
A Novel ZIP4-HDAC4-VEGFA Axis in High-Grade Serous Ovarian Cancer



Abstract
Simple Summary
Abstract
1. Introduction
2. Results
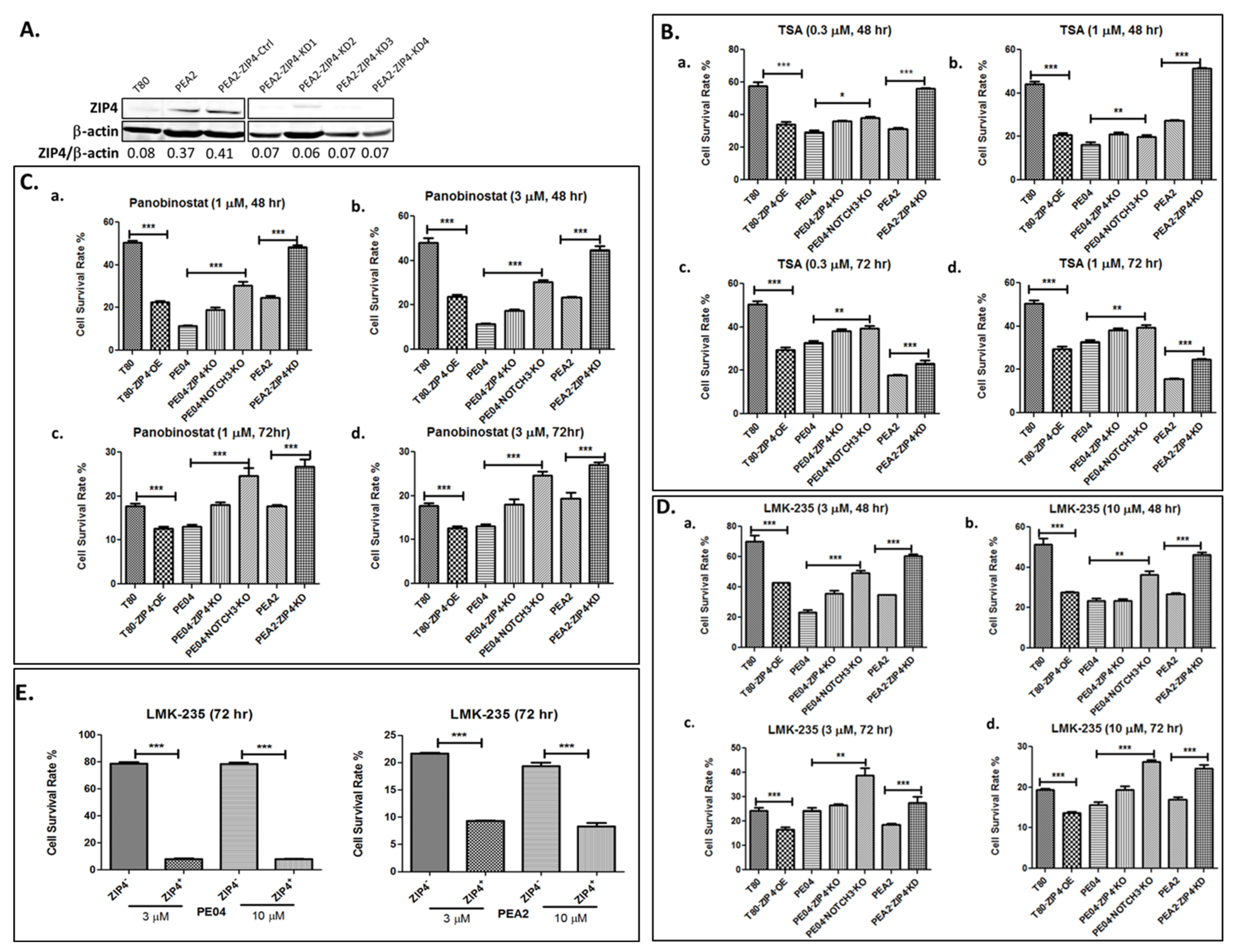
2.1. ZIP4 Sensitized HGSOC Cells to HDACis
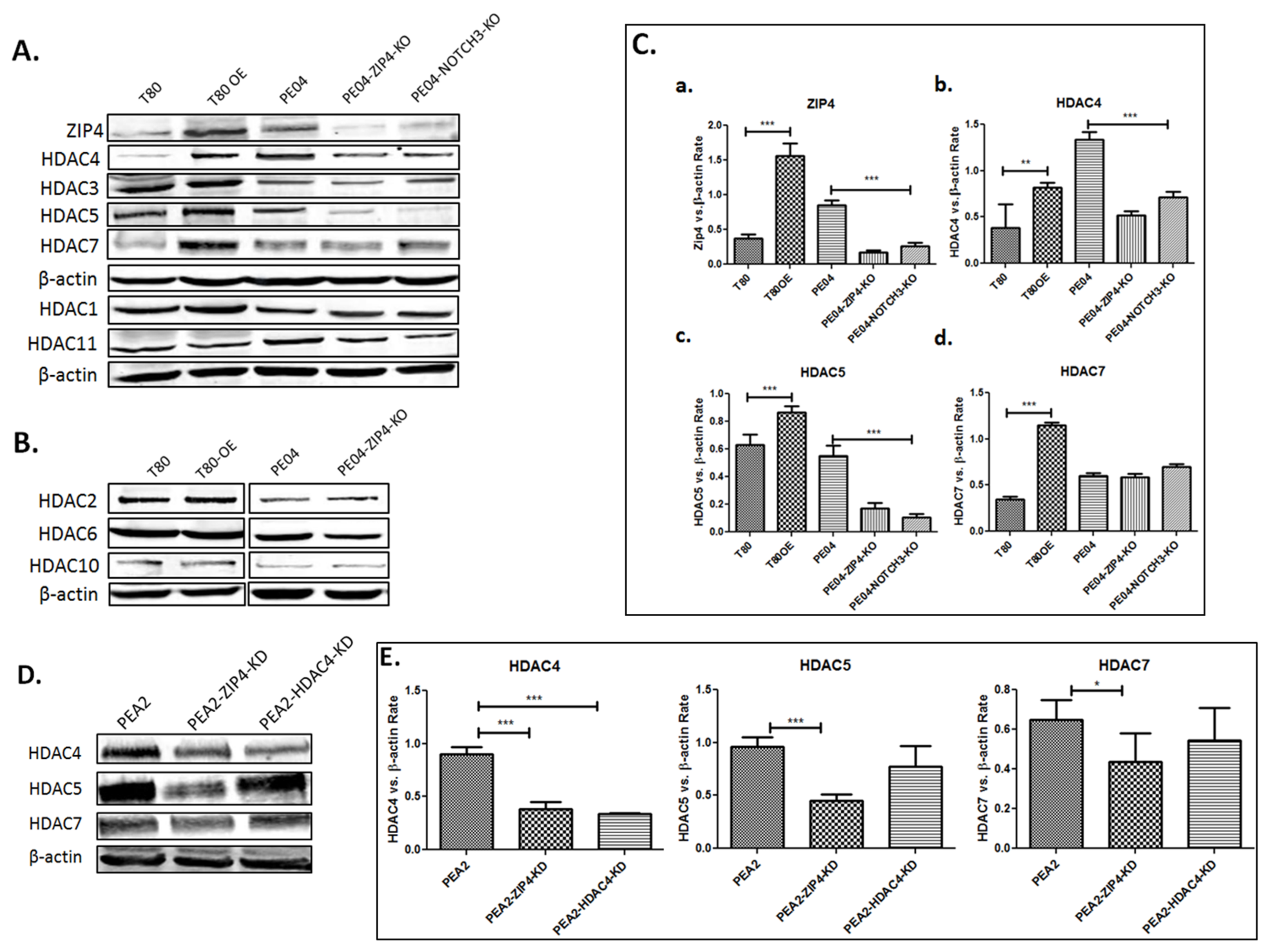
2.2. ZIP4 Induced Upregulation of Class IIa HDACs
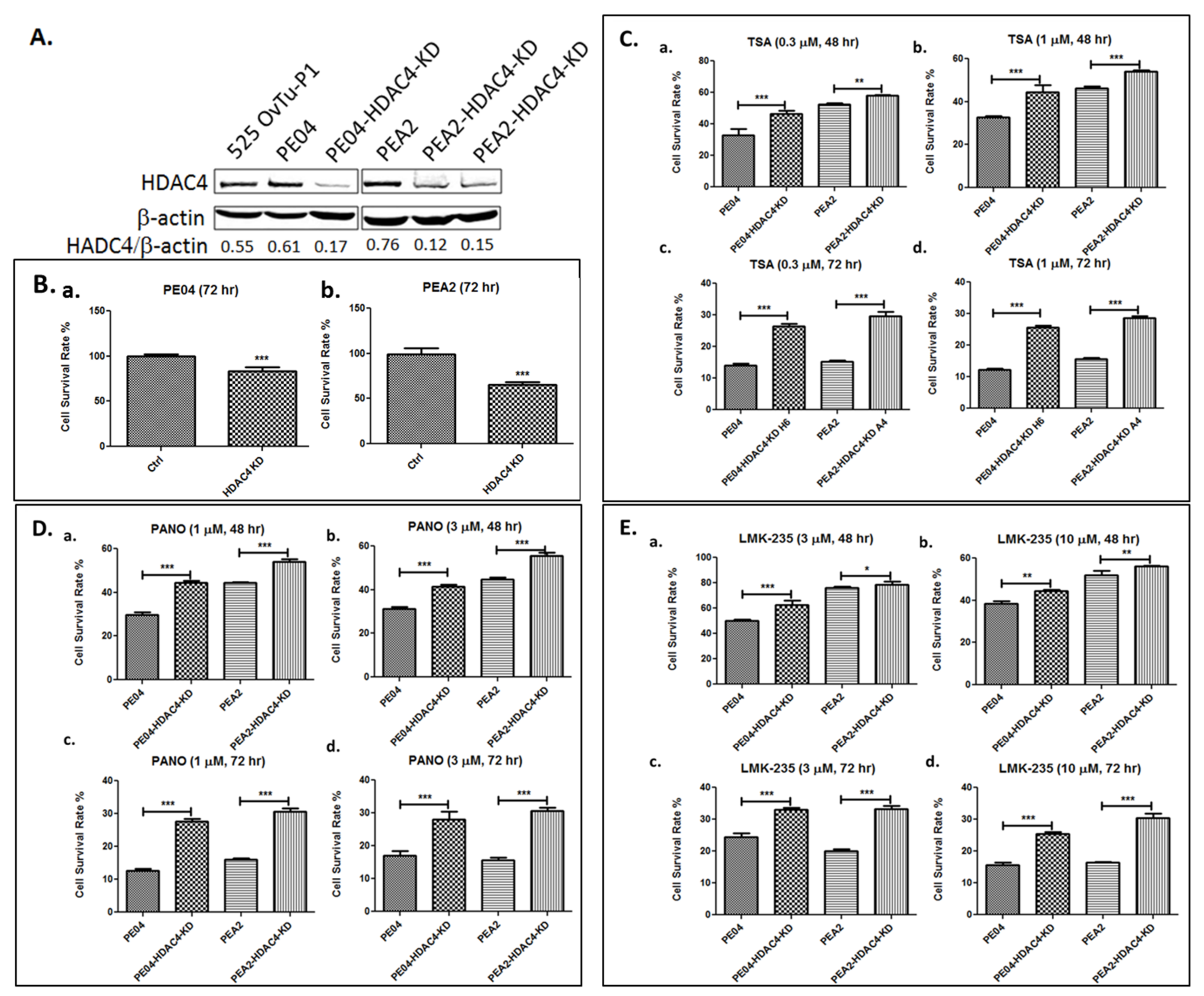
2.3. HDAC4 Played an Important Role in HDACi Sensitization in PE04 and PEA2 Cells
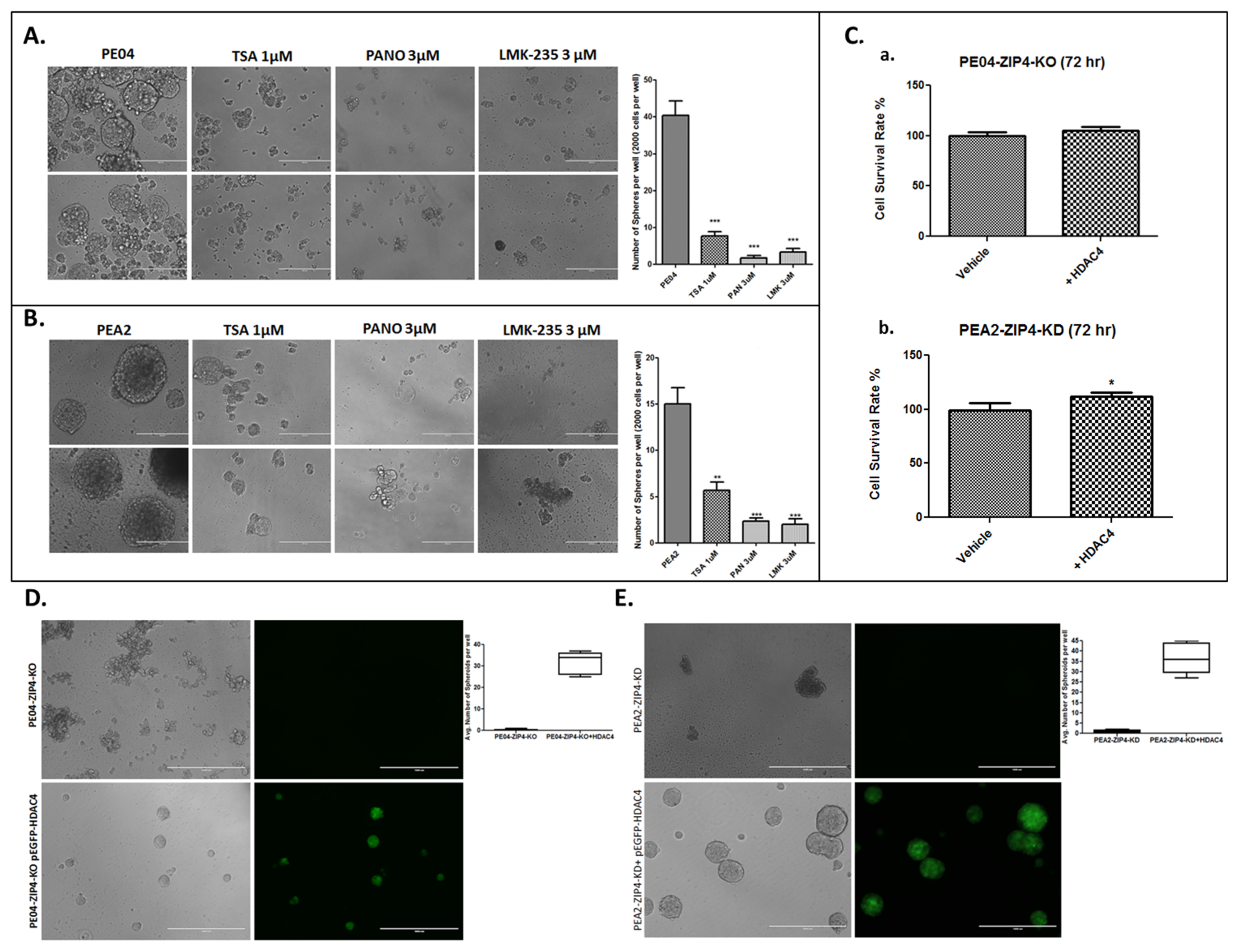
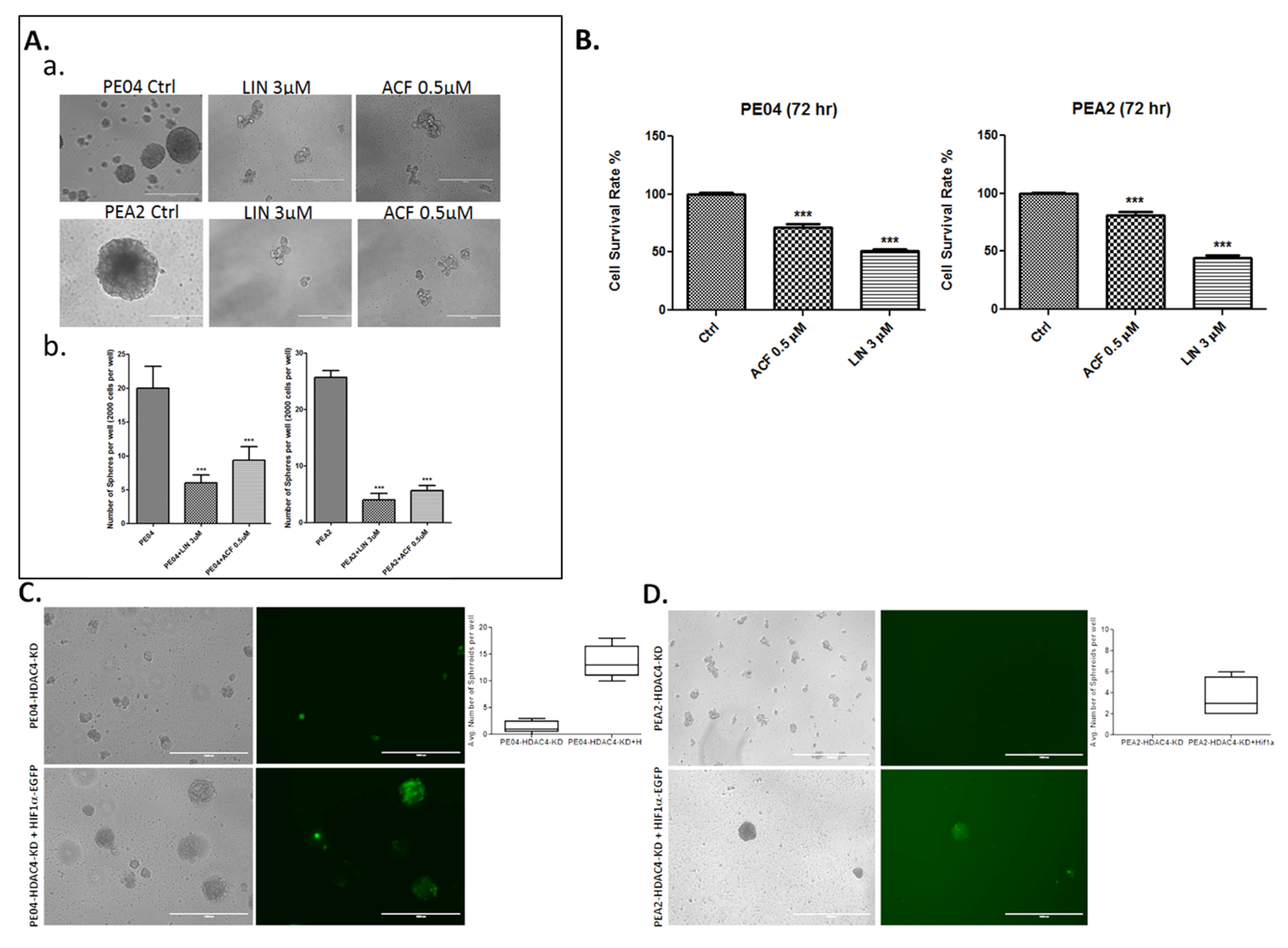
2.4. HDACis and HDAC4-KD Inhibited Spheroid Formation in HGSOC Cells
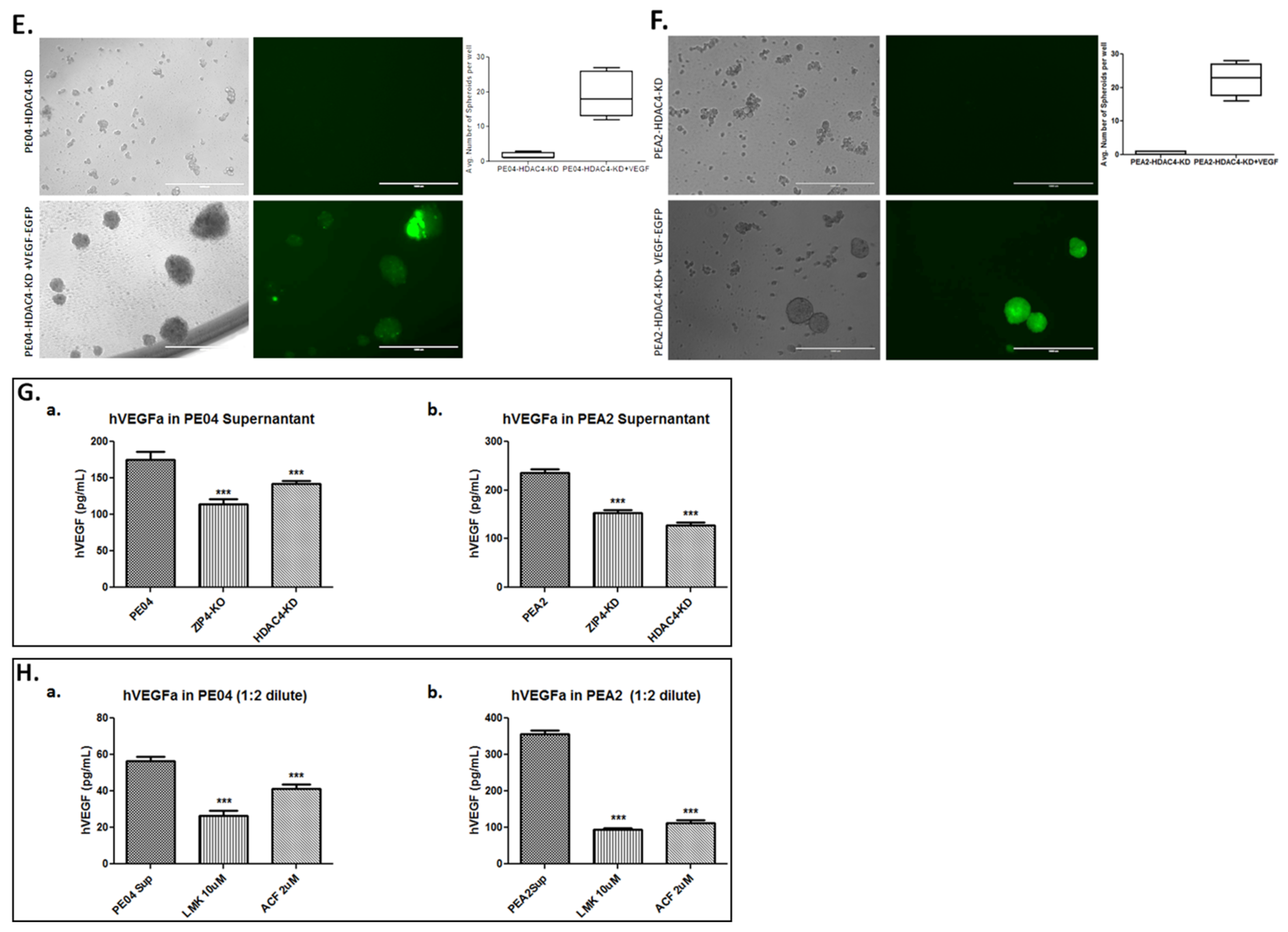
2.5. The Role of the Hypoxia Inducible Factor-1 Alpha (HIF1α) and Endothelial Growth Factor A (VEGFA) in Spheroid Formation in HGSOC Cells
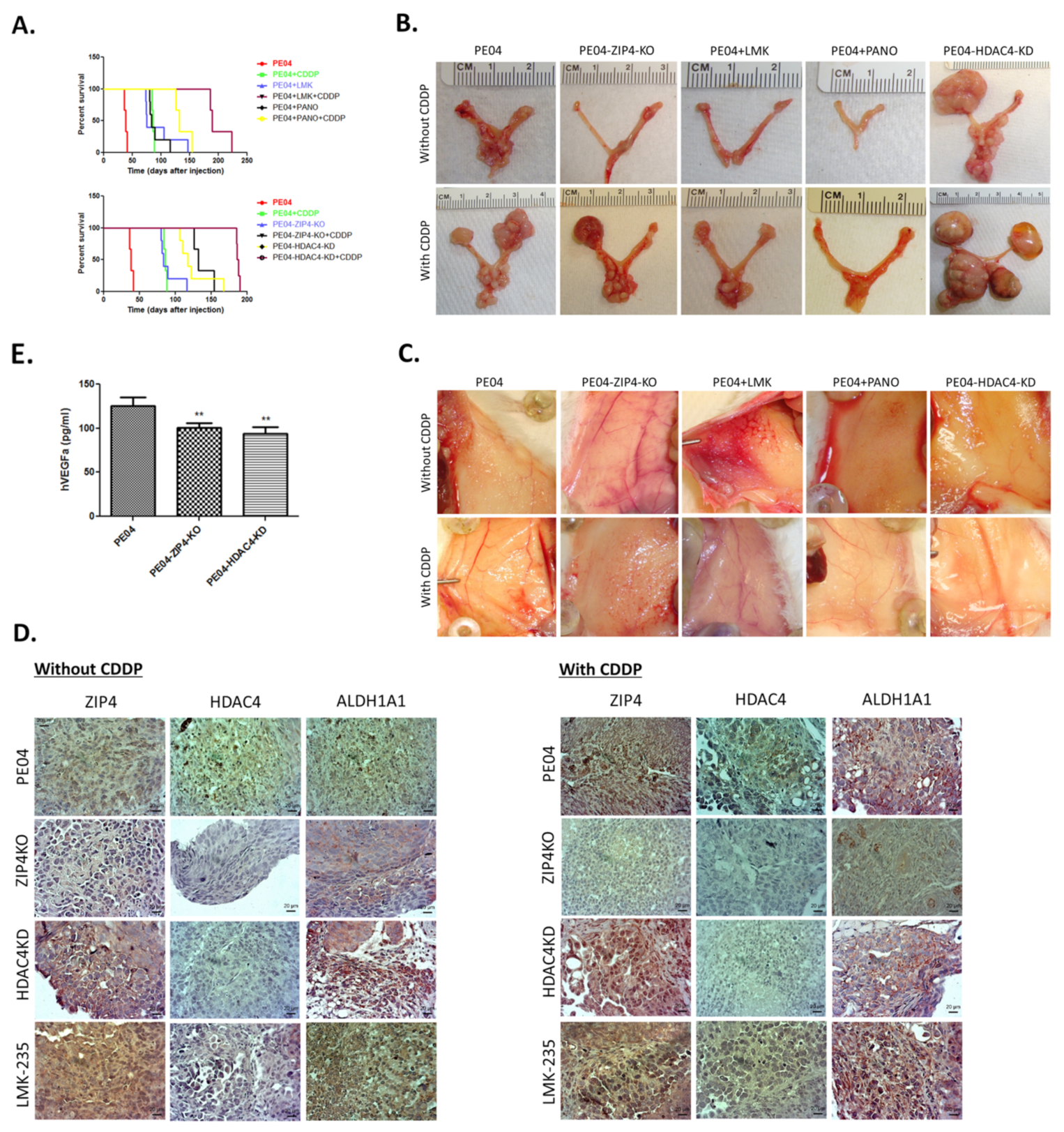
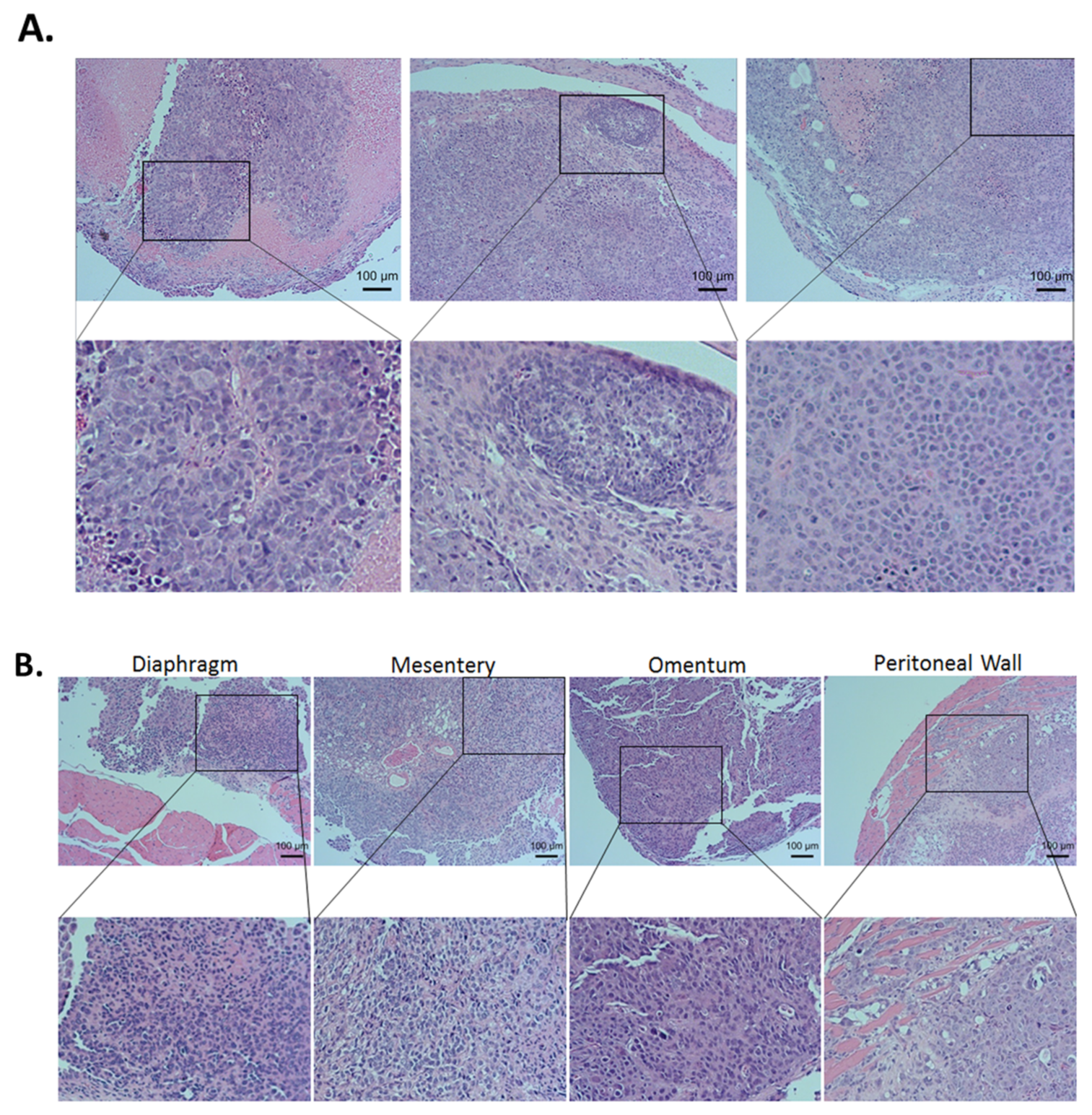
2.6. The Combinational Targeting CSC via the ZIP4-HDAC4 Axis and Non-CSC Using CDDP Was Highly Effective in Blocking Tumorigenesis
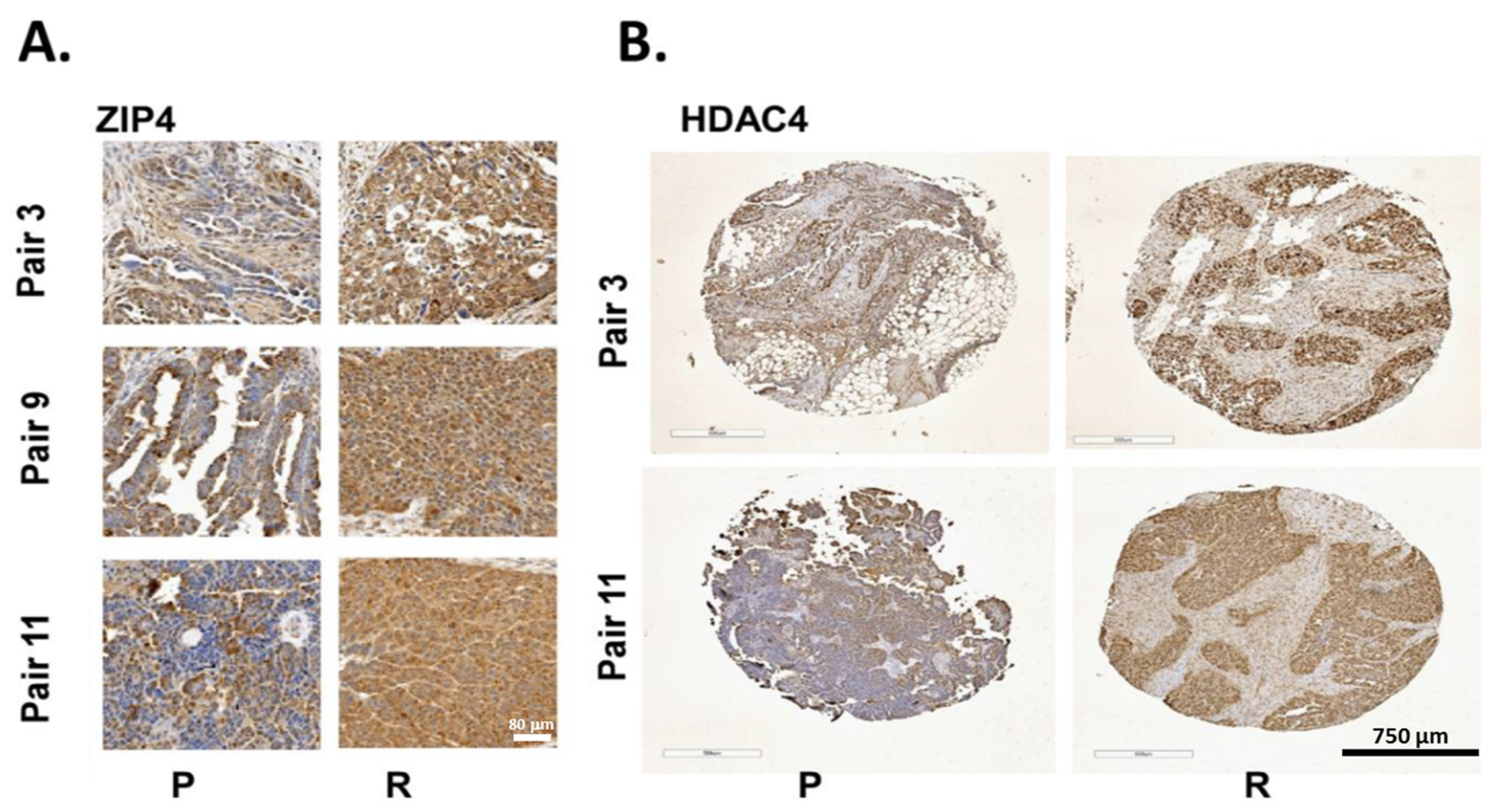
2.7. ZIP4 and HDAC4 Were Upregulated in a Subset of Recurrent vs. Primary Human HGSOC Samples
3. Discussion
4. Materials and Methods
4.1. Reagents, Cell Lines and Culture
4.2. Western Blot Analysis
4.3. Fluorescence-Activated Cell Sorting (FACS) of ZIP4+ Cells
4.4. DNA Transfection and Establishment of Stable Clones
4.5. Spheroids-Formation Assays
4.6. VEGFA ELISA Assays
4.7. Immunohistochemistry (IHC) Staining
4.8. HDACi Cytotoxicity Assay
4.9. Xenograft Mouse Model
4.10. Human HGSOC IHC
4.11. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shih Ie, M.; Kurman, R.J. Ovarian tumorigenesis: A proposed model based on morphological and molecular genetic analysis. Am. J. Pathol. 2004, 164, 1511–1518. [Google Scholar] [CrossRef]
- Ricciardelli, C.; Oehler, M.K. Diverse molecular pathways in ovarian cancer and their clinical significance. Maturitas 2009, 62, 270–275. [Google Scholar] [CrossRef]
- Rojas, V.; Hirshfield, K.M.; Ganesan, S.; Rodriguez-Rodriguez, L. Molecular Characterization of Epithelial Ovarian Cancer: Implications for Diagnosis and Treatment. Int. J. Mol. Sci. 2016, 17, 2113. [Google Scholar] [CrossRef]
- Davidson, B. Biomarkers of drug resistance in ovarian cancer—An update. Expert Rev. Mol. Diagn. 2019, 19, 469–476. [Google Scholar] [CrossRef]
- Moffitt, L.; Karimnia, N.; Stephens, A.; Bilandzic, M. Therapeutic Targeting of Collective Invasion in Ovarian Cancer. Int. J. Mol. Sci. 2019, 20, 1466. [Google Scholar] [CrossRef] [PubMed]
- Lisio, M.A.; Fu, L.; Goyeneche, A.; Gao, Z.H.; Telleria, C. High-Grade Serous Ovarian Cancer: Basic Sciences, Clinical and Therapeutic Standpoints. Int. J. Mol. Sci. 2019, 20, 952. [Google Scholar] [CrossRef] [PubMed]
- Ponti, D.; Costa, A.; Zaffaroni, N.; Pratesi, G.; Petrangolini, G.; Coradini, D.; Pilotti, S.; Pierotti, M.A.; Daidone, M.G. Isolation and in vitro propagation of tumorigenic breast cancer cells with stem/progenitor cell properties. Cancer Res. 2005, 65, 5506–5511. [Google Scholar] [CrossRef]
- Klemba, A.; Purzycka-Olewiecka, J.K.; Wcislo, G.; Czarnecka, A.M.; Lewicki, S.; Lesyng, B.; Szczylik, C.; Kieda, C. Surface markers of cancer stem-like cells of ovarian cancer and their clinical relevance. Contemp. Oncol. (Pozn.) 2018, 22, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Nwani, N.G.; Condello, S.; Wang, Y.; Swetzig, W.M.; Barber, E.; Hurley, T.; Matei, D. A Novel ALDH1A1 Inhibitor Targets Cells with Stem Cell Characteristics in Ovarian Cancer. Cancers 2019, 11, 502. [Google Scholar] [CrossRef]
- Reya, T.; Morrison, S.J.; Clarke, M.F.; Weissman, I.L. Stem cells, cancer, and cancer stem cells. Nature 2001, 414, 105–111. [Google Scholar] [CrossRef]
- Wang, X.; Li, X.; Fu, X.; Bai, M.; Li, X.; Mei, Q.; Nie, J.; Wu, Z.; Han, W. Eliminating ovarian cancer stem cells: A potential therapeutic target for ovarian cancer chemoresistance. Curr. Protein Pept. Sci. 2015, 16, 270–278. [Google Scholar] [CrossRef]
- Al-Alem, L.F.; Pandya, U.M.; Baker, A.T.; Bellio, C.; Zarrella, B.D.; Clark, J.; DiGloria, C.M.; Rueda, B.R. Ovarian cancer stem cells: What progress have we made? Int. J. Biochem. Cell Biol. 2019, 107, 92–103. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615. [Google Scholar] [CrossRef]
