
D-Propranolol Impairs EGFR Trafficking and Destabilizes Mutant p53 Counteracting AKT Signaling and Tumor Malignancy

 , , ,
, , ,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Indirect Immunofluorescence
2.3. EGFR Endocytosis
2.4. Cell Viability and Apoptosis
2.5. Inverted Invasion Assay
2.6. Immunoprecipitation and Immunoblotting
2.7. PA Levels and PKA Activity Biosensors
2.8. TP53 Targeted Sequencing
2.9. Subcutaneous Xenografts
2.10. Statistical Methods
3. Results
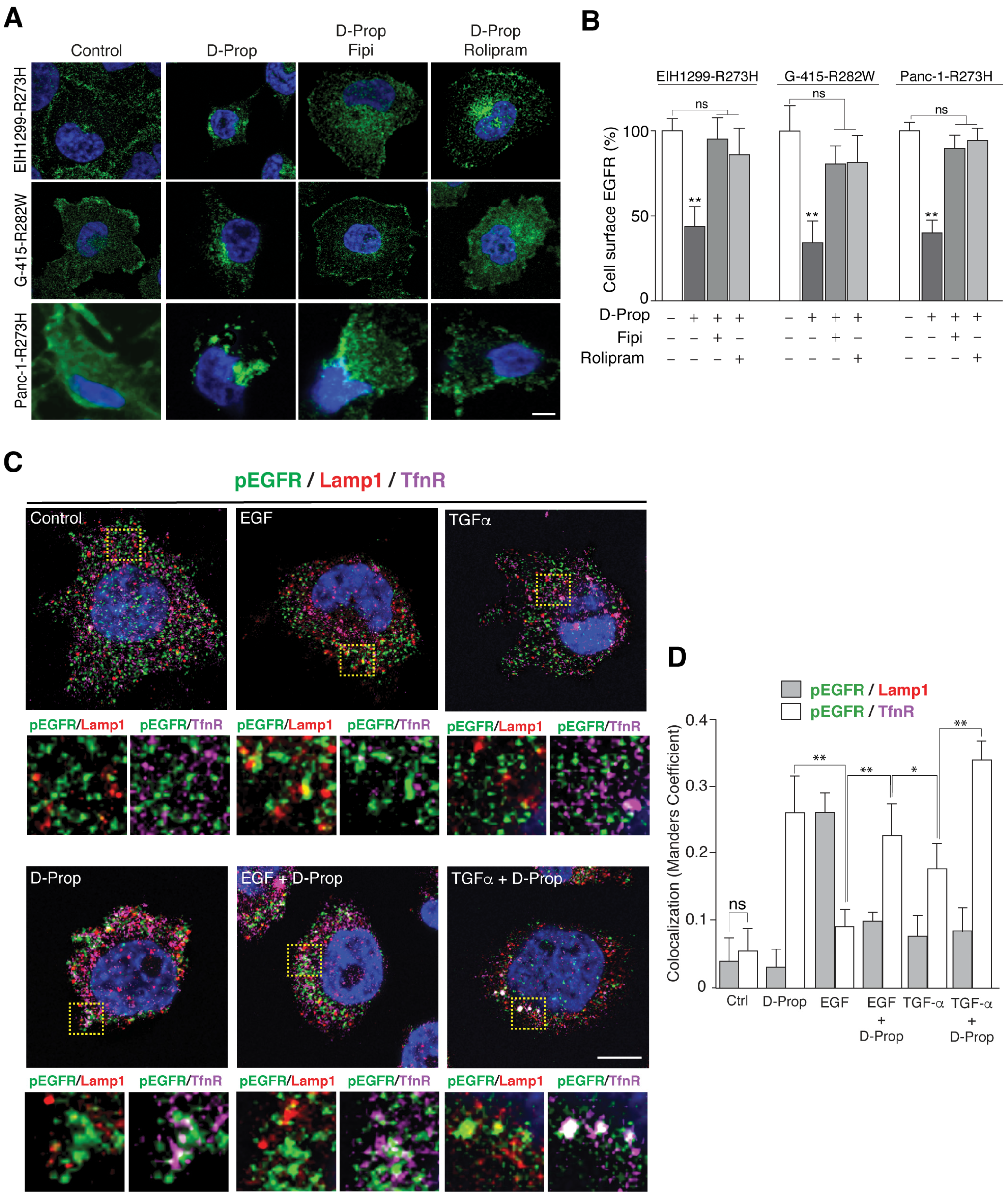
3.1. D-Prop Induces Internalization of EGFR in Tumoral Cells Expressing GOF mutp53
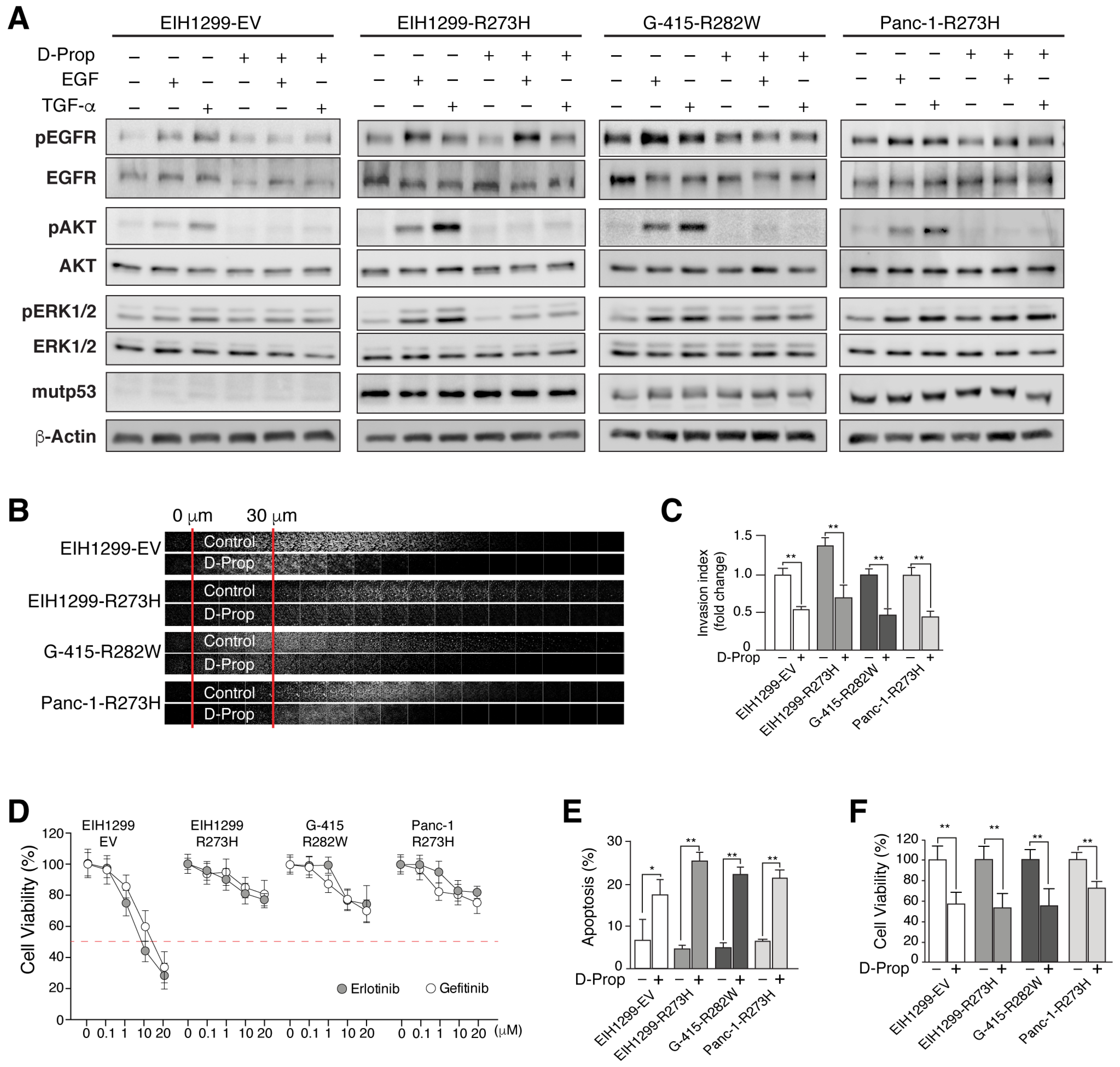
3.2. D-Prop Decreases AKT and ERK Signaling, Invasive Migration and Viability of GOF mutp53-Expressing Cells
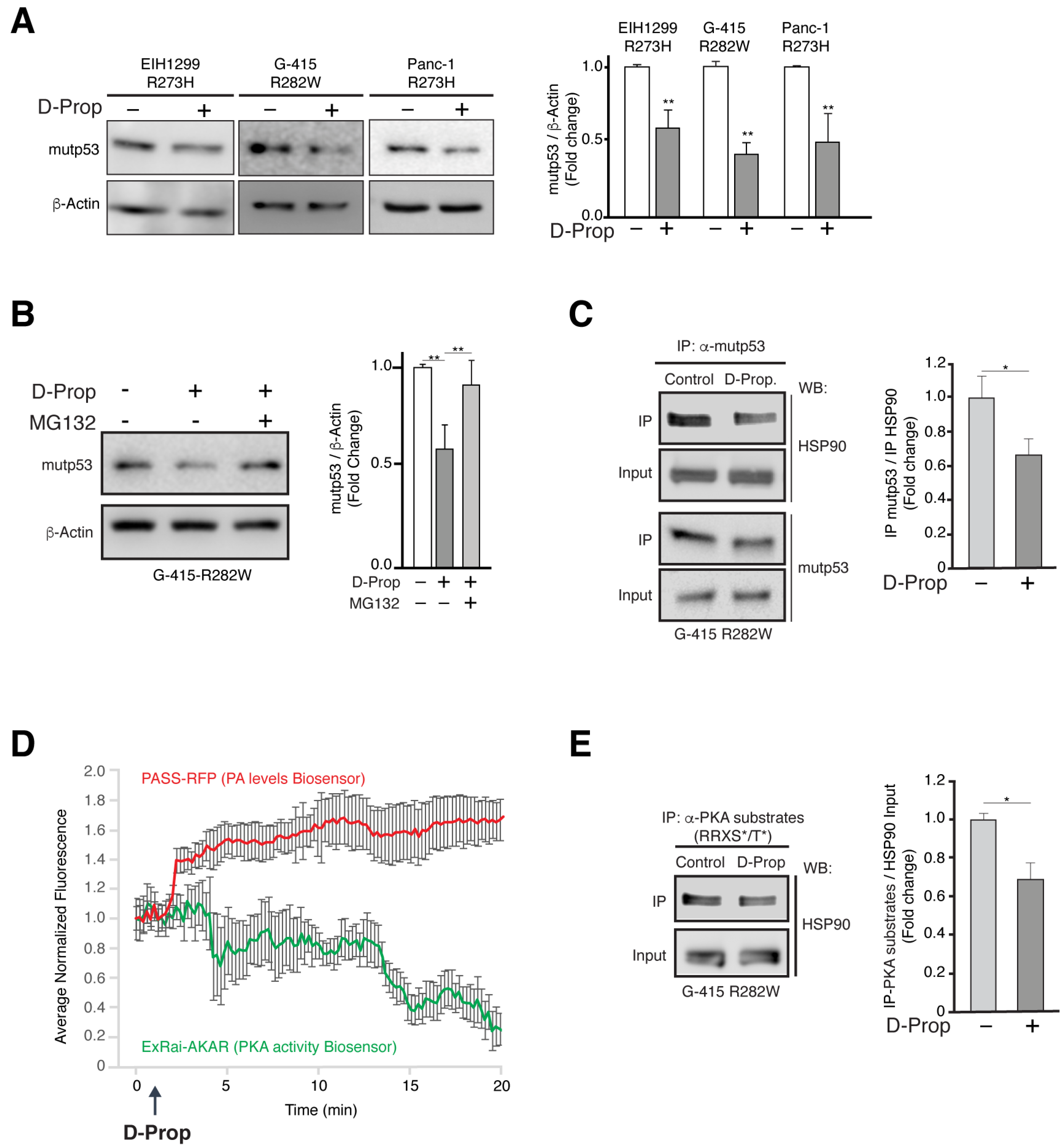
3.3. D-Prop Induces Mutp53 Proteasomal Degradation through PKA Dependent Destabilization of Mutp53-HSP90 Complex
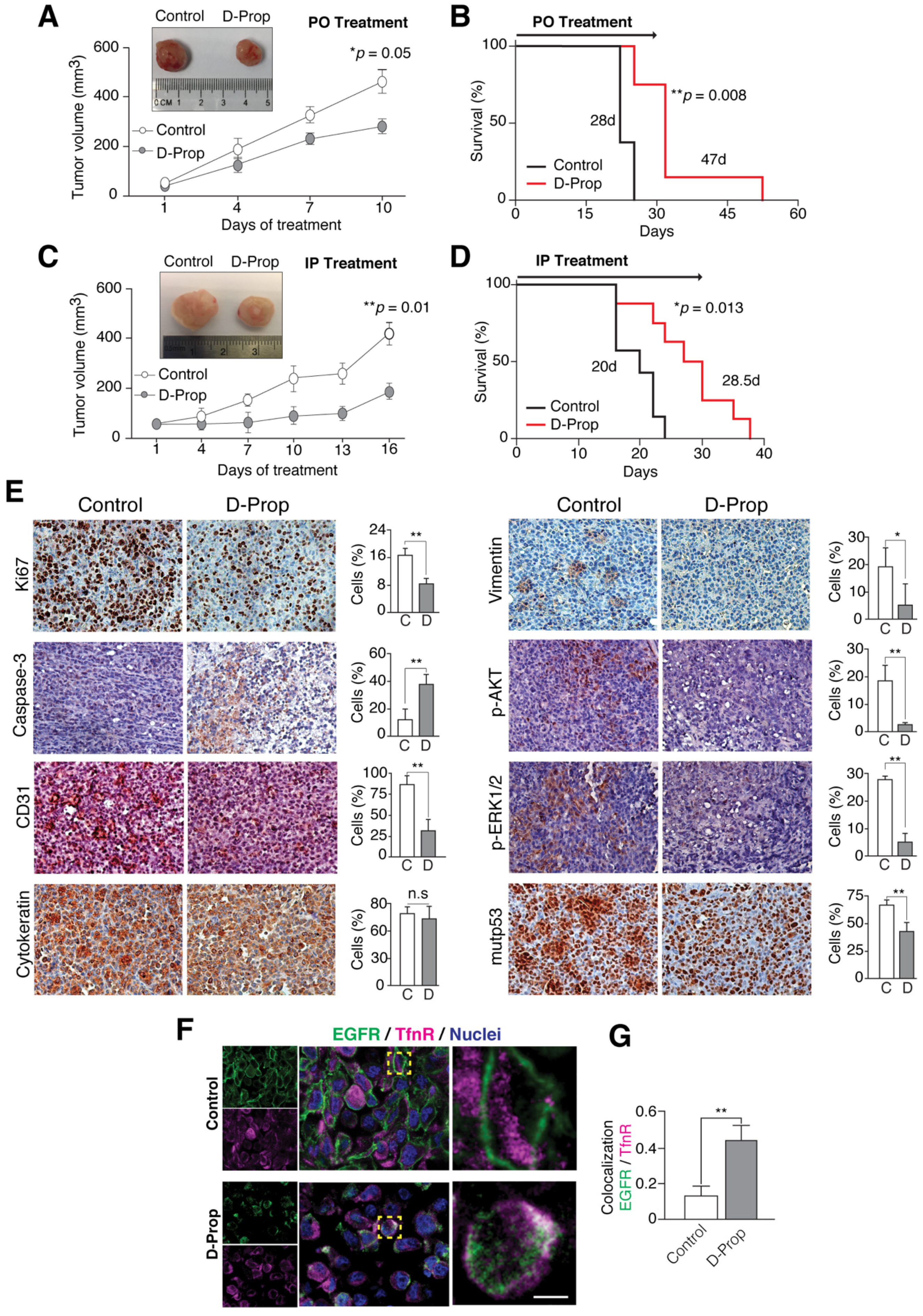
3.4. D-Prop Decreases Tumor Growth and Extends Free-Survival of G-415-R282W Xenograft-Bearing Mice
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
References
- Mendelsohn, J. Targeting the epidermal growth factor receptor for cancer therapy. J. Clin. Oncol. 2002, 20, 1S–13S. [Google Scholar] [PubMed]
- Yarden, Y.; Sliwkowski, M.X. Untangling the ErbB signalling network. Nat. Rev. Mol. Cell Biol. 2001, 2, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Sabapathy, K.; Lane, D. Therapeutic targeting of p53: All mutants are equal, but some mutants are more equal than others. Nat. Rev. Clin. Oncol. 2018, 15, 13–30. [Google Scholar] [CrossRef]
- Muller, P.A.; Vousden, K.H. Mutant p53 in Cancer: New Functions and Therapeutic Opportunities. Cancer Cell 2014, 25, 304–317. [Google Scholar] [CrossRef]
- Goh, L.K.; Sorkin, A. Endocytosis of Receptor Tyrosine Kinases. Cold Spring Harb. Perspect. Biol. 2013, 5, a017459. [Google Scholar] [CrossRef]
- Tomas, A.; Futter, C.; Eden, E.R. EGF receptor trafficking: Consequences for signaling and cancer. Trends Cell Biol. 2014, 24, 26–34. [Google Scholar] [CrossRef]
- Sigismund, S.; Avanzato, D.; Lanzetti, L. Emerging functions of the EGFR in cancer. Mol. Oncol. 2018, 12, 3–20. [Google Scholar] [CrossRef]
- Mellman, I.; Yarden, Y. Endocytosis and Cancer. Cold Spring Harb. Perspect. Biol. 2013, 5, a016949. [Google Scholar] [CrossRef]
- Di Fiore, P.P.; Von Zastrow, M. Endocytosis, Signaling, and Beyond. Cold Spring Harb. Perspect. Biol. 2014, 6, a016865. [Google Scholar] [CrossRef]
- Schulz-Heddergott, R.; Moll, U.M. Gain-of-Function (GOF) Mutant p53 as Actionable Therapeutic Target. Cancers 2018, 10, 188. [Google Scholar] [CrossRef]
