
Anastasis: Return Journey from Cell Death


{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. A Few Historical Remarks
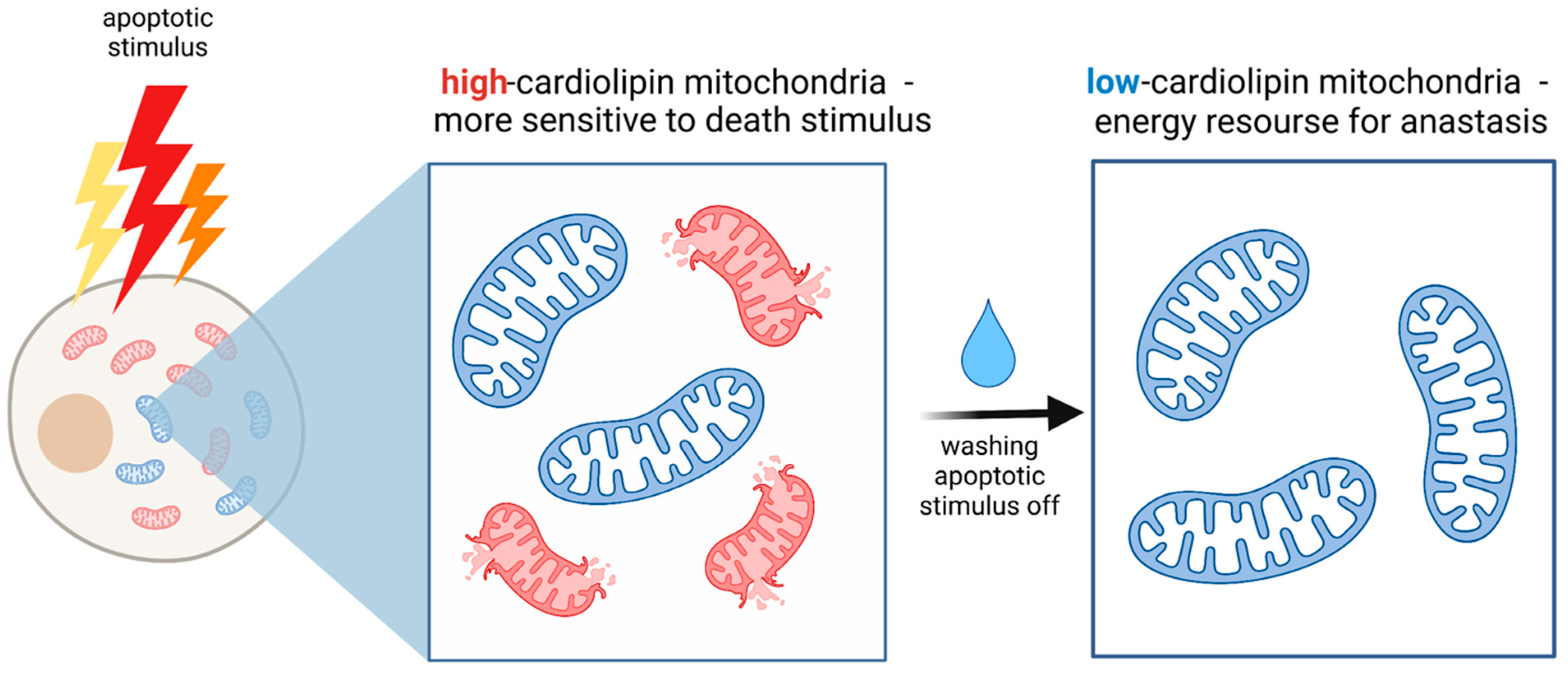
3. A Mitochondria-Anastasis Link
4. Role of Transcription and Post-Transcriptional Modifications in Anastasis
5. Double Role of Caspases
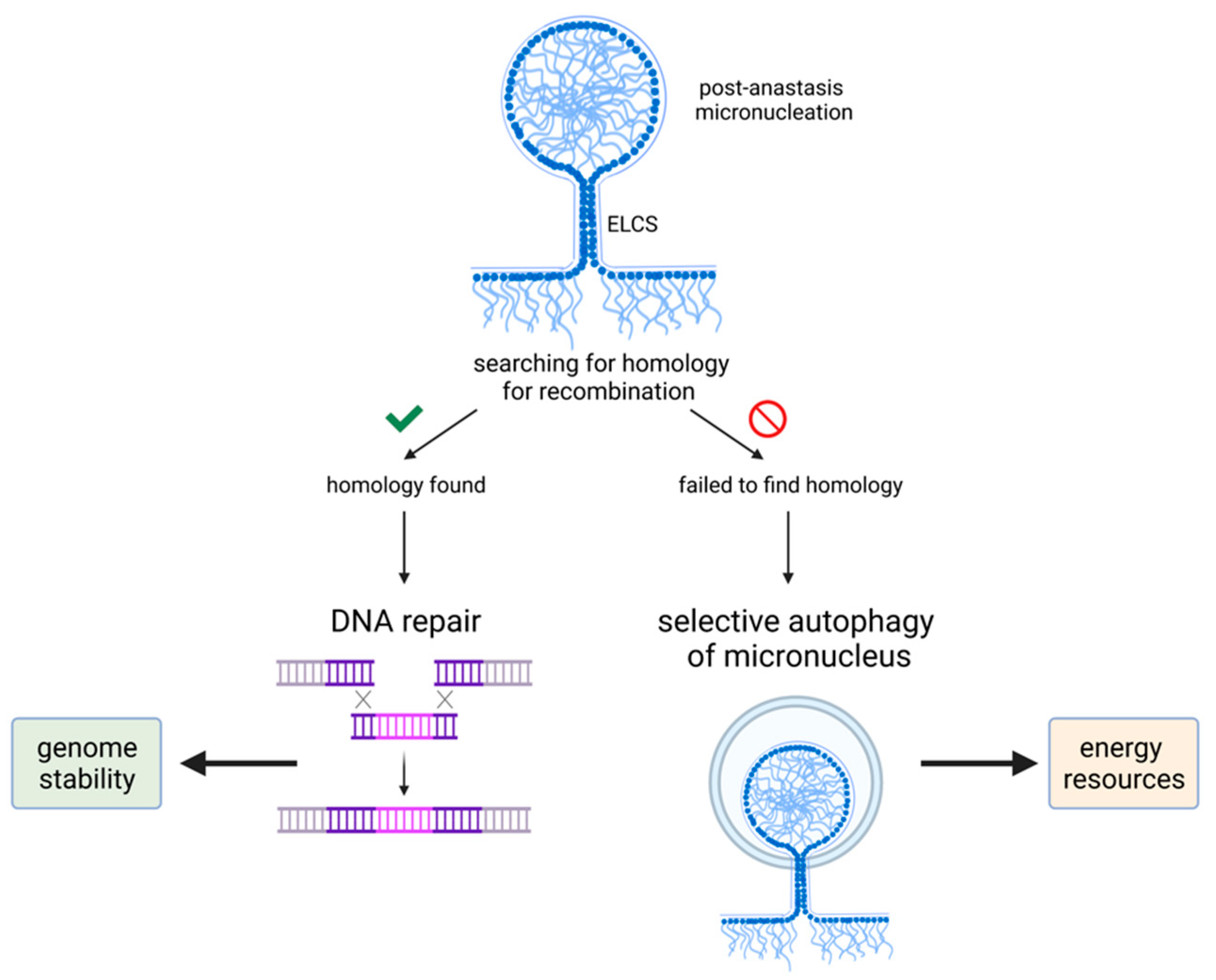
6. A Role of Nuclear Changes in Anastasis
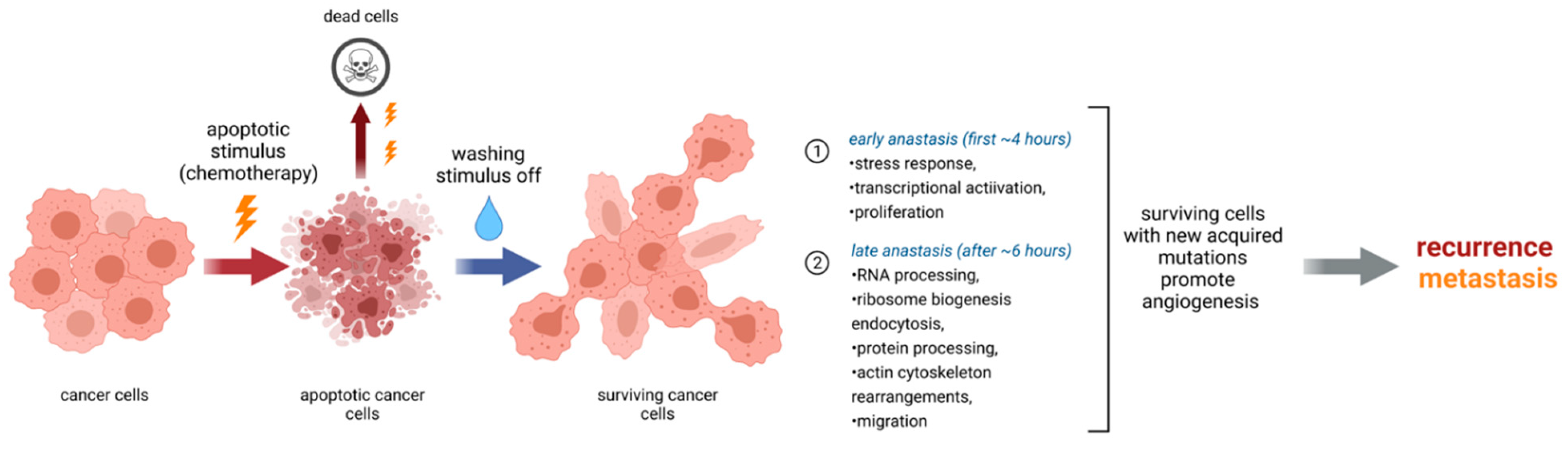
7. Anastasis and Other Forms of Cell Death
8. Detection of Anastasis
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Reed, J.C. Apoptosis-targeted therapies for cancer. Cancer Cell 2003, 3, 17–22. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Igney, F.H.; Krammer, P.H. Death and anti-death: Tumour resistance to apoptosis. Nat. Rev. Cancer 2002, 2, 277–288. [Google Scholar] [CrossRef]
- Green, D.R. The Coming Decade of Cell Death Research: Five Riddles. Cell 2019, 177, 1094–1107. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.L.; Tang, H.M.; Mak, K.H.; Hu, S.; Wang, S.S.; Wong, K.M.; Wong, C.S.T.; Wu, H.Y.; Law, H.T.; Liu, K.; et al. Cell survival, DNA damage, and oncogenic transformation after a transient and reversible apoptotic response. Mol. Biol. Cell 2012, 23, 2240–2252. [Google Scholar] [CrossRef] [PubMed]
- Haider, N.; Narula, N.; Narula, J. Apoptosis in heart failure represents programmed cell survival, not death, of cardiomyocytes and likelihood of reverse remodeling. J. Card. Fail. 2002, 8, S512–S517. [Google Scholar] [CrossRef] [PubMed]
- Pulkki, A.S.; Voipio-Pulkki, L.M. Significance of myocytes with positive DNA in situ nick end-labeling (TUNEL) in hearts with dilated cardiomyopathy. Circulation 2000, 101, e239. [Google Scholar] [CrossRef] [PubMed]
- Narula, J.; Haider, N.; Arbustini, E.; Chandrashekhar, Y. Mechanisms of Disease: Apoptosis in heart failure—seeing hope in death. Nat. Clin. Pract. Cardiovasc. Med. 2006, 3, 681–688. [Google Scholar] [CrossRef]
- Albeck, J.G.; Burke, J.M.; Aldridge, B.B.; Zhang, M.; Lauffenburger, D.A.; Sorger, P.K. Quantitative Analysis of Pathways Controlling Extrinsic Apoptosis in Single Cells. Mol. Cell 2008, 30, 11–25. [Google Scholar] [CrossRef]
- Lovric, M.M.; Hawkins, C.J. TRAIL treatment provokes mutations in surviving cells. Oncogene 2010, 29, 5048–5060. [Google Scholar] [CrossRef]
- Gama, V.; Deshmukh, M. Life after MOMP. Mol. Cell 2015, 58, 199–201. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; He, Y.; Li, F.; Huang, Q.; Kato, T.A.; Hall, R.P.; Li, C.Y. Caspase-3 Promotes Genetic Instability and Carcinogenesis. Mol. Cell 2015, 58, 284–296. [Google Scholar] [CrossRef]
- Ding, A.X.; Sun, G.; Argaw, Y.G.; Wong, J.O.; Easwaran, S.; Montell, D.J. CasExpress reveals widespread and diverse patterns of cell survival of caspase-3 activation during development in vivo. eLife 2016, 5, e10936. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, U.; Groscurth, P. Morphological features of cell death. News Physiol. Sci. 2004, 19, 124–128. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z.; El-Deiry, W.S. Overview of cell death signaling pathways. Cancer Biol. Ther. 2005, 4, 147–171. [Google Scholar] [CrossRef]
