Profiling Anti-Apoptotic BCL-xL Protein Expression in Glioblastoma Tumorspheres


 , and
, and
Simple Summary
Abstract
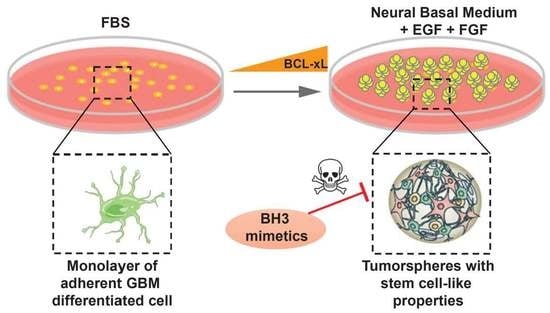
1. Introduction
2. Results
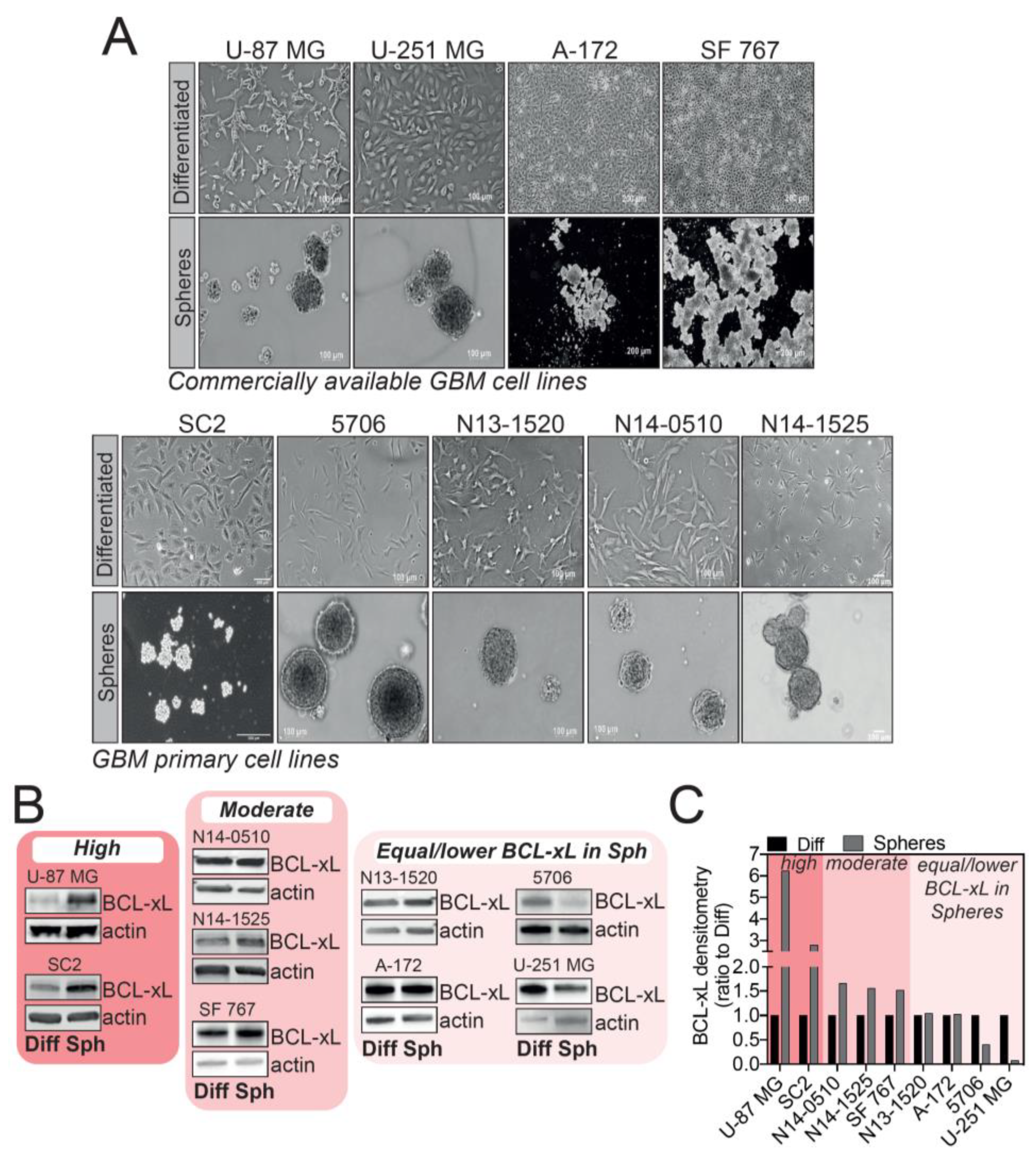
2.1. High Level of Diversity in BCL-xL Expression in Tumorspheres Compared to Differentiated GBM Cells
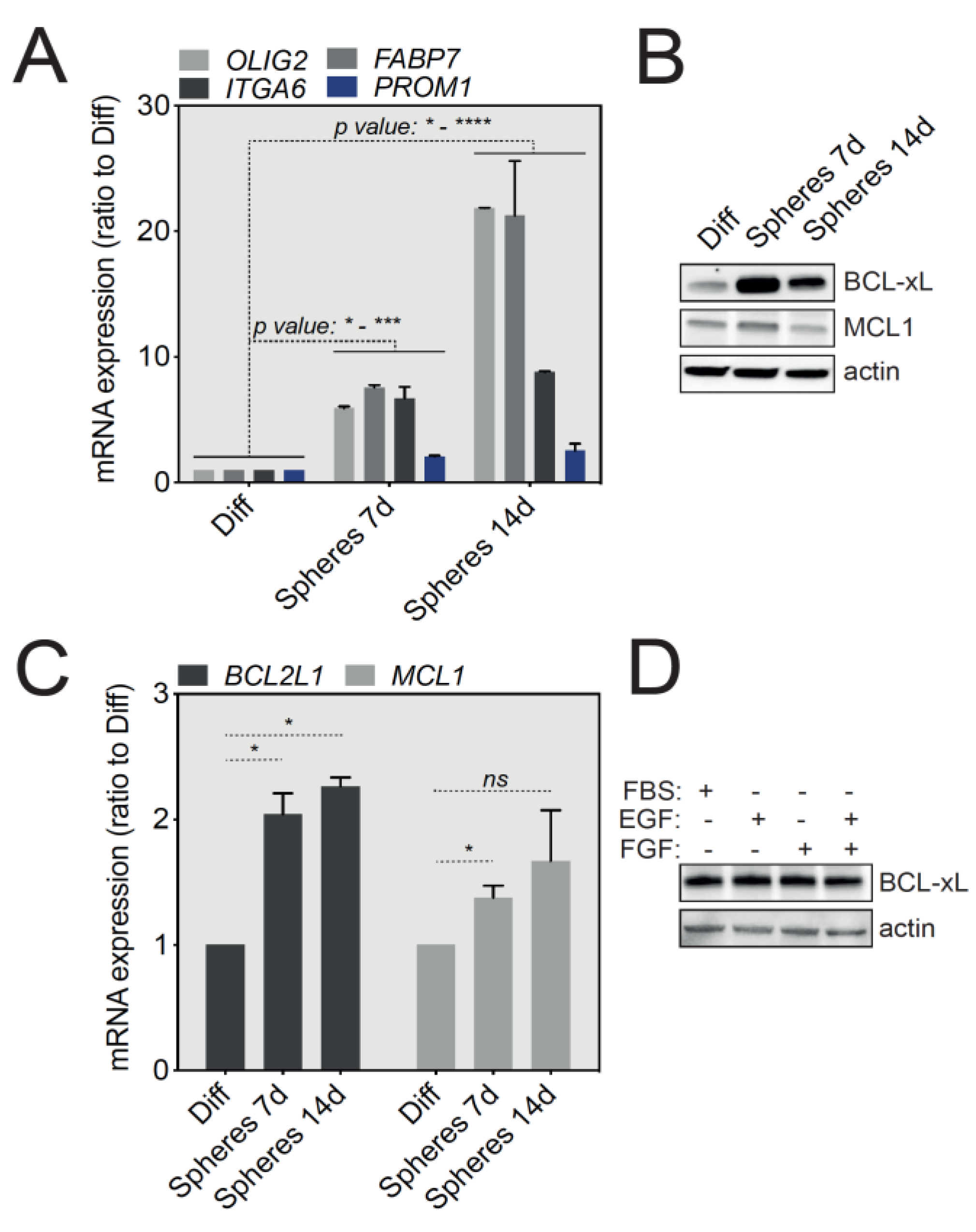
2.2. U-87 MG and SC2 GBM Cell-Derived Tumorspheres Upregulate the Anti-Apoptotic Protein BCL-xL
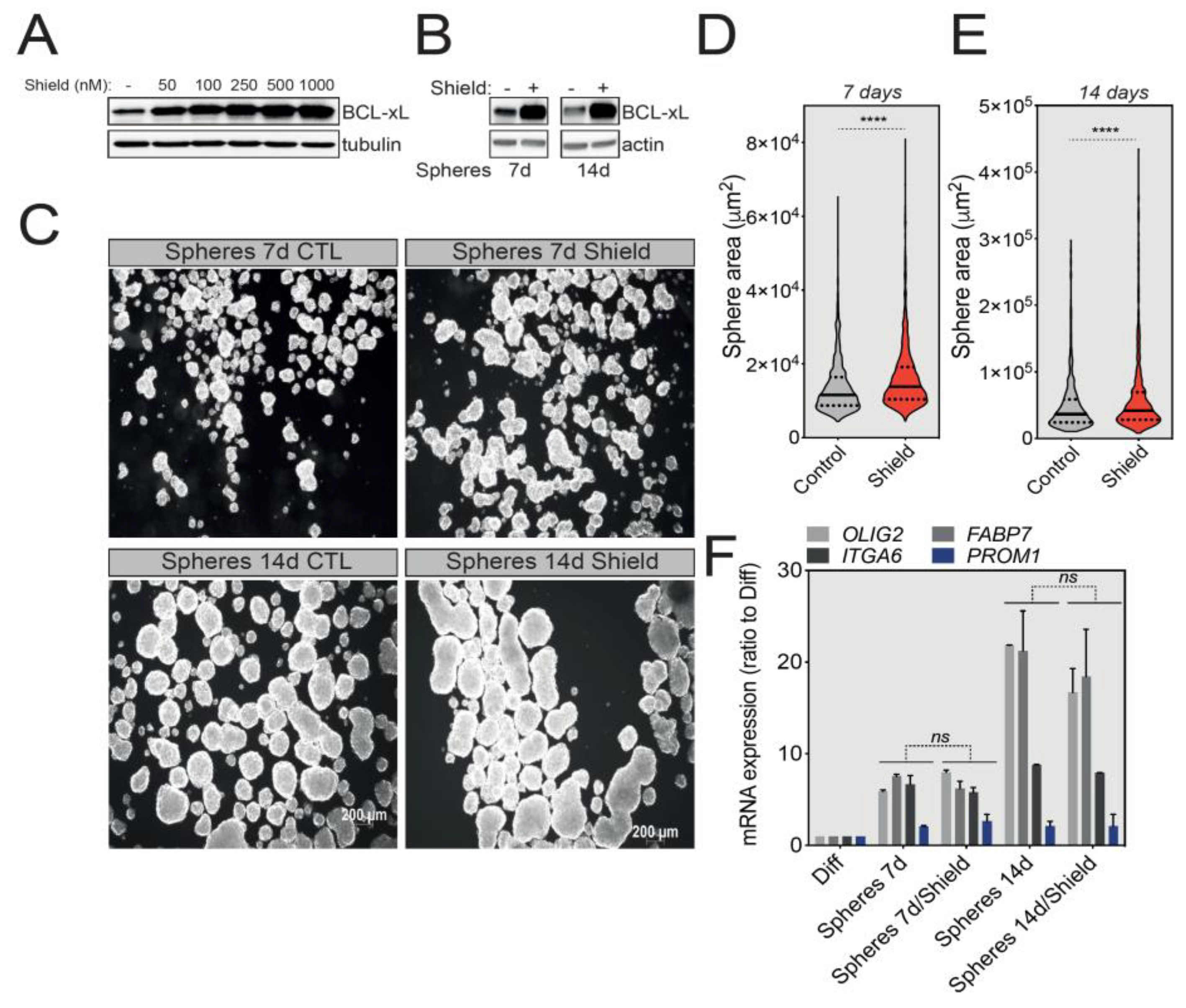
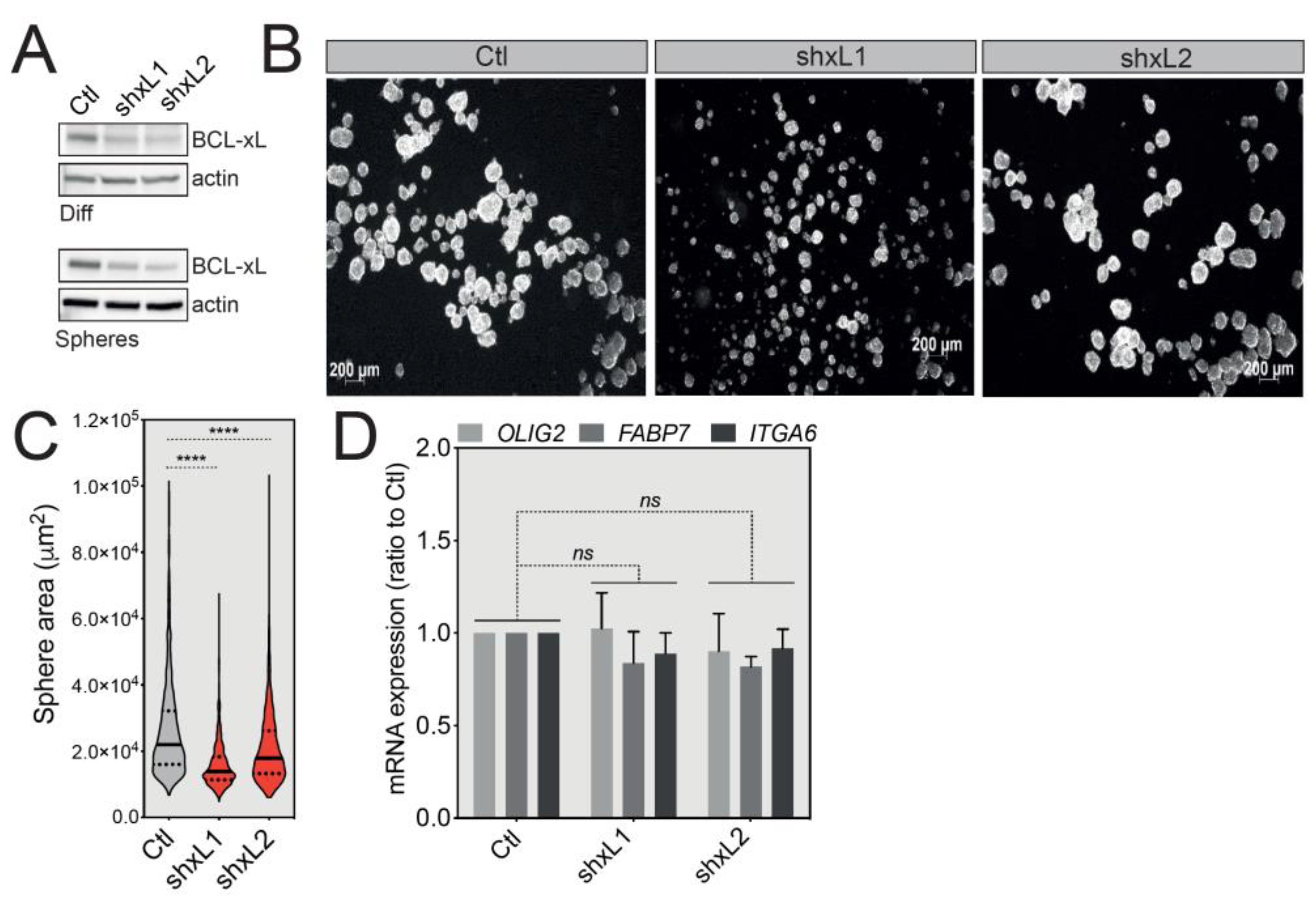
2.3. BCL-xL Regulates the Size of U-87 MG Tumorspheres
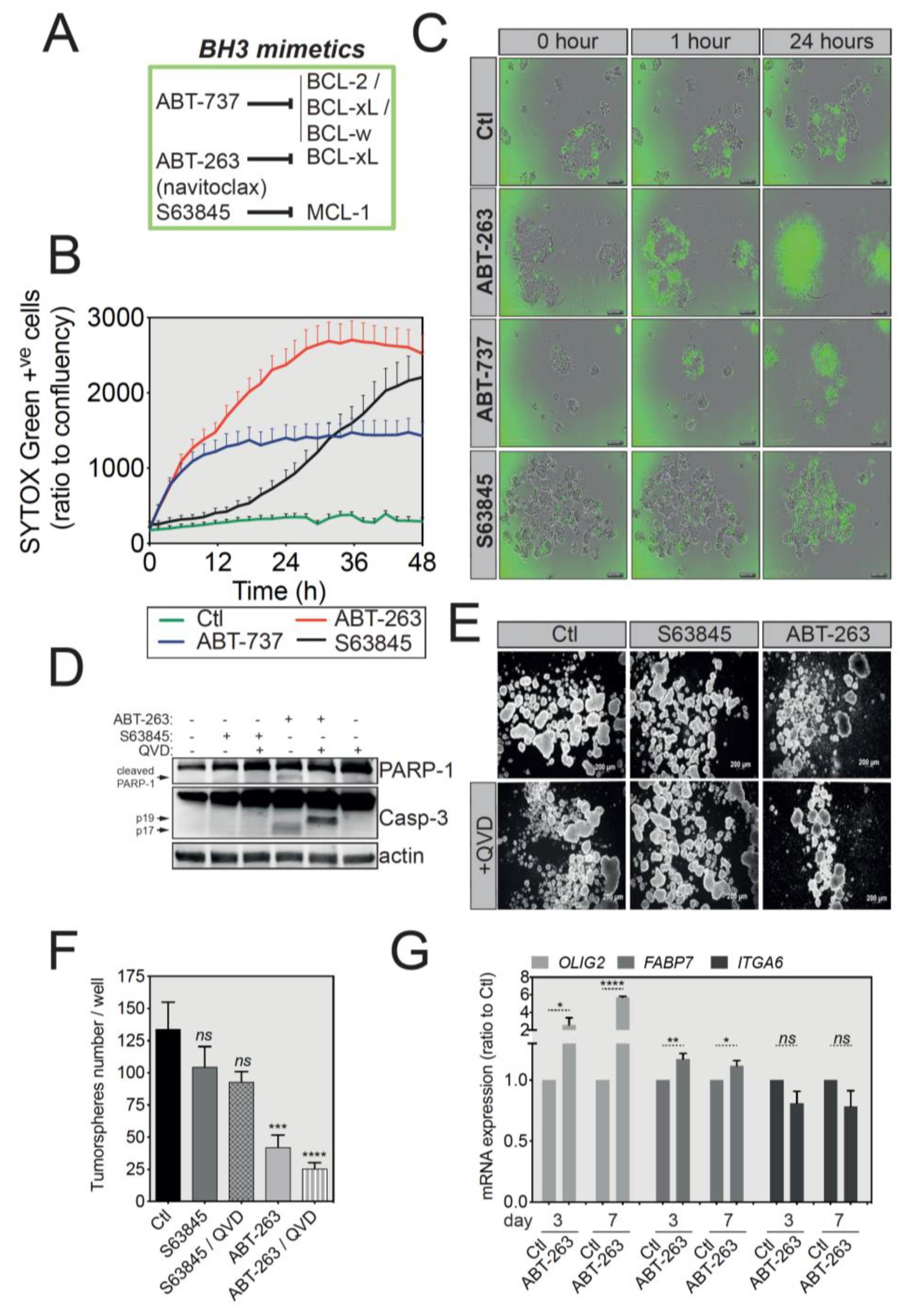
2.4. BCL-xL Upregulation Sensitizes GBM Tumorspheres to BH3 Mimetics-Induced Cell Death
3. Discussion
4. Materials and Methods
4.1. Cell Lines
4.2. Reagents
4.3. Stable Cell Line Generation
4.4. Western Blotting
4.5. Quantitative RT-PCR
4.6. Apoptosis Assay
4.7. Image Analysis
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ostrom, Q.T.; Gittleman, H.; de Blank, P.M.; Finlay, J.L.; Gurney, J.G.; McKean-Cowdin, R.; Stearns, D.S.; Wolff, J.E.; Liu, M.; Wolinsky, Y.; et al. American Brain Tumor Association Adolescent and Young Adult Primary Brain and Central Nervous System Tumors Diagnosed in the United States in 2008–2012. Neuro-Oncology 2016, 18 (Suppl. 1), i1–i50. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Hegi, M.E.; Mason, W.P.; van den Bent, M.J.; Taphoorn, M.J.; Janzer, R.C.; Ludwin, S.K.; Allgeier, A.; Fisher, B.; Belanger, K.; et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009, 10, 459–466. [Google Scholar] [CrossRef]
