, V.P.; McPhun, V.; Matthaei, K.I.; Hulett, M.; Parish, C.R. Bystander B cells rapidly acquire antigen receptors from activated B cells by membrane transfer. Proc. Natl. Acad. Sci. USA 2008, 105, 4259–4264. [Google Scholar] [CrossRef] [Green Version]
- Chastagner, P.; Loria, F.; Vargas, J.Y.; Tois, J.; I Diamond, M.; Okafo, G.; Brou, C.; Zurzolo, C. Fate and propagation of endogenously formed Tau aggregates in neuronal cells. EMBO Mol. Med. 2020, 12, e12025. [Google Scholar] [CrossRef] [PubMed]
- Lou, E.; Fujisawa, S.; Morozov, A.; Barlas, A.; Romin, Y.; Dogan, Y.; Gholami, S.; Moreira, A.L.; Manova-Todorova, K.; Moore, M.A.S. Tunneling Nanotubes Provide a Unique Conduit for Intercellular Transfer of Cellular Contents in Human Malignant Pleural Mesothelioma. PLoS ONE 2012, 7, e33093. [Google Scholar] [CrossRef] [Green Version]
- Kretschmer, A.; Zhang, F.; Somasekharan, S.P.; Tse, C.; Leachman, L.; Gleave, A.; Li, B.; Asmaro, I.; Huang, T.; Kotula, L.; et al. Stress-induced tunneling nanotubes support treatment adaptation in prostate cancer. Sci. Rep. 2019, 9, 7826. [Google Scholar] [CrossRef]
- Chinnery, H.R.; Pearlman, E.; McMenamin, P.G. Cutting Edge: Membrane Nanotubes In Vivo: A Feature of MHC Class II+ Cells in the Mouse Cornea. J. Immunol. 2008, 180, 5779–5783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goodman, S.; Naphade, S.; Khan, M.; Sharma, J.; Cherqui, S. Macrophage polarization impacts tunneling nanotube formation and intercellular organelle trafficking. Sci. Rep. 2019, 9, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jackson, M.V.; Morrison, T.J.; Doherty, D.F.; McAuley, D.; Matthay, M.A.; Kissenpfennig, A.; O’Kane, C.; Krasnodembskaya, A.D. Mitochondrial Transfer via Tunneling Nanotubes is an Important Mechanism by Which Mesenchymal Stem Cells Enhance Macrophage Phagocytosis in the In Vitro and In Vivo Models of ARDS. Stem Cells 2016, 34, 2210–2223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerdes, H.-H.; Rustom, A.; Wang, X. Tunneling nanotubes, an emerging intercellular communication route in development. Mech. Dev. 2013, 130, 381–387. [Google Scholar] [CrossRef] [PubMed]
- Abounit, S.; Zurzolo, C. Wiring through tunneling nanotubes—From electrical signals to organelle transfer. J. Cell Sci. 2012, 125, 1089–1098. [Google Scholar] [CrossRef] [Green Version]
- Davis, D.M.; Sowinski, S. Membrane nanotubes: Dynamic long-distance connections between animal cells. Nat. Rev. Mol. Cell Biol. 2008, 9, 431–436. [Google Scholar] [CrossRef] [PubMed]
- Thayanithy, V.; Babatunde, V.; Dickson, E.L.; Wong, P.; Oh, S.; Ke, X.; Barlas, A.; Fujisawa, S.; Romin, Y.; Moreira, A.L.; et al. Tumor exosomes induce tunneling nanotubes in lipid raft-enriched regions of human mesothelioma cells. Exp. Cell Res. 2014, 323, 178–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kabaso, D.; Lokar, M.; Kralj-Iglic, V.; Veranic, P.; Iglic, A. Temperature and cholera toxin B are factors that influence formation of membrane nanotubes in RT4 and T24 urothelial cancer cell lines. Int. J. Nanomed. 2011, 6, 495–509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lokar, M.; Iglič, A.; Veranič, P. Protruding membrane nanotubes: Attachment of tubular protrusions to adjacent cells by several anchoring junctions. Protoplasma 2010, 246, 81–87. [Google Scholar] [CrossRef]
- Okafo, G.; Prevedel, L.; Eugenin, E. Tunneling nanotubes (TNT) mediate long-range gap junctional communication: Implications for HIV cell to cell spread. Sci. Rep. 2017, 7, 1–9. [Google Scholar] [CrossRef]