- Kalkavan, H.; Green, D.R. MOMP, cell suicide as a BCL-2 family business. Cell Death Differ. 2018, 25, 46–55. [Google Scholar] [CrossRef] [PubMed]
- Sun, G.; Guzman, E.; Balasanyan, V.; Conner, C.M.; Wong, K.; Zhou, H.R.; Kosik, K.S.; Montell, D.J. A molecular signature for anastasis, recovery from the brink of apoptotic cell death. J. Cell Biol. 2017, 216, 3355–3368. [Google Scholar] [CrossRef]
- Ichim, G.; Lopez, J.; Ahmed, S.U.; Muthalagu, N.; Giampazolias, E.; Delgado, M.E.; Haller, M.; Riley, J.S.; Mason, S.M.; Athineos, D.; et al. Limited Mitochondrial Permeabilization Causes DNA Damage and Genomic Instability in the Absence of Cell Death. Mol. Cell 2015, 57, 860–872. [Google Scholar] [CrossRef]
- Tait, S.W.G.; Parsons, M.J.; Llambi, F.; Bouchier-Hayes, L.; Connell, S.; Muñoz-Pinedo, C.; Green, D.R. Resistance to caspase-independent cell death requires persistence of intact mitochondria. Dev. Cell 2010, 18, 802–813. [Google Scholar] [CrossRef]
- Tait, S.W.G.; Green, D.R. Mitochondria and cell death: Outer membrane permeabilization and beyond. Nat. Rev. Mol. Cell Biol. 2010, 11, 621–632. [Google Scholar] [CrossRef]
- Daniele, T.; Hurbain, I.; Vago, R.; Casari, G.; Raposo, G.; Tacchetti, C.; Schiaffino, M.V. Mitochondria and melanosomes establish physical contacts modulated by Mfn2 and involved in organelle biogenesis. Curr. Biol. 2014, 24, 393–403. [Google Scholar] [CrossRef]
- Dzeja, P.P.; Bortolon, R.; Perez-Terzic, C.; Holmuhamedov, E.L.; Terzic, A. Energetic communication between mitochondria and nucleus directed by catalyzed phosphotransfer. Proc. Natl. Acad. Sci. USA 2002, 99, 10156–10161. [Google Scholar] [CrossRef] [PubMed]
- Collins, T.J.; Berridge, M.J.; Lipp, P.; Bootman, M.D. Mitochondria are morphologically and functionally heterogeneous within cells. EMBO J. 2002, 21, 1616–1627. [Google Scholar] [CrossRef]
- Kuznetsov, A.V.; Margreiter, R. Heterogeneity of mitochondria and mitochondrial function within cells as another level of mitochondrial complexity. Int. J. Mol. Sci. 2009, 10, 1911–1929. [Google Scholar] [CrossRef] [PubMed]
- Aryaman, J.; Johnston, I.G.; Jones, N.S. Mitochondrial heterogeneity. Front. Genet. 2019, 9, 718. [Google Scholar] [CrossRef] [PubMed]
- Schug, Z.T.; Gottlieb, E. Cardiolipin acts as a mitochondrial signalling platform to launch apoptosis. Biochim. Biophys. Acta Biomembr. 2009, 1788, 2022–2031. [Google Scholar] [CrossRef]
- Sorice, M.; Manganelli, V.; Matarrese, P.; Tinari, A.; Misasi, R.; Malorni, W.; Garofalo, T. Cardiolipin-enriched raft-like microdomains are essential activating platforms for apoptotic signals on mitochondria. FEBS Lett. 2009, 583, 2447–2450. [Google Scholar] [CrossRef]
- Kim, T.H.; Zhao, Y.; Ding, W.X.; Shin, J.N.; He, X.; Seo, Y.W.; Chen, J.; Rabinowich, H.; Amoscato, A.A.; Yin, X.M. Bid-cardiolipin interaction at mitochondrial contact site contributes to mitochondrial cristae reorganization and cytochrome c release. Mol. Biol. Cell 2004, 15, 3061–3072. [Google Scholar] [CrossRef]