- Chaul-Barbosa, C.; Marques, D.F. How We Treat Recurrent Glioblastoma Today and Current Evidence. Curr. Oncol. Rep. 2019, 21, 94. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.P.; Tirosh, I.; Trombetta, J.J.; Shalek, A.K.; Gillespie, S.M.; Wakimoto, H.; Cahill, D.P.; Nahed, B.V.; Curry, W.T.; Martuza, R.L.; et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 2014, 344, 1396–1401. [Google Scholar] [CrossRef] [PubMed]
- Dirkse, A.; Golebiewska, A.; Buder, T.; Nazarov, P.V.; Muller, A.; Poovathingal, S.; Brons, N.H.C.; Leite, S.; Sauvageot, N.; Sarkisjan, D.; et al. Stem cell-associated heterogeneity in Glioblastoma results from intrinsic tumor plasticity shaped by the microenvironment. Nat. Commun. 2019, 10, 1787. [Google Scholar] [CrossRef]
- Lathia, J.D.; Mack, S.C.; Mulkearns-Hubert, E.E.; Valentim, C.L.; Rich, J.N. Cancer stem cells in glioblastoma. Genes Dev. 2015, 29, 1203–1217. [Google Scholar] [CrossRef]
- Chen, J.; Li, Y.; Yu, T.S.; McKay, R.M.; Burns, D.K.; Kernie, S.G.; Parada, L.F. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature 2012, 488, 522–526. [Google Scholar] [CrossRef]
- Pavon, L.F.; Marti, L.C.; Sibov, T.T.; Malheiros, S.M.; Brandt, R.A.; Cavalheiro, S.; Gamarra, L.F. In vitro Analysis of Neurospheres Derived from Glioblastoma Primary Culture: A Novel Methodology Paradigm. Front. Neurol. 2014, 4, 214. [Google Scholar] [CrossRef]
- Caragher, S.; Chalmers, A.J.; Gomez-Roman, N. Glioblastoma’s Next Top Model: Novel Culture Systems for Brain Cancer Radiotherapy Research. Cancers 2019, 11, 44. [Google Scholar] [CrossRef]
- Horsman, M.R.; Vaupel, P. Pathophysiological Basis for the Formation of the Tumor Microenvironment. Front. Oncol. 2016, 6, 66. [Google Scholar] [CrossRef]