- Mantovani, F.; Collavin, L.; Del Sal, G. Mutant p53 as a guardian of the cancer cell. Cell Death Differ. 2019, 26, 199–212. [Google Scholar] [CrossRef]
- Weinstein, I.B.; Joe, A.K. Mechanisms of Disease: Oncogene addiction—A rationale for molecular targeting in cancer therapy. Nat. Clin. Pr. Oncol. 2006, 3, 448–457. [Google Scholar] [CrossRef]
- Di Agostino, S.; Fontemaggi, G.; Strano, S.; Blandino, G.; D’Orazi, G. Targeting mutant p53 in cancer: The latest insights. J. Exp. Clin. Cancer Res. 2019, 38, 1–3. [Google Scholar] [CrossRef]
- Xu, M.J.; Johnson, D.E.; Grandis, J.R. EGFR-targeted therapies in the post-genomic era. Cancer Metastasis Rev. 2017, 36, 463–473. [Google Scholar] [CrossRef] [PubMed]
- Yarden, Y.; Pines, G. The ERBB network: At last, cancer therapy meets systems biology. Nat. Rev. Cancer 2012, 12, 553–563. [Google Scholar] [CrossRef] [PubMed]
- Levine, A.J. Targeting Therapies for the p53 Protein in Cancer Treatments. Annu. Rev. Cancer Biol. 2019, 3, 21–34. [Google Scholar] [CrossRef]
- Tan, X.; Lambert, P.F.; Rapraeger, A.C.; Anderson, R.A. Stress-Induced EGFR Trafficking: Mechanisms, Functions, and Therapeutic Implications. Trends Cell Biol. 2016, 26, 352–366. [Google Scholar] [CrossRef] [PubMed]
- Sigismund, S.; Scita, G. The ‘endocytic matrix reloaded’ and its impact on the plasticity of migratory strategies. Curr. Opin. Cell Biol. 2018, 54, 9–17. [Google Scholar] [CrossRef]
- Muller, P.A.J.; Caswell, P.T.; Doyle, B.; Iwanicki, M.P.; Tan, E.H.; Karim, S.; Lukashchuk, N.; Gillespie, D.A.; Ludwig, R.L.; Gosselin, P.; et al. Mutant p53 Drives Invasion by Promoting Integrin Recycling. Cell 2009, 139, 1327–1341. [Google Scholar] [CrossRef]
- Shaughnessy, R.; Retamal, C.; Oyanadel, C.; Norambuena, A.; López, A.; Bravo-Zehnder, M.; Montecino, F.J.; Metz, C.; Soza, A.; González, A. Epidermal growth factor receptor endocytic traffic perturbation by phosphatidate phosphohydrolase inhibition: New strategy against cancer. FEBS J. 2014, 281, 2172–2189. [Google Scholar] [CrossRef]
- Muller, P.A.J.; Vousden, K.H.; Norman, J.C. p53 and its mutants in tumor cell migration and invasion. J. Cell Biol. 2011, 192, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Schlessinger, J. Receptor Tyrosine Kinases: Legacy of the First Two Decades. Cold Spring Harb. Perspect. Biol. 2014, 6, a008912. [Google Scholar] [CrossRef] [PubMed]
- Schmid, S.L. Reciprocal regulation of signaling and endocytosis: Implications for the evolving cancer cell. J. Cell Biol. 2017, 216, 2623–2632. [Google Scholar] [CrossRef]