- Keyvani, V.; Farshchian, M.; Esmaeili, S.A.; Yari, H.; Moghbeli, M.; Nezhad, S.K.; Abbaszadegan, M.R. Ovarian cancer stem cells and targeted therapy. J. Ovarian Res. 2019, 12, 120. [Google Scholar] [CrossRef]
- Donahue, T.; Hines, O.J. The ZIP4 pathway in pancreatic cancer. Cancer Biol. Ther. 2010, 9, 243–245. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Fan, Q.; Cai, Q.; Li, P.; Wang, W.; Wang, J.; Gerry, E.; Wang, T.L.; Shih, I.M.; Nephew, K.P.; Xu, Y. The novel ZIP4 regulation and its role in ovarian cancer. Oncotarget 2017, 8, 90090–90107. [Google Scholar] [CrossRef] [PubMed]
- Fan, Q.; Zhang, W.; Emerson, R.E.; Xu, Y. ZIP4 Is a Novel Cancer Stem Cell Marker in High-Grade Serous Ovarian Cancer. Cancers 2020, 12, 3692. [Google Scholar] [CrossRef]
- Li, M.; Zhang, Y.; Bharadwaj, U.; Zhai, Q.J.; Ahern, C.H.; Fisher, W.E.; Brunicardi, F.C.; Logsdon, C.D.; Chen, C.; Yao, Q. Down-regulation of ZIP4 by RNA interference inhibits pancreatic cancer growth and increases the survival of nude mice with pancreatic cancer xenografts. Clin. Cancer Res. 2009, 15, 5993–6001. [Google Scholar] [CrossRef]
- Li, M.; Zhang, Y.; Liu, Z.; Bharadwaj, U.; Wang, H.; Wang, X.; Zhang, S.; Liuzzi, J.P.; Chang, S.M.; Cousins, R.J.; et al. Aberrant expression of zinc transporter ZIP4 (SLC39A4) significantly contributes to human pancreatic cancer pathogenesis and progression. Proc. Natl. Acad. Sci. USA 2007, 104, 18636–18641. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Chen, Y.; Wang, Y.; Yang, J.; Zhu, V.F.; Liu, Y.; Cui, X.; Chen, L.; Yan, W.; Jiang, T.; et al. ZIP4 is a novel molecular marker for glioma. Neuro Oncol. 2013, 15, 1008–1016. [Google Scholar] [CrossRef]
- Shen, H.; Qin, H.H.; Long, J.G.; Wang, F.D. [Effect of zinc on mRNA expression of ZIP4 in Caco2 cells]. Wei Sheng Yan Jiu 2006, 35, 426–427. [Google Scholar] [PubMed]
- Weaver, B.P.; Zhang, Y.; Hiscox, S.; Guo, G.L.; Apte, U.; Taylor, K.M.; Sheline, C.T.; Wang, L.; Andrews, G.K. Zip4 (Slc39a4) expression is activated in hepatocellular carcinomas and functions to repress apoptosis, enhance cell cycle and increase migration. PLoS ONE 2010, 5, e13158. [Google Scholar] [CrossRef]
- Xu, C.; Wallace, M.B.; Yang, J.; Jiang, L.; Zhai, Q.; Zhang, Y.; Hong, C.; Chen, Y.; Frank, T.S.; Stauffer, J.A.; et al. ZIP4 is a novel diagnostic and prognostic marker in human pancreatic cancer: A systemic comparison between EUS-FNA and surgical specimens. Curr. Mol. Med. 2014, 14, 309–315. [Google Scholar] [CrossRef]
- Xu, X.; Guo, H.J.; Xie, H.Y.; Li, J.; Zhuang, R.Z.; Ling, Q.; Zhou, L.; Wei, X.Y.; Liu, Z.K.; Ding, S.M.; et al. ZIP4, a novel determinant of tumor invasion in hepatocellular carcinoma, contributes to tumor recurrence after liver transplantation. Int. J. Biol. Sci. 2014, 10, 245–256. [Google Scholar] [CrossRef]
- Zeng, Q.; Liu, Y.M.; Liu, J.; Han, J.; Guo, J.X.; Lu, S.; Huang, X.M.; Yi, P.; Lang, J.Y.; Zhang, P.; et al. Inhibition of ZIP4 reverses epithelial-to-mesenchymal transition and enhances the radiosensitivity in human nasopharyngeal carcinoma cells. Cell Death Dis. 2019, 10, 588. [Google Scholar] [CrossRef]
- Zhang, Q.; Sun, X.; Yang, J.; Ding, H.; LeBrun, D.; Ding, K.; Houchen, C.W.; Postier, R.G.; Ambrose, C.G.; Li, Z.; et al. ZIP4 silencing improves bone loss in pancreatic cancer. Oncotarget 2015, 6, 26041–26051. [Google Scholar] [CrossRef]
- Zhang, Y.; Bharadwaj, U.; Logsdon, C.D.; Chen, C.; Yao, Q.; Li, M. ZIP4 regulates pancreatic cancer cell growth by activating IL-6/STAT3 pathway through zinc finger transcription factor CREB. Clin. Cancer Res. 2010, 16, 1423–1430. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, C.; Yao, Q.; Li, M. ZIP4 upregulates the expression of neuropilin-1, vascular endothelial growth factor, and matrix metalloproteases in pancreatic cancer cell lines and xenografts. Cancer Biol. Ther. 2010, 9, 236–242. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yang, J.; Cui, X.; Chen, Y.; Zhu, V.F.; Hagan, J.P.; Wang, H.; Yu, X.; Hodges, S.E.; Fang, J.; et al. A novel epigenetic CREB-miR-373 axis mediates ZIP4-induced pancreatic cancer growth. EMBO Mol. Med. 2013, 5, 1322–1334. [Google Scholar] [CrossRef] [PubMed]