- Bock, F.J.; Tait, S.W.G. Mitochondria as multifaceted regulators of cell death. Nat. Rev. Mol. Cell Biol. 2020, 21, 85–100. [Google Scholar] [CrossRef] [PubMed]
- Dickens, L.S.; Powley, I.R.; Hughes, M.A.; MacFarlane, M. The ‘complexities’ of life and death: Death receptor signalling platforms. Exp. Cell Res. 2012, 318, 1269–1277. [Google Scholar] [CrossRef] [PubMed]
- Campbell, K.J.; Tait, S.W.G. Targeting BCL-2 regulated apoptosis in cancer. Open Biol. 2018, 8, 180002. [Google Scholar] [CrossRef] [PubMed]
- Ichim, G.; Tait, S.W. A fate worse than death: Apoptosis as an oncogenic process. Nat. Rev. Cancer 2016, 16, 539–548. [Google Scholar] [CrossRef]
- Fulda, S. Cell death-based treatment of glioblastoma. Cell Death Dis. 2018, 9, 121. [Google Scholar] [CrossRef]
- Daniele, S.; Costa, B.; Zappelli, E.; Da Pozzo, E.; Sestito, S.; Nesi, G.; Campiglia, P.; Marinelli, L.; Novellino, E.; Rapposelli, S.; et al. Combined inhibition of AKT/mTOR and MDM2 enhances Glioblastoma Multiforme cell apoptosis and differentiation of cancer stem cells. Sci. Rep. 2015, 5, 9956. [Google Scholar] [CrossRef]
- Ziegler, D.S.; Wright, R.D.; Kesari, S.; Lemieux, M.E.; Tran, M.A.; Jain, M.; Zawel, L.; Kung, A.L. Resistance of human glioblastoma multiforme cells to growth factor inhibitors is overcome by blockade of inhibitor of apoptosis proteins. J. Clin. Investig. 2008, 118, 3109–3122. [Google Scholar] [CrossRef]
- Strik, H.; Deininger, M.; Streffer, J.; Grote, E.; Wickboldt, J.; Dichgans, J.; Weller, M.; Meyermann, R. BCL-2 family protein expression in initial and recurrent glioblastomas: Modulation by radiochemotherapy. J. Neurol. Neurosurg. Psychiatry 1999, 67, 763–768. [Google Scholar] [CrossRef]