- Sigismund, S.; Argenzio, E.; Tosoni, D.; Cavallaro, E.; Polo, S.; Di Fiore, P.P. Clathrin-Mediated Internalization Is Essential for Sustained EGFR Signaling but Dispensable for Degradation. Dev. Cell 2008, 15, 209–219. [Google Scholar] [CrossRef]
- Francavilla, C.; Papetti, M.; Rigbolt, K.T.G.; Pedersen, A.-K.; Sigurdsson, J.O.; Cazzamali, G.; Karemore, G.; Blagoev, B.; Olsen, J.V. Multilayered proteomics reveals molecular switches dictating ligand-dependent EGFR trafficking. Nat. Struct. Mol. Biol. 2016, 23, 608–618. [Google Scholar] [CrossRef]
- Pinilla-Macua, I.; Grassart, A.; Duvvuri, U.; Watkins, S.C.; Sorkin, A. EGF receptor signaling, phosphorylation, ubiquitylation and endocytosis in tumors in vivo. eLife 2017, 6, e31993. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, Y.; Takiguchi, S.; Ito, S.; Itoh, K. EGF-stimulated AKT activation is mediated by EGFR recycling via an early endocytic pathway in a gefitinib-resistant human lung cancer cell line. Int. J. Oncol. 2015, 46, 1721–1729. [Google Scholar] [CrossRef]
- Lien, E.C.; Dibble, C.C.; Toker, A. PI3K signaling in cancer: Beyond AKT. Curr. Opin. Cell Biol. 2017, 45, 62–71. [Google Scholar] [CrossRef]
- Lavoie, H.; Gagnon, J.; Therrien, M. ERK signalling: A master regulator of cell behaviour, life and fate. Nat. Rev. Mol. Cell Biol. 2020, 21, 607–632. [Google Scholar] [CrossRef]
- Muller, P.; Hrstka, R.; Coomber, D.; Lane, D.; Vojtesek, B. Chaperone-dependent stabilization and degradation of p53 mutants. Oncogene 2008, 27, 3371–3383. [Google Scholar] [CrossRef]
- Wawrzynow, B.; Zylicz, A.; Zylicz, M. Chaperoning the guardian of the genome. The two-faced role of molecular chaperones in p53 tumor suppressor action. Biochim. Biophys. Acta (BBA) Bioenerg. 2018, 1869, 161–174. [Google Scholar] [CrossRef]
- Kastenhuber, E.R.; Lowe, S.W. Putting p53 in Context. Cell 2017, 170, 1062–1078. [Google Scholar] [CrossRef] [PubMed]
- Levine, A.J. p53: 800 million years of evolution and 40 years of discovery. Nat. Rev. Cancer 2020, 20, 471–480. [Google Scholar] [CrossRef] [PubMed]
- Alexandrova, E.M.; Yallowitz, A.R.; Li, D.; Xu, S.; Schulz, R.; Proia, D.A.; Lozano, G.; Dobbelstein, M.; Moll, U.M. Improving survival by exploiting tumour dependence on stabilized mutant p53 for treatment. Nat. Cell Biol. 2015, 523, 352–356. [Google Scholar] [CrossRef] [PubMed]
- Lakoduk, A.M.; Roudot, P.; Mettlen, M.; Grossman, H.M.; Schmid, S.L.; Chen, P.-H. Mutant p53 amplifies a dynamin-1/APPL1 endosome feedback loop that regulates recycling and migration. J. Cell Biol. 2019, 218, 1928–1942. [Google Scholar] [CrossRef] [PubMed]