- Khalifat, N.; Fournier, J.B.; Angelova, M.I.; Puff, N. Lipid packing variations induced by pH in cardiolipin-containing bilayers: The driving force for the cristae-like shape instability. Biochim. Biophys. Acta Biomembr. 2011, 1808, 2724–2733. [Google Scholar] [CrossRef]
- Tadato, B.; Heymann, J.A.W.; Song, Z.; Hinshaw, J.E.; Chan, D.C. OPA1 disease alleles causing dominant optic atrophy have defects in cardiolipin-stimulated GTP hydrolysis and membrane tubulation. Hum. Mol. Genet. 2010, 19, 2113–2122. [Google Scholar] [CrossRef]
- Saunders, J.E.; Beeson, C.C.; Schnellmann, R.G. Characterization of functionally distinct mitochondrial subpopulations. J. Bioenerg. Biomembr. 2013, 45, 87–99. [Google Scholar] [CrossRef]
- Rowland, A.A.; Voeltz, G.K. Endoplasmic reticulum-mitochondria contacts: Function of the junction. Nat. Rev. Mol. Cell Biol. 2012, 13, 607–615. [Google Scholar] [CrossRef] [PubMed]
- Prudent, J.; McBride, H.M. The mitochondria–endoplasmic reticulum contact sites: A signalling platform for cell death. Curr. Opin. Cell Biol. 2017, 47, 52–63. [Google Scholar] [CrossRef]
- Doghman-Bouguerra, M.; Lalli, E. ER-mitochondria interactions: Both strength and weakness within cancer cells. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 650–662. [Google Scholar] [CrossRef] [PubMed]
- Hajnóczky, G.; Csordás, G.; Das, S.; Garcia-Perez, C.; Saotome, M.; Sinha Roy, S.; Yi, M. Mitochondrial calcium signalling and cell death: Approaches for assessing the role of mitochondrial Ca2+ uptake in apoptosis. Cell Calcium 2006, 40, 553–560. [Google Scholar] [CrossRef]
- Bravo, R.; Vicencio, J.M.; Parra, V.; Troncoso, R.; Munoz, J.P.; Bui, M.; Quiroga, C.; Rodriguez, A.E.; Verdejo, H.E.; Ferreira, J.; et al. Increased ER-mitochondrial coupling promotes mitochondrial respiration and bioenergetics during early phases of ER stress. J. Cell Sci. 2011, 124, 2143–2152. [Google Scholar] [CrossRef] [PubMed]
- Vannuvel, K.; Renard, P.; Raes, M.; Arnould, T. Functional and morphological impact of ER stress on mitochondria. J. Cell. Physiol. 2013, 228, 1802–1818. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Guo, Y.; Tang, J.; Jiang, J.; Chen, Z. New insights into the roles of CHOP-induced apoptosis in ER stress. Acta Biochim. Biophys. Sin. 2014, 46, 629–640. [Google Scholar] [CrossRef] [PubMed]
- Manalo, R.V.M. Anastasis and the ER stress response: Solving the paradox of the unfolded protein response in cancer. Med. Hypotheses 2017, 109, 25–27. [Google Scholar] [CrossRef]
- Tang, H.M.; Talbot, C.C., Jr.; Fung, M.C.; Tang, H.L. Molecular signature of anastasis for reversal of apoptosis. F1000Research 2017, 6, 43. [Google Scholar] [CrossRef] [PubMed]
- Katsuno, Y.; Lamouille, S.; Derynck, R. TGF-β signaling and epithelial-mesenchymal transition in cancer progression. Curr. Opin. Oncol. 2013, 25, 76–84. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Chen, S.; Zeng, J. TGF-β signaling: A complex role in tumorigenesis (Review). Mol. Med. Rep. 2018, 17, 699–704. [Google Scholar] [CrossRef] [PubMed]