- Dubois, F.; Jean-Jacques, B.; Roberge, H.; Bénard, M.; Galas, L.; Schapman, D.; Elie, N.; Goux, D.; Keller, M.; Maille, E.; et al. A role for RASSF1A in tunneling nanotube formation between cells through GEFH1/Rab11 pathway control. Cell Commun. Signal. 2018, 16, 66. [Google Scholar] [CrossRef] [Green Version]
- Bénard, M.; Schapman, D.; Lebon, A.; Monterroso, B.; Bellenger, M.; Le Foll, F.; Pasquier, J.; Vaudry, H.; Vaudry, D.; Galas, L. Structural and functional analysis of tunneling nanotubes (TnTs) usinggCW STED andgconfocal approaches. Biol. Cell 2015, 107, 419–425. [Google Scholar] [CrossRef]
- Pasquier, J.; Galas, L.; Boulange-Lecomte, C.; Rioult, D.; Bultelle, F.; Magal, P.; Webb, G.; Le Foll, F. Different Modalities of Intercellular Membrane Exchanges Mediate Cell-to-cell P-glycoprotein Transfers in MCF-7 Breast Cancer Cells. J. Biol. Chem. 2012, 287, 7374–7387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Cui, J.; Sun, X.; Zhang, Y. Tunneling-nanotube development in astrocytes depends on p53 activation. Cell Death Differ. 2011, 18, 732–742. [Google Scholar] [CrossRef] [Green Version]
- Sherer, N.M.; Lehmann, M.J.; Soto, L.F.J.; Horensavitz, C.; Pypaert, M.; Mothes, W. Retroviruses can establish filopodial bridges for efficient cell-to-cell transmission. Nat. Cell Biol. 2007, 9, 310–315. [Google Scholar] [CrossRef] [Green Version]
- Kimura, S.; Hase, K.; Ohno, H. The molecular basis of induction and formation of tunneling nanotubes. Cell Tissue 2013, 352, 67–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desir, S.; Dickson, E.L.; Vogel, R.; Thayanithy, V.; Wong, P.; Teoh, D.; Geller, M.A.; Steer, C.J.; Subramanian, S.; Lou, E. Tunneling nanotube formation is stimulated by hypoxia in ovarian cancer cells. Oncotarget 2016, 7, 43150–43161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Gerdes, H.-H. Transfer of mitochondria via tunneling nanotubes rescues apoptotic PC12 cells. Cell Death Differ. 2015, 22, 1181–1191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osswald, M.; Jung, E.; Sahm, F.; Solecki, G.; Venkataramani, V.; Blaes, J.; Weil, S.; Horstmann, H.; Wiestler, B.; Syed, M.; et al. Brain tumour cells interconnect to a functional and resistant network. Nat. Cell Biol. 2015, 528, 93–98. [Google Scholar] [CrossRef]
- Reindl, J.; Shevtsov, M.; Dollinger, G.; Stangl, S.; Multhoff, G. Membrane Hsp70-supported cell-to-cell connections via tunneling nanotubes revealed by live-cell STED nanoscopy. Cell Stress Chaperones 2019, 24, 213–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmad, T.; Mukherjee, S.; Pattnaik, B.; Kumar, M.; Singh, S.; Rehman, R.; Tiwari, B.K.; Jha, K.A.; Barhanpurkar, A.P.; Wani, M.R.; et al. Miro1 regulates intercellular mitochondrial transport & enhances mesenchymal stem cell rescue efficacy. EMBO J. 2014, 33, 994–1010. [Google Scholar] [CrossRef]
- Abounit, S.; Bousset, L.; Loria, F.; Zhu, S.; de Chaumont, F.; Pieri, L.; Olivo-Marin, J.; Melki, R.; Zurzolo, C. Tunneling nanotubes spread fibrillar α-synuclein by intercellular trafficking of lysosomes. EMBO J. 2016, 35, 2120–2138. [Google Scholar] [CrossRef]
- Bhat, S.; Ljubojevic, N.; Zhu, S.; Fukuda, M.; Echard, A.; Zurzolo, C. Rab35 and its effectors promote formation of tunneling nanotubes in neuronal cells. Sci. Rep. 2020, 10, 1–14. [Google Scholar] [CrossRef]
- Schiller, C.; Huber, J.; Diakopoulos, K.; Weiss, E.H. Tunneling nanotubes enable intercellular transfer of MHC class I molecules. Hum. Immunol. 2013, 74, 412–416. [Google Scholar] [CrossRef]
