
Identification and Functional Characterization of Novel MYC-Regulated Long Noncoding RNAs in Group 3 Medulloblastoma


 ,
,  , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Generation of D283-OMO Stable Cell Line
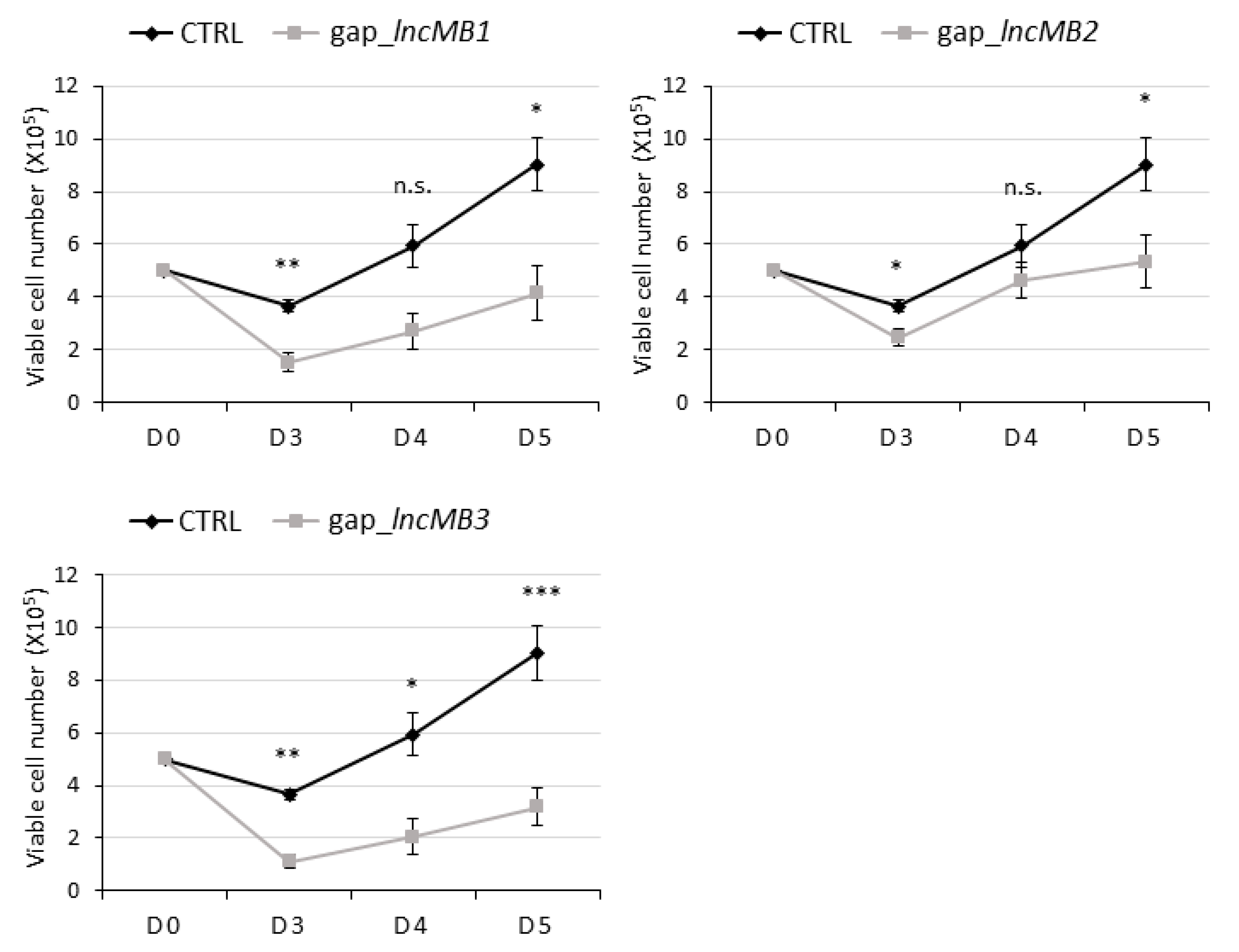
2.3. Viable Cell Number Evaluation
2.4. LNA GapmeR Transfections
2.5. RNA Extraction and Analysis
2.6. RNA-Seq and Bioinformatic Analysis
2.7. Chromatin Immunoprecipitation (ChIP) Assay
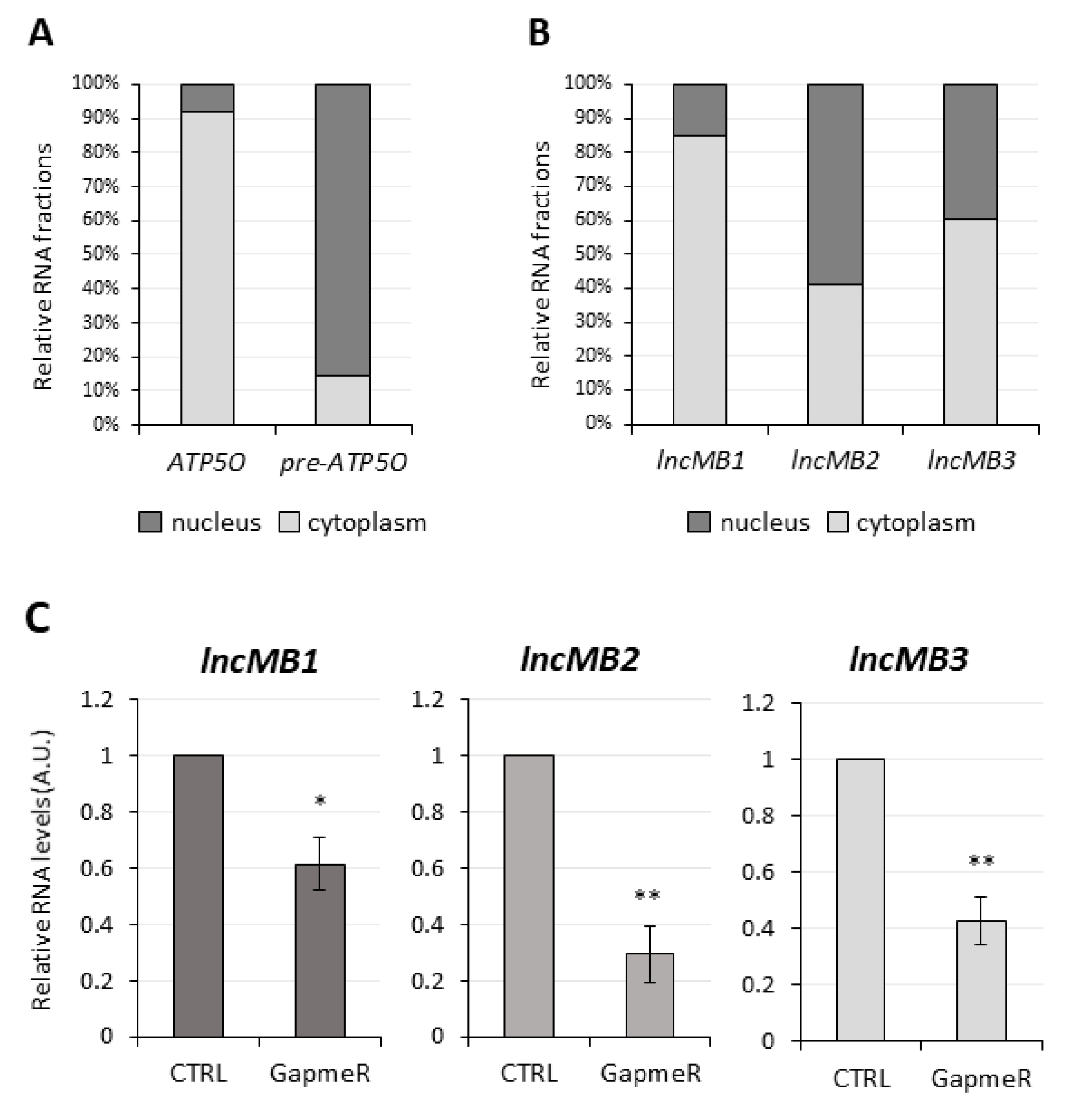
2.8. Cell Fractionation
2.9. Immunoblotting
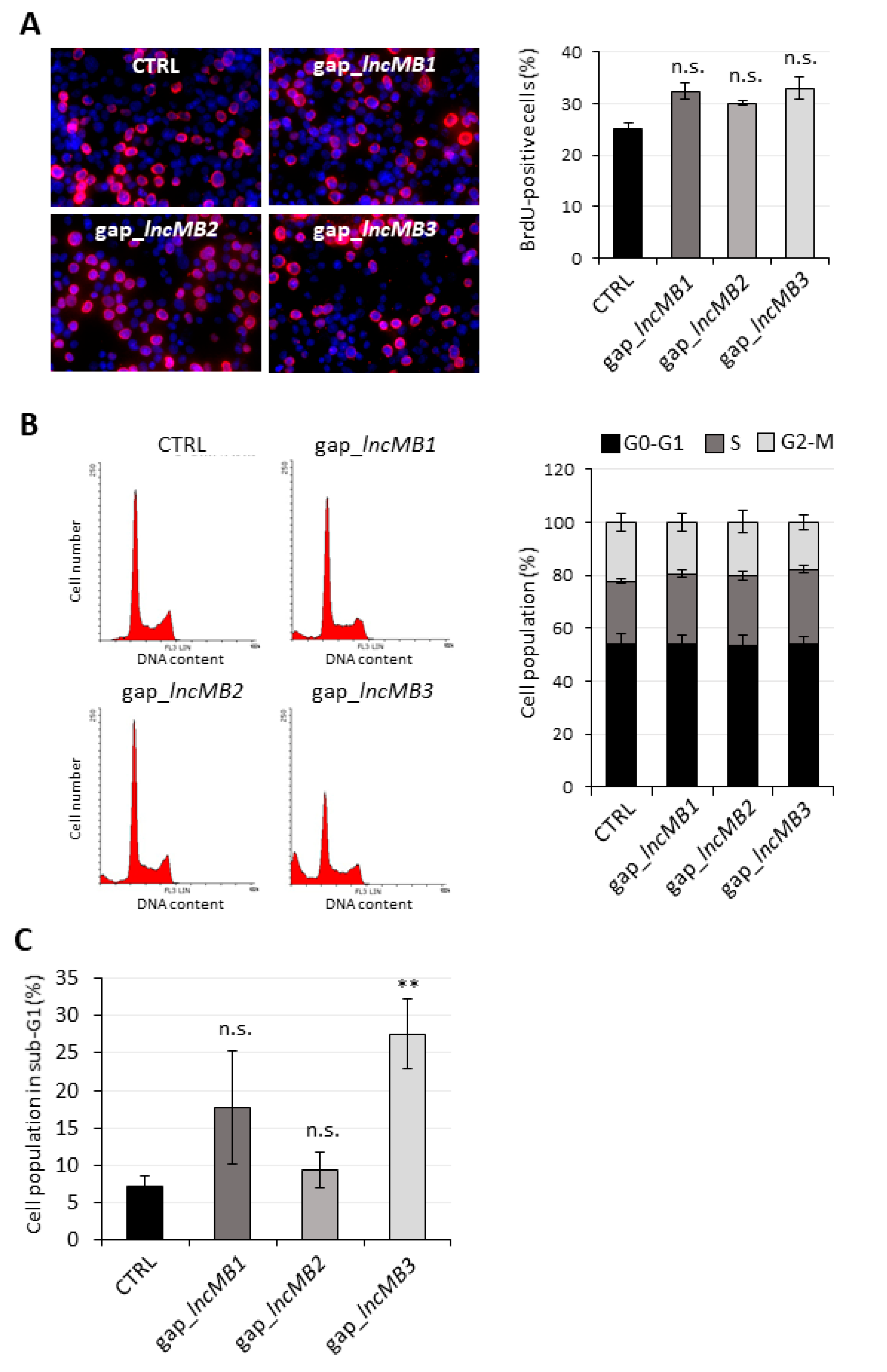
2.10. Flow Cytometry Analyses
2.11. BrdU Assay
2.12. Migration and Invasion Assays
2.13. Cell Pictures
2.14. Statistical Analyses
2.15. Oligonucleotides
3. Results
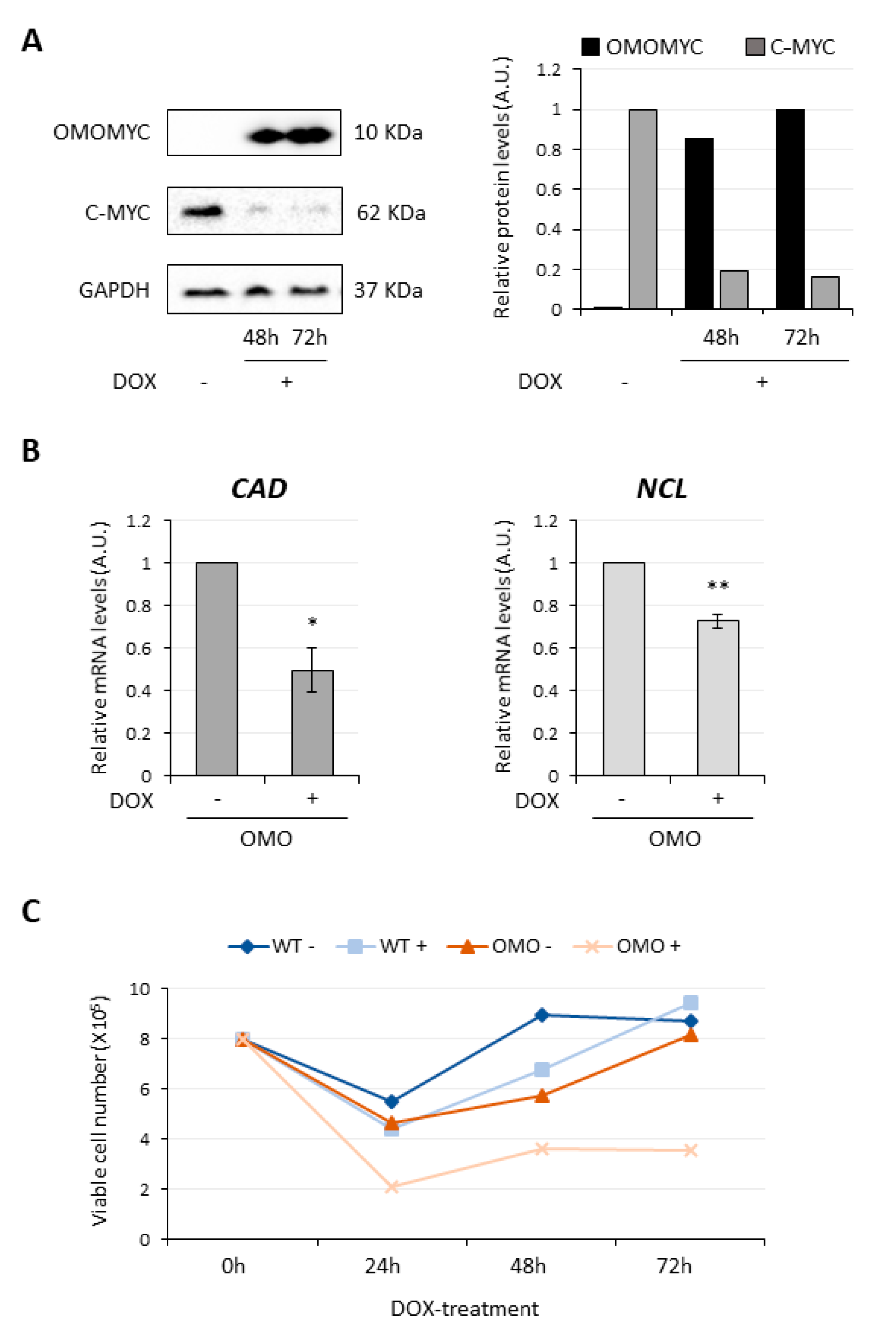
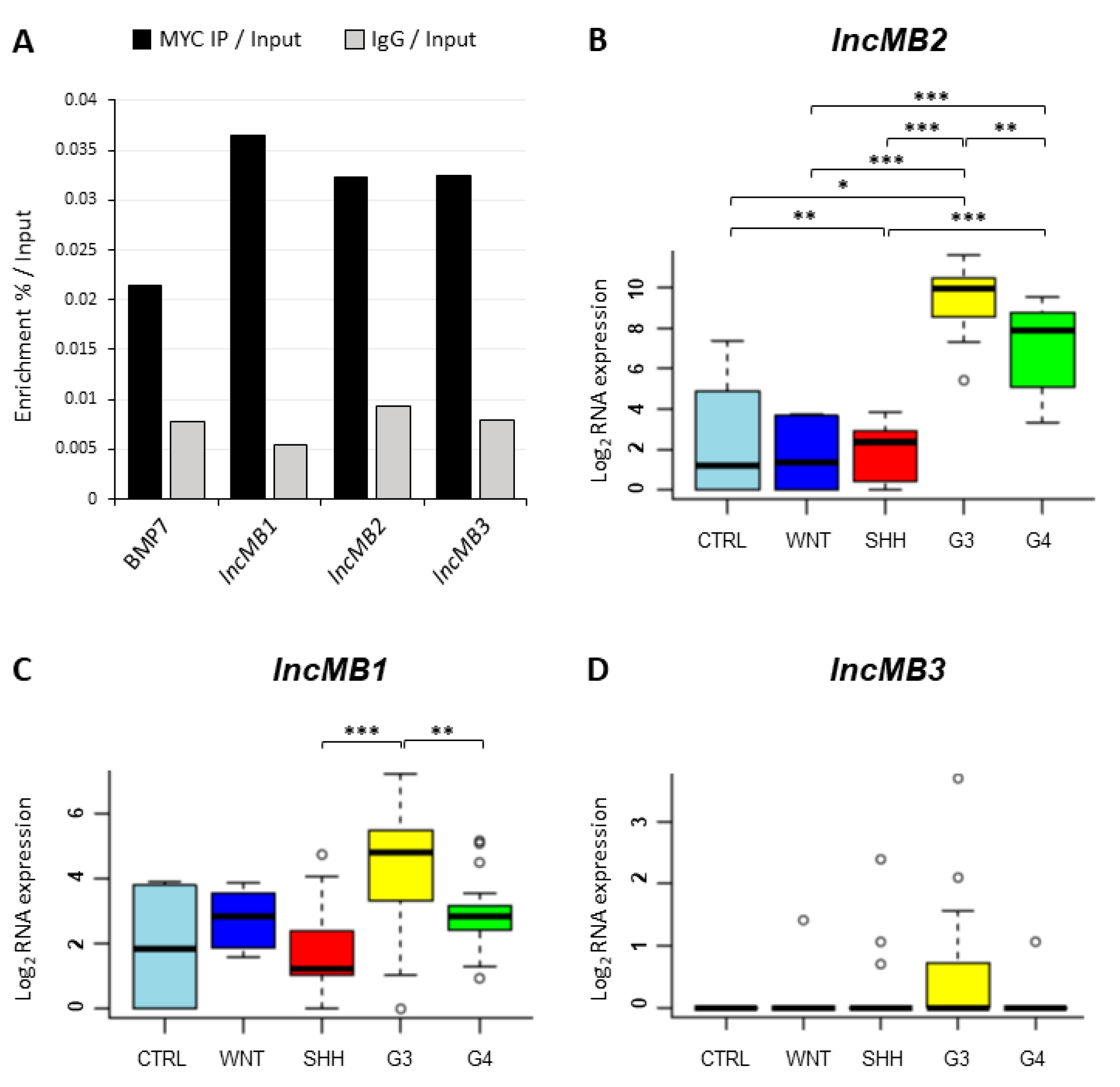
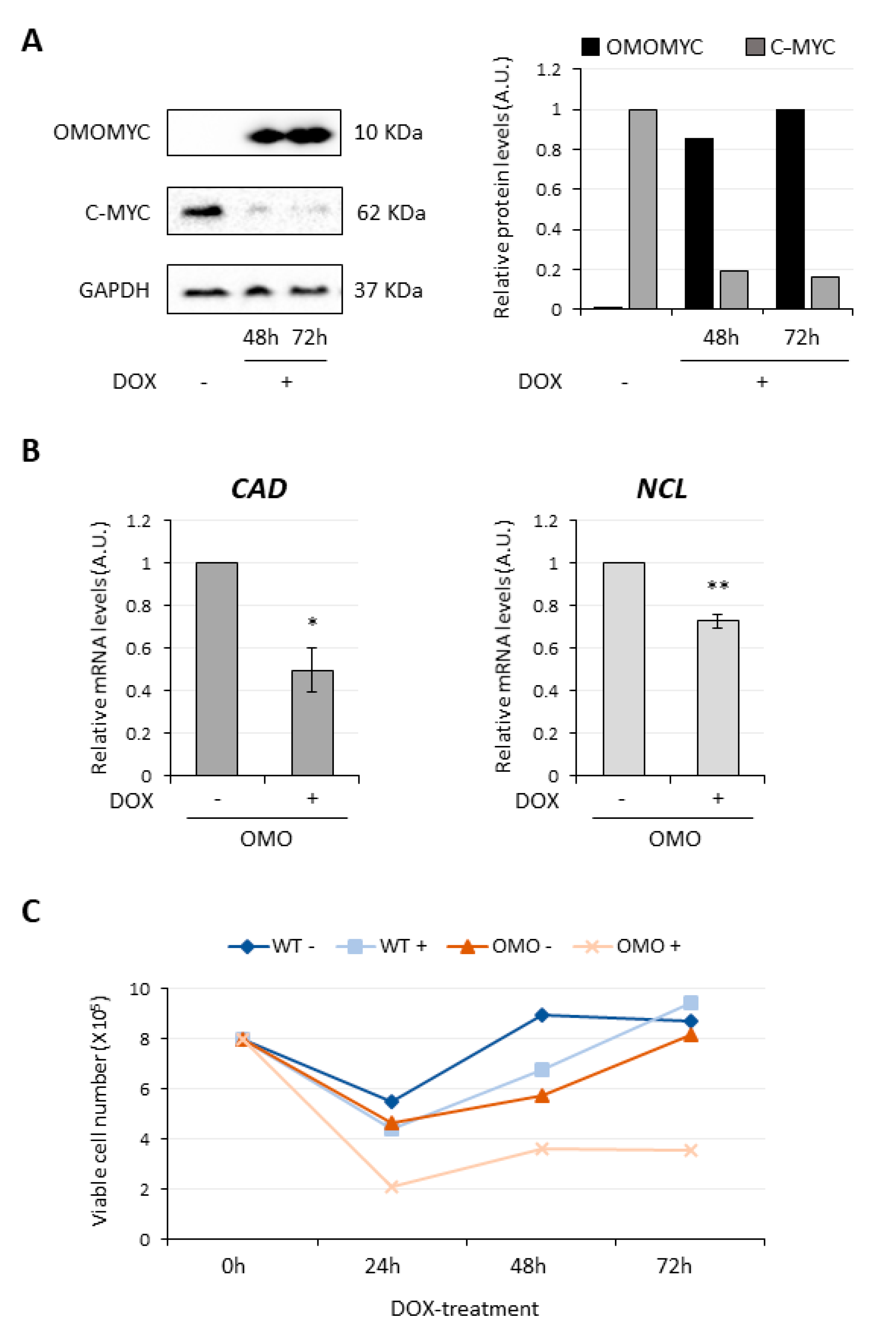
3.1. OMOMYC Is Able to Inhibit MYC Activity in D283 MED Cells
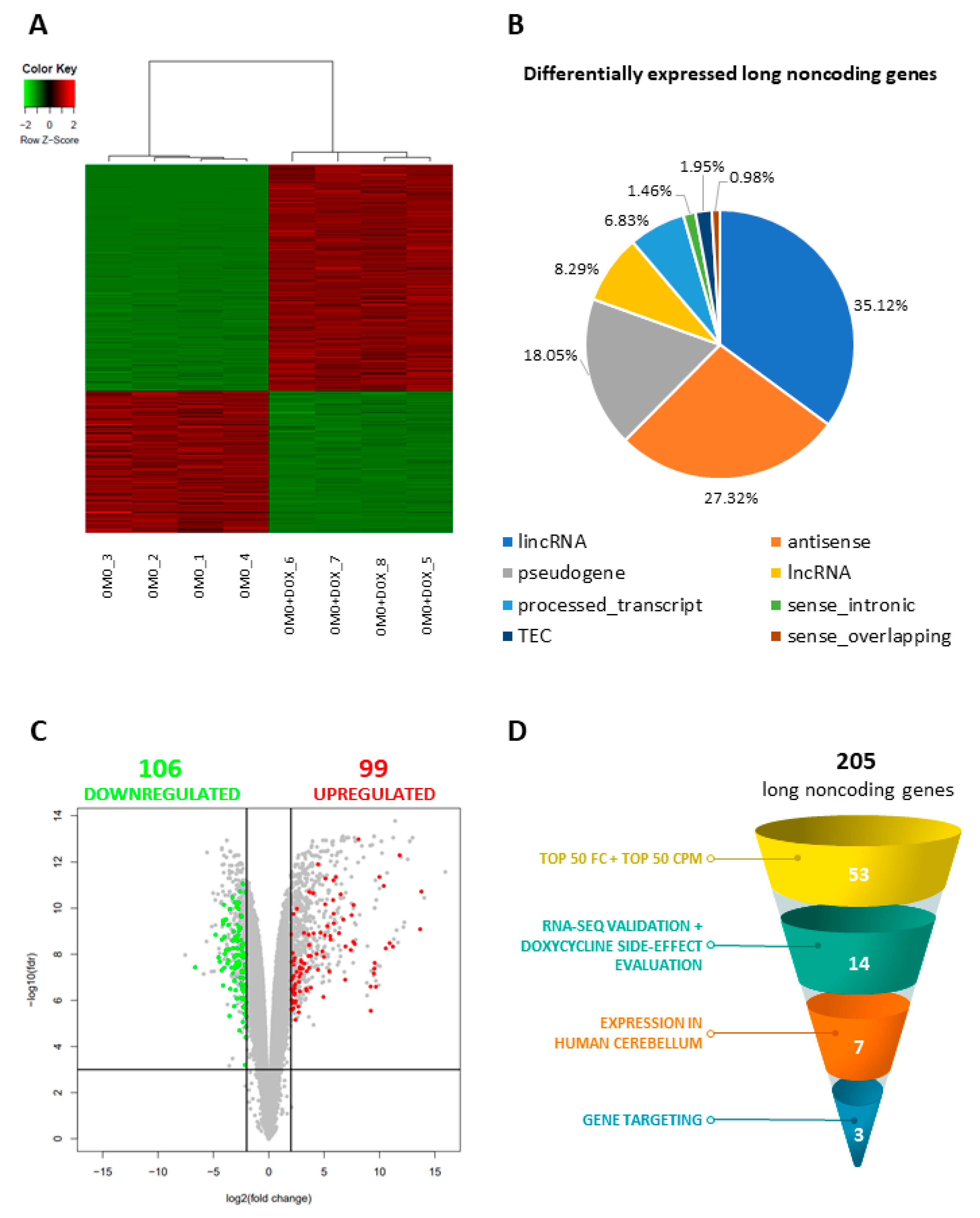
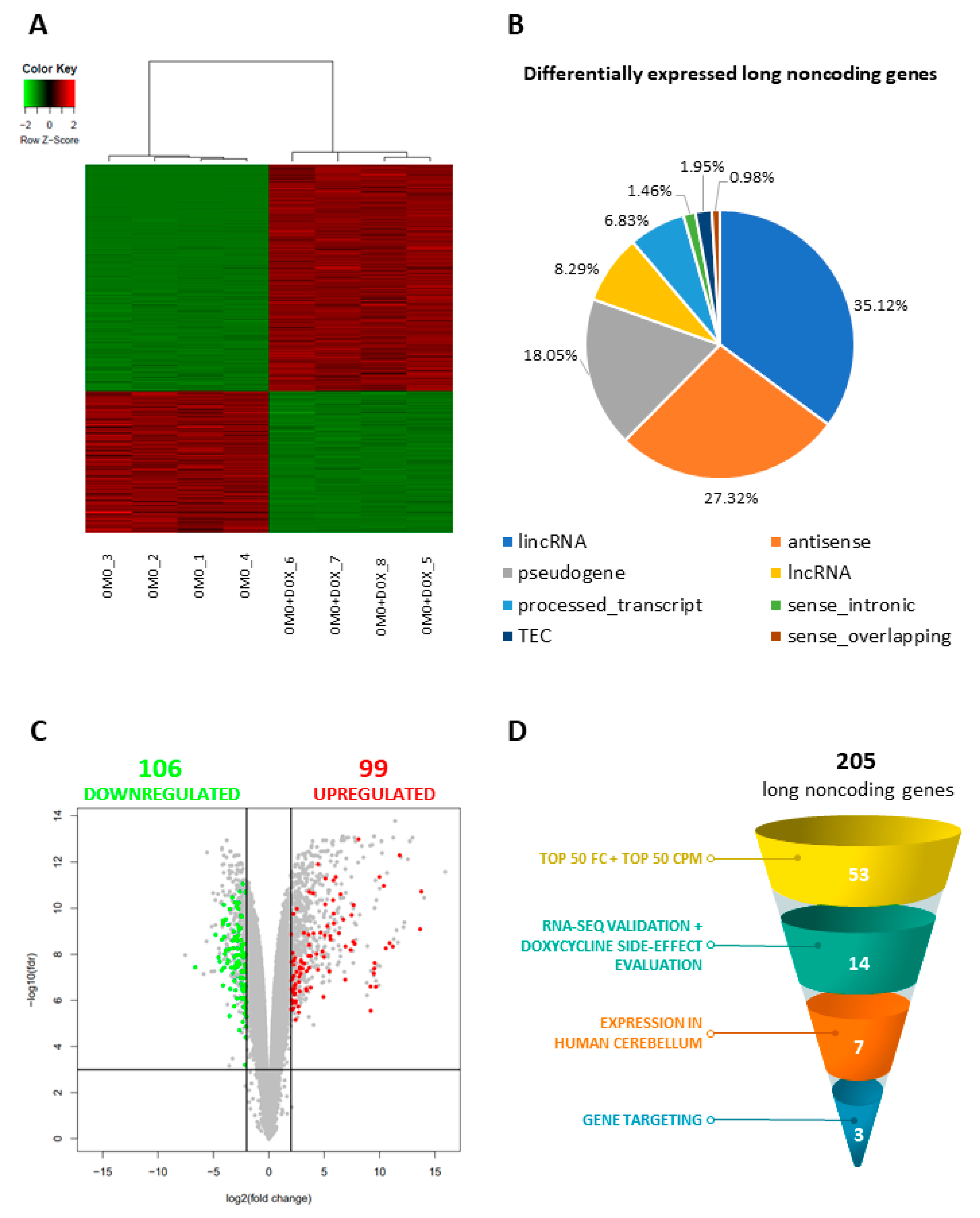
3.2. The Inhibition of MYC Function Alters D283 MED Transcriptome
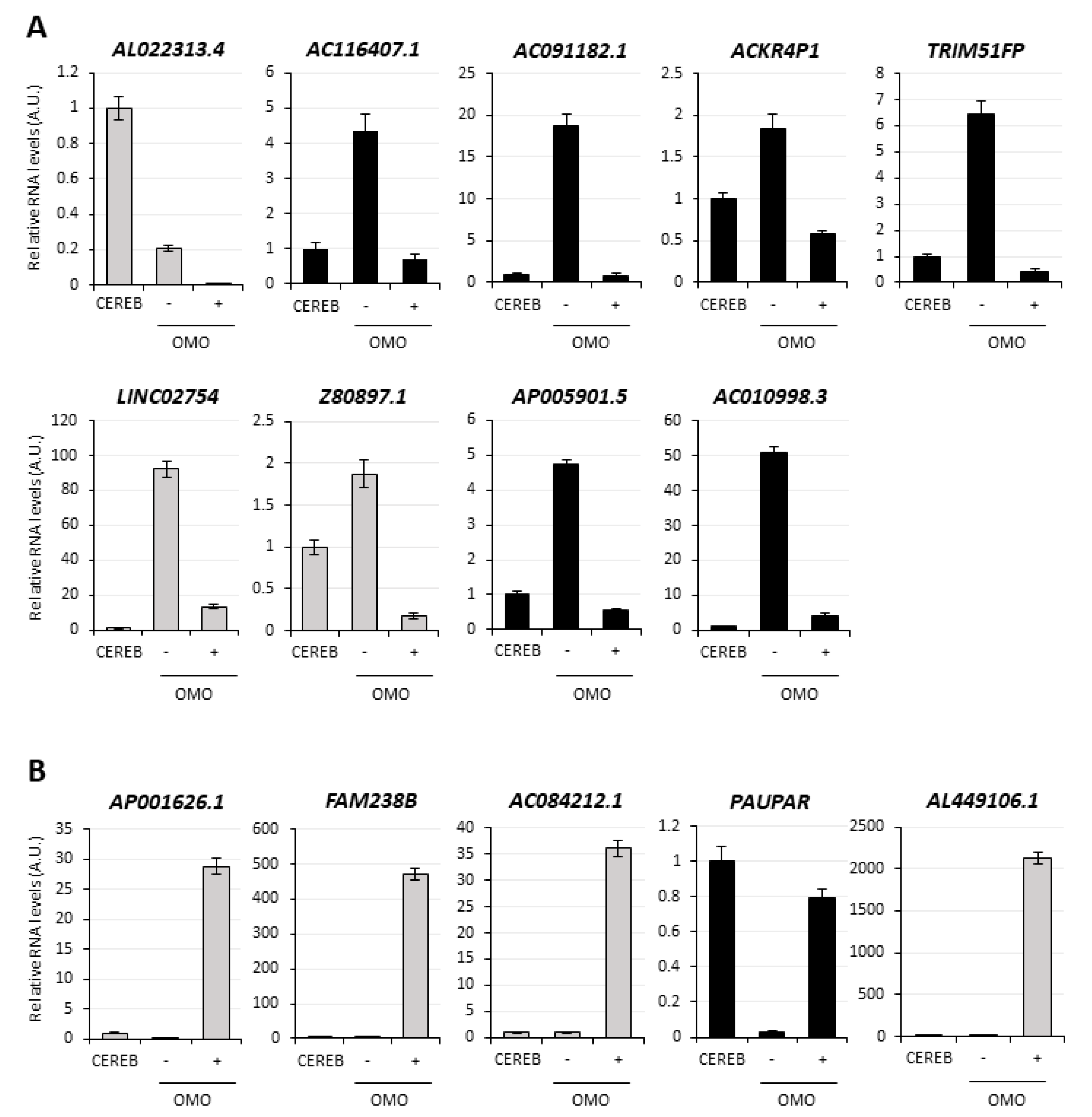
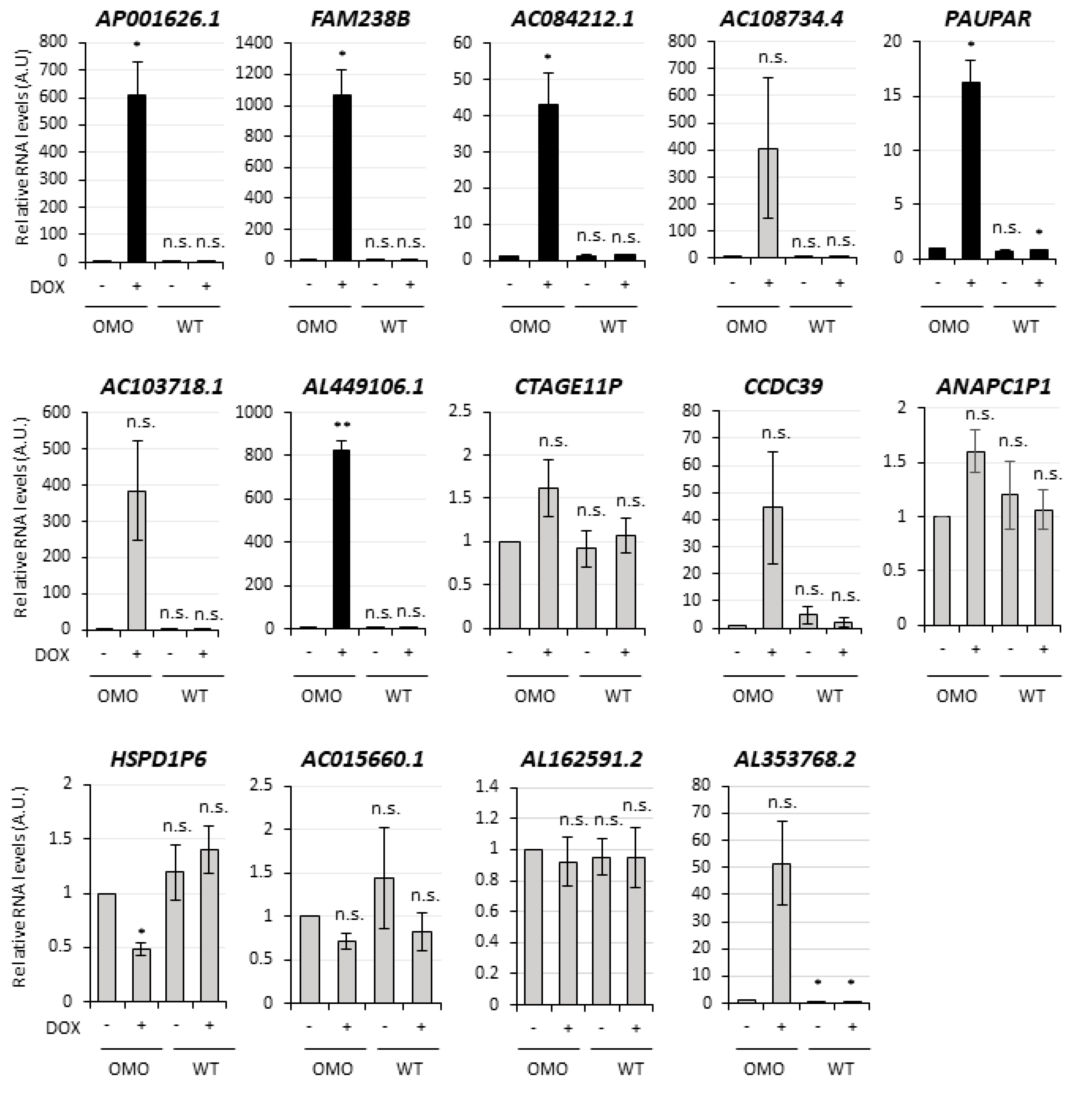
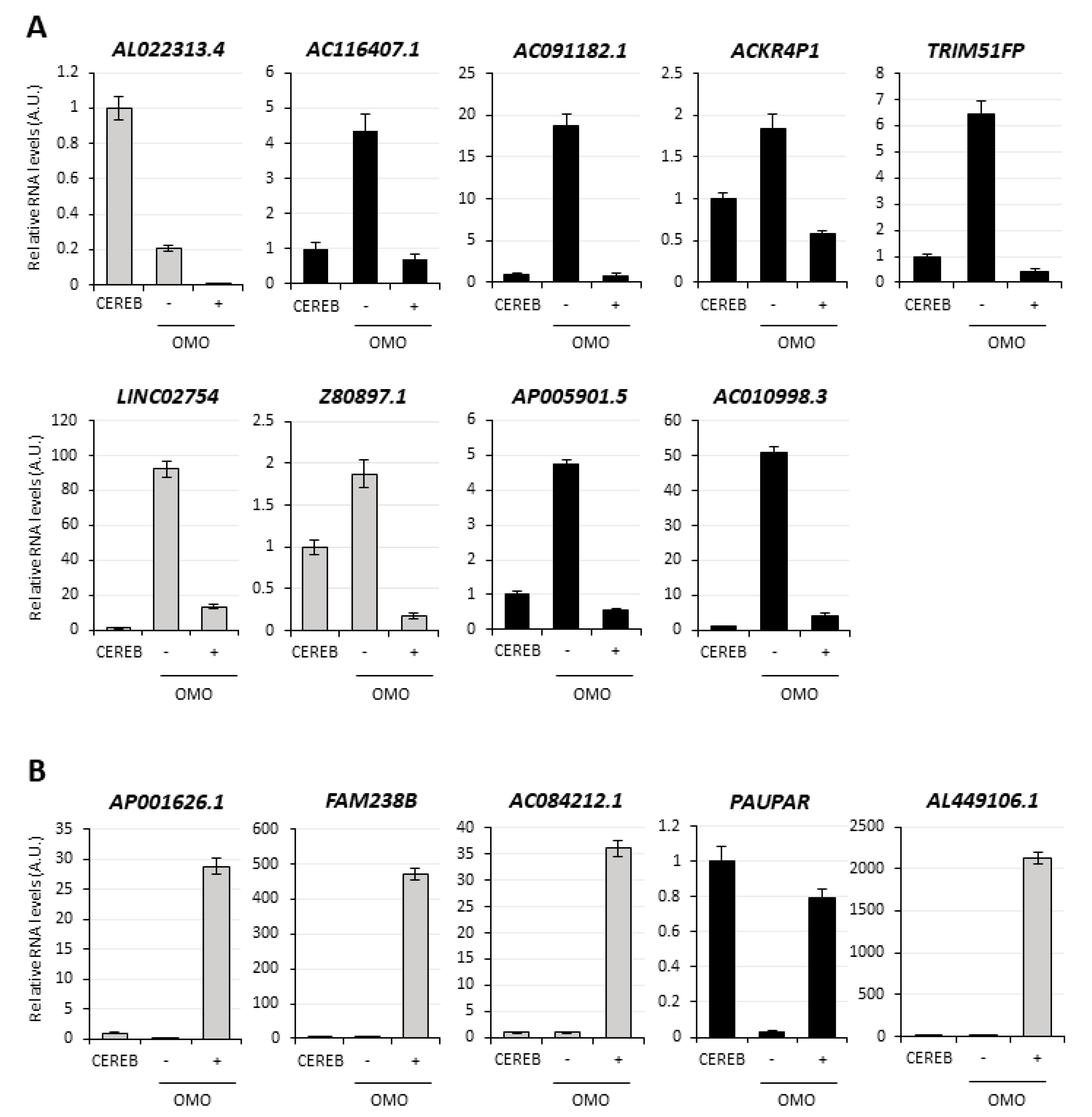
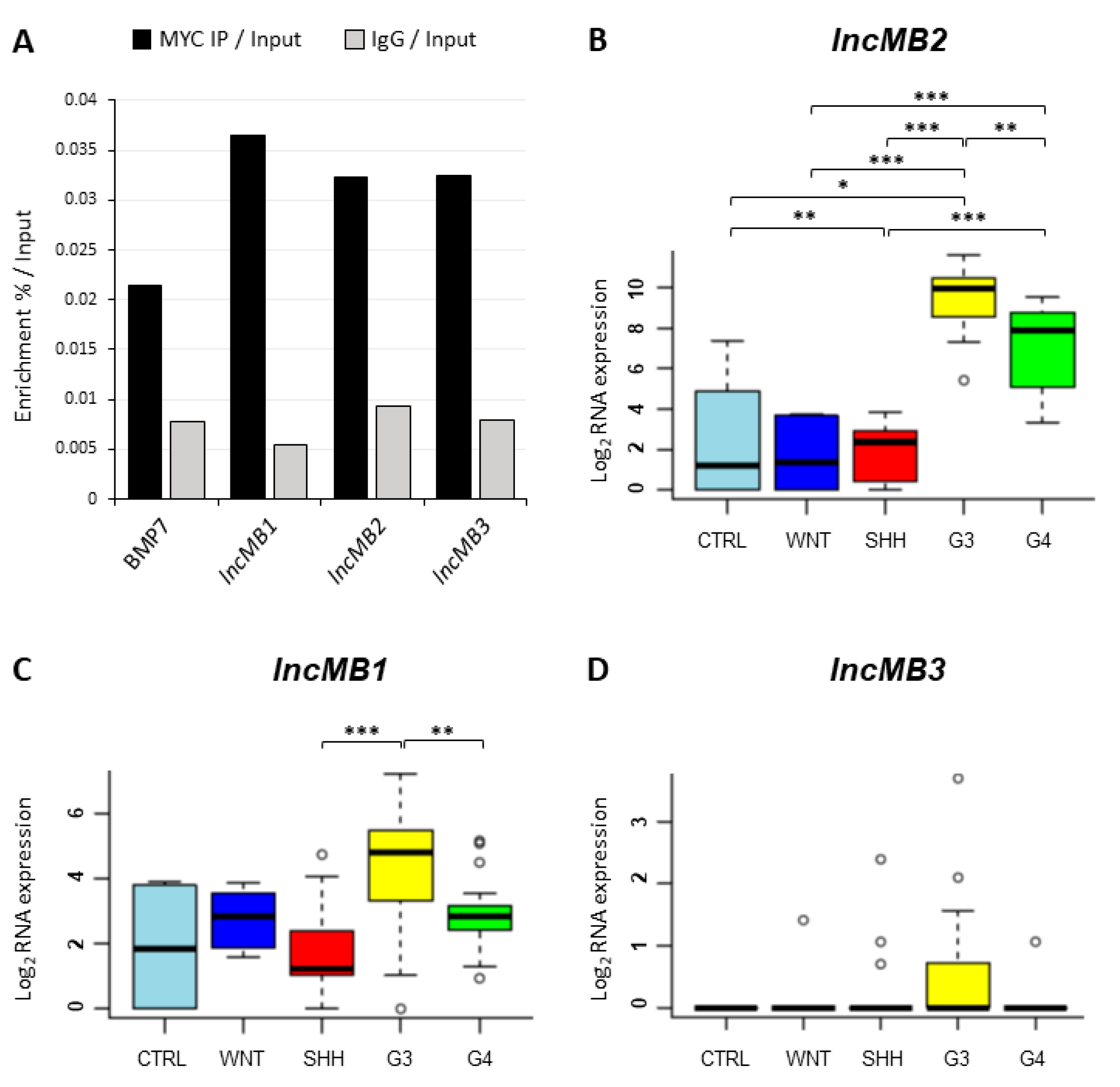
3.3. MYC-Dependent lncRNAs May Represent Potential Oncogenes or Oncosuppressors
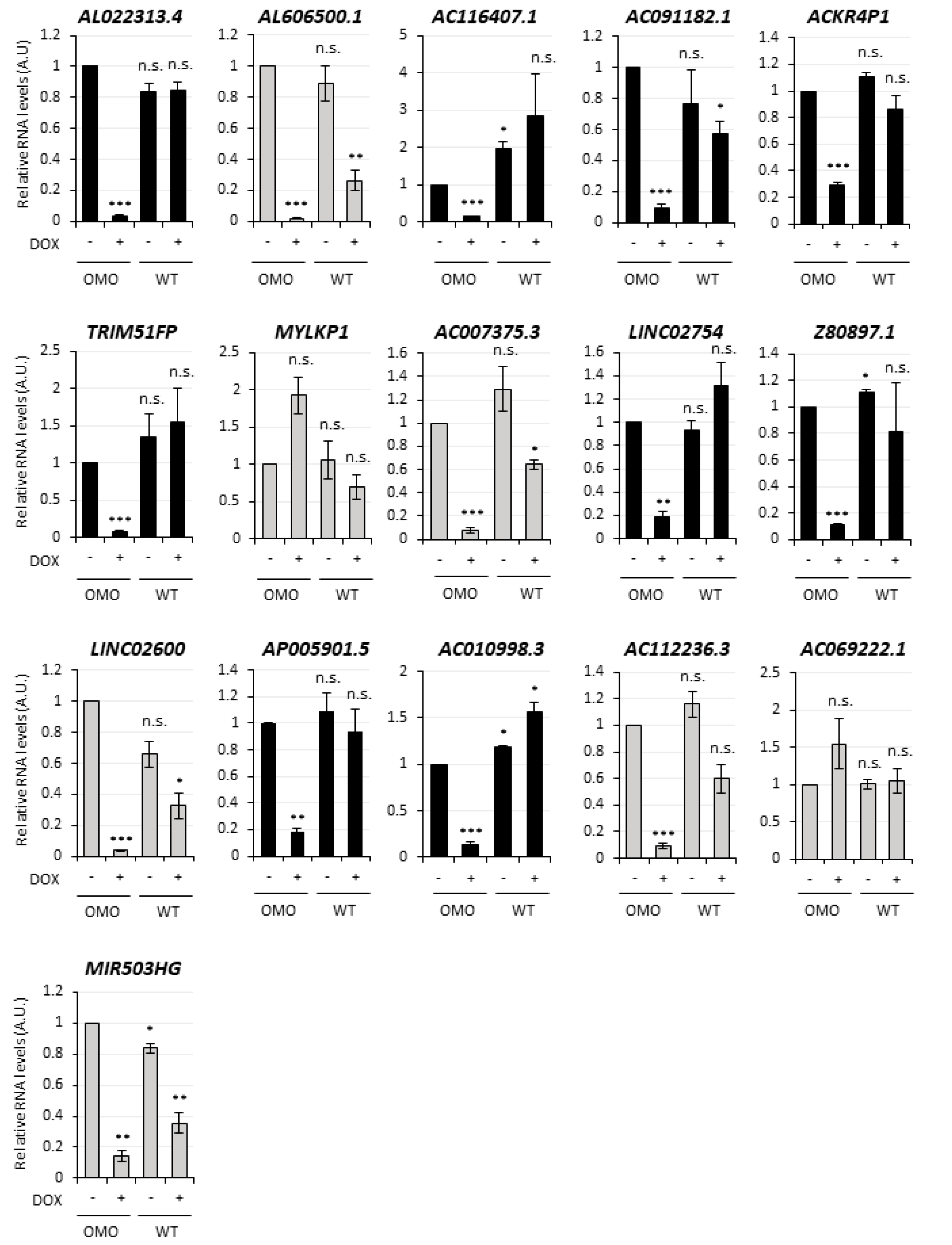
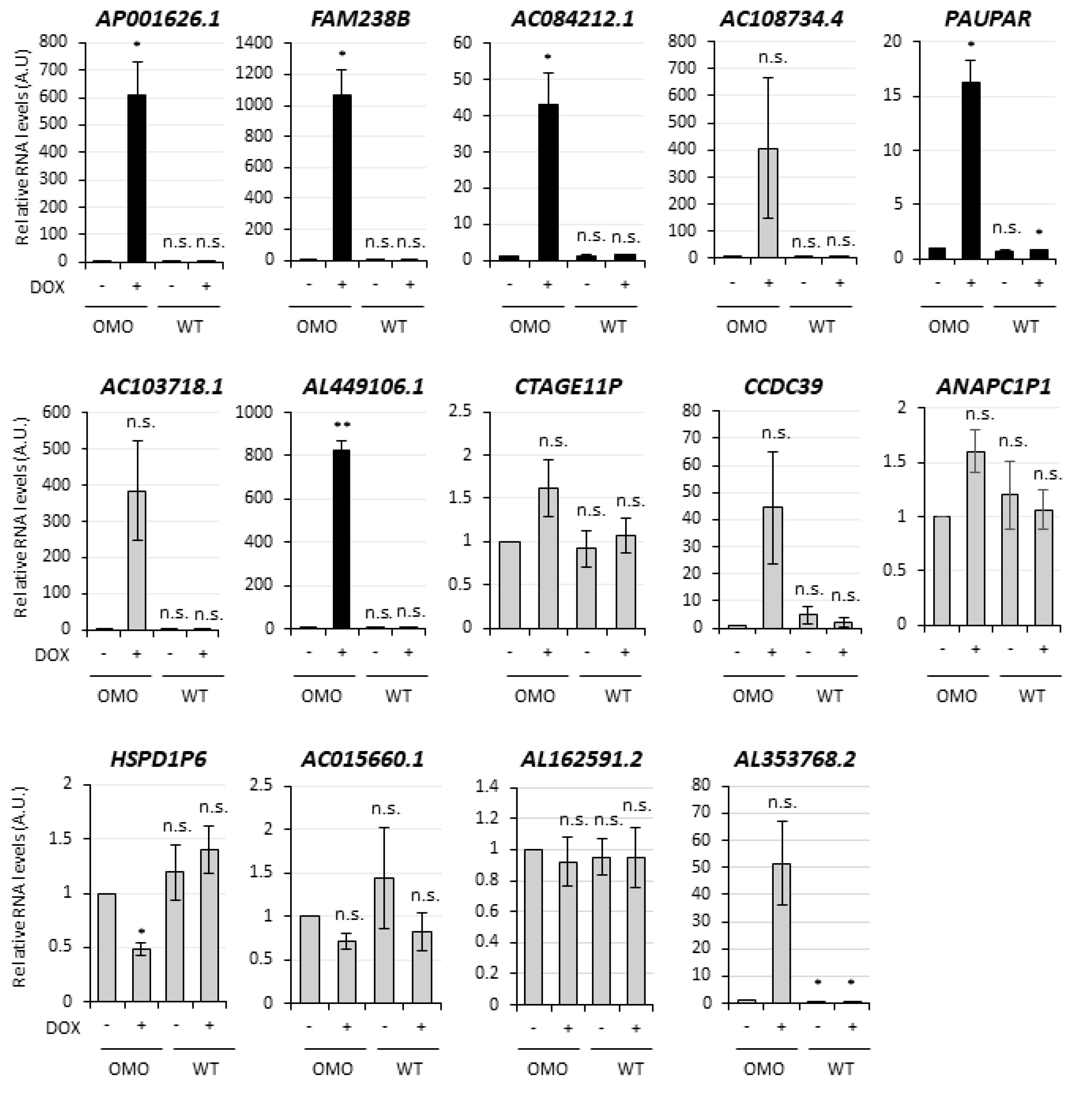
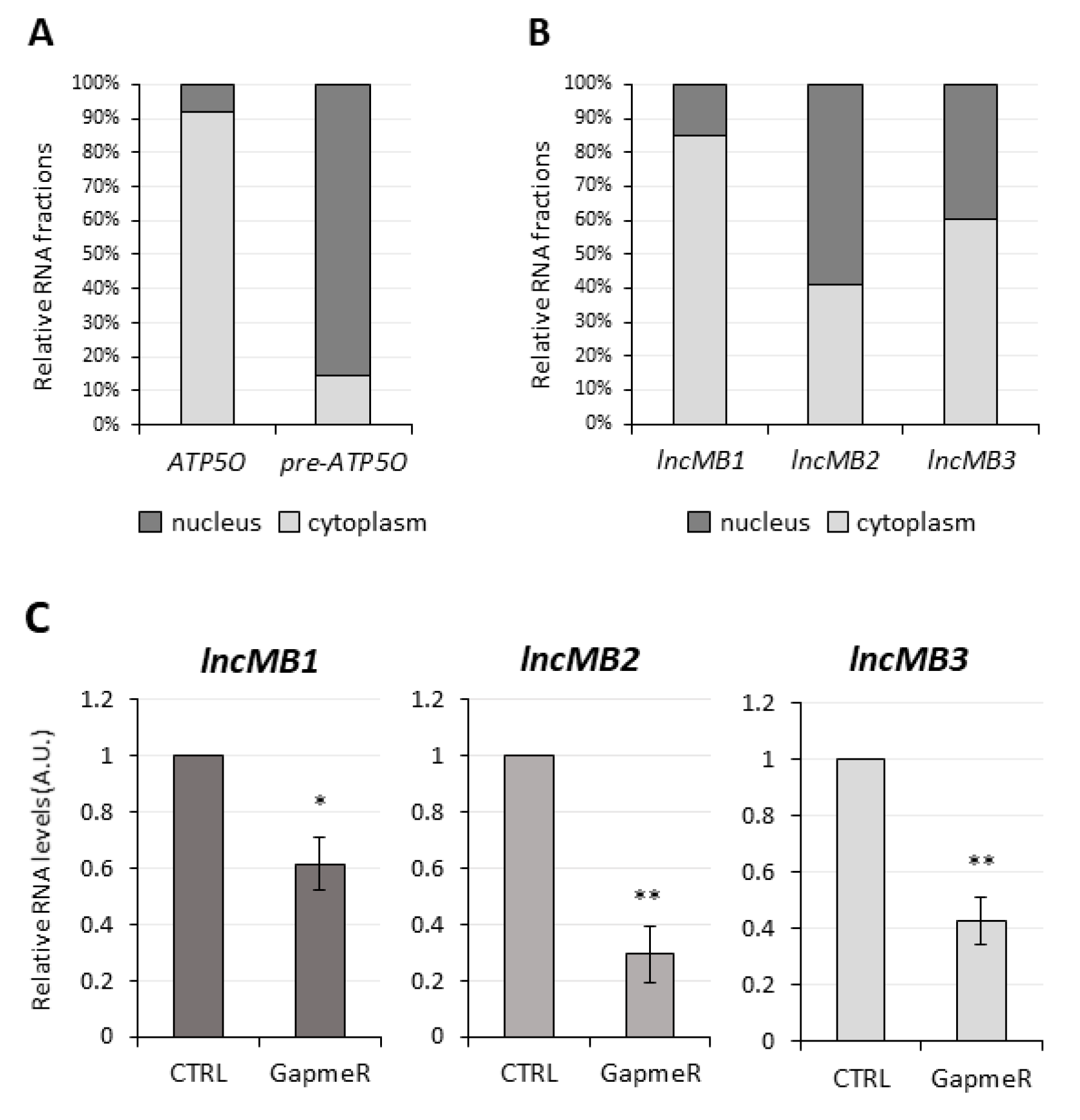
3.4. Selection of lncMB1, lncMB2 and lncMB3 for Functional Analyses
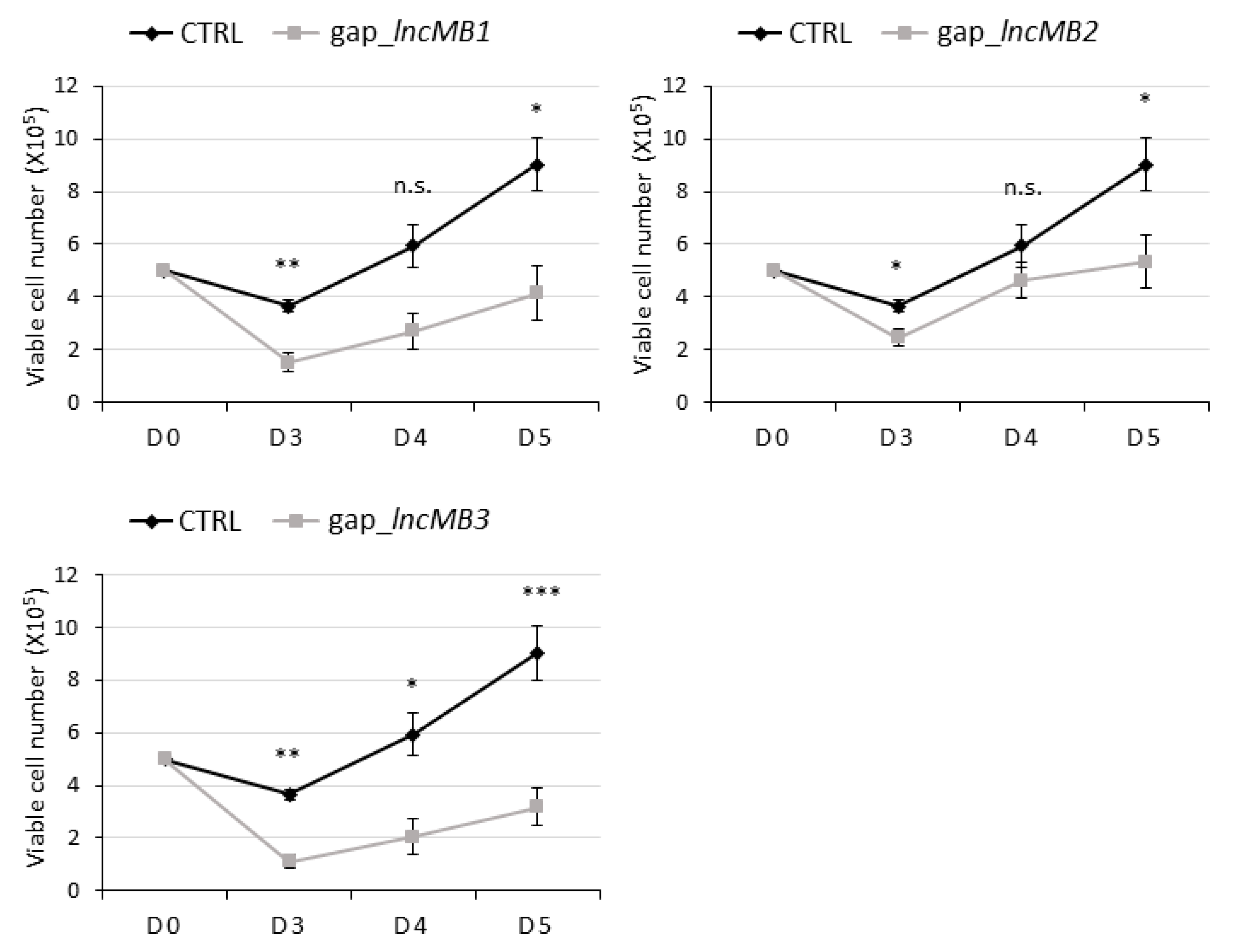
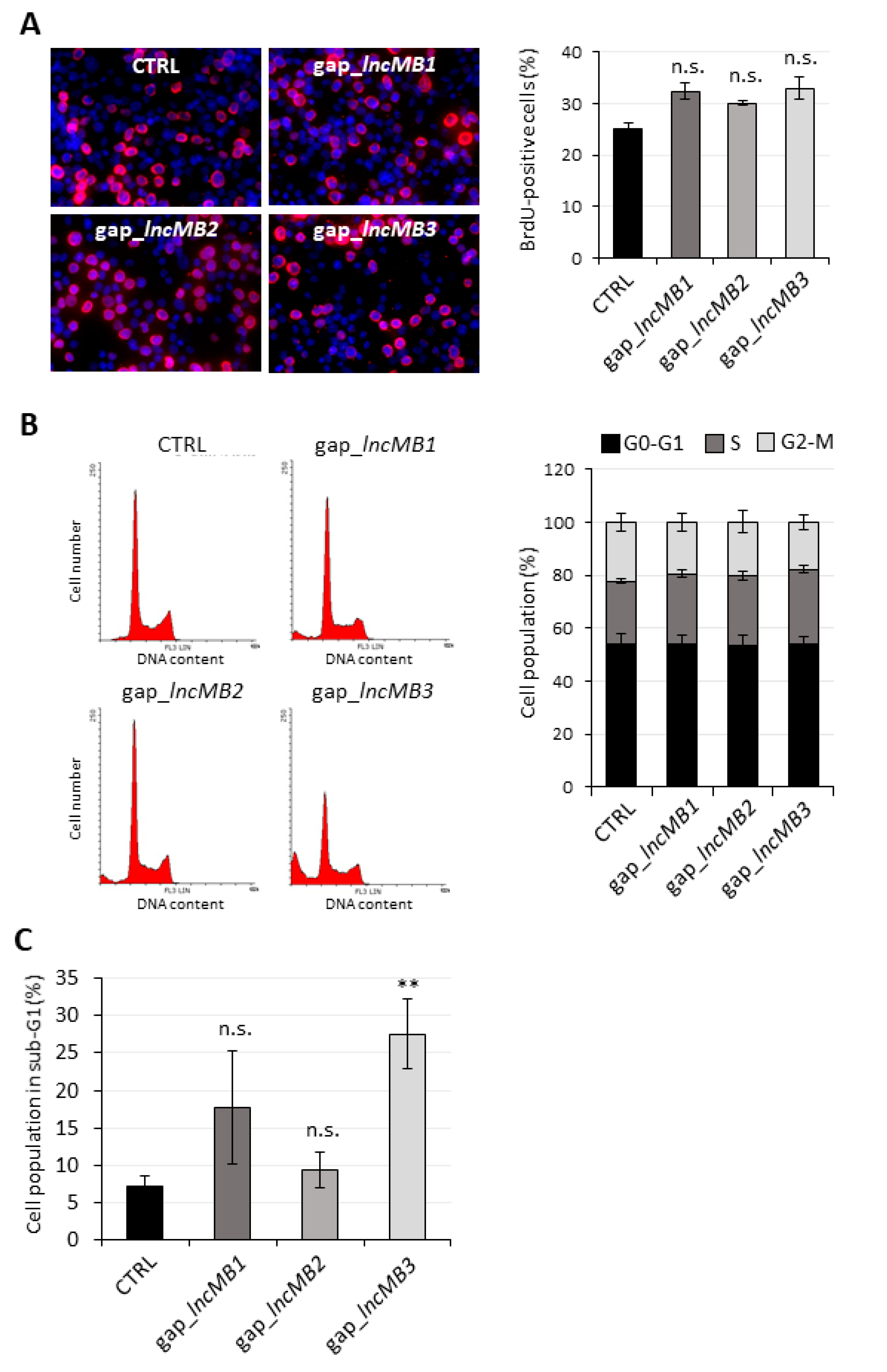
3.5. Antisense Targeting of lncMB1, lncMB2 and lncMB3 Affects D283 MED Cell Survival
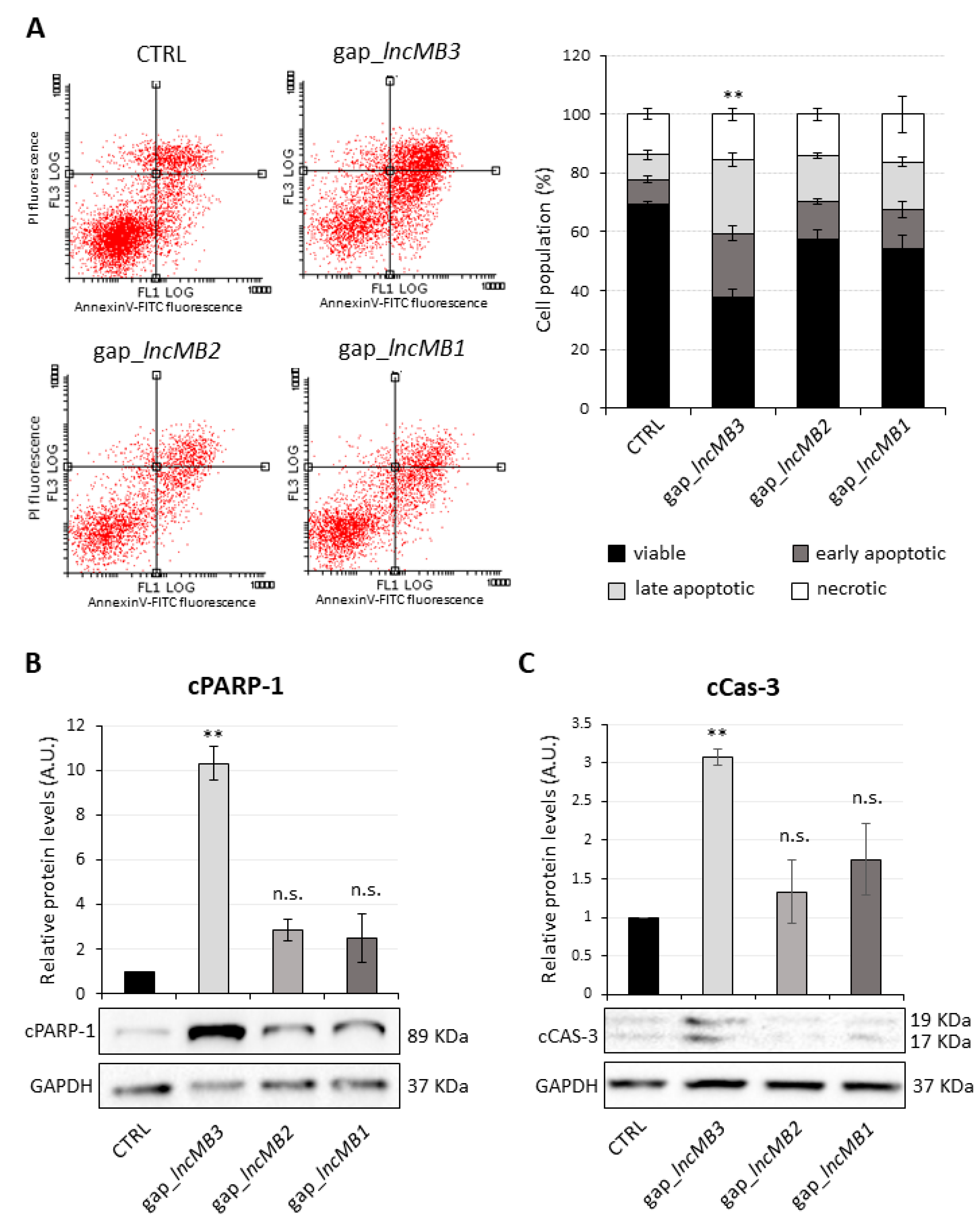
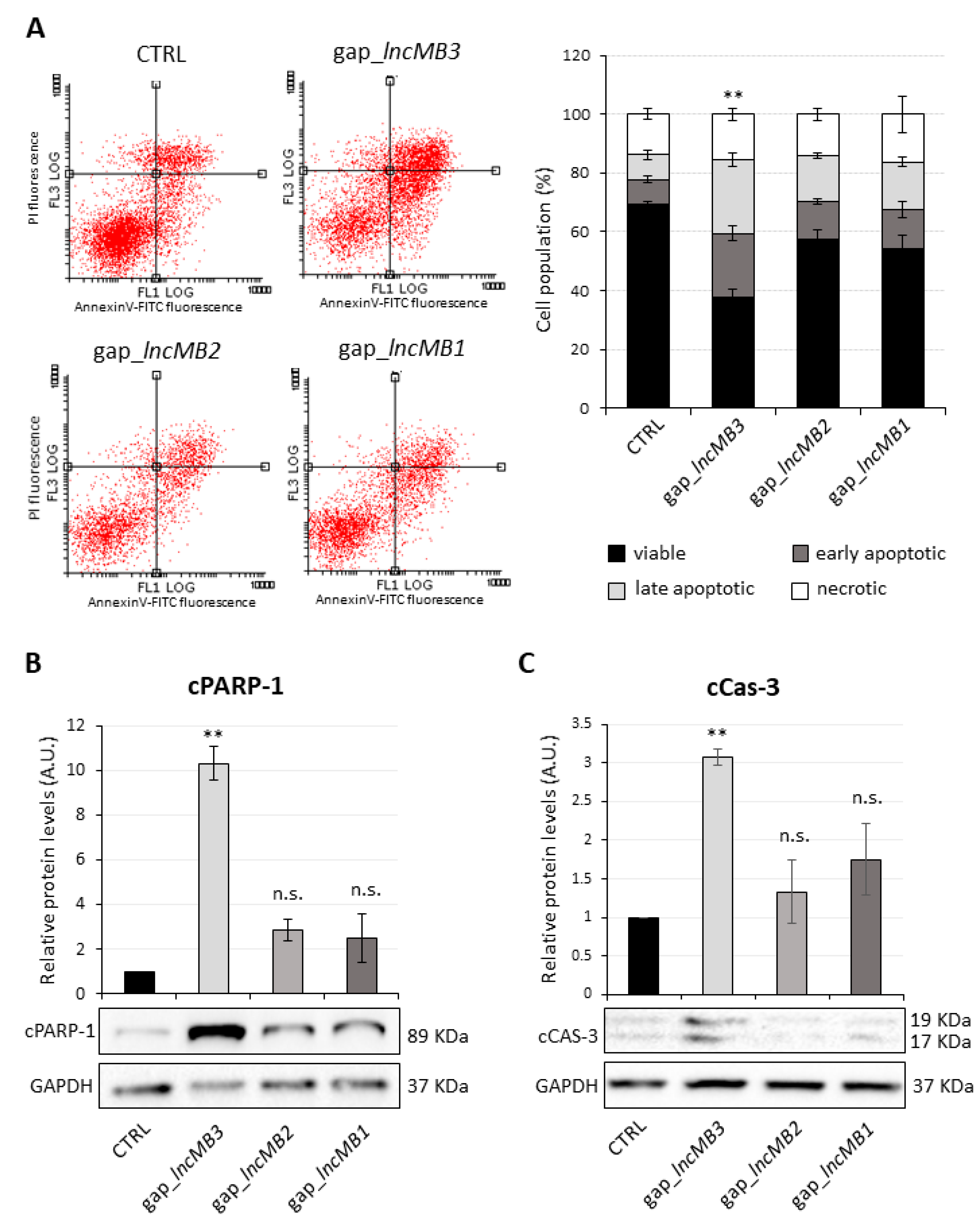
3.6. LncMB3 Counteracts Apoptosis
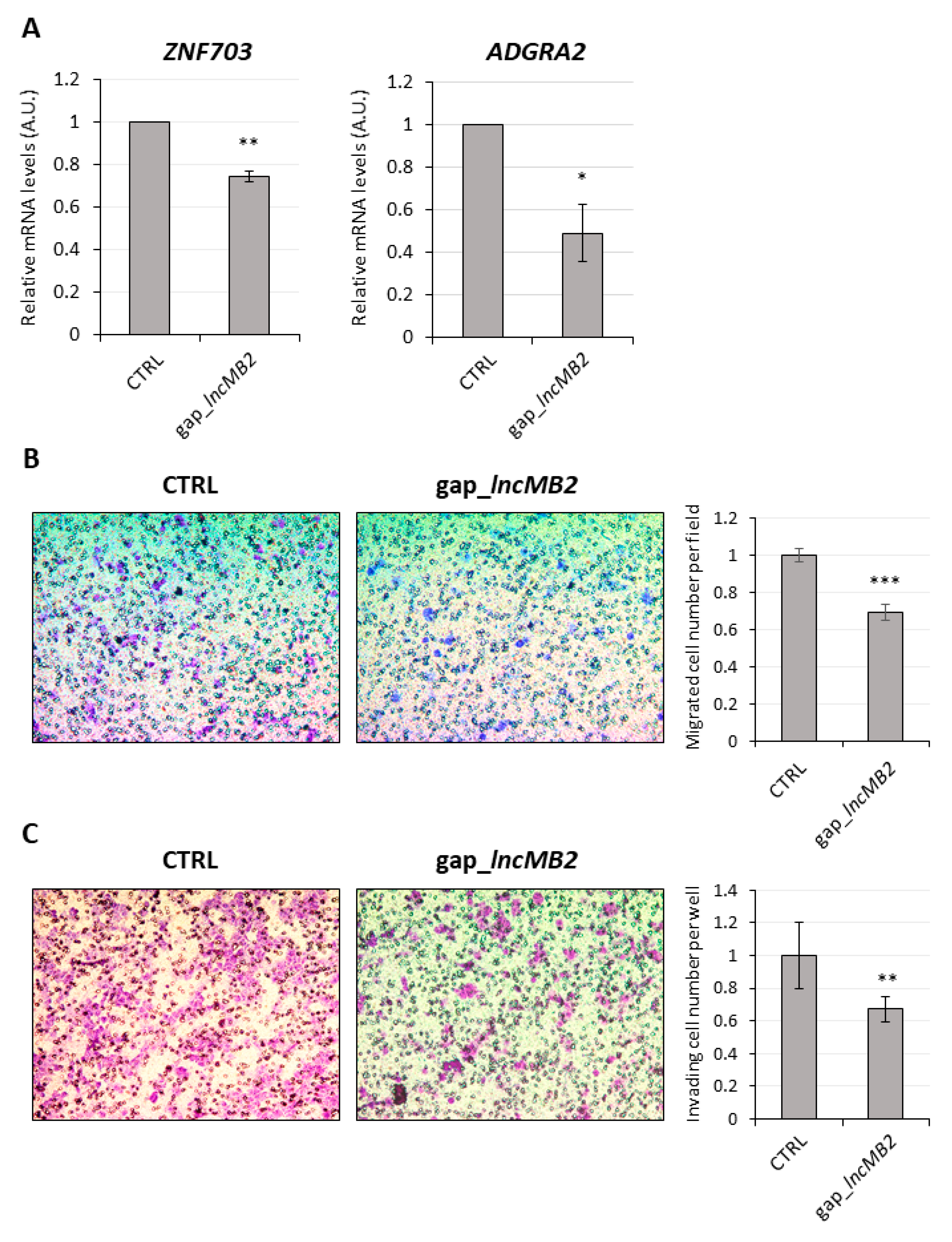
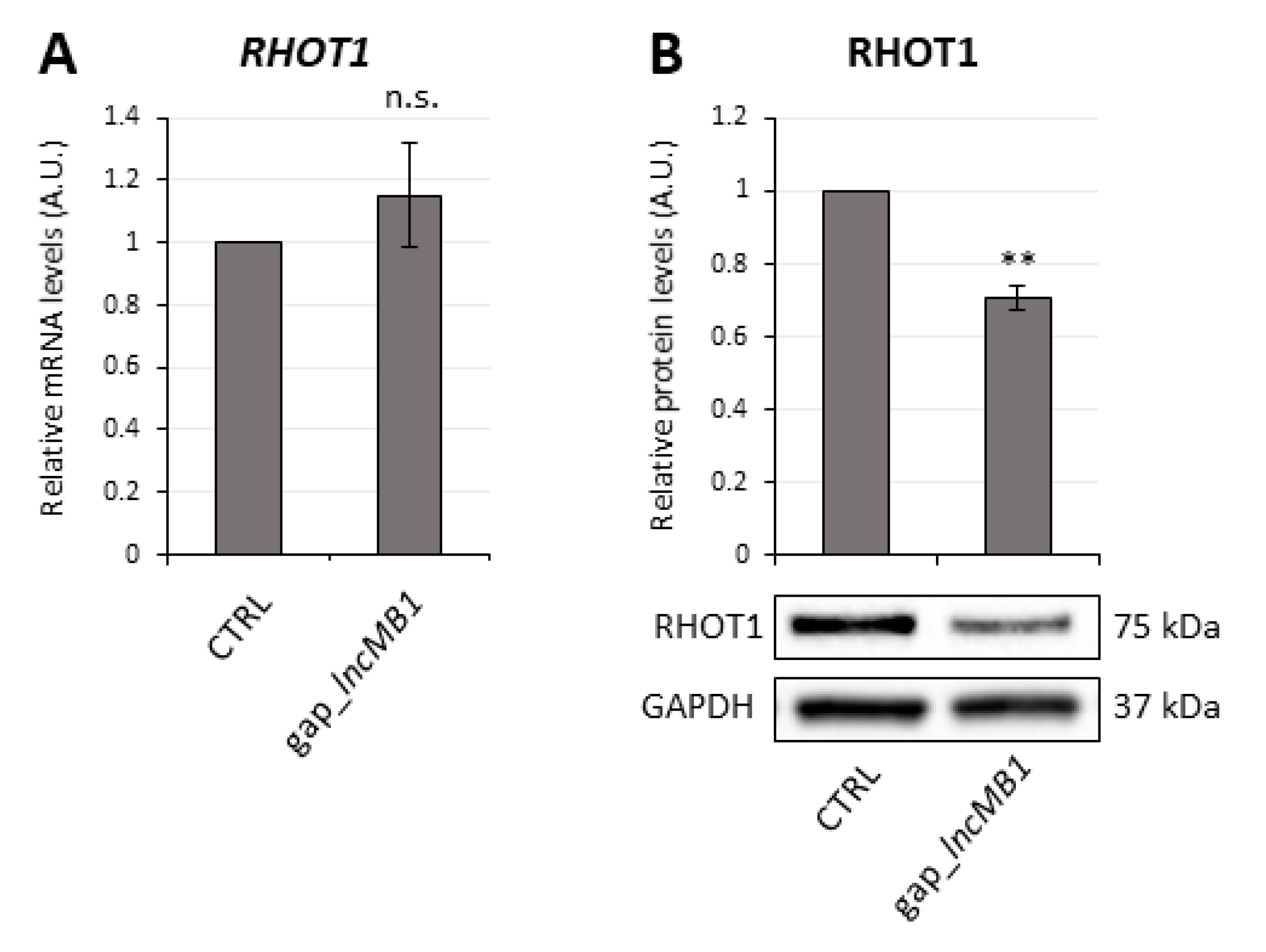
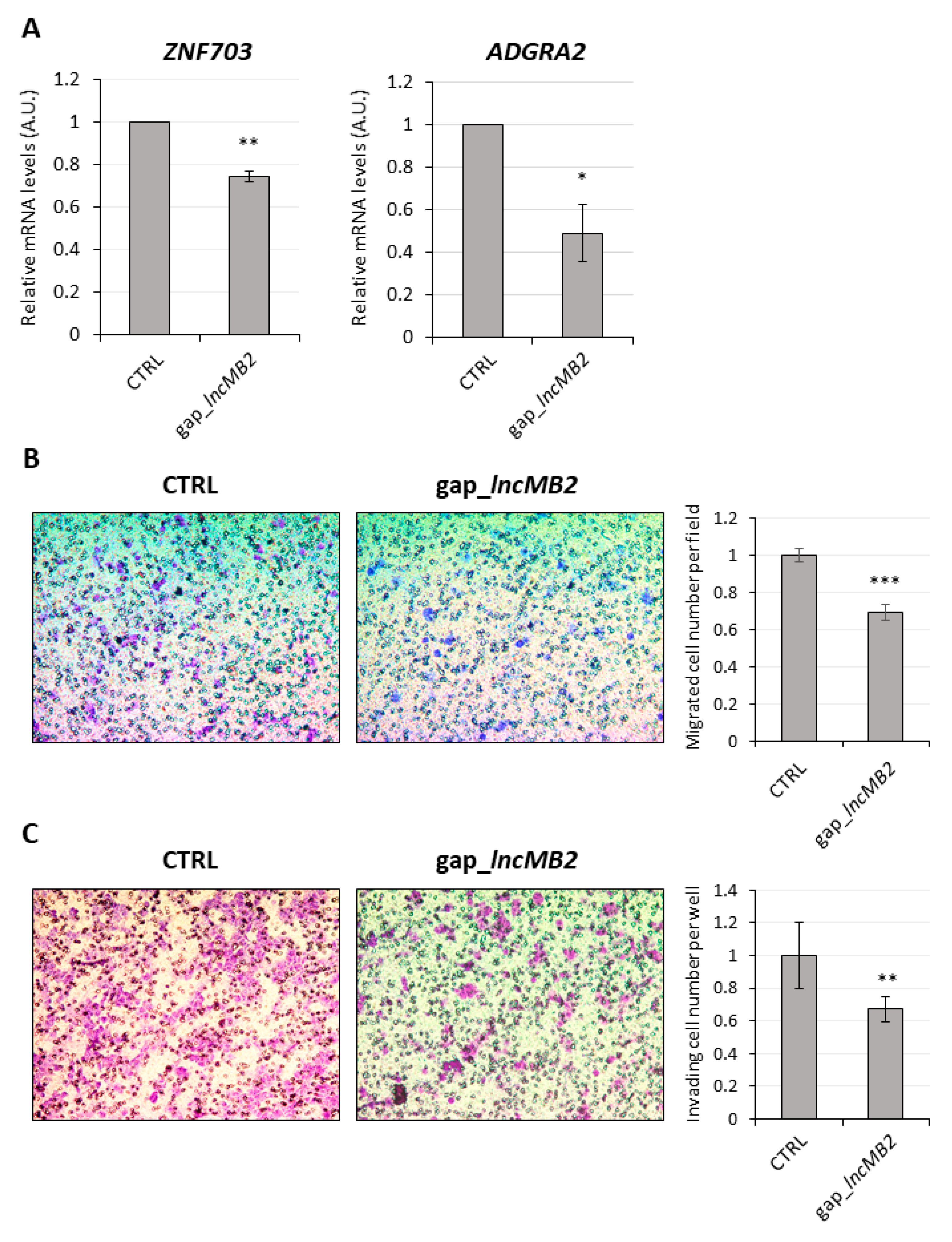
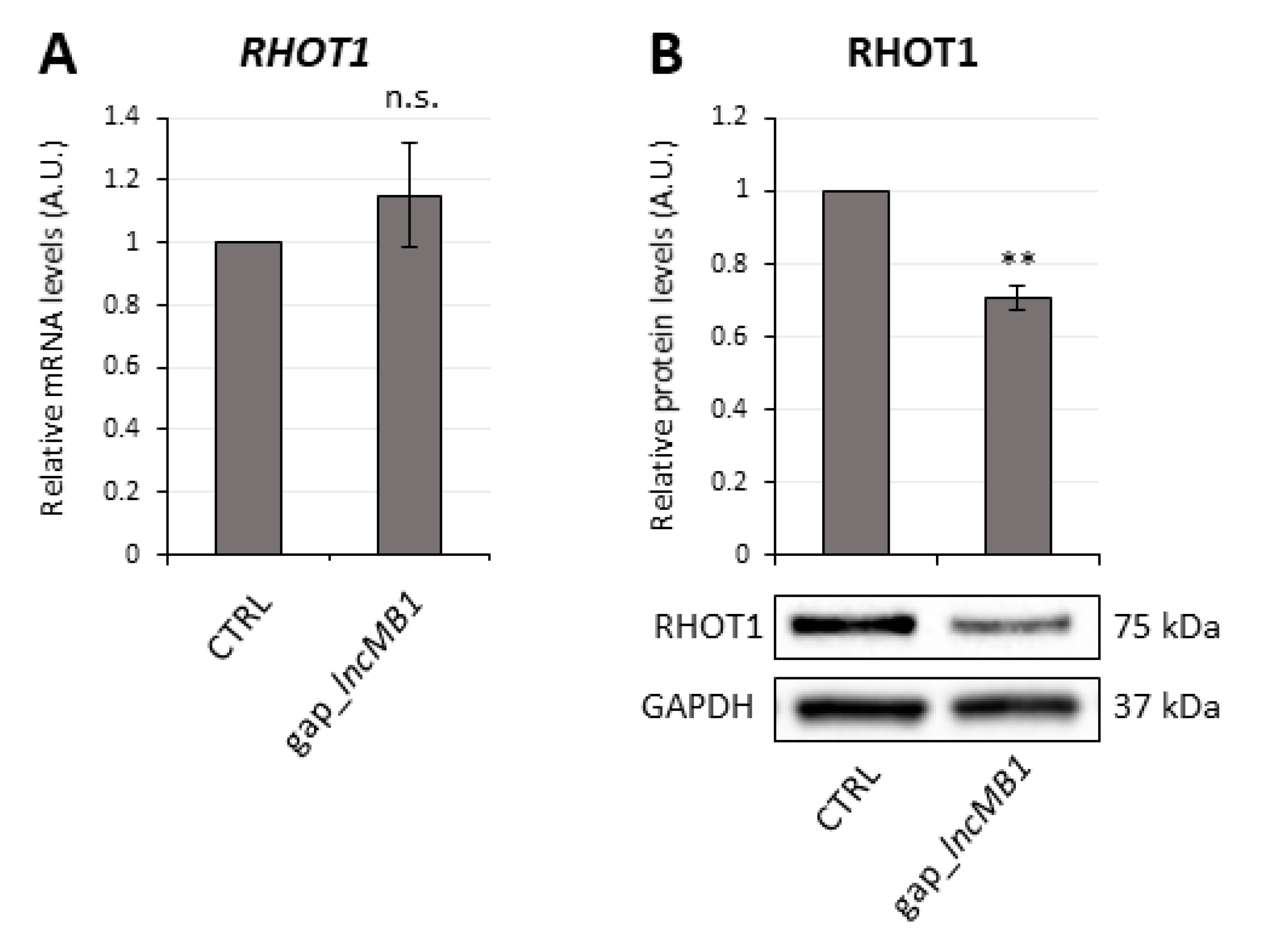
3.7. LncMB2 and lncMB1 Control ZNF703 and RHOT1 Gene Expression, Respectively
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Northcott, P.A.; Buchhalter, I.; Morrissy, S.; Hovestadt, V.; Weischenfeldt, J.; Ehrenberger, T.; Gröbner, S.; Segura-Wang, M.; Zichner, T.; Rudneva, V.; et al. The whole-genome landscape of medulloblastoma subtypes. Nat. Cell Biol. 2017, 547, 311–317. [Google Scholar] [CrossRef] [Green Version]
- Cavalli, F.M.; Remke, M.; Rampasek, L.; Peacock, J.; Shih, D.J.H.; Luu, B.; Garzia, L.; Torchia, J.; Nor, C.; Morrissy, S.; et al. Intertumoral Heterogeneity within Medulloblastoma Subgroups. Cancer Cell 2017, 31, 737–754.e6. [Google Scholar] [CrossRef] [Green Version]
