
Differential Effects of Combined ATR/WEE1 Inhibition in Cancer Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
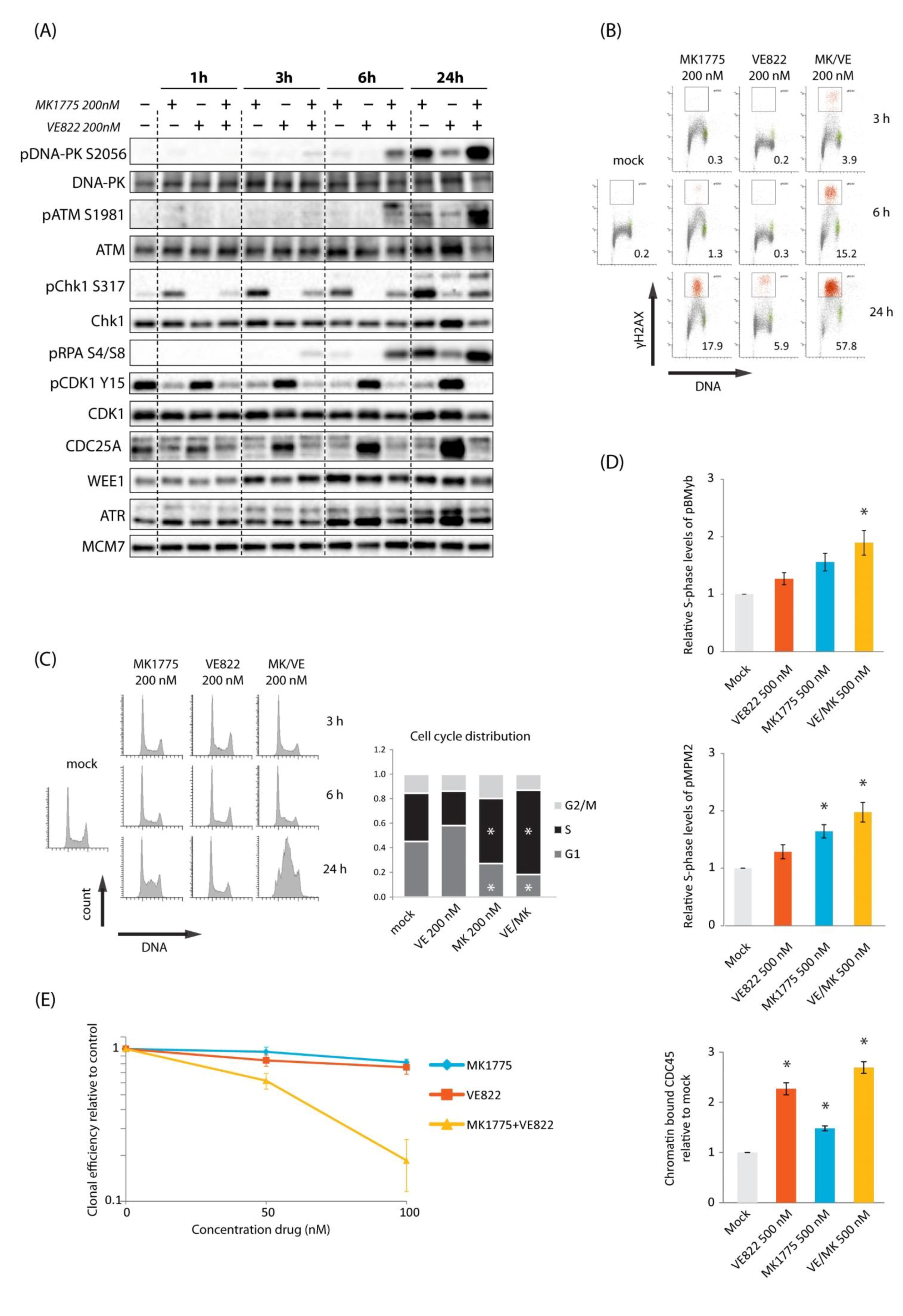
2.1. Combined Inhibition of WEE1 and ATR Gives Synergistic Induction of DNA Damage in S-Phase Accompanied by Synergistic Cell Killing in U2OS Cells
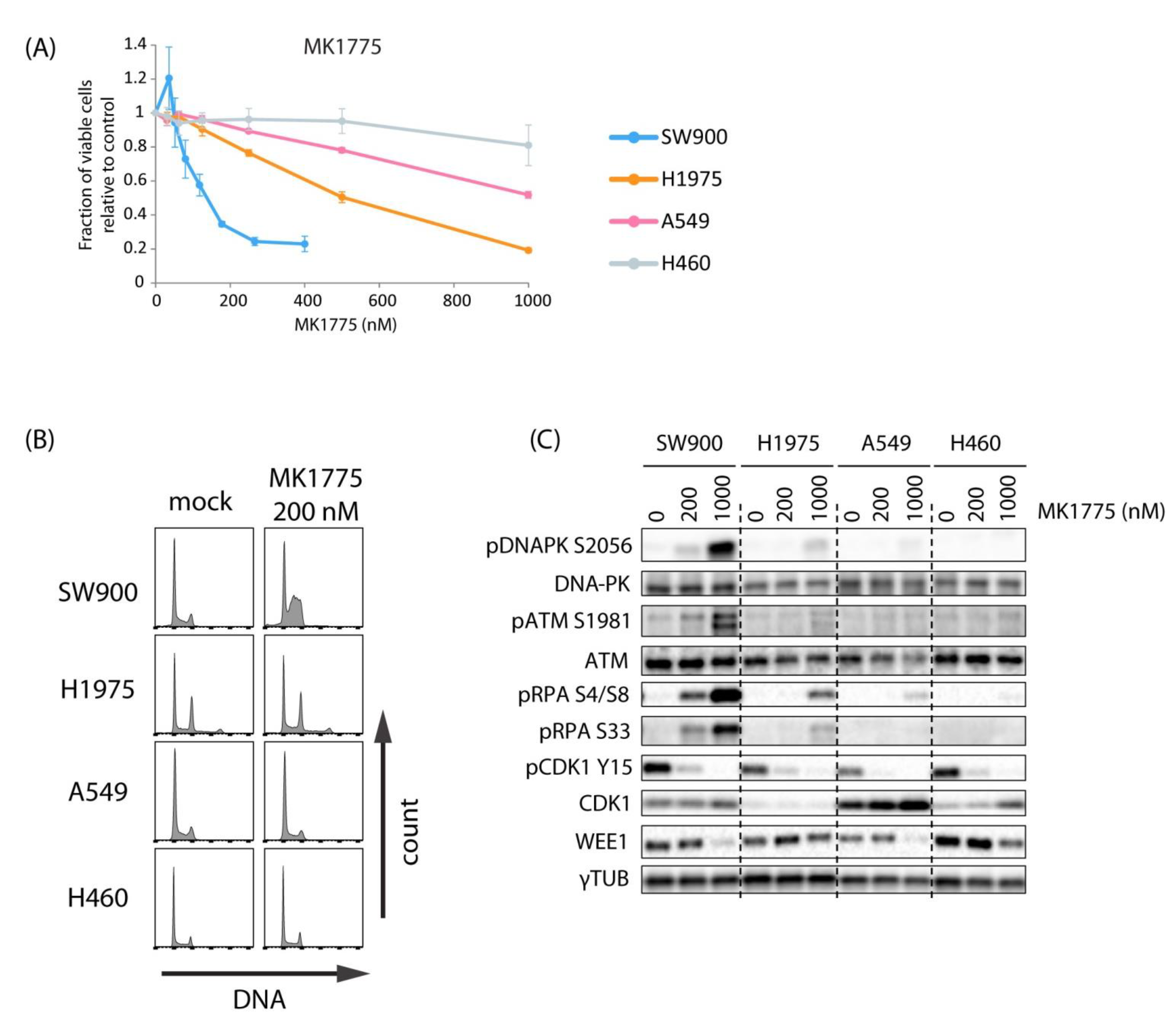
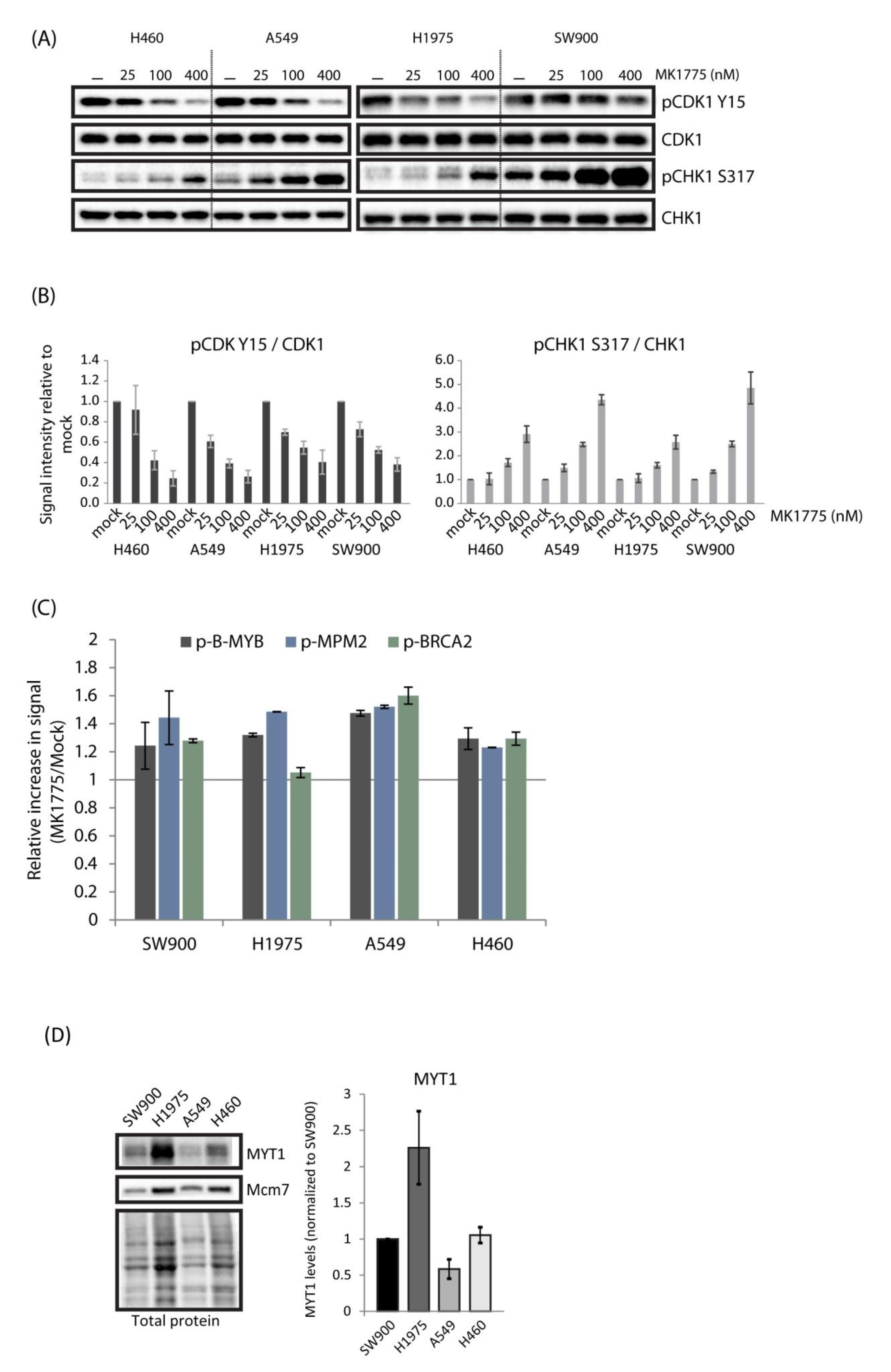
2.2. Lung Cancer Cell Lines H460, A549, H1975, and SW900 Show Large Differences in Sensitivity to the WEE1 Inhibitor Despite a Similar Induction of CDK Activity
2.3. Combined Inhibition of WEE1 and ATR Gives Variable Effects in the Lung Cancer Cell Lines
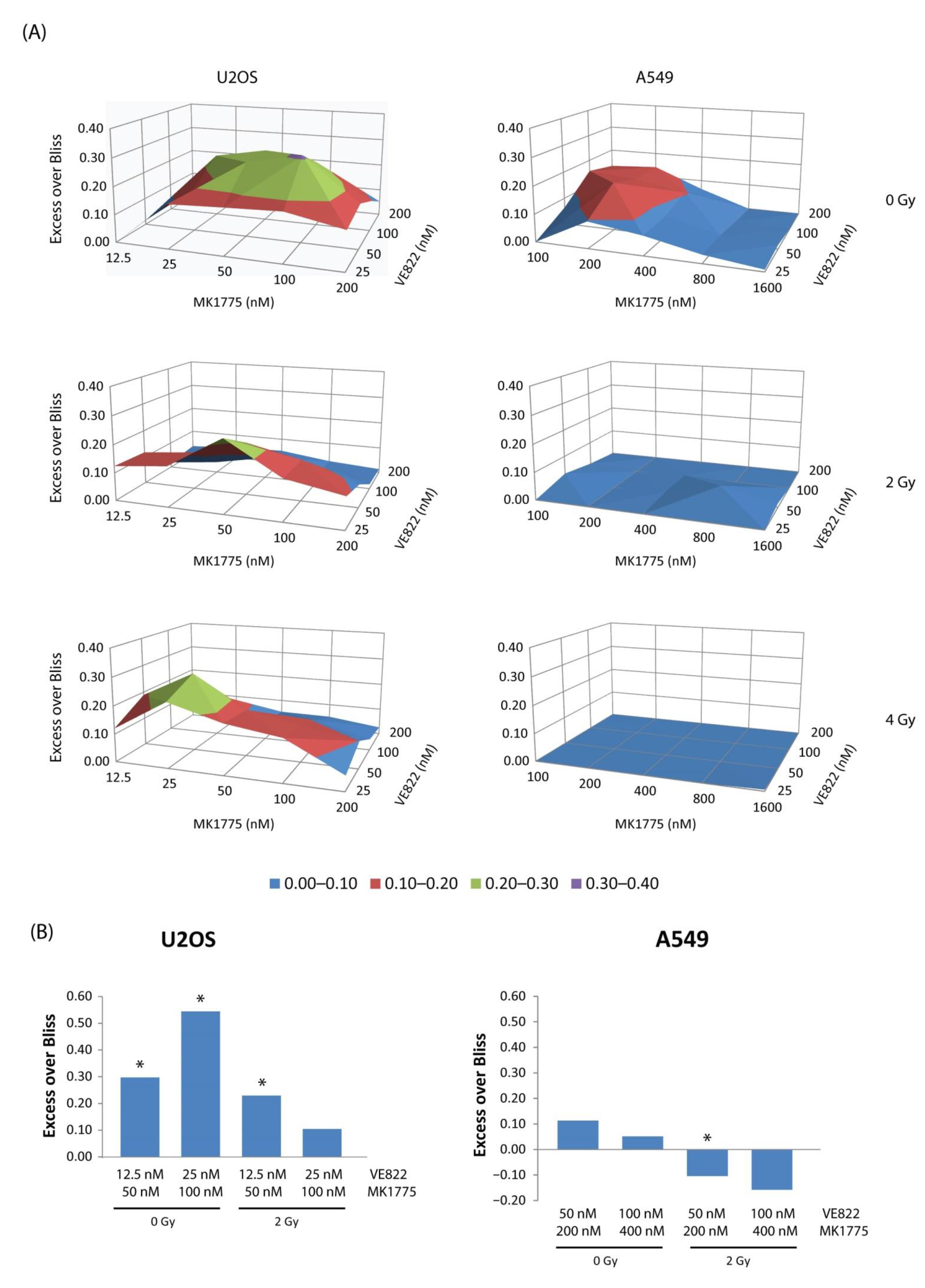
2.4. Less Synergy Is Observed between WEE1/ATR Inhibitors When Cells Are Co-Treated with Radiation
2.5. No Correlation Is Found between Expression Levels of Biomarkers Associated with WEE1 and ATR Inhibitor Sensitivity and Observed Differences in Sensitivity in Lung Cancer and U2OS Cells
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Drug Treatments
4.2. Flow Cytometry Analysis of CDK Targets, Chromatin-Bound CDC45 and MUS81, DNA Damage, and Cell Cycle Distribution
4.3. Viability Assay (CellTiter-Glo)
4.4. Clonogenic Survival Assays
4.5. Immunoblotting
4.6. siRNA Transfection
4.7. DNA Fiber Assay
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pilie, P.G.; Tang, C.; Mills, G.B.; Yap, T.A. State-of-the-art strategies for targeting the DNA damage response in cancer. Nat. Rev. Clin. Oncol. 2019, 16, 81–104. [Google Scholar] [CrossRef] [PubMed]
- Cimprich, K.A.; Cortez, D. ATR: An essential regulator of genome integrity. Nat. Rev. Mol. Cell Biol. 2008, 9, 616–627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lecona, E.; Fernandez-Capetillo, O. Targeting ATR in cancer. Nat. Rev. Cancer 2018, 18, 586–595. [Google Scholar] [CrossRef] [PubMed]
- Syljuåsen, R.G.; Hasvold, G.; Hauge, S.; Helland, Å. Targeting lung cancer through inhibition of checkpoint kinases. Front. Genet. 2015, 6, 70. [Google Scholar] [PubMed] [Green Version]