- Sanaei, M.; Kavoosi, F. Histone Deacetylases and Histone Deacetylase Inhibitors: Molecular Mechanisms of Action in Various Cancers. Adv. Biomed. Res. 2019, 8, 63. [Google Scholar] [PubMed]
- Roca, M.S.; Di Gennaro, E.; Budillon, A. Implication for Cancer Stem Cells in Solid Cancer Chemo-Resistance: Promising Therapeutic Strategies Based on the Use of HDAC Inhibitors. J. Clin. Med. 2019, 8, 912. [Google Scholar] [CrossRef]
- Wang, Z.; Qin, G.; Zhao, T.C. HDAC4: Mechanism of regulation and biological functions. Epigenomics 2014, 6, 139–150. [Google Scholar] [CrossRef]
- Wang, Y.; Cardenas, H.; Fang, F.; Condello, S.; Taverna, P.; Segar, M.; Liu, Y.; Nephew, K.P.; Matei, D. Epigenetic targeting of ovarian cancer stem cells. Cancer Res. 2014, 74, 4922–4936. [Google Scholar] [CrossRef]
- Smith, H.J.; Straughn, J.M.; Buchsbaum, D.J.; Arend, R.C. Epigenetic therapy for the treatment of epithelial ovarian cancer: A clinical review. Gynecol. Oncol. Rep. 2017, 20, 81–86. [Google Scholar] [CrossRef]
- Kim, M.G.; Pak, J.H.; Choi, W.H.; Park, J.Y.; Nam, J.H.; Kim, J.H. The relationship between cisplatin resistance and histone deacetylase isoform overexpression in epithelial ovarian cancer cell lines. J. Gynecol. Oncol. 2012, 23, 182–189. [Google Scholar] [CrossRef] [PubMed]
- Mrkvicova, A.; Chmelarova, M.; Peterova, E.; Havelek, R.; Baranova, I.; Kazimirova, P.; Rudolf, E.; Rezacova, M. The effect of sodium butyrate and cisplatin on expression of EMT markers. PLoS ONE 2019, 14, e0210889. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Xu, X.; Liu, H.; Hu, X.; Zhang, W.; Ye, M.; Zhu, X. Prognosis Analysis of Histone Deacetylases mRNA Expression in Ovarian Cancer Patients. J. Cancer 2018, 9, 4547–4555. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Qi, Z.; Yin, H.; Yang, G. Interaction between p53 and Ras signaling controls cisplatin resistance via HDAC4- and HIF-1alpha-mediated regulation of apoptosis and autophagy. Theranostics 2019, 9, 1096–1114. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.F.; Wei, A.M.; Kou, Q.; Zhu, Q.Y.; Zhang, L. Histone deacetylase 4 increases progressive epithelial ovarian cancer cells via repression of p21 on fibrillar collagen matrices. Oncol. Rep. 2016, 35, 948–954. [Google Scholar] [CrossRef] [PubMed]
- Stronach, E.A.; Alfraidi, A.; Rama, N.; Datler, C.; Studd, J.B.; Agarwal, R.; Guney, T.G.; Gourley, C.; Hennessy, B.T.; Mills, G.B.; et al. HDAC4-regulated STAT1 activation mediates platinum resistance in ovarian cancer. Cancer Res. 2011, 71, 4412–4422. [Google Scholar] [CrossRef] [PubMed]
- Davidson, B. Recently identified drug resistance biomarkers in ovarian cancer. Expert Rev. Mol. Diagn. 2016, 16, 569–578. [Google Scholar] [CrossRef] [PubMed]
- Mottet, D.; Pirotte, S.; Lamour, V.; Hagedorn, M.; Javerzat, S.; Bikfalvi, A.; Bellahcene, A.; Verdin, E.; Castronovo, V. HDAC4 represses p21(WAF1/Cip1) expression in human cancer cells through a Sp1-dependent, p53-independent mechanism. Oncogene 2009, 28, 243–256. [Google Scholar] [CrossRef]
- Jeon, H.S.; Ahn, M.Y.; Park, J.H.; Kim, T.H.; Chun, P.; Kim, W.H.; Kim, J.; Moon, H.R.; Jung, J.H.; Kim, H.S. Anticancer effects of the MHY218 novel hydroxamic acid-derived histone deacetylase inhibitor in human ovarian cancer cells. Int. J. Oncol. 2010, 37, 419–428. [Google Scholar] [PubMed]
- Ahn, M.Y.; Kang, D.O.; Na, Y.J.; Yoon, S.; Choi, W.S.; Kang, K.W.; Chung, H.Y.; Jung, J.H.; Min do, S.; Kim, H.S. Histone deacetylase inhibitor, apicidin, inhibits human ovarian cancer cell migration via class II histone deacetylase 4 silencing. Cancer Lett. 2012, 325, 189–199. [Google Scholar] [CrossRef] [PubMed]
- De Cecco, L.; Berardi, M.; Sommariva, M.; Cataldo, A.; Canevari, S.; Mezzanzanica, D.; Iorio, M.V.; Tagliabue, E.; Balsari, A. Increased sensitivity to chemotherapy induced by CpG-ODN treatment is mediated by microRNA modulation. PLoS ONE 2013, 8, e58849. [Google Scholar] [CrossRef]
- Zhang, Y.; Ren, Y.J.; Guo, L.C.; Ji, C.; Hu, J.; Zhang, H.H.; Xu, Q.H.; Zhu, W.D.; Ming, Z.J.; Yuan, Y.S.; et al. Nucleus accumbens-associated protein-1 promotes glycolysis and survival of hypoxic tumor cells via the HDAC4-HIF-1alpha axis. Oncogene 2017, 36, 4171–4181. [Google Scholar] [CrossRef]