- Northcott, P.A.; Shih, D.J.H.; Remke, M.; Cho, Y.-J.; Kool, M.; Hawkins, C.; Eberhart, C.G.; Dubuc, A.; Guettouche, T.; Cardentey, Y.; et al. Rapid, reliable, and reproducible molecular sub-grouping of clinical medulloblastoma samples. Acta Neuropathol. 2011, 123, 615–626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, M.D.; Northcott, P.A.; Korshunov, A.; Remke, M.; Cho, Y.-J.; Clifford, S.C.; Eberhart, C.G.; Parsons, D.W.; Rutkowski, S.; Gajjar, A.; et al. Molecular subgroups of medulloblastoma: The current consensus. Acta Neuropathol. 2011, 123, 465–472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clifford, S.C.; Lusher, M.E.; Lindsey, J.C.; Langdon, J.A.; Gilbertson, R.J.; Straughton, D.; Ellison, D.W. Wnt/Wingless Pathway Activation and Chromosome 6 Loss Characterise a Distinct Molecular Sub-Group of Medulloblastomas Associated with a Favourable Prognosis. Cell Cycle 2006, 5, 2666–2670. [Google Scholar] [CrossRef] [Green Version]
- Cho, Y.-J.; Tsherniak, A.; Tamayo, P.; Santagata, S.; Ligon, A.; Greulich, H.; Berhoukim, R.; Amani, V.; Goumnerova, L.; Eberhart, C.G.; et al. Integrative Genomic Analysis of Medulloblastoma Identifies a Molecular Subgroup That Drives Poor Clinical Outcome. J. Clin. Oncol. 2011, 29, 1424–1430. [Google Scholar] [CrossRef]
- Kool, M.; Koster, J.; Bunt, J.; Hasselt, N.E.; Lakeman, A.; Van Sluis, P.; Troost, D.; Meeteren, N.S.-V.; Caron, H.N.; Cloos, J.; et al. Integrated Genomics Identifies Five Medulloblastoma Subtypes with Distinct Genetic Profiles, Pathway Signatures and Clinicopathological Features. PLoS ONE 2008, 3, e3088. [Google Scholar] [CrossRef]
- Robinson, G.; Parker, M.; Kranenburg, T.; Lu, C.; Chen, X.; Ding, L.; Phoenix, T.N.; Hedlund, E.; Wei, L.; Zhu, X.; et al. Novel mutations target distinct subgroups of medulloblastoma. Nat. Cell Biol. 2012, 488, 43–48. [Google Scholar] [CrossRef] [Green Version]
- Ballabio, C.; Anderle, M.; Gianesello, M.; Lago, C.; Miele, E.; Cardano, M.; Aiello, G.; Piazza, S.; Caron, D.; Gianno, F.; et al. Modeling medulloblastoma in vivo and with human cerebellar organoids. Nat. Commun. 2020, 11, 1–18. [Google Scholar] [CrossRef]
- Oster, S.K.; Ho, C.S.; Soucie, E.; Penn, L. The myc Oncogene: Omplex. Adv. Cancer Res. 2002, 84, 81–154. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Fang, H.; Zhang, J.; Gu, Y. Targeting “undruggable” c-Myc protein by synthetic lethality. Front. Med. 2021, 1–10. [Google Scholar] [CrossRef]