- Beck, H.; Nähse-Kumpf, V.; Larsen, M.S.; O’Hanlon, K.A.; Patzke, S.; Holmberg, C.; Mejlvang, J.; Groth, A.; Nielsen, O.; Syljuåsen, R.G.; et al. Cyclin-dependent kinase suppression by WEE1 kinase protects the genome through control of replication initiation and nucleotide consumption. Mol. Cell Biol. 2012, 32, 4226–4236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirai, H.; Iwasawa, Y.; Okada, M.; Arai, T.; Nishibata, T.; Kobayashi, M.; Kimura, T.; Kaneko, N.; Ohtani, J.; Yamanaka, K.; et al. Small-molecule inhibition of Wee1 kinase by MK-1775 selectively sensitizes p53-deficient tumor cells to DNA-damaging agents. Mol. Cancer Ther. 2009, 8, 2992–3000. [Google Scholar] [CrossRef] [Green Version]
- Parker, L.L.; Piwnica-Worms, H. Inactivation of the p34cdc2-cyclin B complex by the human WEE1 tyrosine kinase. Science 1992, 257, 1955–1957. [Google Scholar] [CrossRef]
- Watanabe, N.; Broome, M.; Hunter, T. Regulation of the human WEE1Hu CDK tyrosine 15-kinase during the cell cycle. EMBO J. 1995, 14, 1878–1891. [Google Scholar] [CrossRef]
- Mir, S.E.; De Witt Hamer, P.C.; Krawczyk, P.M.; Balaj, L.; Claes, A.; Niers, J.M.; Van Tilborg, A.A.; Zwinderman, A.H.; Geerts, D.; Kaspers, G.J.; et al. In silico analysis of kinase expression identifies WEE1 as a gatekeeper against mitotic catastrophe in glioblastoma. Cancer Cell 2010, 18, 244–257. [Google Scholar] [CrossRef] [Green Version]
- Beck, H.; Nähse, V.; Larsen, M.S.; Groth, P.; Clancy, T.; Lees, M.; Jørgensen, M.; Helleday, T.; Syljuåsen, R.G.; Sørensen, C.S. Regulators of cyclin-dependent kinases are crucial for maintaining genome integrity in S phase. J. Cell Biol. 2010, 188, 629–638. [Google Scholar] [CrossRef]
- Dominguez-Kelly, R.; Martin, Y.; Koundrioukoff, S.; Tanenbaum, M.E.; Smits, V.A.; Medema, R.H.; Debatisse, M.; Freire, R. Wee1 controls genomic stability during replication by regulating the Mus81-Eme1 endonuclease. J. Cell Biol. 2011, 194, 567–579. [Google Scholar] [CrossRef]
- Hauge, S.; Naucke, C.; Hasvold, G.; Joel, M.; Rødland, G.E.; Juzenas, P.; Stokke, T.; Syljuåsen, R.G. Combined inhibition of Wee1 and Chk1 gives synergistic DNA damage in S-phase due to distinct regulation of CDK activity and CDC45 loading. Oncotarget 2017, 8, 10966–10979. [Google Scholar] [CrossRef] [Green Version]
- Carrassa, L.; Chila, R.; Lupi, M.; Ricci, F.; Celenza, C.; Mazzoletti, M.; Broggini, M.; Damia, G. Combined inhibition of Chk1 and Wee1: In vitro synergistic effect translates to tumor growth inhibition in vivo. Cell Cycle 2012, 11, 2507–2517. [Google Scholar] [CrossRef] [Green Version]