- Geng, H.; Harvey, C.T.; Pittsenbarger, J.; Liu, Q.; Beer, T.M.; Xue, C.; Qian, D.Z. HDAC4 protein regulates HIF1alpha protein lysine acetylation and cancer cell response to hypoxia. J. Biol. Chem. 2011, 286, 38095–38102. [Google Scholar] [CrossRef]
- Kim, K.S.; Sengupta, S.; Berk, M.; Kwak, Y.G.; Escobar, P.F.; Belinson, J.; Mok, S.C.; Xu, Y. Hypoxia enhances lysophosphatidic acid responsiveness in ovarian cancer cells and lysophosphatidic acid induces ovarian tumor metastasis in vivo. Cancer Res. 2006, 66, 7983–7990. [Google Scholar] [CrossRef]
- Nakai, H.; Watanabe, Y.; Ueda, H.; Hoshiai, H. Hypoxia inducible factor 1-alpha expression as a factor predictive of efficacy of taxane/platinum chemotherapy in advanced primary epithelial ovarian cancer. Cancer Lett. 2007, 251, 164–167. [Google Scholar] [CrossRef]
- Gomez-Roman, N.; Sahasrabudhe, N.M.; McGregor, F.; Chalmers, A.J.; Cassidy, J.; Plumb, J. Hypoxia-inducible factor 1 alpha is required for the tumourigenic and aggressive phenotype associated with Rab25 expression in ovarian cancer. Oncotarget 2016, 7, 22650–22664. [Google Scholar] [CrossRef]
- Lindemann, R.K.; Gabrielli, B.; Johnstone, R.W. Histone-deacetylase inhibitors for the treatment of cancer. Cell Cycle 2004, 3, 779–788. [Google Scholar] [CrossRef]
- Huang, Z.; Zhou, W.; Li, Y.; Cao, M.; Wang, T.; Ma, Y.; Guo, Q.; Wang, X.; Zhang, C.; Zhang, C.; et al. Novel hybrid molecule overcomes the limited response of solid tumours to HDAC inhibitors via suppressing JAK1-STAT3-BCL2 signalling. Theranostics 2018, 8, 4995–5011. [Google Scholar] [CrossRef]
- Khabele, D. The therapeutic potential of class I selective histone deacetylase inhibitors in ovarian cancer. Front. Oncol. 2014, 4, 111. [Google Scholar] [CrossRef] [PubMed]
- Benoit, Y.D. Identification of Novel Molecules Targeting Cancer Stem Cells. Methods Mol. Biol. 2018, 1765, 333–347. [Google Scholar] [PubMed]
- Chae, Y.C.; Kim, J.H. Cancer stem cell metabolism: Target for cancer therapy. BMB Rep. 2018, 51, 319–326. [Google Scholar] [CrossRef] [PubMed]
- Najafi, M.; Farhood, B.; Mortezaee, K. Cancer stem cells (CSCs) in cancer progression and therapy. J. Cell Physiol. 2019, 234, 8381–8395. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Liu, L.; Zhang, T.; Zhu, Y.L.; Qiu, F.; Wu, X.G.; Wang, X.L.; Hu, F.Q.; Huang, J. Oxaliplatin-incorporated micelles eliminate both cancer stem-like and bulk cell populations in colorectal cancer. Int. J. Nanomed. 2011, 6, 3207–3218. [Google Scholar]
- Sulaiman, A.; Sulaiman, B.; Khouri, L.; McGarry, S.; Nessim, C.; Arnaout, A.; Li, X.; Addison, C.; Dimitroulakos, J.; Wang, L. Both bulk and cancer stem cell subpopulations in triple-negative breast cancer are susceptible to Wnt, HDAC, and ERalpha coinhibition. FEBS Lett. 2016, 590, 4606–4616. [Google Scholar] [CrossRef]
- Ma, Y.Y.; Lin, H.; Moh, J.S.; Chen, K.D.; Wang, I.W.; Ou, Y.C.; You, Y.S.; Lung, C.C. Low-dose LBH589 increases the sensitivity of cisplatin to cisplatin-resistant ovarian cancer cells. Taiwan J. Obstet. Gynecol. 2011, 50, 165–171. [Google Scholar] [CrossRef][Green Version]
- Ong, P.S.; Wang, X.Q.; Lin, H.S.; Chan, S.Y.; Ho, P.C. Synergistic effects of suberoylanilide hydroxamic acid combined with cisplatin causing cell cycle arrest independent apoptosis in platinum-resistant ovarian cancer cells. Int. J. Oncol. 2012, 40, 1705–1713. [Google Scholar]
- Meng, F.; Sun, G.; Zhong, M.; Yu, Y.; Brewer, M.A. Anticancer efficacy of cisplatin and trichostatin A or 5-aza-2′-deoxycytidine on ovarian cancer. Br. J. Cancer 2013, 108, 579–586. [Google Scholar] [CrossRef]
- Bandolik, J.J.; Hamacher, A.; Schrenk, C.; Weishaupt, R.; Kassack, M.U. Class I-Histone Deacetylase (HDAC) Inhibition is Superior to pan-HDAC Inhibition in Modulating Cisplatin Potency in High Grade Serous Ovarian Cancer Cell Lines. Int. J. Mol. Sci. 2019, 20, 3052. [Google Scholar] [CrossRef]
