
Lineage Plasticity in Cancer: The Tale of a Skin-Walker

 and
and 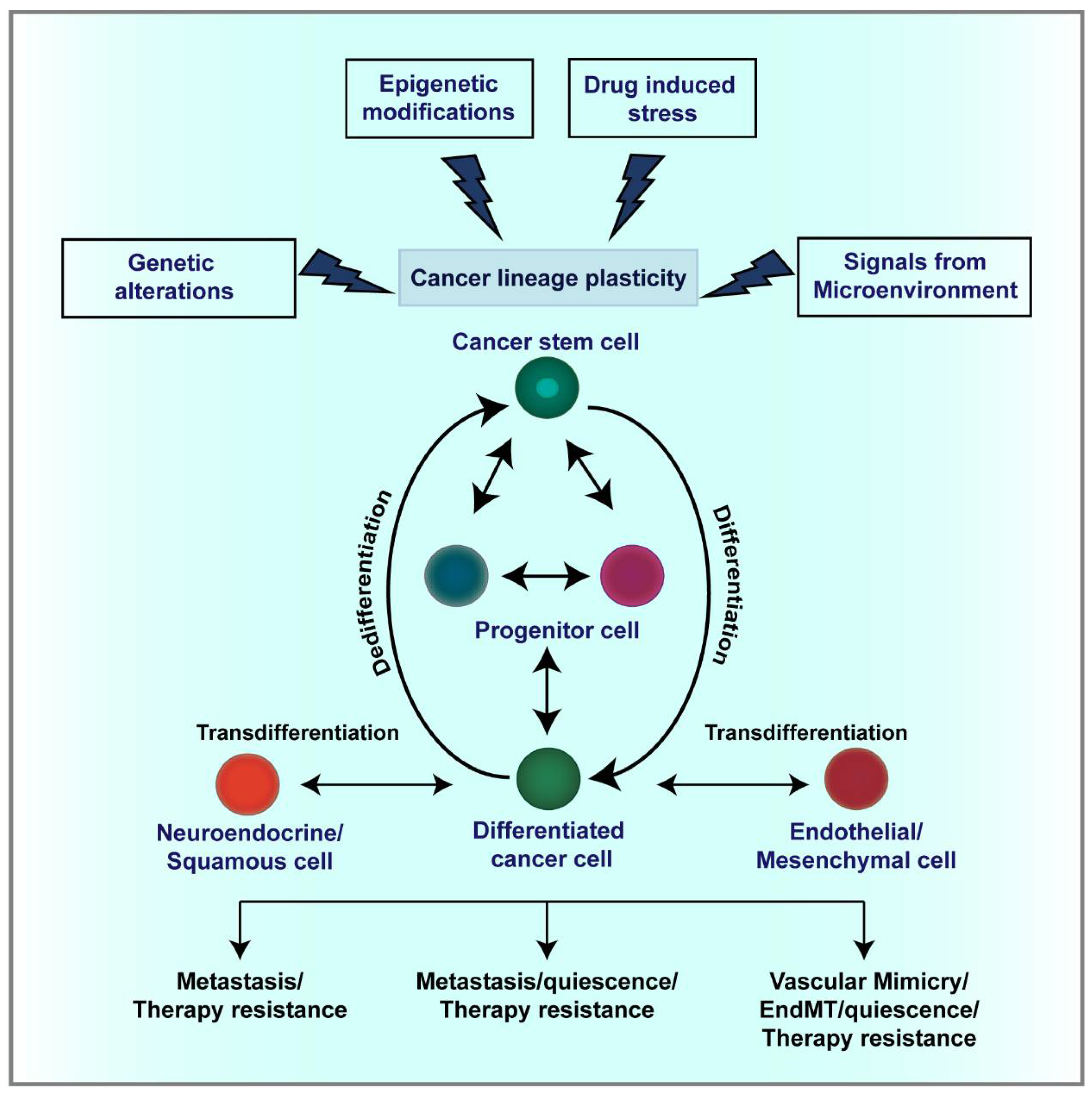{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Lineage Plasticity in Normal Development and Tissue Repair
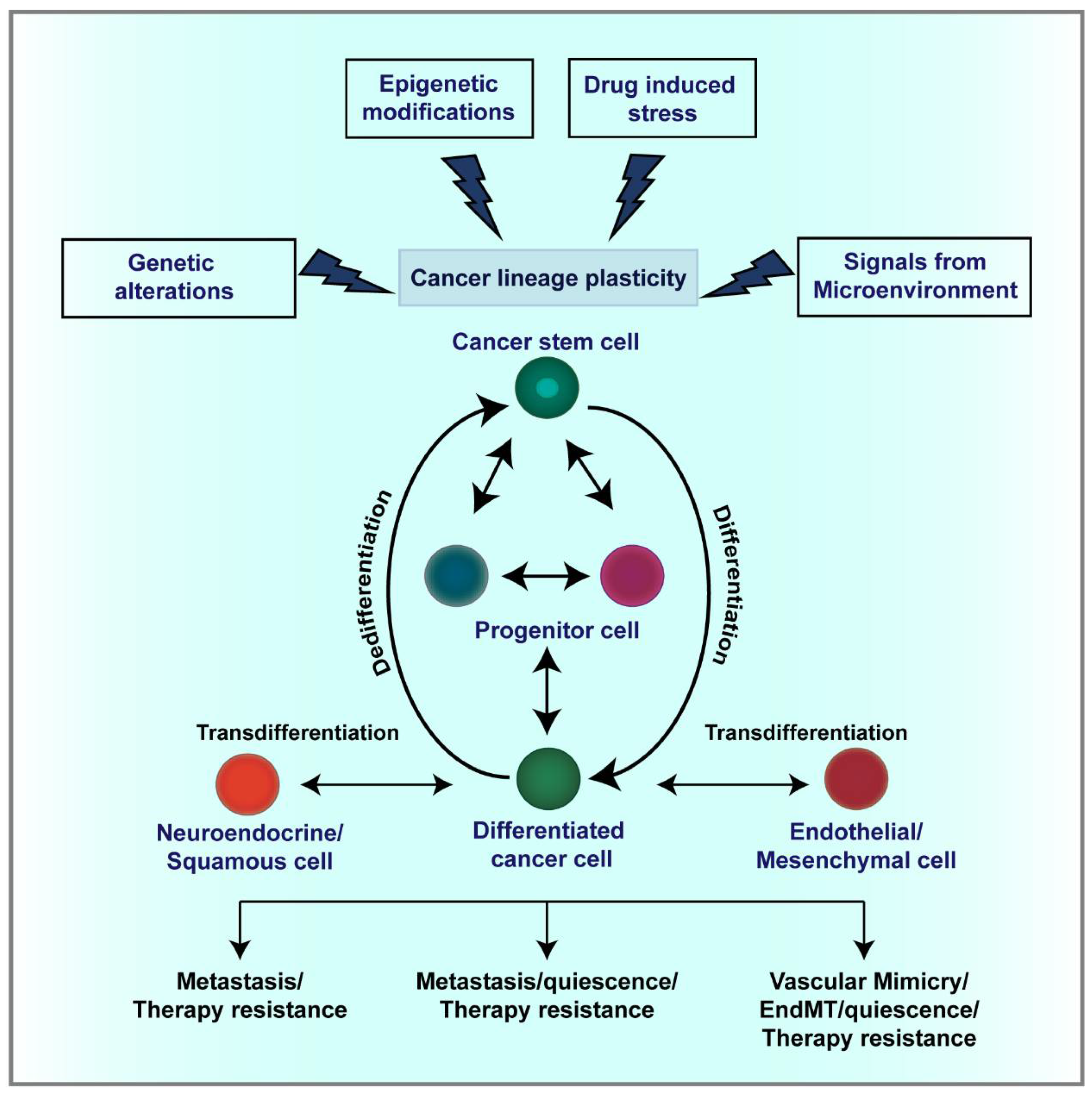
3. Cellular and Lineage Plasticity in Cancer
3.1. CSC Plasticity
3.2. Endothelial Trans-Differentiation and Vasculogenic Mimicry
3.3. Epithelial-Mesenchymal Plasticity
3.4. Lineage Plasticity in Leukemia
3.5. Lineage Plasticity and Therapeutic Resistance
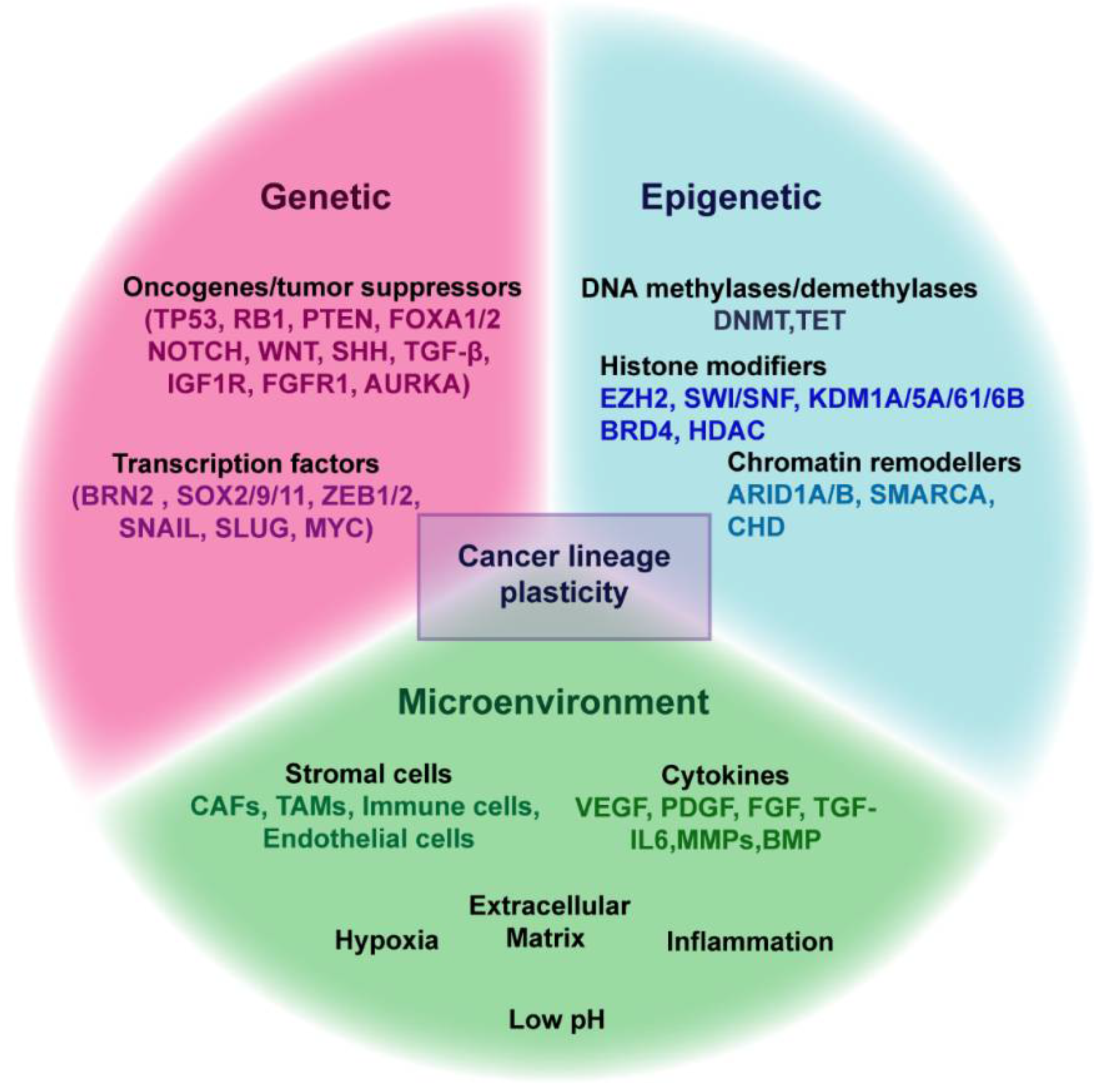
4. Mechanisms Regulating Lineage Plasticity in Cancer
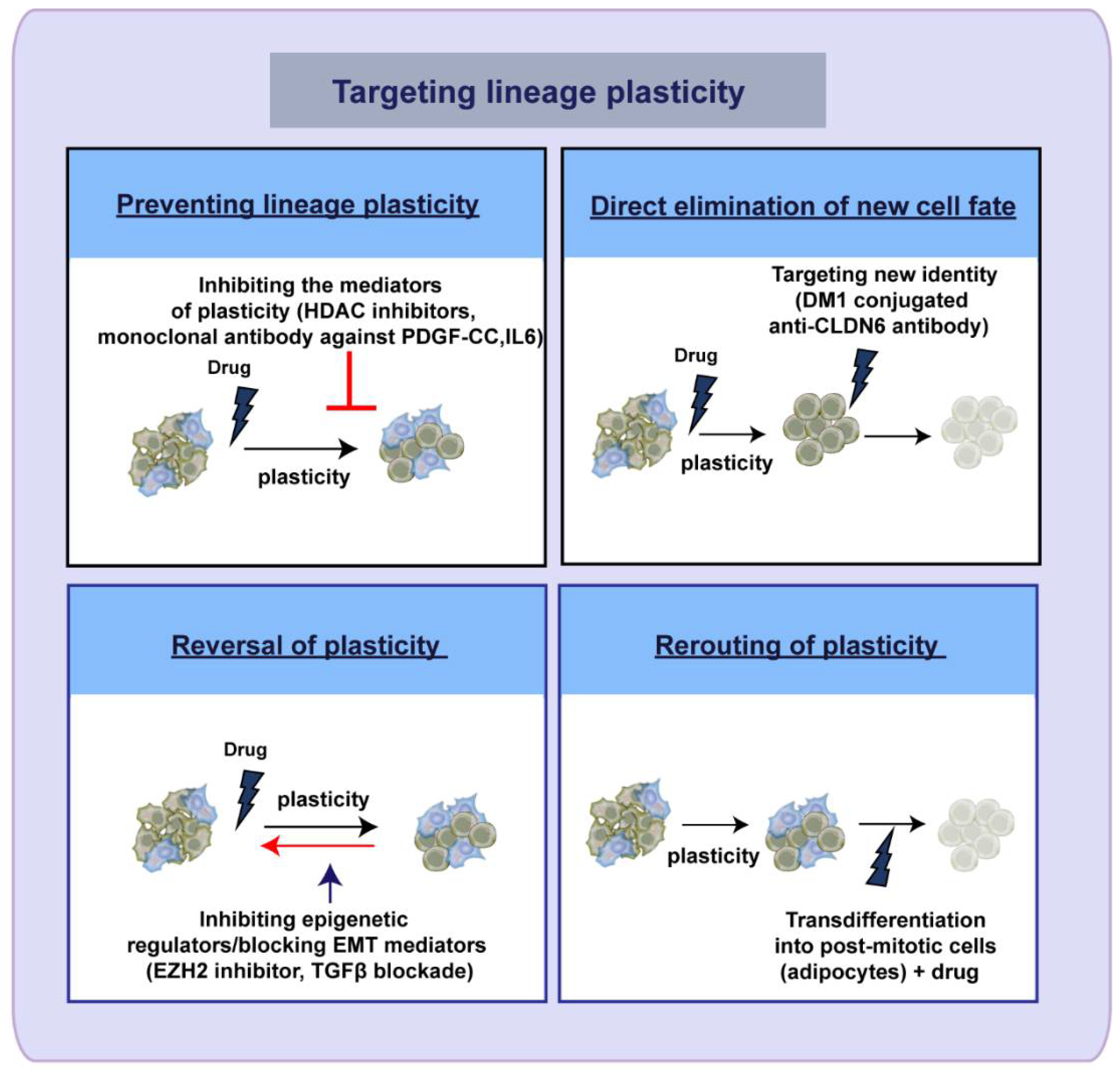
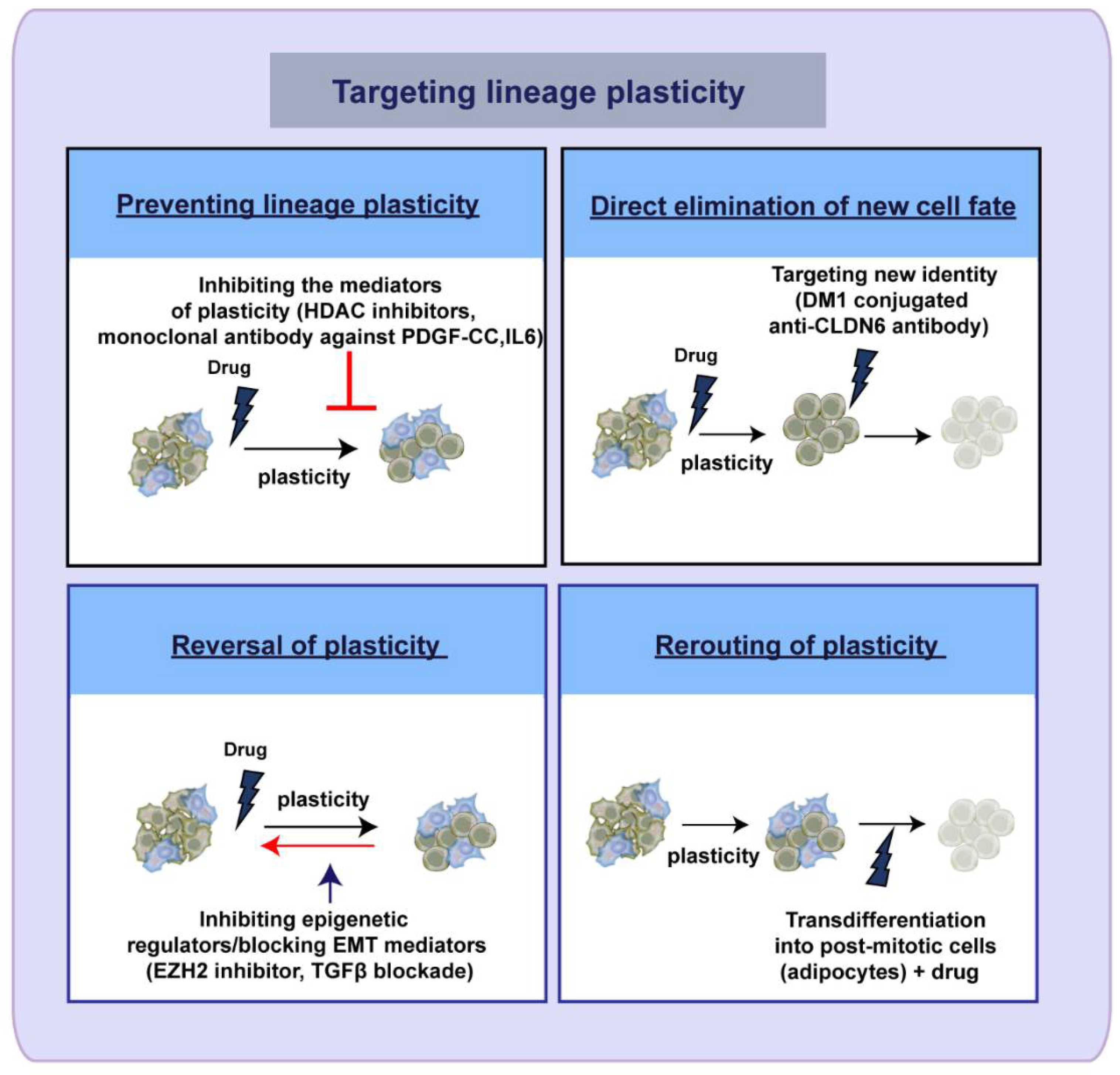
5. Therapeutic Targeting of Lineage Plasticity—Taming the Shape Shifter
Author Contributions
Funding
Conflicts of Interest
References
- Sebe-Pedros, A.; Chomsky, E.; Pang, K.; Lara-Astiaso, D.; Gaiti, F.; Mukamel, Z.; Amit, I.; Hejnol, A.; Degnan, B.M.; Tanay, A. Early metazoan cell type diversity and the evolution of multicellular gene regulation. Nat. Ecol. Evol. 2018, 2, 1176–1188. [Google Scholar] [CrossRef]