- Chaudhuri, L.; Vincelette, N.D.; Koh, B.D.; Naylor, R.M.; Flatten, K.S.; Peterson, K.L.; McNally, A.; Gojo, I.; Karp, J.E.; Mesa, R.A.; et al. CHK1 and WEE1 inhibition combine synergistically to enhance therapeutic efficacy in acute myeloid leukemia ex vivo. Haematologica 2014, 99, 688–696. [Google Scholar] [CrossRef]
- Ghelli Luserna Di Rora, A.; Bocconcelli, M.; Ferrari, A.; Terragna, C.; Bruno, S.; Imbrogno, E.; Beeharry, N.; Robustelli, V.; Ghetti, M.; Napolitano, R.; et al. Synergism Through WEE1 and CHK1 Inhibition in Acute Lymphoblastic Leukemia. Cancers 2019, 11, 1654. [Google Scholar] [CrossRef] [Green Version]
- Magnussen, G.I.; Emilsen, E.; Giller Fleten, K.; Engesaeter, B.; Nähse-Kumpf, V.; Fjaer, R.; Slipicevic, A.; Florenes, V.A. Combined inhibition of the cell cycle related proteins Wee1 and Chk1/2 induces synergistic anti-cancer effect in melanoma. BMC Cancer 2015, 15, 462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russell, M.R.; Levin, K.; Rader, J.; Belcastro, L.; Li, Y.; Martinez, D.; Pawel, B.; Shumway, S.D.; Maris, J.M.; Cole, K.A. Combination therapy targeting the Chk1 and Wee1 kinases shows therapeutic efficacy in neuroblastoma. Cancer Res. 2013, 73, 776–784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koh, S.B.; Wallez, Y.; Dunlop, C.R.; Bernaldo de Quiros Fernandez, S.; Bapiro, T.E.; Richards, F.M.; Jodrell, D.I. Mechanistic Distinctions between CHK1 and WEE1 Inhibition Guide the Scheduling of Triple Therapy with Gemcitabine. Cancer Res. 2018, 78, 3054–3066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bukhari, A.B.; Lewis, C.W.; Pearce, J.J.; Luong, D.; Chan, G.K.; Gamper, A.M. Inhibiting Wee1 and ATR kinases produces tumor-selective synthetic lethality and suppresses metastasis. J. Clin. Investig. 2019, 129, 1329–1344. [Google Scholar] [CrossRef]
- Jin, J.; Fang, H.; Yang, F.; Ji, W.; Guan, N.; Sun, Z.; Shi, Y.; Zhou, G.; Guan, X. Combined Inhibition of ATR and WEE1 as a Novel Therapeutic Strategy in Triple-Negative Breast Cancer. Neoplasia 2018, 20, 478–488. [Google Scholar] [CrossRef] [PubMed]
- Qi, W.; Xu, X.; Wang, M.; Li, X.; Wang, C.; Sun, L.; Zhao, D.; Sun, L. Inhibition of Wee1 sensitizes AML cells to ATR inhibitor VE-822-induced DNA damage and apoptosis. Biochem. Pharmacol. 2019, 164, 273–282. [Google Scholar] [CrossRef]