- Pareja, F.; Macleod, D.; Shu, C.; Crary, J.F.; Canoll, P.D.; Ross, A.H.; Siegelin, M.D. PI3K and Bcl-2 inhibition primes glioblastoma cells to apoptosis through downregulation of Mcl-1 and Phospho-BAD. Mol. Cancer Res. 2014, 12, 987–1001. [Google Scholar] [CrossRef]
- Jiang, Z.; Zheng, X.; Rich, K.M. Down-regulation of Bcl-2 and Bcl-xL expression with bispecific antisense treatment in glioblastoma cell lines induce cell death. J. Neurochem. 2003, 84, 273–281. [Google Scholar] [CrossRef]
- Trisciuoglio, D.; Tupone, M.G.; Desideri, M.; Di Martile, M.; Gabellini, C.; Buglioni, S.; Pallocca, M.; Alessandrini, G.; D’Aguanno, S.; Del Bufalo, D. BCL-XL overexpression promotes tumor progression-associated properties. Cell Death Dis. 2017, 8, 3216. [Google Scholar] [CrossRef] [PubMed]
- Merino, D.; Kelly, G.L.; Lessene, G.; Wei, A.H.; Roberts, A.W.; Strasser, A. BH3-Mimetic Drugs: Blazing the Trail for New Cancer Medicines. Cancer Cell 2018, 34, 879–891. [Google Scholar] [CrossRef] [PubMed]
- Todt, F.; Cakir, Z.; Reichenbach, F.; Emschermann, F.; Lauterwasser, J.; Kaiser, A.; Ichim, G.; Tait, S.W.; Frank, S.; Langer, H.F.; et al. Differential retrotranslocation of mitochondrial Bax and Bak. EMBO J. 2015, 34, 67–80. [Google Scholar] [CrossRef] [PubMed]
- Mirzayans, R.; Andrais, B.; Murray, D. Viability Assessment Following Anticancer Treatment Requires Single-Cell Visualization. Cancers 2018, 10, 255. [Google Scholar] [CrossRef] [PubMed]
- Husmann, M. Vital dyes and virtual deaths. Cell Death Differ. 2013, 20, 963. [Google Scholar] [CrossRef] [PubMed]