- Shackleton, M. Normal stem cells and cancer stem cells: Similar and different. Semin. Cancer Biol. 2010, 20, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Van Neerven, S.M.; Tieken, M.; Vermeulen, L.; Bijlsma, M.F. Bidirectional interconversion of stem and non-stem cancer cell populations: A reassessment of theoretical models for tumor heterogeneity. Mol. Cell Oncol. 2016, 3, e1098791. [Google Scholar] [CrossRef] [PubMed]
- Pisco, A.O.; Huang, S. Non-genetic cancer cell plasticity and therapy-induced stemness in tumour relapse: ‘What does not kill me strengthens me’. Br. J. Cancer 2015, 112, 1725–1732. [Google Scholar] [CrossRef] [PubMed]
- Biddle, A.; Gammon, L.; Liang, X.; Costea, D.E.; Mackenzie, I.C. Phenotypic Plasticity Determines Cancer Stem Cell Therapeutic Resistance in Oral Squamous Cell Carcinoma. EBioMedicine 2016, 4, 138–145. [Google Scholar] [CrossRef] [PubMed]
- Ayob, A.Z.; Ramasamy, T.S. Cancer stem cells as key drivers of tumour progression. J. Biomed. Sci. 2018, 25, 20. [Google Scholar] [CrossRef]
- Hajizadeh, F.; Okoye, I.; Esmaily, M.; Ghasemi Chaleshtari, M.; Masjedi, A.; Azizi, G.; Irandoust, M.; Ghalamfarsa, G.; Jadidi-Niaragh, F. Hypoxia inducible factors in the tumor microenvironment as therapeutic targets of cancer stem cells. Life Sci. 2019, 237, 116952. [Google Scholar] [CrossRef]
- Clottes, E. [Hypoxia-inducible factor 1: Regulation, involvement in carcinogenesis and target for anticancer therapy]. Bull. Cancer 2005, 92, 119–127. [Google Scholar]
- Jun, J.C.; Rathore, A.; Younas, H.; Gilkes, D.; Polotsky, V.Y. Hypoxia-Inducible Factors and Cancer. Curr. Sleep Med. Rep. 2017, 3, 1–10. [Google Scholar] [CrossRef]
- Pezzuto, A.; Carico, E. Role of HIF-1 in Cancer Progression: Novel Insights. A Review. Curr. Mol. Med. 2018, 18, 343–351. [Google Scholar] [CrossRef]
- Mesiano, S.; Ferrara, N.; Jaffe, R.B. Role of vascular endothelial growth factor in ovarian cancer: Inhibition of ascites formation by immunoneutralization. Am. J. Pathol. 1998, 153, 1249–1256. [Google Scholar] [CrossRef]
- Dalal, V.; Kumar, R.; Kumar, S.; Sharma, A.; Kumar, L.; Sharma, J.B.; Roy, K.K.; Singh, N.; Vanamail, P. Biomarker potential of IL-6 and VEGF-A in ascitic fluid of epithelial ovarian cancer patients. Clin. Chim. Acta 2018, 482, 27–32. [Google Scholar] [CrossRef]
- Chen, S.S.; Michael, A.; Butler-Manuel, S.A. Advances in the treatment of ovarian cancer: A potential role of antiinflammatory phytochemicals. Discov. Med. 2012, 13, 7–17. [Google Scholar] [PubMed]
- Fang, X.; Gaudette, D.; Furui, T.; Mao, M.; Estrella, V.; Eder, A.; Pustilnik, T.; Sasagawa, T.; Lapushin, R.; Yu, S.; et al. Lysophospholipid growth factors in the initiation, progression, metastases, and management of ovarian cancer. Ann. N. Y. Acad. Sci. 2000, 905, 188–208. [Google Scholar] [CrossRef] [PubMed]
- Westermann, A.M.; Beijnen, J.H.; Moolenaar, W.H.; Rodenhuis, S. Growth factors in human ovarian cancer. Cancer Treat. Rev. 1997, 23, 113–131. [Google Scholar] [CrossRef]
- Hazelton, D.A.; Hamilton, T.C. Vascular endothelial growth factor in ovarian cancer. Curr. Oncol. Rep. 1999, 1, 59–63. [Google Scholar] [CrossRef] [PubMed]
- Bamias, A.; Pignata, S.; Pujade-Lauraine, E. Angiogenesis: A promising therapeutic target for ovarian cancer. Crit. Rev. Oncol. Hematol. 2012, 84, 314–326. [Google Scholar] [CrossRef]
- Smolle, E.; Taucher, V.; Haybaeck, J. Malignant ascites in ovarian cancer and the role of targeted therapeutics. Anticancer Res. 2014, 34, 1553–1561. [Google Scholar]
- Dong, Z.; Yu, C.; Rezhiya, K.; Gulijiahan, A.; Wang, X. Downregulation of miR-146a promotes tumorigenesis of cervical cancer stem cells via VEGF/CDC42/PAK1 signaling pathway. Artif. Cells Nanomed. Biotechnol. 2019, 47, 3711–3719. [Google Scholar] [CrossRef]
- Mercurio, A.M. VEGF/Neuropilin Signaling in Cancer Stem Cells. Int. J. Mol. Sci. 2019, 20, 490. [Google Scholar] [CrossRef]
- Vera, N.; Acuna-Gallardo, S.; Grunenwald, F.; Caceres-Verschae, A.; Realini, O.; Acuna, R.; Lladser, A.; Illanes, S.E.; Varas-Godoy, M. Small Extracellular Vesicles Released from Ovarian Cancer Spheroids in Response to Cisplatin Promote the Pro-Tumorigenic Activity of Mesenchymal Stem Cells. Int. J. Mol. Sci. 2019, 20, 4972. [Google Scholar] [CrossRef]