- Young, L.A.; O’Connor, L.O.; de Renty, C.; Veldman-Jones, M.H.; Dorval, T.; Wilson, Z.; Jones, D.R.; Lawson, D.; Odedra, R.; Maya-Mendoza, A.; et al. Differential Activity of ATR and WEE1 Inhibitors in a Highly Sensitive Subpopulation of DLBCL Linked to Replication Stress. Cancer Res. 2019, 79, 3762–3775. [Google Scholar] [CrossRef] [Green Version]
- Nam, A.R.; Jin, M.H.; Bang, J.H.; Oh, K.S.; Seo, H.R.; Oh, D.Y.; Bang, Y.J. Inhibition of ATR Increases the Sensitivity to WEE1 Inhibitor in Biliary Tract Cancer. Cancer Res. Treat. 2020, 52, 945–956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sørensen, C.S.; Syljuåsen, R.G.; Falck, J.; Schroeder, T.; Ronnstrand, L.; Khanna, K.K.; Zhou, B.B.; Bartek, J.; Lukas, J. Chk1 regulates the S phase checkpoint by coupling the physiological turnover and ionizing radiation-induced accelerated proteolysis of Cdc25A. Cancer Cell 2003, 3, 247–258. [Google Scholar] [CrossRef] [Green Version]
- Sørensen, C.S.; Syljuåsen, R.G.; Lukas, J.; Bartek, J. ATR, Claspin and the Rad9-Rad1-Hus1 complex regulate Chk1 and Cdc25A in the absence of DNA damage. Cell Cycle 2004, 3, 941–945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guertin, A.D.; Li, J.; Liu, Y.; Hurd, M.S.; Schuller, A.G.; Long, B.; Hirsch, H.A.; Feldman, I.; Benita, Y.; Toniatti, C.; et al. Preclinical evaluation of the WEE1 inhibitor MK-1775 as single-agent anticancer therapy. Mol. Cancer Ther. 2013, 12, 1442–1452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis, C.W.; Bukhari, A.B.; Xiao, E.J.; Choi, W.S.; Smith, J.D.; Homola, E.; Mackey, J.R.; Campbell, S.D.; Gamper, A.M.; Chan, G.K. Upregulation of Myt1 Promotes Acquired Resistance of Cancer Cells to Wee1 Inhibition. Cancer Res. 2019, 79, 5971–5985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bridges, K.A.; Hirai, H.; Buser, C.A.; Brooks, C.; Liu, H.; Buchholz, T.A.; Molkentine, J.M.; Mason, K.A.; Meyn, R.E. MK-1775, a novel Wee1 kinase inhibitor, radiosensitizes p53-defective human tumor cells. Clin. Cancer Res. 2011, 17, 5638–5648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dillon, M.T.; Barker, H.E.; Pedersen, M.; Hafsi, H.; Bhide, S.A.; Newbold, K.L.; Nutting, C.M.; McLaughlin, M.; Harrington, K.J. Radiosensitization by the ATR Inhibitor AZD6738 through Generation of Acentric Micronuclei. Mol. Cancer Ther. 2017, 16, 25–34. [Google Scholar] [CrossRef] [Green Version]