- Le Magnen, C.; Shen, M.M.; Abate-Shen, C. Lineage Plasticity in Cancer Progression and Treatment. Annu. Rev. Cancer Biol. 2018, 2, 271–289. [Google Scholar] [CrossRef]
- Waddington, C.H. The Strategy of the Genes: A Discussion of Some Aspects of Theoretical Biology; Allen & Unwin: London, UK, 1957. [Google Scholar]
- Jopling, C.; Boue, S.; Izpisua Belmonte, J.C. Dedifferentiation, transdifferentiation and reprogramming: Three routes to regeneration. Nat. Rev. Mol. Cell Biol. 2011, 12, 79–89. [Google Scholar] [CrossRef]
- Merrell, A.J.; Stanger, B.Z. Adult cell plasticity in vivo: De-differentiation and transdifferentiation are back in style. Nat. Rev. Mol. Cell Biol. 2016, 17, 413–425. [Google Scholar] [CrossRef] [PubMed]
- Varga, J.; Greten, F.R. Cell plasticity in epithelial homeostasis and tumorigenesis. Nat. Cell Biol. 2017, 19, 1133–1141. [Google Scholar] [CrossRef] [PubMed]
- Wei, G.; Schubiger, G.; Harder, F.; Muller, A.M. Stem cell plasticity in mammals and transdetermination in Drosophila: Common themes? Stem Cells 2000, 18, 409–414. [Google Scholar] [CrossRef] [PubMed]