- Sheng, W.J.; Jiang, H.; Wu, D.L.; Zheng, J.H. Early responses of the STAT3 pathway to platinum drugs are associated with cisplatin resistance in epithelial ovarian cancer. Braz. J. Med. Biol. Res. 2013, 46, 650–658. [Google Scholar] [CrossRef] [PubMed]
- Jackson, A.L.; Eisenhauer, E.L.; Herzog, T.J. Emerging therapies: Angiogenesis inhibitors for ovarian cancer. Expert Opin. Emerg. Drugs 2015, 20, 331–346. [Google Scholar] [CrossRef]
- Monk, B.J.; Minion, L.E.; Coleman, R.L. Anti-angiogenic agents in ovarian cancer: Past, present, and future. Ann. Oncol. 2016, 27 (Suppl. 1), i33–i39. [Google Scholar] [CrossRef] [PubMed]
- Shoji, T.; Eto, H.; Sato, T.; Soma, R.; Fukagawa, D.; Tomabechi, H.; Takatori, E.; Nagasawa, T.; Sato, S.; Kagabu, M.; et al. A New Therapeutic Strategy for Recurrent Ovarian Cancer-Bevacizumab beyond Progressive Disease. Healthcare 2019, 7, 109. [Google Scholar] [CrossRef]
- Guan, L.Y.; Lu, Y. New developments in molecular targeted therapy of ovarian cancer. Discov. Med. 2018, 26, 219–229. [Google Scholar] [PubMed]
- Lewis, A.D.; Hayes, J.D.; Wolf, C.R. Glutathione and glutathione-dependent enzymes in ovarian adenocarcinoma cell lines derived from a patient before and after the onset of drug resistance: Intrinsic differences and cell cycle effects. Carcinogenesis 1988, 9, 1283–1287. [Google Scholar] [CrossRef]
- Sakai, W.; Swisher, E.M.; Jacquemont, C.; Chandramohan, K.V.; Couch, F.J.; Langdon, S.P.; Wurz, K.; Higgins, J.; Villegas, E.; Taniguchi, T. Functional restoration of BRCA2 protein by secondary BRCA2 mutations in BRCA2-mutated ovarian carcinoma. Cancer Res. 2009, 69, 6381–6386. [Google Scholar] [CrossRef]
- Marek, L.; Hamacher, A.; Hansen, F.K.; Kuna, K.; Gohlke, H.; Kassack, M.U.; Kurz, T. Histone deacetylase (HDAC) inhibitors with a novel connecting unit linker region reveal a selectivity profile for HDAC4 and HDAC5 with improved activity against chemoresistant cancer cells. J. Med. Chem. 2013, 56, 427–436. [Google Scholar] [CrossRef]
- Mehta, P.; Novak, C.; Raghavan, S.; Ward, M.; Mehta, G. Self-Renewal and CSCs In Vitro Enrichment: Growth as Floating Spheres. Methods Mol. Biol. 2018, 1692, 61–75. [Google Scholar] [PubMed]
- Sato, M.; Kawana, K.; Adachi, K.; Fujimoto, A.; Yoshida, M.; Nakamura, H.; Nishida, H.; Inoue, T.; Taguchi, A.; Ogishima, J.; et al. Detachment from the primary site and suspension in ascites as the initial step in metabolic reprogramming and metastasis to the omentum in ovarian cancer. Oncol. Lett. 2018, 15, 1357–1361. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Balch, C.; Chan, M.W.; Lai, H.C.; Matei, D.; Schilder, J.M.; Yan, P.S.; Huang, T.H.; Nephew, K.P. Identification and characterization of ovarian cancer-initiating cells from primary human tumors. Cancer Res. 2008, 68, 4311–4320. [Google Scholar] [CrossRef]
- Wang, Y.; Miao, Z.; Ren, G.; Xu, Y.; Cheng, Z. A novel Affibody bioconjugate for dual-modality imaging of ovarian cancer. Chem. Commun. (Camb.) 2014, 50, 12832–12835. [Google Scholar] [CrossRef] [PubMed]
- Wiechert, A.; Saygin, C.; Thiagarajan, P.S.; Rao, V.S.; Hale, J.S.; Gupta, N.; Hitomi, M.; Nagaraj, A.B.; DiFeo, A.; Lathia, J.D.; et al. Cisplatin induces stemness in ovarian cancer. Oncotarget 2016, 7, 30511–30522. [Google Scholar] [CrossRef]
- Yoshihara, K.; Tajima, A.; Komata, D.; Yamamoto, T.; Kodama, S.; Fujiwara, H.; Suzuki, M.; Onishi, Y.; Hatae, M.; Sueyoshi, K.; et al. Gene expression profiling of advanced-stage serous ovarian cancers distinguishes novel subclasses and implicates ZEB2 in tumor progression and prognosis. Cancer Sci. 2009, 100, 1421–1428. [Google Scholar] [CrossRef]
- Kim, M.J.; Kim, A.R.; Jeong, J.Y.; Kim, K.I.; Kim, T.H.; Lee, C.; Chung, K.; Ko, Y.H.; An, H.J. Correlation of ALDH1 and Notch3 Expression: Clinical implication in Ovarian Carcinomas. J. Cancer 2017, 8, 3331–3342. [Google Scholar] [CrossRef]
- Yokoyama, Y.; Zhu, H.; Lee, J.H.; Kossenkov, A.V.; Wu, S.Y.; Wickramasinghe, J.M.; Yin, X.; Palozola, K.C.; Gardini, A.; Showe, L.C.; et al. BET Inhibitors Suppress ALDH Activity by Targeting ALDH1A1 Super-Enhancer in Ovarian Cancer. Cancer Res. 2016, 76, 6320–6330. [Google Scholar] [CrossRef]