- Sowinski, S.; Jolly, C.; Berninghausen, O.; Purbhoo, M.A.; Chauveau, A.; Köhler, K.; Oddos, S.; Eissmann, P.; Brodsky, F.; Hopkins, C.; et al. Membrane nanotubes physically connect T cells over long distances presenting a novel route for HIV-1 transmission. Nat. Cell Biol. 2008, 10, 211–219. [Google Scholar] [CrossRef] [PubMed]
- Panasiuk, M.; Rychłowski, M.; Derewońko, N.; Bieńkowska-Szewczyk, K. Tunneling Nanotubes as a Novel Route of Cell-to-Cell Spread of Herpesviruses. J. Virol. 2018, 92, e00090-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, J.J.; Yang, W.M.; Li, F.; Zhu, W.; Chen, Z. Tunneling Nanotubes Mediated microRNA-155 Intercellular Transportation Promotes Bladder Cancer Cells’ Invasive and Proliferative Capacity. Int. J. Nanomed. 2019, 14, 9731–9743. [Google Scholar] [CrossRef] [Green Version]
- Austefjord, M.W.; Gerdes, H.-H.; Wang, X. Tunneling nanotubes: Diversity in morphology and structure. Commun. Integr. Biol. 2014, 7, e27934. [Google Scholar] [CrossRef] [PubMed]
- Armulik, A.; Abramsson, A.; Betsholtz, C. Endothelial/Pericyte Interactions. Circ. Res. 2005, 97, 512–523. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Liu, X.; Qiu, Y.; Shi, Y.; Cai, J.; Wang, B.; Wei, X.; Ke, Q.; Sui, X.; Wang, Y.; et al. Cell adhesion-mediated mitochondria transfer contributes to mesenchymal stem cell-induced chemoresistance on T cell acute lymphoblastic leukemia cells. J. Hematol. Oncol. 2018, 11, 1–13. [Google Scholar] [CrossRef]
- Desir, S.; Wong, P.; Turbyville, T.; Chen, D.; Shetty, M.; Clark, C.; Zhai, E.; Romin, Y.; Manova-Todorova, K.; Starr, T.K.; et al. Intercellular Transfer of Oncogenic KRAS via Tunneling Nanotubes Introduces Intracellular Mutational Heterogeneity in Colon Cancer Cells. Cancers 2019, 11, 892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antanavičiūtė, I.; Šimatonis, L.; Ulčinas, O.; Gadeikytė, A.; Abakevičienė, B.; Tamulevičius, S.; Mikalayeva, V.; Skeberdis, V.A.; Stankevičius, E.; Tamulevičius, T. Femtosecond laser micro-machined polyimide films for cell scaffold applications. J. Tissue Eng. Regen. Med. 2018, 12, e760–e773. [Google Scholar] [CrossRef]
- Matejka, N.; Reindl, J. Perspectives of cellular communication through tunneling nanotubes in cancer cells and the connection to radiation effects. Radiat. Oncol. 2019, 14, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Pinto, G.; Saenz-De-Santa-Maria, I.; Chastagner, P.; Perthame, E.; Delmas, C.; Toulas, C.; Moyal-Jonathan-Cohen, E.; Brou, C.; Zurzolo, C. Patient-derived glioblastoma stem cells transfer mitochondria through tunneling nanotubes in tumor organoids. Biochem. J. 2021, 478, 21–39. [Google Scholar] [CrossRef] [PubMed]
- Civita, P.; Leite, D.M.; Pilkington, G.J. Pre-Clinical Drug Testing in 2D and 3D Human In Vitro Models of Glioblastoma Incorporating Non-Neoplastic Astrocytes: Tunneling Nano Tubules and Mitochondrial Transfer Modulates Cell Behavior and Therapeutic Response. Int. J. Mol. Sci. 2019, 20, 6017. [Google Scholar] [CrossRef] [Green Version]
- Formicola, B.; D’Aloia, A.; Magro, R.D.; Stucchi, S.; Rigolio, R.; Ceriani, M.; Re, F. Differential Exchange of Multifunctional Liposomes Between Glioblastoma Cells and Healthy Astrocytes via Tunneling Nanotubes. Front. Bioeng. Biotechnol. 2019, 7, 403. [Google Scholar] [CrossRef] [Green Version]
- Matejka, N.; Reindl, J. Influence of α-Particle Radiation on Intercellular Communication Networks of Tunneling Nanotubes in U87 Glioblastoma Cells. Front. Oncol. 2020, 10, 1691. [Google Scholar] [CrossRef]