- Norambuena, A.; Metz, C.; Jung, J.E.; Silva, A.; Otero, C.; Cancino, J.; Retamal, C.; Valenzuela, J.C.; Soza, A.; González, A. Phosphatidic Acid Induces Ligand-independent Epidermal Growth Factor Receptor Endocytic Traffic through PDE4 Activation. Mol. Biol. Cell 2010, 21, 2916–2929. [Google Scholar] [CrossRef] [PubMed]
- Grange, M.; Sette, C.; Cuomo, M.; Conti, M.; Lagarde, M.; Prigent, A.-F.; Némoz, G. The cAMP-specific Phosphodiesterase PDE4D3 Is Regulated by Phosphatidic Acid Binding. J. Biol. Chem. 2000, 275, 33379–33387. [Google Scholar] [CrossRef]
- Salazar, G.; González, A. Novel Mechanism for Regulation of Epidermal Growth Factor Receptor Endocytosis Revealed by Protein Kinase a Inhibition. Mol. Biol. Cell 2002, 13, 1677–1693. [Google Scholar] [CrossRef]
- Houslay, M.D.; Milligan, G. Tailoring cAMP-signalling responses through isoform multiplicity. Trends Biochem. Sci. 1997, 22, 217–224. [Google Scholar] [CrossRef]
- Alexander, R.W.; Williams, L.T.; Lefkowitz, R.J. Identification of cardiac beta-adrenergic receptors by (minus) [3H]alprenolol binding. Proc. Natl. Acad. Sci. USA 1975, 72, 1564–1568. [Google Scholar] [CrossRef]
- Murray, K.T.; Reilly, C.; Koshakji, R.P.; Roden, D.M.; Lineberry, M.D.; Wood, A.J.; Siddoway, L.A.; Barbey, J.T.; Woosley, R.L. Suppression of ventricular arrhythmias in man by d-propranolol independent of beta-adrenergic receptor blockade. J. Clin. Investig. 1990, 85, 836–842. [Google Scholar] [CrossRef]
- Dagar, M.; Singh, J.P.; Dagar, G.; Tyagi, R.K.; Bagchi, G. Phosphorylation of HSP90 by protein kinase A is essential for the nuclear translocation of androgen receptor. J. Biol. Chem. 2019, 294, 8699–8710. [Google Scholar] [CrossRef]
- Muller, P.A.J.; Trinidad, A.G.; Timpson, P.; Morton, J.; Zanivan, S.; Van Den Berghe, P.V.E.; Nixon, C.; Karim, S.A.; Caswell, P.T.; Noll, J.; et al. Mutant p53 enhances MET trafficking and signalling to drive cell scattering and invasion. Oncogene 2012, 32, 1252–1265. [Google Scholar] [CrossRef]
- Bolte, S.; Cordelières, F. A guided tour into subcellular colocalization analysis in light microscopy. J. Microsc. 2006, 224, 213–232. [Google Scholar] [CrossRef]
- Macanas-Pirard, P.; Broekhuizen, R.; González, A.; Oyanadel, C.; Ernst, D.M.; García, P.; Montecinos, V.P.; Court, F.; Ocqueteau, M.; Ramirez, P.; et al. Resistance of leukemia cells to cytarabine chemotherapy is mediated by bone marrow stroma, involves cell-surface equilibrative nucleoside transporter-1 removal and correlates with patient outcome. Oncotarget 2017, 8, 23073–23086. [Google Scholar] [CrossRef]
- Hennigan, R.F.; Hawker, K.L.; Ozanne, B.W.; Hennigan, R.F.; Hawker, K.L.; Ozanne, B.W. Fos-transformation activates genes associated with invasion. Oncogene 1994, 9, 3591–3600. [Google Scholar]