- Hadorn, E.; Gsell, R.; Schultz, J. Stability of a position-effect variegation in normal and transdetermined larval blastemas from Drosophila melanogaster. Proc. Natl. Acad. Sci. USA 1970, 65, 633–637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, C.N.; Burke, Z.D.; Tosh, D. Transdifferentiation, metaplasia and tissue regeneration. Organogenesis 2004, 1, 36–44. [Google Scholar] [CrossRef] [Green Version]
- Pesaresi, M.; Sebastian-Perez, R.; Cosma, M.P. Dedifferentiation, transdifferentiation and cell fusion: In vivo reprogramming strategies for regenerative medicine. FEBS J. 2019, 286, 1074–1093. [Google Scholar] [CrossRef] [Green Version]
- Shenoy, S. Cell plasticity in cancer: A complex interplay of genetic, epigenetic mechanisms and tumor micro-environment. Surg. Oncol. 2020, 34, 154–162. [Google Scholar] [CrossRef]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gurdon, J.B. Adult frogs derived from the nuclei of single somatic cells. Dev. Biol. 1962, 4, 256–273. [Google Scholar] [CrossRef]
- Davis, R.L.; Weintraub, H.; Lassar, A.B. Expression of a single transfected cDNA converts fibroblasts to myoblasts. Cell 1987, 51, 987–1000. [Google Scholar] [CrossRef]
- Johnston, R.J., Jr.; Desplan, C. Stochastic mechanisms of cell fate specification that yield random or robust outcomes. Annu. Rev. Cell Dev. Biol. 2010, 26, 689–719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tata, P.R.; Rajagopal, J. Cellular plasticity: 1712 to the present day. Curr. Opin. Cell Biol. 2016, 43, 46–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flavahan, W.A.; Gaskell, E.; Bernstein, B.E. Epigenetic plasticity and the hallmarks of cancer. Science 2017, 357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwon, O.J.; Zhang, L.; Ittmann, M.M.; Xin, L. Prostatic inflammation enhances basal-to-luminal differentiation and accelerates initiation of prostate cancer with a basal cell origin. Proc. Natl. Acad. Sci. USA 2014, 111, E592–E600. [Google Scholar] [CrossRef] [Green Version]
- Sahoo, S.; Mishra, A.; Diehl, A.M.; Jolly, M.K. Dynamics of a hepatocyte-cholangiocyte decision-making gene regulatory network during liver development and regeneration. Biorxiv Prepr 2021. Available online: https://www.biorxiv.org/content/10.1101/2021.04.22.440352v1 (accessed on 25 April 2021).