- Tan, N.; Malek, M.; Zha, J.; Yue, P.; Kassees, R.; Berry, L.; Fairbrother, W.J.; Sampath, D.; Belmont, L.D. Navitoclax enhances the efficacy of taxanes in non-small cell lung cancer models. Clin. Cancer Res. 2011, 17, 1394–1404. [Google Scholar] [CrossRef] [PubMed]
- Wong, M.; Tan, N.; Zha, J.; Peale, F.V.; Yue, P.; Fairbrother, W.J.; Belmont, L.D. Navitoclax (ABT-263) reduces Bcl-x(L)-mediated chemoresistance in ovarian cancer models. Mol. Cancer Ther. 2012, 11, 1026–1035. [Google Scholar] [CrossRef]
- Lee, J.; Kotliarova, S.; Kotliarov, Y.; Li, A.; Su, Q.; Donin, N.M.; Pastorino, S.; Purow, B.W.; Christopher, N.; Zhang, W.; et al. Tumor stem cells derived from glioblastomas cultured in bFGF and EGF more closely mirror the phenotype and genotype of primary tumors than do serum-cultured cell lines. Cancer Cell 2006, 9, 391–403. [Google Scholar] [CrossRef]
- Pastrana, E.; Silva-Vargas, V.; Doetsch, F. Eyes wide open: A critical review of sphere-formation as an assay for stem cells. Cell Stem Cell 2011, 8, 486–498. [Google Scholar] [CrossRef]
- Chen, R.; Nishimura, M.C.; Bumbaca, S.M.; Kharbanda, S.; Forrest, W.F.; Kasman, I.M.; Greve, J.M.; Soriano, R.H.; Gilmour, L.L.; Rivers, C.S.; et al. A hierarchy of self-renewing tumor-initiating cell types in glioblastoma. Cancer Cell 2010, 17, 362–375. [Google Scholar] [CrossRef]
- Singh, S.K.; Hawkins, C.; Clarke, I.D.; Squire, J.A.; Bayani, J.; Hide, T.; Henkelman, R.M.; Cusimano, M.D.; Dirks, P.B. Identification of human brain tumour initiating cells. Nature 2004, 432, 396–401. [Google Scholar] [CrossRef] [PubMed]
- Kemper, K.; Sprick, M.R.; de Bree, M.; Scopelliti, A.; Vermeulen, L.; Hoek, M.; Zeilstra, J.; Pals, S.T.; Mehmet, H.; Stassi, G.; et al. The AC133 epitope, but not the CD133 protein, is lost upon cancer stem cell differentiation. Cancer Res. 2010, 70, 719–729. [Google Scholar] [CrossRef] [PubMed]