- Van Dang, C. Enigmatic MYC Conducts an Unfolding Systems Biology Symphony. Genes Cancer 2010, 1, 526–531. [Google Scholar] [CrossRef]
- Dang, C.V.; O’Donnell, K.A.; Zeller, K.I.; Nguyen, T.; Osthus, R.C.; Li, F. The c-Myc target gene network. Semin. Cancer Biol. 2006, 16, 253–264. [Google Scholar] [CrossRef]
- Patel, J.H.; Loboda, A.P.; Showe, M.K.; Showe, L.C.; McMahon, S.B. Analysis of genomic targets reveals complex functions of MYC. Nat. Rev. Cancer 2004, 4, 562–568. [Google Scholar] [CrossRef] [PubMed]
- Smith, K.; Dalton, S. Myc transcription factors: Key regulators behind establishment and maintenance of pluripotency. Regen. Med. 2010, 5, 947–959. [Google Scholar] [CrossRef] [Green Version]
- Whitfield, J.; Beaulieu, M.-E.; Soucek, L. Strategies to Inhibit Myc and Their Clinical Applicability. Front. Cell Dev. Biol. 2017, 5, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Massó-Vallés, D.; Soucek, L. Blocking Myc to Treat Cancer: Reflecting on Two Decades of Omomyc. Cells 2020, 9, 883. [Google Scholar] [CrossRef] [Green Version]
- Dang, C.V. MYC on the Path to Cancer. Cell 2012, 149, 22–35. [Google Scholar] [CrossRef] [Green Version]
- Laneve, P.; Caffarelli, E. The Non-coding Side of Medulloblastoma. Front. Cell Dev. Biol. 2020, 8, 275. [Google Scholar] [CrossRef]
- Kesherwani, V.; Shukla, M.; Coulter, D.W.; Sharp, J.G.; Joshi, S.S.; Chaturvedi, N.K. Long non-coding RNA profiling of pediatric Medulloblastoma. BMC Med Genom. 2020, 13, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Roussel, M.F.; Robinson, G. Role of MYC in Medulloblastoma. Cold Spring Harb. Perspect. Med. 2013, 3, a014308. [Google Scholar] [CrossRef] [Green Version]
- Takwi, A.A.L.; Li, Y.; Buscaglia, L.E.B.; Zhang, J.; Choudhury, S.; Park, A.K.; Liu, M.; Young, K.H.; Park, W.-Y.; Martin, R.C.G.; et al. A statin-regulated microRNA represses human c-Myc expression and function. EMBO Mol. Med. 2012, 4, 896–909. [Google Scholar] [CrossRef] [PubMed]
- Soucek, L.; Helmer-Citterich, M.; Sacco, A.; Jucker, R.; Cesareni, G.; Nasi, S. Design and properties of a Myc derivative that efficiently homodimerizes. Oncogene 1998, 17, 2463–2472. [Google Scholar] [CrossRef] [Green Version]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2012, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [Green Version]
- Robinson, M.D.; McCarthy, D.; Smyth, G. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2009, 26, 139–140. [Google Scholar] [CrossRef] [Green Version]