- Dunne, V.; Ghita, M.; Small, D.M.; Coffey, C.B.M.; Weldon, S.; Taggart, C.C.; Osman, S.O.; McGarry, C.K.; Prise, K.M.; Hanna, G.G.; et al. Inhibition of ataxia telangiectasia related-3 (ATR) improves therapeutic index in preclinical models of non-small cell lung cancer (NSCLC) radiotherapy. Radiother. Oncol. 2017, 124, 475–481. [Google Scholar] [CrossRef] [Green Version]
- Fokas, E.; Prevo, R.; Pollard, J.R.; Reaper, P.M.; Charlton, P.A.; Cornelissen, B.; Vallis, K.A.; Hammond, E.M.; Olcina, M.M.; Gillies McKenna, W.; et al. Targeting ATR in vivo using the novel inhibitor VE-822 results in selective sensitization of pancreatic tumors to radiation. Cell Death Dis. 2012, 3, e441. [Google Scholar] [CrossRef] [Green Version]
- Hauge, S.; Eek Mariampillai, A.; Rødland, G.E.; Bay, L.T.E.; Landsverk, H.B.L.; Syljuåsen, R.G. Expanding roles of cell cycle checkpoint inhibitors in radiation oncology. Int. J. Radiat. Biol. 2021, in press. [Google Scholar] [CrossRef]
- Lee, Y.Y.; Cho, Y.J.; Shin, S.W.; Choi, C.; Ryu, J.Y.; Jeon, H.K.; Choi, J.J.; Hwang, J.R.; Choi, C.H.; Kim, T.J.; et al. Anti-Tumor Effects of Wee1 Kinase Inhibitor with Radiotherapy in Human Cervical Cancer. Sci. Rep. 2019, 9, 15394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Shen, C.; Pettit, C.J.; Li, T.; Hu, A.J.; Miller, E.D.; Zhang, J.; Lin, S.H.; Williams, T.M. Wee1 Kinase Inhibitor AZD1775 Effectively Sensitizes Esophageal Cancer to Radiotherapy. Clin. Cancer Res. 2020, 26, 3740–3750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobbelstein, M.; Sorensen, C.S. Exploiting replicative stress to treat cancer. Nat. Rev. Drug Discov. 2015, 14, 405–423. [Google Scholar] [CrossRef] [PubMed]
- Ng, N.; Purshouse, K.; Foskolou, I.P.; Olcina, M.M.; Hammond, E.M. Challenges to DNA replication in hypoxic conditions. FEBS J. 2018, 285, 1563–1571. [Google Scholar] [CrossRef] [Green Version]
- Li, C.Y.; Nagasawa, H.; Dahlberg, W.K.; Little, J.B. Diminished capacity for p53 in mediating a radiation-induced G1 arrest in established human tumor cell lines. Oncogene 1995, 11, 1885–1892. [Google Scholar]