- Macdonald, E.; Brown, L.; Selvais, A.; Liu, H.; Waring, T.; Newman, D.; Bithell, J.; Grimes, D.; Urbé, S.; Clague, M.J.; et al. HRS–WASH axis governs actin-mediated endosomal recycling and cell invasion. J. Cell Biol. 2018, 217, 2549–2564. [Google Scholar] [CrossRef] [PubMed]
- Oyanadel, C.; Holmes, C.; Pardo, E.; Retamal, C.; Shaughnessy, R.; Smith, P.; Cortés, P.; Bravo-Zehnder, M.; Metz, C.; Feuerhake, T.; et al. Galectin-8 induces partial epithelial–mesenchymal transition with invasive tumorigenic capabilities involving a FAK/EGFR/proteasome pathway in Madin–Darby canine kidney cells. Mol. Biol. Cell 2018, 29, 557–574. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Wang, Z.; Lu, M.; Yonekubo, Y.; Liang, X.; Zhang, Y.; Wu, P.; Zhou, Y.; Grinstein, S.; Hancock, J.; et al. Temporal Production of the Signaling Lipid Phosphatidic Acid by Phospholipase D2 Determines the Output of Extracellular Signal-Regulated Kinase Signaling in Cancer Cells. Mol. Cell. Biol. 2014, 34, 84–95. [Google Scholar] [CrossRef]
- Mehta, S.; Zhang, Y.; Roth, R.H.; Zhang, J.-F.; Mo, A.; Tenner, B.; Huganir, R.L. Single-fluorophore biosensors for sensitive and multiplexed detection of signalling activities. Nat. Cell Biol. 2018, 20, 1215–1225. [Google Scholar] [CrossRef] [PubMed]
- Bernier, M.; Catazaro, J.; Singh, N.S.; Wnorowski, A.; Boguszewska-Czubara, A.; Jozwiak, K.; Powers, R.; Wainer, I.W. GPR55 receptor antagonist decreases glycolytic activity in PANC-1 pancreatic cancer cell line and tumor xenografts. Int. J. Cancer 2017, 141, 2131–2142. [Google Scholar] [CrossRef]
- Weber, H.; Leal, P.; Stein, S.; Kunkel, H.; García, P.; Bizama, C.; Espinoza, J.; Riquelme, I.; Nervi, B.; Araya-Orostica, J.; et al. Rapamycin and WYE-354 suppress human gallbladder cancer xenografts in mice. Oncotarget 2015, 6, 31877–31888. [Google Scholar] [CrossRef]
- Baugh, E.H.; Ke, H.; Levine, A.J.; Bonneau, R.A.; Chan, C.S. Why are there hotspot mutations in the TP53 gene in human cancers? Cell Death Differ. 2018, 25, 154–160. [Google Scholar] [CrossRef] [PubMed]
- Walerych, D.; Lisek, K.; Del Sal, G. Mutant p53: One, No One, and One Hundred Thousand. Front. Oncol. 2015, 5, 289. [Google Scholar] [CrossRef] [PubMed]
- Muller, P.A.J.; Vousden, K.H. p53 mutations in cancer. Nat. Cell Biol. 2013, 15, 2–8. [Google Scholar] [CrossRef] [PubMed]
- Iaea, D.B.; Maxfield, F.R. Membrane order in the plasma membrane and endocytic recycling compartment. PLoS ONE 2017, 12, e0188041. [Google Scholar] [CrossRef][Green Version]