- Lathia, J.D.; Gallagher, J.; Heddleston, J.M.; Wang, J.; Eyler, C.E.; Macswords, J.; Wu, Q.; Vasanji, A.; McLendon, R.E.; Hjelmeland, A.B.; et al. Integrin alpha 6 regulates glioblastoma stem cells. Cell Stem Cell 2010, 6, 421–432. [Google Scholar] [CrossRef] [PubMed]
- Grad, J.M.; Zeng, X.R.; Boise, L.H. Regulation of Bcl-xL: A little bit of this and a little bit of STAT. Curr. Opin. Oncol. 2000, 12, 543–549. [Google Scholar] [CrossRef]
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef]
- Gross, A.; Katz, S.G. Non-apoptotic functions of BCL-2 family proteins. Cell Death Differ. 2017, 24, 1348–1358. [Google Scholar] [CrossRef]
- Liwak, U.; Jordan, L.E.; Von-Holt, S.D.; Singh, P.; Hanson, J.E.; Lorimer, I.A.; Roncaroli, F.; Holcik, M. Loss of PDCD4 contributes to enhanced chemoresistance in Glioblastoma multiforme through de-repression of Bcl-xL translation. Oncotarget 2013, 4, 1365–1372. [Google Scholar] [CrossRef]
- Tagscherer, K.E.; Fassl, A.; Campos, B.; Farhadi, M.; Kraemer, A.; Böck, B.C.; Macher-Goeppinger, S.; Radlwimmer, B.; Wiestler, O.D.; Herold-Mende, C.; et al. Apoptosis-based treatment of glioblastomas with ABT-737, a novel small molecule inhibitor of Bcl-2 family proteins. Oncogene 2008, 27, 6646–6656. [Google Scholar] [CrossRef]
- Tagscherer, K.E.; Fassl, A.; Sinkovic, T.; Combs, S.E.; Roth, W. p53-dependent regulation of Mcl-1 contributes to synergistic cell death by ionizing radiation and the Bcl-2/Bcl-XL inhibitor ABT-737. Apoptosis 2012, 17, 187–199. [Google Scholar] [CrossRef]