- Vincent, T.; Neve, E.P.A.; Johnson, J.R.; Kukalev, A.; Rojo, F.; Albanell, J.; Pietras, K.; Virtanen, I.; Philipson, L.; Leopold, P.L.; et al. A SNAIL1-SMAD3/4 transcriptional repressor complex promotes TGF-β mediated epithelial-mesenchymal transition. Nat. Cell Biol. 2009, 11, 943–950. [Google Scholar] [CrossRef] [PubMed]
- Fan, F.; Samuel, S.; Evans, K.W.; Lu, J.; Xia, L.; Zhou, Y.; Sceusi, E.; Tozzi, F.; Ye, X.C.; Mani, S.A.; et al. Overexpression of Snail induces epithelial-mesenchymal transition and a cancer stem cell-like phenotype in human colorectal cancer cells. Cancer Med. 2012, 1, 5–16. [Google Scholar] [CrossRef]
- Wang, Y.; Shi, J.; Chai, K.; Ying, X.; Zhou, B. The Role of Snail in EMT and Tumorigenesis. Curr. Cancer Drug Targets 2014, 13, 963–972. [Google Scholar] [CrossRef]
- Nawneet, K.K.; Swati, P.J.; Alok, V.J.; Avinash, D.G.; Prasad, D.C.; Rahul, Y.D.; Sharmila, A.B. Snail and Slug Mediate Radioresistance and Chemoresistance by Antagonizing p53-Mediated Apoptosis and Acquiring a Stem-Like Phenotype in Ovarian Cancer Cells. Stem Cells 2009, 27, 2059–2068. [Google Scholar]
- Vandromme, M. Regulation of transcription factor localization: Fine-tuning of gene expression. Trends Biochem. Sci. 1996, 21, 59–64. [Google Scholar] [CrossRef]
- Turner, J.G.; Dawson, J.; Sullivan, D.M. Nuclear export of proteins and drug resistance in cancer. Biochem. Pharmacol. 2012, 83, 1021–1032. [Google Scholar] [CrossRef]
- Seervi, M.; Sumi, S.; Chandrasekharan, A.; Sharma, A.K.; SanthoshKumar, T.R. Molecular profiling of anastatic cancer cells: Potential role of the nuclear export pathway. Cell. Oncol. 2019, 42, 645–661. [Google Scholar] [CrossRef]
- Muqbil, I.; Wu, J.; Aboukameel, A.; Mohammad, R.M.; Azmi, A.S. Snail nuclear transport: The gateways regulating epithelial-to-mesenchymal transition? Semin. Cancer Biol. 2014, 27, 39–45. [Google Scholar] [CrossRef] [PubMed]
- De Falco, S. The discovery of placenta growth factor and its biological activity. Exp. Mol. Med. 2012, 44, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Barquilla, A.; Pasquale, E.B. Eph receptors and ephrins: Therapeutic opportunities. Annu. Rev. Pharmacol. Toxicol. 2015, 55, 467–487. [Google Scholar] [CrossRef]
- Deryugina, E.I.; Quigley, J.P. Matrix metalloproteinases and tumor metastasis. Cancer Metastasis Rev. 2006, 25, 9–34. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2010, 120, 1786. [Google Scholar] [CrossRef]
- Hauer, M.H.; Seeber, A.; Singh, V.; Thierry, R.; Sack, R.; Amitai, A.; Kryzhanovska, M.; Eglinger, J.; Holcman, D.; Owen-Hughes, T.; et al. Histone degradation in response to DNA damage enhances chromatin dynamics and recombination rates. Nat. Struct. Mol. Biol. 2017, 24, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Zimmer, C.; Fabre, E. Chromatin mobility upon DNA damage: State of the art and remaining questions. Curr. Genet. 2019, 65, 1–9. [Google Scholar] [CrossRef]