- Deng, X.; Zhang, X.; Li, W.; Feng, R.X.; Li, L.; Yi, G.R.; Zhang, X.N.; Yin, C.; Yu, H.Y.; Zhang, J.P.; et al. Chronic Liver Injury Induces Conversion of Biliary Epithelial Cells into Hepatocytes. Cell Stem Cell 2018, 23, 114–122.e3. [Google Scholar] [CrossRef] [Green Version]
- Tarlow, B.D.; Pelz, C.; Naugler, W.E.; Wakefield, L.; Wilson, E.M.; Finegold, M.J.; Grompe, M. Bipotential adult liver progenitors are derived from chronically injured mature hepatocytes. Cell Stem Cell 2014, 15, 605–618. [Google Scholar] [CrossRef] [Green Version]
- Thorel, F.; Nepote, V.; Avril, I.; Kohno, K.; Desgraz, R.; Chera, S.; Herrera, P.L. Conversion of adult pancreatic alpha-cells to beta-cells after extreme beta-cell loss. Nature 2010, 464, 1149–1154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chera, S.; Baronnier, D.; Ghila, L.; Cigliola, V.; Jensen, J.N.; Gu, G.; Furuyama, K.; Thorel, F.; Gribble, F.M.; Reimann, F.; et al. Diabetes recovery by age-dependent conversion of pancreatic delta-cells into insulin producers. Nature 2014, 514, 503–507. [Google Scholar] [CrossRef] [PubMed]
- van Es, J.H.; Sato, T.; van de Wetering, M.; Lyubimova, A.; Yee Nee, A.N.; Gregorieff, A.; Sasaki, N.; Zeinstra, L.; van den Born, M.; Korving, J.; et al. Dll1+ secretory progenitor cells revert to stem cells upon crypt damage. Nat. Cell Biol. 2012, 14, 1099–1104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ninche, N.; Kwak, M.; Ghazizadeh, S. Diverse epithelial cell populations contribute to the regeneration of secretory units in injured salivary glands. Development 2020, 147. [Google Scholar] [CrossRef]
- Ge, Y.; Fuchs, E. Stretching the limits: From homeostasis to stem cell plasticity in wound healing and cancer. Nat. Rev. Genet. 2018, 19, 311–325. [Google Scholar] [CrossRef]
- Blanpain, C.; Fuchs, E. Stem cell plasticity. Plasticity of epithelial stem cells in tissue regeneration. Science 2014, 344, 1242281. [Google Scholar] [CrossRef] [Green Version]
- Christin, J.R.; Wang, C.; Chung, C.Y.; Liu, Y.; Dravis, C.; Tang, W.; Oktay, M.H.; Wahl, G.M.; Guo, W. Stem Cell Determinant SOX9 Promotes Lineage Plasticity and Progression in Basal-like Breast Cancer. Cell Rep. 2020, 31, 107742. [Google Scholar] [CrossRef]
- Kusoglu, A.; Biray Avci, C. Cancer stem cells: A brief review of the current status. Gene 2019, 681, 80–85. [Google Scholar] [CrossRef]
- Thankamony, A.P.; Saxena, K.; Murali, R.; Jolly, M.K.; Nair, R. Cancer Stem Cell Plasticity—A Deadly Deal. Front. Mol. Biosci. 2020, 7, 79. [Google Scholar] [CrossRef]
- Chaffer, C.L.; Brueckmann, I.; Scheel, C.; Kaestli, A.J.; Wiggins, P.A.; Rodrigues, L.O.; Brooks, M.; Reinhardt, F.; Su, Y.; Polyak, K.; et al. Normal and neoplastic nonstem cells can spontaneously convert to a stem-like state. Proc. Natl. Acad. Sci. USA 2011, 108, 7950–7955. [Google Scholar] [CrossRef] [Green Version]
- Gupta, P.B.; Pastushenko, I.; Skibinski, A.; Blanpain, C.; Kuperwasser, C. Phenotypic Plasticity: Driver of Cancer Initiation, Progression, and Therapy Resistance. Cell Stem Cell 2019, 24, 65–78. [Google Scholar] [CrossRef] [Green Version]
- Chaffer, C.L.; Marjanovic, N.D.; Lee, T.; Bell, G.; Kleer, C.G.; Reinhardt, F.; D’Alessio, A.C.; Young, R.A.; Weinberg, R.A. Poised chromatin at the ZEB1 promoter enables breast cancer cell plasticity and enhances tumorigenicity. Cell 2013, 154, 61–74. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Cong, Y.; Wang, D.; Sun, Y.; Deng, L.; Liu, Y.; Martin-Trevino, R.; Shang, L.; McDermott, S.P.; Landis, M.D.; et al. Breast cancer stem cells transition between epithelial and mesenchymal states reflective of their normal counterparts. Stem Cell Rep. 2014, 2, 78–91. [Google Scholar] [CrossRef]
- Wang, J.; Sakariassen, P.O.; Tsinkalovsky, O.; Immervoll, H.; Boe, S.O.; Svendsen, A.; Prestegarden, L.; Rosland, G.; Thorsen, F.; Stuhr, L.; et al. CD133 negative glioma cells form tumors in nude rats and give rise to CD133 positive cells. Int. J. Cancer 2008, 122, 761–768. [Google Scholar] [CrossRef]
- Dirkse, A.; Golebiewska, A.; Buder, T.; Nazarov, P.V.; Muller, A.; Poovathingal, S.; Brons, N.H.C.; Leite, S.; Sauvageot, N.; Sarkisjan, D.; et al. Stem cell-associated heterogeneity in Glioblastoma results from intrinsic tumor plasticity shaped by the microenvironment. Nat. Commun. 2019, 10, 1787. [Google Scholar] [CrossRef] [PubMed]
- Roesch, A.; Fukunaga-Kalabis, M.; Schmidt, E.C.; Zabierowski, S.E.; Brafford, P.A.; Vultur, A.; Basu, D.; Gimotty, P.; Vogt, T.; Herlyn, M. A temporarily distinct subpopulation of slow-cycling melanoma cells is required for continuous tumor growth. Cell 2010, 141, 583–594. [Google Scholar] [CrossRef] [Green Version]
- Quintana, E.; Shackleton, M.; Foster, H.R.; Fullen, D.R.; Sabel, M.S.; Johnson, T.M.; Morrison, S.J. Phenotypic heterogeneity among tumorigenic melanoma cells from patients that is reversible and not hierarchically organized. Cancer Cell 2010, 18, 510–523. [Google Scholar] [CrossRef] [Green Version]
- Poli, V.; Fagnocchi, L.; Zippo, A. Tumorigenic Cell Reprogramming and Cancer Plasticity: Interplay between Signaling, Microenvironment, and Epigenetics. Stem Cells Int. 2018, 2018, 4598195. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Dong, J.; Haiech, J.; Kilhoffer, M.C.; Zeniou, M. Cancer Stem Cell Quiescence and Plasticity as Major Challenges in Cancer Therapy. Stem Cells Int. 2016, 2016, 1740936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vinogradova, T.V.; Chernov, I.P.; Monastyrskaya, G.S.; Kondratyeva, L.G.; Sverdlov, E.D. Cancer Stem Cells: Plasticity Works against Therapy. Acta Nat. 2015, 7, 46–55. [Google Scholar] [CrossRef]
- Das, P.K.; Pillai, S.; Rakib, M.A.; Khanam, J.A.; Gopalan, V.; Lam, A.K.Y.; Islam, F. Plasticity of Cancer Stem Cell: Origin and Role in Disease Progression and Therapy Resistance. Stem Cell Rev. Rep. 2020, 16, 397–412. [Google Scholar] [CrossRef]