- Chefetz, I.; Grimley, E.; Yang, K.; Hong, L.; Vinogradova, E.V.; Suciu, R.; Kovalenko, I.; Karnak, D.; Morgan, C.A.; Chtcherbinine, M.; et al. A Pan-ALDH1A Inhibitor Induces Necroptosis in Ovarian Cancer Stem-like Cells. Cell Rep. 2019, 26, 3061–3075.e6. [Google Scholar] [CrossRef]
- Liu, L.; Cai, S.; Han, C.; Banerjee, A.; Wu, D.; Cui, T.; Xie, G.; Zhang, J.; Zhang, X.; McLaughlin, E.; et al. ALDH1A1 Contributes to PARP Inhibitor Resistance via Enhancing DNA Repair in BRCA2(-/-) Ovarian Cancer Cells. Mol. Cancer Ther. 2020, 19, 199–210. [Google Scholar] [CrossRef]
- Dinavahi, S.S.; Gowda, R.; Gowda, K.; Bazewicz, C.G.; Chirasani, V.R.; Battu, M.B.; Berg, A.; Dokholyan, N.V.; Amin, S.; Robertson, G.P. Development of a Novel Multi-Isoform ALDH Inhibitor Effective as an Antimelanoma Agent. Mol. Cancer Ther. 2020, 19, 447–459. [Google Scholar]
- Sulaiman, A.; McGarry, S.; El-Sahli, S.; Li, L.; Chambers, J.; Phan, A.; Cote, M.; Cron, G.O.; Alain, T.; Le, Y.; et al. Co-targeting Bulk Tumor and CSCs in Clinically Translatable TNBC Patient-Derived Xenografts via Combination Nanotherapy. Mol. Cancer Ther. 2019, 18, 1755–1764. [Google Scholar] [CrossRef]
- Bezuidenhout, N.; Shoshan, M. A Shifty Target: Tumor-Initiating Cells and Their Metabolism. Int. J. Mol. Sci. 2019, 20, 5370. [Google Scholar] [CrossRef] [PubMed]
- Narayan, S.; Ramisetti, S.; Jaiswal, A.S.; Law, B.K.; Singh-Pillay, A.; Singh, P.; Amin, S.; Sharma, A.K. ASR352, A potent anticancer agent: Synthesis, preliminary SAR, and biological activities against colorectal cancer bulk, 5-fluorouracil/oxaliplatin resistant and stem cells. Eur. J. Med. Chem. 2019, 161, 456–467. [Google Scholar] [CrossRef] [PubMed]
- Sridharan, S.; Howard, C.M.; Tilley, A.M.C.; Subramaniyan, B.; Tiwari, A.K.; Ruch, R.J.; Raman, D. Novel and Alternative Targets Against Breast Cancer Stemness to Combat Chemoresistance. Front. Oncol. 2019, 9, 1003. [Google Scholar] [CrossRef]
- Kim, O.; Park, E.Y.; Kwon, S.Y.; Shin, S.; Emerson, R.E.; Shin, Y.H.; DeMayo, F.J.; Lydon, J.P.; Coffey, D.M.; Hawkins, S.M.; et al. Targeting progesterone signaling prevents metastatic ovarian cancer. Proc. Natl. Acad. Sci. USA 2020, 117, 31993–32004. [Google Scholar] [CrossRef]
- Lin, P.C.; Hsieh, H.Y.; Chu, P.C.; Chen, C.S. Therapeutic Opportunities of Targeting Histone Deacetylase Isoforms to Eradicate Cancer Stem Cells. Int. J. Mol. Sci. 2018, 19, 1939. [Google Scholar] [CrossRef]
- Wang, L.; Liu, X.; Ren, Y.; Zhang, J.; Chen, J.; Zhou, W.; Guo, W.; Wang, X.; Chen, H.; Li, M.; et al. Cisplatin-enriching cancer stem cells confer multidrug resistance in non-small cell lung cancer via enhancing TRIB1/HDAC activity. Cell Death Dis. 2017, 8, e2746. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.H.; Wu, S.Y.; Huang, Y.J.; Wei, P.L.; Wu, A.T.; Chao, T.Y. The identification and validation of Trichosstatin A as a potential inhibitor of colon tumorigenesis and colon cancer stem-like cells. Am. J. Cancer Res. 2017, 7, 1227–1237. [Google Scholar] [PubMed]
- Hsieh, H.Y.; Chuang, H.C.; Shen, F.H.; Detroja, K.; Hsin, L.W.; Chen, C.S. Targeting breast cancer stem cells by novel HDAC3-selective inhibitors. Eur. J. Med. Chem. 2017, 140, 42–51. [Google Scholar] [CrossRef]
- Liu, N.; Li, S.; Wu, N.; Cho, K.S. Acetylation and deacetylation in cancer stem-like cells. Oncotarget 2017, 8, 89315–89325. [Google Scholar] [CrossRef]
- Su, Y.H.; Huang, W.C.; Huang, T.H.; Huang, Y.J.; Sue, Y.K.; Huynh, T.T.; Hsiao, M.; Liu, T.Z.; Wu, A.T.; Lin, C.M. Folate deficient tumor microenvironment promotes epithelial-to-mesenchymal transition and cancer stem-like phenotypes. Oncotarget 2016, 7, 33246–33256. [Google Scholar] [CrossRef]
- Scicchitano, B.M.; Sorrentino, S.; Proietti, G.; Lama, G.; Dobrowolny, G.; Catizone, A.; Binda, E.; Larocca, L.M.; Sica, G. Levetiracetam enhances the temozolomide effect on glioblastoma stem cell proliferation and apoptosis. Cancer Cell Int. 2018, 18, 136. [Google Scholar] [CrossRef]
- Kaowinn, S.; Kaewpiboon, C.; Koh, S.S.; Kramer, O.H.; Chung, Y.H. STAT1HDAC4 signaling induces epithelialmesenchymal transition and sphere formation of cancer cells overexpressing the oncogene, CUG2. Oncol. Rep. 2018, 40, 2619–2627. [Google Scholar]