- Yang, M.C.; Loh, J.K.; Li, Y.Y.; Huang, W.S.; Chou, C.H.; Cheng, J.T.; Wang, Y.T.; Lieu, A.S.; Howng, S.L.; Hong, Y.R.; et al. Bcl2L12 with a BH3-like domain in regulating apoptosis and TMZ-induced autophagy: A prospective combination of ABT-737 and TMZ for treating glioma. Int. J. Oncol. 2015, 46, 1304–1316. [Google Scholar] [CrossRef]
- Li, F.; Huang, Q.; Chen, J.; Peng, Y.; Roop, D.R.; Bedford, J.S.; Li, C.Y. Apoptotic cells activate the “phoenix rising” pathway to promote wound healing and tissue regeneration. Sci. Signal. 2010, 3, ra13. [Google Scholar] [CrossRef] [PubMed]
- Bhola, P.D.; Ahmed, E.; Guerriero, J.L.; Sicinska, E.; Su, E.; Lavrova, E.; Ni, J.; Chipashvili, O.; Hagan, T.; Pioso, M.S.; et al. High-throughput dynamic BH3 profiling may quickly and accurately predict effective therapies in solid tumors. Sci. Signal. 2020, 13. [Google Scholar] [CrossRef] [PubMed]
- Salvucci, M.; Zakaria, Z.; Carberry, S.; Tivnan, A.; Seifert, V.; Kögel, D.; Murphy, B.M.; Prehn, J.H.M. System-based approaches as prognostic tools for glioblastoma. BMC Cancer 2019, 19, 1092. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.; Chen, Z.; Tang, L.H.; Fang, Y.; Shin, S.J.; Panarelli, N.C.; Chen, Y.T.; Li, Y.; Jiang, X.; Du, Y.N. Bcl-xL promotes metastasis independent of its anti-apoptotic activity. Nat. Commun. 2016, 7, 10384. [Google Scholar] [CrossRef] [PubMed]
- Ho, J.N.; Kang, G.Y.; Lee, S.S.; Kim, J.; Bae, I.H.; Hwang, S.G.; Um, H.D. Bcl-XL and STAT3 mediate malignant actions of gamma-irradiation in lung cancer cells. Cancer Sci. 2010, 101, 1417–1423. [Google Scholar] [CrossRef]