- Luo, Z.; Dong, X.; Yu, J.; Xia, Y.; Berry, K.P.; Rao, R.; Xu, L.; Xue, P.; Chen, T.; Lin, Y.; et al. Genomic and Transcriptomic Analyses Reveals ZNF124 as a Critical Regulator in Highly Aggressive Medulloblastomas. Front. Cell Dev. Biol. 2021, 9. [Google Scholar] [CrossRef]
- Ecker, J.; Thatikonda, V.; Sigismondo, G.; Selt, F.; Valinciute, G.; Oehme, I.; Müller, C.; Buhl, J.L.; Ridinger, J.; Usta, D.; et al. Reduced chromatin binding of MYC is a key effect of HDAC inhibition in MYC amplified medulloblastoma. Neuro-Oncology 2020, 23, 226–239. [Google Scholar] [CrossRef]
- Ferrara, M.; Sessa, G.; Fiore, M.; Bernard, F.; A Asteriti, I.; Cundari, E.; Colotti, G.; Ferla, S.; Desideri, M.; Buglioni, S.; et al. Small molecules targeted to the microtubule–Hec1 interaction inhibit cancer cell growth through microtubule stabilization. Oncogene 2017, 37, 231–240. [Google Scholar] [CrossRef] [Green Version]
- Friedman, H.S.; Burger, P.C.; Bigner, S.H.; Trojanowski, J.Q.; Wikstrand, C.J.; Halperin, E.C.; Bigner, D.D. Establishment and Characterization of the Human Medulloblastoma Cell Line and Transplantable Xenograft D283 Med. J. Neuropathol. Exp. Neurol. 1985, 44, 592–605. [Google Scholar] [CrossRef]
- Ivanov, D.P.; Coyle, B.; Walker, D.A.; Grabowska, A.M. In vitro models of medulloblastoma: Choosing the right tool for the job. J. Biotechnol. 2016, 236, 10–25. [Google Scholar] [CrossRef]
- Siu, I.-M.; Lal, A.; Blankenship, J.R.; Aldosari, N.; Riggins, G.J. c-Myc Promoter Activation in Medulloblastoma. Cancer Res. 2003, 63, 4773–4776. [Google Scholar]
- Galardi, S.; Savino, M.; Scagnoli, F.; Pellegatta, S.; Pisati, F.; Zambelli, F.; Illi, B.; Annibali, D.; Beji, S.; Orecchini, E.; et al. Resetting cancer stem cell regulatory nodes upon MYC inhibition. EMBO Rep. 2016, 17, 1872–1889. [Google Scholar] [CrossRef] [Green Version]
- Demma, M.J.; Mapelli, C.; Sun, A.; Bodea, S.; Ruprecht, B.; Javaid, S.; Wiswell, D.; Muise, E.; Chen, S.; Zelina, J.; et al. Omomyc Reveals New Mechanisms To Inhibit the MYC Oncogene. Mol. Cell. Biol. 2019, 39. [Google Scholar] [CrossRef] [Green Version]
- Fiaschetti, G.; Castelletti, D.; Zoller, S.; Schramm, A.; Schroeder, C.; Nagaishi, M.; Stearns, D.; Mittelbronn, M.; Eggert, A.; Westermann, F.; et al. Bone morphogenetic protein-7 is a MYC target with prosurvival functions in childhood medulloblastoma. Oncogene 2011, 30, 2823–2835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castanotto, D.; Lin, M.; Kowolik, C.; Wang, L.; Ren, X.-Q.; Soifer, H.S.; Koch, T.; Hansen, B.R.; Oerum, H.; Armstrong, B.; et al. A cytoplasmic pathway for gapmer antisense oligonucleotide-mediated gene silencing in mammalian cells. Nucleic Acids Res. 2015, 43, 9350–9361. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.-H.; Sun, H.; Nichols, J.G.; Crooke, S.T. RNase H1-Dependent Antisense Oligonucleotides Are Robustly Active in Directing RNA Cleavage in Both the Cytoplasm and the Nucleus. Mol. Ther. 2017, 25, 2075–2092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plesca, D.; Mazumder, S.; Almasan, A. Chapter 6 DNA Damage Response and Apoptosis. Methods Enzymol. 2008, 446, 107–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaufmann, S.H.; Desnoyers, S.; Ottaviano, Y.; Davidson, N.E.; Poirier, G.G. Specific Proteolytic Cleavage of Poly(ADP-ribose) Polymerase: An Early Marker of Chemotherapy-induced Apoptosis. Cancer Res. 1993, 53, 3976–3985. [Google Scholar] [PubMed]
- Tewari, M.; Quan, L.T.; O’Rourke, K.; Desnoyers, S.; Zeng, Z.; Beidler, D.R.; Poirier, G.G.; Salvesen, G.S.; Dixit, V.M. Yama/CPP32β, a mammalian homolog of CED-3, is a CrmA-inhibitable protease that cleaves the death substrate poly(ADP-ribose) polymerase. Cell 1995, 81, 801–809. [Google Scholar] [CrossRef] [Green Version]
- Pistritto, G.; Trisciuoglio, D.; Ceci, C.; Garufi, A.; D’Orazi, G. Apoptosis as anticancer mechanism: Function and dysfunction of its modulators and targeted therapeutic strategies. Aging 2016, 8, 603–619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, G.; Ma, F.; Zhong, M.; Fang, L.; Peng, Y.; Xin, X.; Zhong, J.; Yuan, F.; Gu, H.; Zhu, W.; et al. ZNF703 acts as an oncogene that promotes progression in gastric cancer. Oncol. Rep. 2014, 31, 1877–1882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanhollebeke, B.; Stone, O.; Bostaille, N.; Cho, C.; Zhou, Y.Y.; Maquet, E.; Gauquier, A.; Cabochette, P.; Fukuhara, S.S.; Mochizuki, N.N.; et al. Tip cell-specific requirement for an atypical Gpr124- and Reck-dependent Wnt/β-catenin pathway during brain angiogenesis. eLife 2015, 4, e06489. [Google Scholar] [CrossRef] [PubMed]
- Northcott, P.A.; Jones, D.T.W.; Kool, M.; Robinson, G.; Gilbertson, R.J.; Cho, Y.-J.; Pomeroy, S.L.; Korshunov, A.; Lichter, P.; Taylor, M.D.; et al. Medulloblastomics: The end of the beginning. Nat. Rev. Cancer 2012, 12, 818–834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Northcott, P.A.; Korshunov, A.; Witt, H.; Hielscher, T.; Eberhart, C.G.; Mack, S.C.; Bouffet, E.; Clifford, S.C.; Hawkins, C.E.; French, P.; et al. Medulloblastoma Comprises Four Distinct Molecular Variants. J. Clin. Oncol. 2011, 29, 1408–1414. [Google Scholar] [CrossRef] [PubMed]