- Raj, A.T.; Kheur, S.; Bhonde, R.; Gupta, A.A.; Patil, V.R.; Kharat, A. Potential role of anastasis in cancer initiation and progression. Apoptosis 2019, 24, 383–384. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; So, C.; Lam, H.M.; Fung, M.C.; Tsang, S.Y. Apoptosis Reversal Promotes Cancer Stem Cell-Like Cell Formation. Neoplasia 2018, 20, 295–303. [Google Scholar] [CrossRef] [PubMed]
- Algeciras-Schimnich, A.; Barnhart, B.C.; Peter, M.E. Apoptosis-independent functions of killer caspases. Curr. Opin. Cell Biol. 2002, 14, 721–726. [Google Scholar] [CrossRef]
- Shi, J.; Zhao, Y.; Wang, Y.; Gao, W.; Ding, J.; Li, P.; Hu, L.; Shao, F. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature 2014, 514, 187–192. [Google Scholar] [CrossRef]
- Li, Z.; Jo, J.; Jia, J.M.; Lo, S.C.; Whitcomb, D.J.; Jiao, S.; Cho, K.; Sheng, M. Caspase-3 activation via mitochondria is required for long-term depression and AMPA receptor internalization. Cell 2010, 141, 859–871. [Google Scholar] [CrossRef]
- Hyman, B.T.; Yuan, J. Apoptotic and non-apoptotic roles of caspases in neuronal physiology and pathophysiology. Nat. Rev. Neurosci. 2012, 13, 395–406. [Google Scholar] [CrossRef]
- Kaiser, W.J.; Upton, J.W.; Long, A.B.; Livingston-Rosanoff, D.; Daley-Bauer, L.P.; Hakem, R.; Caspary, T.; Mocarski, E.S. RIP3 mediates the embryonic lethality of caspase-8-deficient mice. Nature 2011, 471, 368–373. [Google Scholar] [CrossRef]
- De Calignon, A.; Fox, L.M.; Pitstick, R.; Carlson, G.A.; Bacskai, B.J.; Spires-Jones, T.L.; Hyman, B.T. Caspase activation precedes and leads to tangles. Nature 2010, 464, 1201–1204. [Google Scholar] [CrossRef]
- Tang, H.M.; Tang, H.L. Anastasis: Recovery from the brink of cell death. R. Soc. Open Sci. 2018, 5, 180442. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Li, F.; Liu, X.; Li, W.; Shi, W.; Liu, F.F.; O’Sullivan, B.; He, Z.; Peng, Y.; Tan, A.C.; et al. Caspase 3-mediated stimulation of tumor cell repopulation during cancer radiotherapy. Nat. Med. 2011, 17, 860–866. [Google Scholar] [CrossRef]
- Greenhough, A.; Smartt, H.J.M.; Moore, A.E.; Roberts, H.R.; Williams, A.C.; Paraskeva, C.; Kaidi, A. The COX-2/PGE2 pathway: Key roles in the hallmarks of cancer and adaptation to the tumour microenvironment. Carcinogenesis 2009, 30, 377–386. [Google Scholar] [CrossRef]
- Liu, B.; Qu, L.; Yan, S. Cyclooxygenase-2 promotes tumor growth and suppresses tumor immunity. Cancer Cell Int. 2015, 15, 106. [Google Scholar] [CrossRef]
- Borowicz, S.; Van Scoyk, M.; Avasarala, S.; Karuppusamy Rathinam, M.K.; Tauler, J.; Bikkavilli, R.K.; Winn, R.A. The soft agar colony formation assay. J. Vis. Exp. 2014, 92, 51998. [Google Scholar] [CrossRef] [PubMed]
- Berthenet, K.; Castillo Ferrer, C.; Fanfone, D.; Popgeorgiev, N.; Neves, D.; Bertolino, P.; Gibert, B.; Hernandez-Vargas, H.; Ichim, G. Failed Apoptosis Enhances Melanoma Cancer Cell Aggressiveness. Cell Rep. 2020, 31, 107731. [Google Scholar] [CrossRef] [PubMed]
- Yue, X.; Hannon, G.J.; Hannon, Z.; Zhang, H.; David, C.; Ryuji, K.; David, B. P21 Is a Universal Inhibitor of Cyclin Kinases. Nature 1993, 363, 701–704. [Google Scholar]
- Georgakilas, A.G.; Martin, O.A.; Bonner, W.M. p21: A Two-Faced Genome Guardian. Trends Mol. Med. 2017, 23, 310–319. [Google Scholar] [CrossRef] [PubMed]