- Ma, C.X.; Janetka, J.W.; Piwnica-Worms, H. Death by releasing the breaks: CHK1 inhibitors as cancer therapeutics. Trends Mol. Med. 2011, 17, 88–96. [Google Scholar] [CrossRef] [PubMed]
- Dent, P. Investigational CHK1 inhibitors in early phase clinical trials for the treatment of cancer. Expert Opin. Investig. Drugs 2019, 28, 1095–1100. [Google Scholar] [CrossRef]
- Baldin, V.; Ducommun, B. Subcellular localisation of human wee1 kinase is regulated during the cell cycle. J. Cell Sci. 1995, 108 Pt 6, 2425–2432. [Google Scholar] [CrossRef]
- Buisson, R.; Niraj, J.; Rodrigue, A.; Ho, C.K.; Kreuzer, J.; Foo, T.K.; Hardy, E.J.; Dellaire, G.; Haas, W.; Xia, B.; et al. Coupling of Homologous Recombination and the Checkpoint by ATR. Mol. Cell 2017, 65, 336–346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dubois, J.C.; Yates, M.; Gaudreau-Lapierre, A.; Clement, G.; Cappadocia, L.; Gaudreau, L.; Zou, L.; Marechal, A. A phosphorylation-and-ubiquitylation circuitry driving ATR activation and homologous recombination. Nucleic Acids Res. 2017, 45, 8859–8872. [Google Scholar] [CrossRef] [PubMed]
- Krajewska, M.; Heijink, A.M.; Bisselink, Y.J.; Seinstra, R.I.; Sillje, H.H.; de Vries, E.G.; van Vugt, M.A. Forced activation of Cdk1 via wee1 inhibition impairs homologous recombination. Oncogene 2013, 32, 3001–3008. [Google Scholar] [CrossRef] [PubMed]
- Yazinski, S.A.; Comaills, V.; Buisson, R.; Genois, M.M.; Nguyen, H.D.; Ho, C.K.; Todorova Kwan, T.; Morris, R.; Lauffer, S.; Nussenzweig, A.; et al. ATR inhibition disrupts rewired homologous recombination and fork protection pathways in PARP inhibitor-resistant BRCA-deficient cancer cells. Genes Dev. 2017, 31, 318–332. [Google Scholar] [CrossRef]
- Chen, X.; Low, K.H.; Alexander, A.; Jiang, Y.; Karakas, C.; Hess, K.R.; Carey, J.P.W.; Bui, T.N.; Vijayaraghavan, S.; Evans, K.W.; et al. Cyclin E Overexpression Sensitizes Triple-Negative Breast Cancer to Wee1 Kinase Inhibition. Clin. Cancer Res. 2018, 24, 6594–6610. [Google Scholar] [CrossRef] [Green Version]
- Pfister, S.X.; Markkanen, E.; Jiang, Y.; Sarkar, S.; Woodcock, M.; Orlando, G.; Mavrommati, I.; Pai, C.C.; Zalmas, L.P.; Drobnitzky, N.; et al. Inhibiting WEE1 Selectively Kills Histone H3K36me3-Deficient Cancers by dNTP Starvation. Cancer Cell 2015, 28, 557–568. [Google Scholar] [CrossRef] [Green Version]