- Maxfield, F.R.; McGraw, T.E. Endocytic recycling. Nat. Rev. Mol. Cell Biol. 2004, 5, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Yamashiro, D.; Tycko, B.; Fluss, S.R.; Maxfield, F. Segregation of transferrin to a mildly acidic (pH 6.5) para-golgi compartment in the recycling pathway. Cell 1984, 37, 789–800. [Google Scholar] [CrossRef]
- Sheff, D.R.; Daro, E.A.; Hull, M.; Mellman, I. The Receptor Recycling Pathway Contains Two Distinct Populations of Early Endosomes with Different Sorting Functions. J. Cell Biol. 1999, 145, 123–139. [Google Scholar] [CrossRef]
- Ullrich, O.; Reinsch, S.; Urbe, S.; Zerial, M.; Parton, R. Rab11 regulates recycling through the pericentriolar recycling endosome. J. Cell Biol. 1996, 135, 913–924. [Google Scholar] [CrossRef] [PubMed]
- Ren, M.; Xu, G.; Zeng, J.; De Lemos-Chiarandini, C.; Adesnik, M.; Sabatini, D.D. Hydrolysis of GTP on rab11 is required for the direct delivery of transferrin from the pericentriolar recycling compartment to the cell surface but not from sorting endosomes. Proc. Natl. Acad. Sci. USA 1998, 95, 6187–6192. [Google Scholar] [CrossRef]
- Campa, C.C.; Margaria, J.P.; Derle, A.; Del Giudice, M.; De Santis, M.C.; Gozzelino, L.; Copperi, F.; Bosia, C.; Hirsch, E. Rab11 activity and PtdIns(3)P turnover removes recycling cargo from endosomes. Nat. Chem. Biol. 2018, 14, 801–810. [Google Scholar] [CrossRef]
- Canale, M.; Petracci, E.; Delmonte, A.; Chiadini, E.; Dazzi, C.; Papi, M.; Capelli, L.; Casanova, C.; De Luigi, N.; Mariotti, M.; et al. Impact of TP53 Mutations on Outcome in EGFR-Mutated Patients Treated with First-Line Tyrosine Kinase Inhibitors. Clin. Cancer Res. 2017, 23, 2195–2202. [Google Scholar] [CrossRef]
- Lindner, W.; Rath, M.; Stoschitzky, K.; Semmelrock, H.J. Pharmacokinetic data of propranolol enantiomers in a comparative human study with (S)- and (R,S)-propranolol. Chirality 1989, 1, 10–13. [Google Scholar] [CrossRef]
- Prakash, C.; Koshakji, R.P.; Wood, A.J.J.; Blair, I.A. Simultaneous Determination of Propranolol Enantiomers in Plasma by High-Performance Liquid Chromatography with FIuorescence Detection. J. Pharm. Sci. 1989, 78, 771–775. [Google Scholar] [CrossRef] [PubMed]
- Mills, G.B.; Jurisica, I.; Yarden, Y.; Norman, J.C. Genomic amplicons target vesicle recycling in breast cancer. J. Clin. Investig. 2009, 119, 2123–2127. [Google Scholar] [CrossRef] [PubMed]
- Caswell, P.T.; Chan, M.; Lindsay, A.; McCaffrey, M.W.; Boettiger, D.; Norman, J.C. Rab-coupling protein coordinates recycling of α5β1 integrin and EGFR1 to promote cell migration in 3D microenvironments. J. Cell Biol. 2008, 183, 143–155. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Liu, X.; Datta, A.; Govindarajan, K.; Tam, W.L.; Han, J.; George, J.; Wong, C.; Ramnarayanan, K.; Phua, T.Y.; et al. RCP is a human breast cancer–promoting gene with Ras-activating function. J. Clin. Investig. 2009, 119, 2171–2183. [Google Scholar] [CrossRef]