- Abbas, T.; Dutta, A. P21 in cancer: Intricate networks and multiple activities. Nat. Rev. Cancer 2009, 9, 400–414. [Google Scholar] [CrossRef]
- Suzuki, A.; Tsutomi, Y.; Yamamoto, N.; Shibutani, T.; Akahane, K. Mitochondrial Regulation of Cell Death: Mitochondria Are Essential for Procaspase 3-p21 Complex Formation to Resist Fas-Mediated Cell Death. Mol. Cell. Biol. 1999, 19, 3842–3847. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, A.; Tsutomi, Y.; Akahane, K.; Araki, T.; Miura, M. Resistance to Fas-mediated apoptosis: Activation of caspase 3 is regulated by cell cycle regulator p21(WAF1) and IAP gene family ILP. Oncogene 1998, 17, 931–939. [Google Scholar] [CrossRef] [PubMed]
- Jung, Y.S.; Qian, Y.; Chen, X. Examination of the expanding pathways for the regulation of p21 expression and activity. Cell. Signal. 2010, 22, 1003–1012. [Google Scholar] [CrossRef]
- Zhang, T.; Jiang, T.; Zhang, F.; Li, C.; Zhou, Y.A.; Zhu, Y.F.; Li, X.F. Involvement of p21Waf1/Cip1 cleavage during roscovitine-induced apoptosis in non-small cell lung cancer cells. Oncol. Rep. 2010, 23, 239–245. [Google Scholar] [CrossRef] [PubMed]
- Végran, F.; Boidot, R.; Oudin, C.; Riedinger, J.M.; Bonnetain, F.; Lizard-Nacol, S. Overexpression of caspase-3s splice variant in locally advanced breast carcinoma is associated with poor response to neoadjuvant chemotherapy. Clin. Cancer Res. 2006, 12, 5794–5800. [Google Scholar] [CrossRef]
- Végran, F.; Boidot, R.; Solary, E.; Lizard-Nacol, S. A short caspase-3 isoform inhibits chemotherapy-induced apoptosis by blocking apoptosome assembly. PLoS ONE 2011, 6, e29058. [Google Scholar] [CrossRef]
- Venables, J.P. Aberrant and alternative splicing in cancer. Cancer Res. 2004, 64, 7647–7654. [Google Scholar] [CrossRef]
- Venables, J.P.; Klinck, R.; Koh, C.; Gervais-Bird, J.; Bramard, A.; Inkel, L.; Durand, M.; Couture, S.; Froehlich, U.; Lapointe, E.; et al. Cancer-associated regulation of alternative splicing. Nat. Struct. Mol. Biol. 2009, 16, 670–676. [Google Scholar] [CrossRef]
- Pinan-Lucarre, B.; Gabel, C.V.; Reina, C.P.; Hulme, S.E.; Shevkoplyas, S.S.; Slone, R.D.; Xue, J.; Qiao, Y.; Weisberg, S.; Roodhouse, K.; et al. The core apoptotic executioner proteins CED-3 and CED-4 promote initiation of neuronal regeneration in caenorhabditis elegans. PLoS Biol. 2012, 10, e1001331. [Google Scholar] [CrossRef]
- Lambie, E.J.; Conradt, B. Deadly dowry: How engulfment pathways promote cell killing. Cell Death Differ. 2016, 23, 553–554. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Mishra, N.; Wei, H.; Conradt, B. Caenorhabditis elegans ced-3 caspase is required for asymmetric divisions that generate cells programmed to die. Genetics 2018, 210, 983–998. [Google Scholar] [CrossRef] [PubMed]
- Nagata, S. Apoptotic DNA fragmentation. Exp. Cell Res. 2000, 256, 12–18. [Google Scholar] [CrossRef] [PubMed]
- Arnoult, D.; Gaume, B.; Karbowski, M.; Sharpe, J.C.; Cecconi, F.; Youle, R.J. Mitochondrial release of AIF and EndoG requires caspase activation downstream of Bax/Bak-mediated permeabilization. EMBO J. 2003, 22, 4385–4399. [Google Scholar] [CrossRef]