- Eguizabal, C.; Montserrat, N.; Veiga, A.; Izpisua Belmonte, J.C. Dedifferentiation, transdifferentiation, and reprogramming: Future directions in regenerative medicine. Semin. Reprod. Med. 2013, 31, 82–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, B.; Zhang, D.; Zhao, N.; Zhao, X. Epithelial-to-endothelial transition and cancer stem cells: Two cornerstones of vasculogenic mimicry in malignant tumors. Oncotarget 2017, 8, 30502–30510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, H.; Liu, W.; Liu, X.; Li, Z.; Feng, T.; Xue, Y.; Liu, Y. Advances and Prospects of Vasculogenic Mimicry in Glioma: A Potential New Therapeutic Target? Oncotargets Ther. 2020, 13, 4473–4483. [Google Scholar] [CrossRef] [PubMed]
- Luo, Q.; Wang, J.; Zhao, W.; Peng, Z.; Liu, X.; Li, B.; Zhang, H.; Shan, B.; Zhang, C.; Duan, C. Vasculogenic mimicry in carcinogenesis and clinical applications. J. Hematol. Oncol. 2020, 13, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angara, K.; Borin, T.F.; Arbab, A.S. Vascular Mimicry: A Novel Neovascularization Mechanism Driving Anti-Angiogenic Therapy (AAT) Resistance in Glioblastoma. Transl. Oncol. 2017, 10, 650–660. [Google Scholar] [CrossRef] [PubMed]
- Maniotis, A.J.; Folberg, R.; Hess, A.; Seftor, E.A.; Gardner, L.M.; Pe’er, J.; Trent, J.M.; Meltzer, P.S.; Hendrix, M.J. Vascular channel formation by human melanoma cells in vivo and in vitro: Vasculogenic mimicry. Am. J. Pathol. 1999, 155, 739–752. [Google Scholar] [CrossRef] [Green Version]
- Streubel, B.; Chott, A.; Huber, D.; Exner, M.; Jager, U.; Wagner, O.; Schwarzinger, I. Lymphoma-specific genetic aberrations in microvascular endothelial cells in B-cell lymphomas. N. Engl. J. Med. 2004, 351, 250–259. [Google Scholar] [CrossRef]
- Soda, Y.; Marumoto, T.; Friedmann-Morvinski, D.; Soda, M.; Liu, F.; Michiue, H.; Pastorino, S.; Yang, M.; Hoffman, R.M.; Kesari, S.; et al. Transdifferentiation of glioblastoma cells into vascular endothelial cells. Proc. Natl. Acad. Sci. USA 2011, 108, 4274–4280. [Google Scholar] [CrossRef] [Green Version]
- Bussolati, B.; Grange, C.; Sapino, A.; Camussi, G. Endothelial cell differentiation of human breast tumour stem/progenitor cells. J. Cell. Mol. Med. 2009, 13, 309–319. [Google Scholar] [CrossRef] [Green Version]
- Sharma, N.; Seftor, R.E.; Seftor, E.A.; Gruman, L.M.; Heidger, P.M., Jr.; Cohen, M.B.; Lubaroff, D.M.; Hendrix, M.J. Prostatic tumor cell plasticity involves cooperative interactions of distinct phenotypic subpopulations: Role in vasculogenic mimicry. Prostate 2002, 50, 189–201. [Google Scholar] [CrossRef]
- Ge, H.; Luo, H. Overview of advances in vasculogenic mimicry—A potential target for tumor therapy. Cancer Manag. Res. 2018, 10, 2429–2437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andonegui-Elguera, M.A.; Alfaro-Mora, Y.; Caceres-Gutierrez, R.; Caro-Sanchez, C.H.S.; Herrera, L.A.; Diaz-Chavez, J. An Overview of Vasculogenic Mimicry in Breast Cancer. Front. Oncol. 2020, 10, 220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wechman, S.L.; Emdad, L.; Sarkar, D.; Das, S.K.; Fisher, P.B. Vascular mimicry: Triggers, molecular interactions and in vivo models. Adv. Cancer Res. 2020, 148, 27–67. [Google Scholar] [CrossRef]
- Kim, H.S.; Won, Y.J.; Shim, J.H.; Kim, H.J.; Kim, B.S.; Hong, H.N. Role of EphA2-PI3K signaling in vasculogenic mimicry induced by cancer-associated fibroblasts in gastric cancer cells. Oncol. Lett. 2019, 18, 3031–3038. [Google Scholar] [CrossRef] [PubMed]
- Rong, X.; Huang, B.; Qiu, S.; Li, X.; He, L.; Peng, Y. Tumor-associated macrophages induce vasculogenic mimicry of glioblastoma multiforme through cyclooxygenase-2 activation. Oncotarget 2016, 7, 83976–83986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, X.; Chen, Y.; Jiang, X.; Peng, M.; Liu, Y.; Mo, Y.; Ren, D.; Hua, Y.; Yu, B.; Zhou, Y.; et al. Mechanisms of vasculogenic mimicry in hypoxic tumor microenvironments. Mol. Cancer 2021, 20, 7. [Google Scholar] [CrossRef]
- Zhao, X.; Sun, B.; Liu, T.; Shao, B.; Sun, R.; Zhu, D.; Zhang, Y.; Gu, Q.; Dong, X.; Liu, F.; et al. Long noncoding RNA n339260 promotes vasculogenic mimicry and cancer stem cell development in hepatocellular carcinoma. Cancer Sci. 2018, 109, 3197–3208. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Ding, J.; He, M.; Chen, Y.; Wang, R.; Han, Z.; Xing, E.Z.; Zhang, C.; Yeh, S. Estrogen receptor beta promotes the vasculogenic mimicry (VM) and cell invasion via altering the lncRNA-MALAT1/miR-145-5p/NEDD9 signals in lung cancer. Oncogene 2019, 38, 1225–1238. [Google Scholar] [CrossRef] [PubMed]
- Salinas-Vera, Y.M.; Gallardo-Rincon, D.; Garcia-Vazquez, R.; Hernandez-de la Cruz, O.N.; Marchat, L.A.; Gonzalez-Barrios, J.A.; Ruiz-Garcia, E.; Vazquez-Calzada, C.; Contreras-Sanzon, E.; Resendiz-Hernandez, M.; et al. HypoxamiRs Profiling Identify miR-765 as a Regulator of the Early Stages of Vasculogenic Mimicry in SKOV3 Ovarian Cancer Cells. Front. Oncol. 2019, 9, 381. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Karras, P.; Torres, R.; Rambow, F.; van den Oord, J.; Marine, J.C.; Kos, L. Disseminated Melanoma Cells Transdifferentiate into Endothelial Cells in Intravascular Niches at Metastatic Sites. Cell Rep. 2020, 31, 107765. [Google Scholar] [CrossRef]
- Hendrix, M.J.; Seftor, E.A.; Meltzer, P.S.; Gardner, L.M.; Hess, A.R.; Kirschmann, D.A.; Schatteman, G.C.; Seftor, R.E. Expression and functional significance of VE-cadherin in aggressive human melanoma cells: Role in vasculogenic mimicry. Proc. Natl. Acad. Sci. USA 2001, 98, 8018–8023. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Antin, P.; Berx, G.; Blanpain, C.; Brabletz, T.; Bronner, M.; Campbell, K.; Cano, A.; Casanova, J.; Christofori, G.; et al. Guidelines and definitions for research on epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2020, 21, 341–352. [Google Scholar] [CrossRef] [Green Version]