- Grossmann, D.; Berenguer-Escuder, C.; Chemla, A.; Arena, G.; Krüger, R. The Emerging Role of RHOT1/Miro1 in the Pathogenesis of Parkinson’s Disease. Front. Neurol. 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Schwarz, T.L. The Mechanism of Ca2+-Dependent Regulation of Kinesin-Mediated Mitochondrial Motility. Cell 2009, 136, 163–174. [Google Scholar] [CrossRef] [Green Version]
- Chang, K.T.; Niescier, R.F.; Min, K.-T. Mitochondrial matrix Ca2+ as an intrinsic signal regulating mitochondrial motility in axons. Proc. Natl. Acad. Sci. USA 2011, 108, 15456–15461. [Google Scholar] [CrossRef] [Green Version]
- MacAskill, A.; Rinholm, J.E.; Twelvetrees, A.; Carcamo, I.L.A.; Muir, J.; Fransson, A.; Aspenstrom, P.; Attwell, D.; Kittler, J.T. Miro1 Is a Calcium Sensor for Glutamate Receptor-Dependent Localization of Mitochondria at Synapses. Neuron 2009, 61, 541–555. [Google Scholar] [CrossRef] [Green Version]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.; Zhou, S.; Diaz, L.; Kinzler, K.W. Cancer Genome Landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef] [PubMed]
- Dagogo-Jack, I.; Shaw, A.T. Tumour heterogeneity and resistance to cancer therapies. Nat. Rev. Clin. Oncol. 2017, 15, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Reyna, M.A.; PCAWG Drivers and Functional Interpretation Working Group; Haan, D.; Paczkowska, M.; Verbeke, L.P.C.; Vazquez, M.; Kahraman, A.; Pulido-Tamayo, S.; Barenboim, J.; Wadi, L.; et al. Pathway and network analysis of more than 2500 whole cancer genomes. Nat. Commun. 2020, 11, 729. [Google Scholar] [CrossRef] [PubMed]
- Owens, M.A.; Horten, B.C.; Da Silva, M.M. HER2 Amplification Ratios by Fluorescence In Situ Hybridization and Correlation with Immunohistochemistry in a Cohort of 6556 Breast Cancer Tissues. Clin. Breast Cancer 2004, 5, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Climente-González, H.; Porta-Pardo, E.; Godzik, A.; Eyras, E. The Functional Impact of Alternative Splicing in Cancer. Cell Rep. 2017, 20, 2215–2226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faderl, S.; Talpaz, M.; Estrov, Z.; O’Brien, S.; Kurzrock, R.; Kantarjian, H.M. The Biology of Chronic Myeloid Leukemia. N. Engl. J. Med. 1999, 341, 164–172. [Google Scholar] [CrossRef]
- Statello, L.; Guo, C.-J.; Chen, L.-L.; Huarte, M. Gene regulation by long non-coding RNAs and its biological functions. Nat. Rev. Mol. Cell Biol. 2020, 22, 96–118. [Google Scholar] [CrossRef] [PubMed]