- D’Amelio, M.; Cavallucci, V.; Cecconi, F. Neuronal caspase-3 signaling: Not only cell death. Cell Death Differ. 2010, 17, 1104–1114. [Google Scholar] [CrossRef]
- Cifone, M.A.; Fidler, I.J. Correlation of patterns of anchorage-independent growth with in vivo behavior of cells from a murine fibrosarcoma. Proc. Natl. Acad. Sci. USA 1980, 77, 1039–1043. [Google Scholar] [CrossRef]
- Thacker, J. The RAD51 gene family, genetic instability and cancer. Cancer Lett. 2005, 219, 125–135. [Google Scholar] [CrossRef]
- Haaf, T.; Raderschall, E.; Reddy, G.; Ward, D.C.; Radding, C.M.; Golub, E.I. Sequestration of mammalian Rad51-recombination protein into micronuclei. J. Cell Biol. 1999, 144, 11–20. [Google Scholar] [CrossRef]
- Baumann, P.; Benson, F.E.; West, S.C. Human Rad51 protein promotes ATP-dependent homologous pairing and strand transfer reactions in vitro. Cell 1996, 87, 757–766. [Google Scholar] [CrossRef]
- Mekeel, K.L.; Tang, W.; Kachnic, L.A.; Luo, C.M.; DeFrank, J.S.; Powell, S.N. Inactivation of p53 results in high rates of homologous recombination. Oncogene 1997, 14, 1847–1857. [Google Scholar] [CrossRef]
- Erenpreisa, J.; Salmina, K.; Huna, A.; Kosmacek, E.A.; Cragg, M.S.; Ianzini, F.; Anisimov, A.P. Polyploid tumour cells elicit paradiploid progeny through depolyploidizing divisions and regulated autophagic degradation. Cell Biol. Int. 2011, 35, 687–695. [Google Scholar] [CrossRef]
- Rello-Varona, S.; Lissa, D.; Shen, S.; Niso-Santano, M.; Senovilla, L.; Mariño, G.; Vitale, I.; Jemaà, M.; Harper, F.; Pierron, G.; et al. Autophagic removal of micronuclei. Cell Cycle 2012, 11, 170–176. [Google Scholar] [CrossRef] [PubMed]
- Erenpreisa, J.; Huna, A.; Salmina, K.; Jackson, T.R.; Cragg, M.S. Macroautophagy-aided elimination of chromatin: Sorting of waste, sorting of fate? Autophagy 2012, 8, 1877–1881. [Google Scholar] [CrossRef]
- Olins, D.E.; Olins, A.L. Nuclear envelope-limited chromatin sheets (ELCS) and heterochromatin higher order structure. Chromosoma 2009, 118, 537–548. [Google Scholar] [CrossRef] [PubMed]
- Erenpreisa, J.; Ivanov, A.; Cragg, M.; Selivanova, G.; Illidge, T. Nuclear envelope-limited chromatin sheets are part of mitotic death. Histochem. Cell Biol. 2002, 117, 243–255. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef] [PubMed]
- Ponpuak, M.; Mandell, M.A.; Kimura, T.; Chauhan, S.; Cleyrat, C.; Deretic, V. Secretory autophagy. Curr. Opin. Cell Biol. 2015, 35, 106–116. [Google Scholar] [CrossRef]
- Kimura, T.; Jia, J.; Claude-Taupin, A.; Kumar, S.; Choi, S.W.; Gu, Y.; Mudd, M.; Dupont, N.; Jiang, S.; Peters, R.; et al. Cellular and molecular mechanism for secretory autophagy. Autophagy 2017, 13, 1084–1085. [Google Scholar] [CrossRef]
- Keulers, T.G.; Schaaf, M.B.E.; Rouschop, K.M.A. Autophagy-dependent secretion: Contribution to tumor progression. Front. Oncol. 2016, 6, 251. [Google Scholar] [CrossRef]