- Hay, E.D. An overview of epithelio-mesenchymal transformation. Acta Anat. 1995, 154, 8–20. [Google Scholar] [CrossRef]
- Hay, E.D.; Zuk, A. Transformations between epithelium and mesenchyme: Normal, pathological, and experimentally induced. Am. J. Kidney Dis. Off. J. Natl. Kidney Found. 1995, 26, 678–690. [Google Scholar] [CrossRef]
- Nieto, M.A. Epithelial plasticity: A common theme in embryonic and cancer cells. Science 2013, 342, 1234850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, D.; Li, X.; Bocci, F.; Tripathi, S.; Deng, Y.; Jolly, M.K.; Onuchic, J.N.; Levine, H. Quantifying Cancer Epithelial-Mesenchymal Plasticity and its Association with Stemness and Immune Response. J. Clin. Med. 2019, 8, 725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Somarelli, J.A.; Shetler, S.; Jolly, M.K.; Wang, X.; Bartholf Dewitt, S.; Hish, A.J.; Gilja, S.; Eward, W.C.; Ware, K.E.; Levine, H.; et al. Mesenchymal-Epithelial Transition in Sarcomas Is Controlled by the Combinatorial Expression of MicroRNA 200s and GRHL2. Mol. Cell. Biol. 2016, 36, 2503–2513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez, D.M.; Medici, D. Signaling mechanisms of the epithelial-mesenchymal transition. Sci. Signal. 2014, 7, re8. [Google Scholar] [CrossRef] [Green Version]
- Nieto, M.A.; Huang, R.Y.; Jackson, R.A.; Thiery, J.P. Emt: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef] [Green Version]
- Lu, M.; Jolly, M.K.; Levine, H.; Onuchic, J.N.; Ben-Jacob, E. MicroRNA-based regulation of epithelial-hybrid-mesenchymal fate determination. Proc. Natl. Acad. Sci. USA 2013, 110, 18144–18149. [Google Scholar] [CrossRef] [Green Version]
- Burk, U.; Schubert, J.; Wellner, U.; Schmalhofer, O.; Vincan, E.; Spaderna, S.; Brabletz, T. A reciprocal repression between ZEB1 and members of the miR-200 family promotes EMT and invasion in cancer cells. Embo Rep. 2008, 9, 582–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siemens, H.; Jackstadt, R.; Hunten, S.; Kaller, M.; Menssen, A.; Gotz, U.; Hermeking, H. miR-34 and SNAIL form a double-negative feedback loop to regulate epithelial-mesenchymal transitions. Cell Cycle 2011, 10, 4256–4271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bocci, F.; Tripathi, S.C.; Vilchez Mercedes, S.A.; George, J.T.; Casabar, J.P.; Wong, P.K.; Hanash, S.M.; Levine, H.; Onuchic, J.N.; Jolly, M.K. NRF2 activates a partial epithelial-mesenchymal transition and is maximally present in a hybrid epithelial/mesenchymal phenotype. Integr. Biol. Quant. Biosci. Nano Macro 2019, 11, 251–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watanabe, K.; Villarreal-Ponce, A.; Sun, P.; Salmans, M.L.; Fallahi, M.; Andersen, B.; Dai, X. Mammary morphogenesis and regeneration require the inhibition of EMT at terminal end buds by Ovol2 transcriptional repressor. Dev. Cell 2014, 29, 59–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, D.; Jolly, M.K.; Boareto, M.; Parsana, P.; Mooney, S.M.; Pienta, K.J.; Levine, H.; Ben-Jacob, E. OVOL guides the epithelial-hybrid-mesenchymal transition. Oncotarget 2015, 6, 15436–15448. [Google Scholar] [CrossRef] [Green Version]
- Bocci, F.; Jolly, M.K.; Tripathi, S.C.; Aguilar, M.; Hanash, S.M.; Levine, H.; Onuchic, J.N. Numb prevents a complete epithelial-mesenchymal transition by modulating Notch signalling. J. R. Soc. Interface 2017, 14. [Google Scholar] [CrossRef] [Green Version]
- Hong, T.; Watanabe, K.; Ta, C.H.; Villarreal-Ponce, A.; Nie, Q.; Dai, X. An Ovol2-Zeb1 Mutual Inhibitory Circuit Governs Bidirectional and Multi-step Transition between Epithelial and Mesenchymal States. PLoS Comput. Biol. 2015, 11, e1004569. [Google Scholar] [CrossRef]
- Subbalakshmi, A.R.; Kundnani, D.; Biswas, K.; Ghosh, A.; Hanash, S.M.; Tripathi, S.C.; Jolly, M.K. NFATc Acts as a Non-Canonical Phenotypic Stability Factor for a Hybrid Epithelial/Mesenchymal Phenotype. Front. Oncol. 2020, 10, 553342. [Google Scholar] [CrossRef]
- Jolly, M.K.; Tripathi, S.C.; Jia, D.; Mooney, S.M.; Celiktas, M.; Hanash, S.M.; Mani, S.A.; Pienta, K.J.; Ben-Jacob, E.; Levine, H. Stability of the hybrid epithelial/mesenchymal phenotype. Oncotarget 2016, 7, 27067–27084. [Google Scholar] [CrossRef] [Green Version]
- Subbalakshmi, A.R.; Sahoo, S.; Biswas, K.; Jolly, M.K. A Computational Systems Biology Approach Identifies SLUG as a Mediator of Partial Epithelial-Mesenchymal Transition (EMT). CellsTissuesOrgans 2021, 1–14. [Google Scholar] [CrossRef]
- Pastushenko, I.; Brisebarre, A.; Sifrim, A.; Fioramonti, M.; Revenco, T.; Boumahdi, S.; Van Keymeulen, A.; Brown, D.; Moers, V.; Lemaire, S.; et al. Identification of the tumour transition states occurring during EMT. Nature 2018, 556, 463–468. [Google Scholar] [CrossRef]
- Tripathi, S.; Chakraborty, P.; Levine, H.; Jolly, M.K. A mechanism for epithelial-mesenchymal heterogeneity in a population of cancer cells. PLoS Comput. Biol. 2020, 16, e1007619. [Google Scholar] [CrossRef] [Green Version]
- Jolly, M.K.; Mani, S.A.; Levine, H. Hybrid epithelial/mesenchymal phenotype(s): The ‘fittest’ for metastasis? Biochim. Et Biophys. Acta. Rev. Cancer 2018, 1870, 151–157. [Google Scholar] [CrossRef]
- Saphir, O.; Vass, A. Carcinosarcoma. Am. J. Cancer 1938, 33, 331–361. [Google Scholar]
- Ho, G.Y.; Kyran, E.; Bedo, J.; Wakefield, M.; Ennis, D.; Mirza, H.; Lieschke, E.; Vandenberg, C.J.; Kondrashova, O.; Upstill-Goddard, R.; et al. Ovarian carcinosarcoma genomics and pre-clinical models highlight the N-MYC pathway as a key driver and susceptibility to EMT-targeting therapy. Biorxiv Prepr 2020. Available online: https://www.biorxiv.org/content/10.1101/2020.11.24.396796v1 (accessed on 11 March 2021).