- Ahir, B.K.; Engelhard, H.H.; Lakka, S.S. Tumor Development and Angiogenesis in Adult Brain Tumor: Glioblastoma. Mol. Neurobiol. 2020, 57, 2461–2478. [Google Scholar] [CrossRef] [Green Version]
- Roehlecke, C.; Schmidt, M.H.H. Tunneling Nanotubes and Tumor Microtubes in Cancer. Cancers 2020, 12, 857. [Google Scholar] [CrossRef] [Green Version]
- Weng, Z.; Zhang, B.; Tsilioni, I.; Theoharides, T.C. Nanotube Formation: A Rapid Form of “Alarm Signaling”? Clin. Ther. 2016, 38, 1066–1072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimura, S.; Yamashita, M.; Yamakami-Kimura, M.; Sato, Y.; Yamagata, A.; Kobashigawa, Y.; Inagaki, F.; Amada, T.; Hase, K.; Iwanaga, T.; et al. Distinct Roles for the N- and C-terminal Regions of M-Sec in Plasma Membrane Deformation during Tunneling Nanotube Formation. Sci. Rep. 2016, 6, 33548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luiz, M.T.; Di Filippo, L.D.; Tofani, L.B.; de Araújo, J.T.C.; Dutra, J.A.P.; Marchetti, J.M.; Chorilli, M. Highlights in targeted nanoparticles as a delivery strategy for glioma treatment. Int. J. Pharm. 2021, 604, 120758. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Wang, X. Opportunities and Challenges in Tunneling Nanotubes Research: How Far from Clinical Application? Int. J. Mol. Sci. 2021, 22, 2306. [Google Scholar] [CrossRef]
- Deng, G.; Wu, Z.; Zhou, F.; Dai, C.; Zhao, J.; Kang, Y.; Wang, Q.; Liu, X.; Wang, Y.; Wang, Q. Exchangeability of FITC-SiO2 Nanoparticles Between Cancer Cells Increases the Range of Drug Delivery. J. Biomed. Nanotechnol. 2018, 14, 127–138. [Google Scholar] [CrossRef] [PubMed]
- Franco, S.; Noureddine, A.; Guo, J.; Keth, J.; Paffett, M.L.; Brinker, C.J.; Serda, R.E. Direct Transfer of Mesoporous Silica Nanoparticles between Macrophages and Cancer Cells. Cancers 2020, 12, 2892. [Google Scholar] [CrossRef] [PubMed]
- Winkler, F.; Wick, W. Harmful networks in the brain and beyond: Membrane tubes can connect cancer cells and drive tumor progression and resistance. Science 2018, 359, 1100–1101. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Taiarol, L.; Formicola, B.; Fagioli, S.; Sierri, G.; D’Aloia, A.; Kravicz, M.; Renda, A.; Viale, F.; Dal Magro, R.; Ceriani, M.; et al. The 3.0 Cell Communication: New Insights in the Usefulness of Tunneling Nanotubes for Glioblastoma Treatment. Cancers 2021, 13, 4001. https://doi.org/10.3390/cancers13164001
Taiarol L, Formicola B, Fagioli S, Sierri G, D’Aloia A, Kravicz M, Renda A, Viale F, Dal Magro R, Ceriani M, et al. The 3.0 Cell Communication: New Insights in the Usefulness of Tunneling Nanotubes for Glioblastoma Treatment. Cancers. 2021; 13(16):4001. https://doi.org/10.3390/cancers13164001
Chicago/Turabian StyleTaiarol, Lorenzo, Beatrice Formicola, Stefano Fagioli, Giulia Sierri, Alessia D’Aloia, Marcelo Kravicz, Antonio Renda, Francesca Viale, Roberta Dal Magro, Michela Ceriani, and et al. 2021. "The 3.0 Cell Communication: New Insights in the Usefulness of Tunneling Nanotubes for Glioblastoma Treatment" Cancers 13, no. 16: 4001. https://doi.org/10.3390/cancers13164001
APA StyleTaiarol, L., Formicola, B., Fagioli, S., Sierri, G., D’Aloia, A., Kravicz, M., Renda, A., Viale, F., Dal Magro, R., Ceriani, M., & Re, F. (2021). The 3.0 Cell Communication: New Insights in the Usefulness of Tunneling Nanotubes for Glioblastoma Treatment. Cancers, 13(16), 4001. https://doi.org/10.3390/cancers13164001