- Gugnoni, M.; Sancisi, V.; Manzotti, G.; Gandolfi, G.; Ciarrocchi, A. Autophagy and epithelial–mesenchymal transition: An intricate interplay in cancer. Cell Death Dis. 2016, 7, e2520. [Google Scholar] [CrossRef]
- Gong, Y.N.; Guy, C.; Olauson, H.; Becker, J.U.; Yang, M.; Fitzgerald, P.; Linkermann, A.; Green, D.R. ESCRT-III Acts Downstream of MLKL to Regulate Necroptotic Cell Death and Its Consequences. Cell 2017, 169, 286–300.e16. [Google Scholar] [CrossRef] [PubMed]
- Gong, Y.N.; Guy, C.; Crawford, J.C.; Green, D.R. Biological events and molecular signaling following MLKL activation during necroptosis. Cell Cycle 2017, 16, 1748–1760. [Google Scholar] [CrossRef] [PubMed]
- He, W.T.; Wan, H.; Hu, L.; Chen, P.; Wang, X.; Huang, Z.; Yang, Z.H.; Zhong, C.Q.; Han, J. Gasdermin D is an executor of pyroptosis and required for interleukin-1β secretion. Cell Res. 2015, 25, 1285–1298. [Google Scholar] [CrossRef] [PubMed]
- Sahoo, M.; Ceballos-Olvera, I.; Del Barrio, L.; Re, F. Role of the inflammasome, IL-1β, and IL-18 in bacterial infections. Sci. World J. 2011, 11, 2037–2050. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.M.; Tang, H.L. Cell recovery by reversal of ferroptosis. Biol. Open 2019, 8, bio043182. [Google Scholar] [CrossRef]
- Gudipaty, S.A.; Conner, C.M.; Rosenblatt, J.; Montell, D.J. Unconventional Ways to Live and Die: Cell Death and Survival in Development, Homeostasis, and Disease. Annu. Rev. Cell Dev. Biol. 2018, 34, 311–332. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.L.; Yuen, K.L.; Tang, H.M.; Fung, M.C. Reversibility of apoptosis in cancer cells. Br. J. Cancer 2009, 100, 118–122. [Google Scholar] [CrossRef]
- Tang, H.L.; Tang, H.M.; Fung, M.C.; Hardwick, J.M. In vivo Caspase tracker biosensor system for detecting anastasis and non-apoptotic caspase activity. Sci. Rep. 2015, 5, 9015. [Google Scholar] [CrossRef]
- Tang, H.M.; Fung, M.C.; Tang, H.L. Detecting anastasis in vivo by caspasetracker biosensor. J. Vis. Exp. 2018, 2018. [Google Scholar] [CrossRef] [PubMed]
- Mirzayans, R.; Andrais, B.; Murray, D. Viability assessment following anticancer treatment requires single-cell visualization. Cancers 2018, 10, 255. [Google Scholar] [CrossRef] [PubMed]
- Mirzayans, R.; Murray, D. Intratumor heterogeneity and therapy resistance: Contributions of dormancy, apoptosis reversal (Anastasis) and cell fusion to disease recurrence. Int. J. Mol. Sci. 2020, 21, 1308. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zaitceva, V.; Kopeina, G.S.; Zhivotovsky, B. Anastasis: Return Journey from Cell Death. Cancers 2021, 13, 3671. https://doi.org/10.3390/cancers13153671
Zaitceva V, Kopeina GS, Zhivotovsky B. Anastasis: Return Journey from Cell Death. Cancers. 2021; 13(15):3671. https://doi.org/10.3390/cancers13153671
Chicago/Turabian StyleZaitceva, Victoria, Gelina S. Kopeina, and Boris Zhivotovsky. 2021. "Anastasis: Return Journey from Cell Death" Cancers 13, no. 15: 3671. https://doi.org/10.3390/cancers13153671
APA StyleZaitceva, V., Kopeina, G. S., & Zhivotovsky, B. (2021). Anastasis: Return Journey from Cell Death. Cancers, 13(15), 3671. https://doi.org/10.3390/cancers13153671