- Kopp, F.; Mendell, J.T. Functional Classification and Experimental Dissection of Long Noncoding RNAs. Cell 2018, 172, 393–407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gutschner, T.; Diederichs, S. The hallmarks of cancer. RNA Biol. 2012, 9, 703–719. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Zhang, X.; Chen, W.; Hu, X.; Li, J.; Liu, C. Regulatory roles of long noncoding RNAs implicated in cancer hallmarks. Int. J. Cancer 2019, 146, 906–916. [Google Scholar] [CrossRef] [PubMed]
- Hart, J.; Roberts, T.C.; Weinberg, M.; Morris, K.; Vogt, P.K. MYC regulates the non-coding transcriptome. Oncotarget 2014, 5, 12543–12554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iyer, M.K.; Niknafs, Y.S.; Malik, R.; Singhal, U.; Sahu, A.; Hosono, Y.; Barrette, T.R.; Prensner, J.; Evans, J.R.; Zhao, S.; et al. The landscape of long noncoding RNAs in the human transcriptome. Nat. Genet. 2015, 47, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, A.M.; Chang, H.Y. Long Noncoding RNAs in Cancer Pathways. Cancer Cell 2016, 29, 452–463. [Google Scholar] [CrossRef] [Green Version]
- Laneve, P.; Po, A.; Favia, A.; Legnini, I.; Alfano, V.; Rea, J.; Di Carlo, V.; Bevilacqua, V.; Miele, E.; Mastronuzzi, A.; et al. The long noncoding RNA linc-NeD125 controls the expression of medulloblastoma driver genes by microRNA sponge activity. Oncotarget 2017, 8, 31003–31015. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.; Wu, D.; Gao, L.; Liu, X.; Jin, Y.; Wang, D.; Wang, T.; Li, X. Competing endogenous RNA networks in human cancer: Hypothesis, validation, and perspectives. Oncotarget 2016, 7, 13479–13490. [Google Scholar] [CrossRef] [Green Version]
- Sun, Q.; Hao, Q.; Prasanth, K.V. Nuclear Long Noncoding RNAs: Key Regulators of Gene Expression. Trends Genet. 2018, 34, 142–157. [Google Scholar] [CrossRef]
- Morlando, M.; Fatica, A. Alteration of Epigenetic Regulation by Long Noncoding RNAs in Cancer. Int. J. Mol. Sci. 2018, 19, 570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ostrom, Q.T.; Gittleman, H.; Truitt, G.; Boscia, A.; Kruchko, C.; Barnholtz-Sloan, J. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2011–2015. Neuro-Oncology 2018, 20, iv1–iv86. [Google Scholar] [CrossRef] [Green Version]
- Laneve, P.; Rea, J.; Caffarelli, E. Long Noncoding RNAs: Emerging Players in Medulloblastoma. Front. Pediatr. 2019, 7. [Google Scholar] [CrossRef]