- Cheng, K.W.; Lahad, J.P.; Kuo, W.-L.; Lapuk, A.; Yamada, K.; Auersperg, N.; Liu, J.; Smith-McCune, K.; Lu, K.H.; Fishman, D.; et al. The RAB25 small GTPase determines aggressiveness of ovarian and breast cancers. Nat. Med. 2004, 10, 1251–1256. [Google Scholar] [CrossRef]
- Caswell, P.T.; Vadrevu, S.; Norman, J.C. Integrins: Masters and slaves of endocytic transport. Nat. Rev. Mol. Cell Biol. 2009, 10, 843–853. [Google Scholar] [CrossRef]
- Jacquemet, G.; Green, D.M.; Bridgewater, R.E.; von Kriegsheim, A.; Humphries, M.J.; Norman, J.C.; Caswell, P.T. RCP-driven alpha5beta1 recycling suppresses Rac and promotes RhoA activity via the RacGAP1-IQGAP1 complex. J. Cell Biol. 2013, 202, 917–935. [Google Scholar] [CrossRef]
- Rush, J.S.; Quinalty, L.M.; Engelman, L.; Sherry, D.M.; Ceresa, B.P. Endosomal Accumulation of the Activated Epidermal Growth Factor Receptor (EGFR) Induces Apoptosis. J. Biol. Chem. 2012, 287, 712–722. [Google Scholar] [CrossRef]
- Li, D.; Marchenko, N.; Moll, U.M. SAHA shows preferential cytotoxicity in mutant p53 cancer cells by destabilizing mutant p53 through inhibition of the HDAC6-Hsp90 chaperone axis. Cell Death Differ. 2011, 18, 1904–1913. [Google Scholar] [CrossRef]
- Abdel-Rahman, O.; Elsayed, Z.; Elhalawani, H. Gemcitabine-based chemotherapy for advanced biliary tract carcinomas. Cochrane Database Syst. Rev. 2018, 4, 011746. [Google Scholar] [CrossRef]
- Shaffer, E.; Hundal, R. Gallbladder cancer: Epidemiology and outcome. Clin. Epidemiol. 2014, 6, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Overman, J.; Fontaine, F.; Wylie-Sears, J.; Moustaqil, M.; Huang, L.; Meurer, M.; Chiang, I.K.; Lesieur, E.; Patel, J.; Zuegg, J.; et al. R-propranolol is a small molecule inhibitor of the SOX18 transcription factor in a rare vascular syndrome and hemangioma. eLife 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- Pandey, A.; Stawiski, E.W.; Durinck, S.; Gowda, H.; Goldstein, L.D.; Barbhuiya, M.; Schröder, M.S.; Sreenivasamurthy, S.K.; Kim, S.-W.; Phalke, S.; et al. Integrated genomic analysis reveals mutated ELF3 as a potential gallbladder cancer vaccine candidate. Nat. Commun. 2020, 11, 4225. [Google Scholar] [CrossRef] [PubMed]
- Wardell, C.; Fujita, M.; Yamada, T.; Simbolo, M.; Fassan, M.; Karlic, R.; Polak, P.; Kim, J.; Hatanaka, Y.; Maejima, K.; et al. Genomic characterization of biliary tract cancers identifies driver genes and predisposing mutations. J. Hepatol. 2018, 68, 959–969. [Google Scholar] [CrossRef] [PubMed]