- Song, B.; Wang, Y.; Xi, Y.; Kudo, K.; Bruheim, S.; Botchkina, G.I.; Gavin, E.; Wan, Y.; Formentini, A.; Kornmann, M.; et al. Mechanism of chemoresistance mediated by miR-140 in human osteosarcoma and colon cancer cells. Oncogene 2009, 28, 4065–4074. [Google Scholar] [CrossRef]
- Citron, F.; Fabris, L. Targeting Epigenetic Dependencies in Solid Tumors: Evolutionary Landscape Beyond Germ Layers Origin. Cancers 2020, 12, 682. [Google Scholar] [CrossRef]
- Matei, D.; Nephew, K.P. Epigenetic Attire in Ovarian Cancer: The Emperor’s New Clothes. Cancer Res. 2020, 80, 3775–3785. [Google Scholar] [CrossRef] [PubMed]
- Ho, T.C.S.; Chan, A.H.Y.; Ganesan, A. Thirty Years of HDAC Inhibitors: 2020 Insight and Hindsight. J. Med. Chem. 2020, 63, 12460–12484. [Google Scholar] [CrossRef] [PubMed]
- Cai, Q.; Fan, Q.; Buechlein, A.; Miller, D.; Nephew, K.P.; Liu, S.; Wan, J.; Xu, Y. Changes in mRNA/protein expression and signaling pathways in in vivo passaged mouse ovarian cancer cells. PLoS ONE 2018, 13, e0197404. [Google Scholar] [CrossRef] [PubMed]
- Fan, Q.; Cai, Q.; Xu, Y. LPA Regulates SOX9 in Ovarian Cancer Cells. J. Obstet. Gynecol. 2017, 10, 227. [Google Scholar] [CrossRef]
- Cai, H.; Xu, Y. The role of LPA and YAP signaling in long-term migration of human ovarian cancer cells. Cell Commun. Signal. 2013, 11, 31. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.A.; Hong, J.; Asaka, R.; Asaka, S.; Hsu, F.C.; Suryo Rahmanto, Y.; Jung, J.G.; Chen, Y.W.; Yen, T.T.; Tomaszewski, A.; et al. Inhibition of the MYC-Regulated Glutaminase Metabolic Axis Is an Effective Synthetic Lethal Approach for Treating Chemoresistant Ovarian Cancers. Cancer Res. 2020, 80, 4514–4526. [Google Scholar] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Cell Type | Cell No. | Treatment | Mice No. | Survival Days | Ascites Vol. (mL) | Tumors ≤ 1 mm | Tumors 1–5 mm | Tumors 5–10 mm | Tumors 10–15 mm | Tumor Sites |
|---|---|---|---|---|---|---|---|---|---|---|---|
| PE04 | PE04 | 5 × 106 | None | 3/3 | 40 ± 6 | 13 ± 2 | 36.6 ± 4.5 | 12 ± 2.6 | 2.3 ± 2 | 0 | Ov, Pw, D, Om, M, P, SI |
| PE04 + CDDP | PE04 | 5 × 106 | CDDP (2.5 mg/kg) | 4/4 | 86 ± 3 | 4.7 ± 1.1 | 23.8 ± 4.5 | 11.8 ± 4.8 | 0 | 0 | Ov, Pw, P, Om, M |
| PE04-ZIP4-KO | PE04-ZIP4-KO | 5 × 106 | None | 5/5 | 96 ± 8 | 3.5 ± 0.7 | 15 ± 2 | 7 ± 2 | 0 | 0 | Ov, Pw, D, M |
| PE04-ZIP4-KO+CDDP | PE04-ZIP4-KO | 5 × 106 | CDDP (2.5 mg/kg) | 4/4 | 141 ± 17 | 7.8 ± 2.7 | 22.2 ± 5.9 | 13.6 ± 5.9 | 0 | 0 | Ov, Pw, SI, Om, D, M |
| PE04-PANO | PE04 | 5 × 106 | Panobinostat (20 mg/kg) | 5/5 | 90 ± 15 | 9 ± 3.7 | 20.7 ± 14.9 | 10 ± 7.7 | 2.5 ± 2 | 0 | Pw(1/5), D, M, P, SI |
| PE04-PANO +CDDP | PE04 | 5 × 106 | CDDP (2.5 mg/kg) +Panobinostat (20 mg/kg) | 5/5 | 136 ± 11 | 0.6 ± 0 | 6 ± 3.5 | 0.4 ± 0.2 | 0 | 0 | Pw (2/5), D, P |
| PE04-LMK | PE04 | 5 × 106 | LMK-235 (13 mg/kg) | 5/5 | 95 ± 32 | 8.2 ± 3.8 | 13.8 ± 3.4 | 1.4 ± 1 | 0 | 0 | Pw (2/5), D, M, Panc |
| PE04-LMK+CDDP | PE04 | 5 × 106 | CDDP (2.5 mg/kg) + LMK-235 (13 mg/kg) | 5/5 | 191 ± 19 | 4.2 ± 4 | 13 ± 7.5 | 3.8 ± 3 | 0.5 ± 0.1 | 0 | Ov, Pw (1/5), Om, P |
| PE04-HDAC4-KD | PE04-HDAC4-KD | 5 × 106 | None | 5/5 | 124 ± 25 | 7.1 ± 2.6 | 23.4 ± 11.1 | 13.6 ± 7.1 | 2.2 ± 1.9 | 1.2 ± 1 | Ov, P, D, Om, Sp, SI, L |
| PE04-HDAC4-KD+CDDP | PE04-HDAC4-KD | 5 × 106 | CDDP (2.5 mg/kg) | 5/5 | 187 ± 3 | 3.1 ± 1.4 | 4.7 ± 2.8 | 2.2 ± 2 | 3 ± 1.5 | 4 ± 1.2 | Ov, L, P, D, Om |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fan, Q.; Li, L.; Wang, T.-L.; Emerson, R.E.; Xu, Y. A Novel ZIP4-HDAC4-VEGFA Axis in High-Grade Serous Ovarian Cancer. Cancers 2021, 13, 3821. https://doi.org/10.3390/cancers13153821
Fan Q, Li L, Wang T-L, Emerson RE, Xu Y. A Novel ZIP4-HDAC4-VEGFA Axis in High-Grade Serous Ovarian Cancer. Cancers. 2021; 13(15):3821. https://doi.org/10.3390/cancers13153821
Chicago/Turabian StyleFan, Qipeng, Lihong Li, Tian-Li Wang, Robert E. Emerson, and Yan Xu. 2021. "A Novel ZIP4-HDAC4-VEGFA Axis in High-Grade Serous Ovarian Cancer" Cancers 13, no. 15: 3821. https://doi.org/10.3390/cancers13153821
APA StyleFan, Q., Li, L., Wang, T.-L., Emerson, R. E., & Xu, Y. (2021). A Novel ZIP4-HDAC4-VEGFA Axis in High-Grade Serous Ovarian Cancer. Cancers, 13(15), 3821. https://doi.org/10.3390/cancers13153821