- Hauge, S.; Macurek, L.; Syljuåsen, R.G. p21 limits S phase DNA damage caused by the Wee1 inhibitor MK1775. Cell Cycle 2019, 18, 834–847. [Google Scholar] [CrossRef]
- Kwok, M.; Davies, N.; Agathanggelou, A.; Smith, E.; Oldreive, C.; Petermann, E.; Stewart, G.; Brown, J.; Lau, A.; Pratt, G.; et al. ATR inhibition induces synthetic lethality and overcomes chemoresistance in TP53- or ATM-defective chronic lymphocytic leukemia cells. Blood 2016, 127, 582–595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohni, K.N.; Kavanaugh, G.M.; Cortez, D. ATR pathway inhibition is synthetically lethal in cancer cells with ERCC1 deficiency. Cancer Res. 2014, 74, 2835–2845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruiz, S.; Mayor-Ruiz, C.; Lafarga, V.; Murga, M.; Vega-Sendino, M.; Ortega, S.; Fernandez-Capetillo, O. A Genome-wide CRISPR Screen Identifies CDC25A as a Determinant of Sensitivity to ATR Inhibitors. Mol. Cell 2016, 62, 307–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nayak, S.; Calvo, J.A.; Cong, K.; Peng, M.; Berthiaume, E.; Jackson, J.; Zaino, A.M.; Vindigni, A.; Hadden, M.K.; Cantor, S.B. Inhibition of the translesion synthesis polymerase REV1 exploits replication gaps as a cancer vulnerability. Sci. Adv. 2020, 6, eaaz7808. [Google Scholar] [CrossRef] [PubMed]
- Dillon, M.T.; Bergerhoff, K.F.; Pedersen, M.; Whittock, H.; Crespo-Rodriguez, E.; Patin, E.C.; Pearson, A.; Smith, H.G.; Paget, J.T.E.; Patel, R.R.; et al. ATR Inhibition Potentiates the Radiation-induced Inflammatory Tumor Microenvironment. Clin. Cancer Res. 2019, 25, 3392–3403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, P.; Sun, L.; Robbins, Y.; Clavijo, P.E.; Friedman, J.; Silvin, C.; Van Waes, C.; Cook, J.; Mitchell, J.; Allen, C. Enhancing direct cytotoxicity and response to immune checkpoint blockade following ionizing radiation with Wee1 kinase inhibition. Oncoimmunology 2019, 8, e1638207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vendetti, F.P.; Karukonda, P.; Clump, D.A.; Teo, T.; Lalonde, R.; Nugent, K.; Ballew, M.; Kiesel, B.F.; Beumer, J.H.; Sarkar, S.N.; et al. ATR kinase inhibitor AZD6738 potentiates CD8+ T cell-dependent antitumor activity following radiation. J. Clin. Investig. 2018, 128, 3926–3940. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Sun, L.; Yuan, Z.; Tao, Z. Wee1 kinase inhibitor AZD1775 potentiates CD8+ T cell-dependent antitumour activity via dendritic cell activation following a single high dose of irradiation. Med. Oncol. 2020, 37, 66. [Google Scholar] [CrossRef]