- Howe, R.; Shanks, R.G. Optical Isomers of Propranolol. Nat. Cell Biol. 1966, 210, 1336–1338. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Cao, X. Beta-Adrenergic Signaling in Tumor Immunology and Immunotherapy. Crit. Rev. Immunol. 2019, 39, 93–103. [Google Scholar] [CrossRef] [PubMed]
- Phadke, S.; Clamon, G. Beta blockade as adjunctive breast cancer therapy: A review. Crit. Rev. Oncol. 2019, 138, 173–177. [Google Scholar] [CrossRef] [PubMed]
- De Giorgi, V.; Grazzini, M.; Benemei, S.; Marchionni, N.; Botteri, E.; Pennacchioli, E.; Geppetti, P.; Gandini, S. Propranolol for Off-label Treatment of Patients with Melanoma. JAMA Oncol. 2018, 4, e172908. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Zhou, L.; Xie, N.; Nice, E.C.; Zhang, T.; Cui, Y.; Huang, C. Overcoming cancer therapeutic bottleneck by drug repurposing. Signal Transduct. Target. Ther. 2020, 5, 113. [Google Scholar] [CrossRef]
- Montecinos, V.P.; Godoy, A.; Hinklin, J.; Vethanayagam, R.R.; Smith, G.J. Primary Xenografts of Human Prostate Tissue as a Model to Study Angiogenesis Induced by Reactive Stroma. PLoS ONE 2012, 7, e29623. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barra, J.; Cerda-Infante, J.; Sandoval, L.; Gajardo-Meneses, P.; Henriquez, J.F.; Labarca, M.; Metz, C.; Venegas, J.; Retamal, C.; Oyanadel, C.; et al. D-Propranolol Impairs EGFR Trafficking and Destabilizes Mutant p53 Counteracting AKT Signaling and Tumor Malignancy. Cancers 2021, 13, 3622. https://doi.org/10.3390/cancers13143622
Barra J, Cerda-Infante J, Sandoval L, Gajardo-Meneses P, Henriquez JF, Labarca M, Metz C, Venegas J, Retamal C, Oyanadel C, et al. D-Propranolol Impairs EGFR Trafficking and Destabilizes Mutant p53 Counteracting AKT Signaling and Tumor Malignancy. Cancers. 2021; 13(14):3622. https://doi.org/10.3390/cancers13143622
Chicago/Turabian StyleBarra, Jonathan, Javier Cerda-Infante, Lisette Sandoval, Patricia Gajardo-Meneses, Jenny F. Henriquez, Mariana Labarca, Claudia Metz, Jaime Venegas, Claudio Retamal, Claudia Oyanadel, and et al. 2021. "D-Propranolol Impairs EGFR Trafficking and Destabilizes Mutant p53 Counteracting AKT Signaling and Tumor Malignancy" Cancers 13, no. 14: 3622. https://doi.org/10.3390/cancers13143622
APA StyleBarra, J., Cerda-Infante, J., Sandoval, L., Gajardo-Meneses, P., Henriquez, J. F., Labarca, M., Metz, C., Venegas, J., Retamal, C., Oyanadel, C., Cancino, J., Soza, A., Cuello, M. A., Roa, J. C., Montecinos, V. P., & Gonzalez, A. (2021). D-Propranolol Impairs EGFR Trafficking and Destabilizes Mutant p53 Counteracting AKT Signaling and Tumor Malignancy. Cancers, 13(14), 3622. https://doi.org/10.3390/cancers13143622