- Jung, L.A.; Gebhardt, A.; Koelmel, W.; Ade, C.P.; Walz, S.; Kuper, J.; von Eyss, B.; Letschert, S.; Redel, C.; D’Artista, L.; et al. OmoMYC blunts promoter invasion by oncogenic MYC to inhibit gene expression characteristic of MYC-dependent tumors. Oncogene 2016, 36, 1911–1924. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.; Wang, C.; Hu, Y.; Li, X.; Jin, S.; Liu, O.; Gou, R.; Zhuang, Y.; Guo, Q.; Nie, X.; et al. ZNF703 promotes tumor progression in ovarian cancer by interacting with HE4 and epigenetically regulating PEA15. J. Exp. Clin. Cancer Res. 2020, 39, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Klæstad, E.; Sawicka, J.E.; Engstrøm, M.J.; Ytterhus, B.; Valla, M.; Bofin, A.M. ZNF703 gene copy number and protein expression in breast cancer; associations with proliferation, prognosis and luminal subtypes. Breast Cancer Res. Treat. 2021, 186, 65–77. [Google Scholar] [CrossRef]
- Guo, J.; Luo, C.; Yang, Y.; Dong, J.; Guo, Z.; Yang, J.; Lian, H.; Ye, C.; Liu, M. MiR-491-5p, as a Tumor Suppressor, Prevents Migration and Invasion of Breast Cancer by Targeting ZNF-703 to Regulate AKT/mTOR Pathway. Cancer Manag. Res. 2021, 13, 403–413. [Google Scholar] [CrossRef]
- Friedl, P.; Wolf, K. Plasticity of cell migration: A multiscale tuning model. J. Cell Biol. 2009, 188, 11–19. [Google Scholar] [CrossRef] [Green Version]
- Agarwal, E.; Altman, B.J.; Ho Seo, J.; Bertolini, I.; Ghosh, J.C.; Kaur, A.; Kossenkov, A.V.; Languino, L.R.; Gabrilovich, D.I.; Speicher, D.W.; et al. Myc Regulation of a Mitochondrial Trafficking Network Mediates Tumor Cell Invasion and Metas-tasis. Mol. Cell. Biol. 2019, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rea, J.; Carissimo, A.; Trisciuoglio, D.; Illi, B.; Picard, D.; Remke, M.; Laneve, P.; Caffarelli, E. Identification and Functional Characterization of Novel MYC-Regulated Long Noncoding RNAs in Group 3 Medulloblastoma. Cancers 2021, 13, 3853. https://doi.org/10.3390/cancers13153853
Rea J, Carissimo A, Trisciuoglio D, Illi B, Picard D, Remke M, Laneve P, Caffarelli E. Identification and Functional Characterization of Novel MYC-Regulated Long Noncoding RNAs in Group 3 Medulloblastoma. Cancers. 2021; 13(15):3853. https://doi.org/10.3390/cancers13153853
Chicago/Turabian StyleRea, Jessica, Annamaria Carissimo, Daniela Trisciuoglio, Barbara Illi, Daniel Picard, Marc Remke, Pietro Laneve, and Elisa Caffarelli. 2021. "Identification and Functional Characterization of Novel MYC-Regulated Long Noncoding RNAs in Group 3 Medulloblastoma" Cancers 13, no. 15: 3853. https://doi.org/10.3390/cancers13153853
APA StyleRea, J., Carissimo, A., Trisciuoglio, D., Illi, B., Picard, D., Remke, M., Laneve, P., & Caffarelli, E. (2021). Identification and Functional Characterization of Novel MYC-Regulated Long Noncoding RNAs in Group 3 Medulloblastoma. Cancers, 13(15), 3853. https://doi.org/10.3390/cancers13153853