- Rødland, G.E.; Melhus, K.; Generalov, R.; Gilani, S.; Bertoni, F.; Dahle, J.; Syljuåsen, R.G.; Patzke, S. The Dual Cell Cycle Kinase Inhibitor JNJ-7706621 Reverses Resistance to CD37-Targeted Radioimmunotherapy in Activated B Cell Like Diffuse Large B Cell Lymphoma Cell Lines. Front. Oncol. 2019, 9, 1301. [Google Scholar] [CrossRef]
- Liu, Q.; Wang, M.; Kern, A.M.; Khaled, S.; Han, J.; Yeap, B.Y.; Hong, T.S.; Settleman, J.; Benes, C.H.; Held, K.D.; et al. Adapting a drug screening platform to discover associations of molecular targeted radiosensitizers with genomic biomarkers. Mol. Cancer Res. 2015, 13, 713–720. [Google Scholar] [CrossRef] [Green Version]
- Landsverk, H.B.; Sandquist, L.E.; Bay, L.T.E.; Steurer, B.; Campsteijn, C.; Landsverk, O.J.B.; Marteijn, J.A.; Petermann, E.; Trinkle-Mulcahy, L.; Syljuåsen, R.G. WDR82/PNUTS-PP1 Prevents Transcription-Replication Conflicts by Promoting RNA Polymerase II Degradation on Chromatin. Cell Rep. 2020, 33, 108469. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rødland, G.E.; Hauge, S.; Hasvold, G.; Bay, L.T.E.; Raabe, T.T.H.; Joel, M.; Syljuåsen, R.G. Differential Effects of Combined ATR/WEE1 Inhibition in Cancer Cells. Cancers 2021, 13, 3790. https://doi.org/10.3390/cancers13153790
Rødland GE, Hauge S, Hasvold G, Bay LTE, Raabe TTH, Joel M, Syljuåsen RG. Differential Effects of Combined ATR/WEE1 Inhibition in Cancer Cells. Cancers. 2021; 13(15):3790. https://doi.org/10.3390/cancers13153790
Chicago/Turabian StyleRødland, Gro Elise, Sissel Hauge, Grete Hasvold, Lilli T. E. Bay, Tine T. H. Raabe, Mrinal Joel, and Randi G. Syljuåsen. 2021. "Differential Effects of Combined ATR/WEE1 Inhibition in Cancer Cells" Cancers 13, no. 15: 3790. https://doi.org/10.3390/cancers13153790
APA StyleRødland, G. E., Hauge, S., Hasvold, G., Bay, L. T. E., Raabe, T. T. H., Joel, M., & Syljuåsen, R. G. (2021). Differential Effects of Combined ATR/WEE1 Inhibition in Cancer Cells. Cancers, 13(15), 3790. https://doi.org/10.3390/cancers13153790