- Karpel-Massler, G.; Ishida, C.T.; Zhang, Y.; Halatsch, M.E.; Westhoff, M.A.; Siegelin, M.D. Targeting intrinsic apoptosis and other forms of cell death by BH3-mimetics in glioblastoma. Expert Opin. Drug Discov. 2017, 12, 1031–1040. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene of Interest | Forward Primer (5′-3′) | Reverse Primer (5′-3′) |
|---|---|---|
| BCL2L1 | AAAAGATCTTCCGGGGGCTG | TCTGAAGGGAGAGAAAGAGATTCA |
| ACTB | AGAGCTACGAGCTGCCTGAC | AGCACTGTGTTGGCGTACAG |
| OLIG2 | CCTAAAGGTGCGGATGCTTAT | ATCTGGATGCGATTTGAGGAG |
| FABP7 | AGCTGACCAACAGTCAGAAC | CCGTTGGTTTGGTCACATTTC |
| ITGA6 | TTGGACTCAGGGAAAGGTAT TG | TGCAGACTTCATGTCTCTCTTC |
| PROM1 | CCCAACATCATCCCTGTTCTT | CTGCTGCTAAGCTGTGTACTT |
| MCL1 | CCAAGAAAGCTGCATCGAACCAT | CAGCACATTCCTGATGCCACCT |
| GAPDH | TGCACCACCAACTGCTTAGC | GGCATGGACTGTGGTCATGAG |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fanfone, D.; Idbaih, A.; Mammi, J.; Gabut, M.; Ichim, G. Profiling Anti-Apoptotic BCL-xL Protein Expression in Glioblastoma Tumorspheres. Cancers 2020, 12, 2853. https://doi.org/10.3390/cancers12102853
Fanfone D, Idbaih A, Mammi J, Gabut M, Ichim G. Profiling Anti-Apoptotic BCL-xL Protein Expression in Glioblastoma Tumorspheres. Cancers. 2020; 12(10):2853. https://doi.org/10.3390/cancers12102853
Chicago/Turabian StyleFanfone, Deborah, Ahmed Idbaih, Jade Mammi, Mathieu Gabut, and Gabriel Ichim. 2020. "Profiling Anti-Apoptotic BCL-xL Protein Expression in Glioblastoma Tumorspheres" Cancers 12, no. 10: 2853. https://doi.org/10.3390/cancers12102853
APA StyleFanfone, D., Idbaih, A., Mammi, J., Gabut, M., & Ichim, G. (2020). Profiling Anti-Apoptotic BCL-xL Protein Expression in Glioblastoma Tumorspheres. Cancers, 12(10), 2853. https://doi.org/10.3390/cancers12102853