- McCluggage, W.G. Malignant biphasic uterine tumours: Carcinosarcomas or metaplastic carcinomas? J. Clin. Pathol. 2002, 55, 321–325. [Google Scholar] [CrossRef] [Green Version]
- Inoue, H.; Hashimura, M.; Akiya, M.; Chiba, R.; Saegusa, M. Functional role of ALK-related signal cascades on modulation of epithelial-mesenchymal transition and apoptosis in uterine carcinosarcoma. Mol. Cancer 2017, 16, 37. [Google Scholar] [CrossRef] [Green Version]
- Cuevas, I.C.; Sahoo, S.S.; Kumar, A.; Zhang, H.; Westcott, J.; Aguilar, M.; Cortez, J.D.; Sullivan, S.A.; Xing, C.; Hayes, D.N.; et al. Fbxw7 is a driver of uterine carcinosarcoma by promoting epithelial-mesenchymal transition. Proc. Natl. Acad. Sci. USA 2019, 116, 25880–25890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, N.; Babaei-Jadidi, R.; Lorenzi, F.; Spencer-Dene, B.; Clarke, P.; Domingo, E.; Tulchinsky, E.; Vries, R.G.J.; Kerr, D.; Pan, Y.; et al. An FBXW7-ZEB2 axis links EMT and tumour microenvironment to promote colorectal cancer stem cells and chemoresistance. Oncogenesis 2019, 8, 13. [Google Scholar] [CrossRef] [Green Version]
- Zhao, S.; Bellone, S.; Lopez, S.; Thakral, D.; Schwab, C.; English, D.P.; Black, J.; Cocco, E.; Choi, J.; Zammataro, L.; et al. Mutational landscape of uterine and ovarian carcinosarcomas implicates histone genes in epithelial-mesenchymal transition. Proc. Natl. Acad. Sci. USA 2016, 113, 12238–12243. [Google Scholar] [CrossRef] [Green Version]
- Osakabe, M.; Fukagawa, D.; Sato, C.; Sugimoto, R.; Uesugi, N.; Ishida, K.; Itamochi, H.; Sugiyama, T.; Sugai, T. Immunohistochemical analysis of the epithelial to mesenchymal transition in uterine carcinosarcoma. Int. J. Gynecol. Cancer Off. J. Int. Gynecol. Cancer Soc. 2019, 29, 277–281. [Google Scholar] [CrossRef] [PubMed]
- Arend, R.; Doneza, J.A.; Wright, J.D. Uterine carcinosarcoma. Curr. Opin. Oncol. 2011, 23, 531–536. [Google Scholar] [CrossRef] [PubMed]
- Dwivedi, S.K.D.; Rao, G.; Dey, A.; Buechel, M.; Zhang, Y.; Zhang, M.; Yang, D.; Mukherjee, P.; Bhattacharya, R. Targeting the TGFbeta pathway in uterine carcinosarcoma. Cell Stress 2020, 4, 252–260. [Google Scholar] [CrossRef] [PubMed]
- Ghia, P.; Ferreri, A.M.; Caligaris-Cappio, F. Chronic lymphocytic leukemia. Crit. Rev. Oncol. Hematol. 2007, 64, 234–246. [Google Scholar] [CrossRef] [Green Version]
- Faderl, S.; Talpaz, M.; Estrov, Z.; O’Brien, S.; Kurzrock, R.; Kantarjian, H.M. The biology of chronic myeloid leukemia. N. Engl. J. Med. 1999, 341, 164–172. [Google Scholar] [CrossRef]
- Lowenberg, B.; Downing, J.R.; Burnett, A. Acute myeloid leukemia. N. Engl. J. Med. 1999, 341, 1051–1062. [Google Scholar] [CrossRef] [PubMed]
- Dorantes-Acosta, E.; Pelayo, R. Lineage switching in acute leukemias: A consequence of stem cell plasticity? Bone Marrow Res. 2012, 2012, 406796. [Google Scholar] [CrossRef] [Green Version]
- Park, B.G.; Park, C.J.; Jang, S.; Seo, E.J.; Chi, H.S.; Lee, J.H. Erythroleukemia relapsing as precursor B-cell lymphoblastic leukemia. Korean J. Lab. Med. 2011, 31, 81–85. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Liu, H.; Zhang, S.; Liang, Y.; Xiao, M.; Hao, Y.; Guan, Y. A case report of lineage switch from T-cell acute leukemia to B-cell acute leukemia. Medicine 2020, 99, e22490. [Google Scholar] [CrossRef]
- Somasundaram, R.; Ahsberg, J.; Okuyama, K.; Ungerback, J.; Lilljebjorn, H.; Fioretos, T.; Strid, T.; Sigvardsson, M. Clonal conversion of B lymphoid leukemia reveals cross-lineage transfer of malignant states. Genes Dev. 2016, 30, 2486–2499. [Google Scholar] [CrossRef]
- Aujla, A.; Hanmantgad, M.; Islam, H.; Shakil, F.; Liu, D.; Seiter, K. Lineage switch from T-cell lymphoblastic leukemia/lymphoma to acute myeloid leukemia and back to T-cell lymphoblastic leukemia/lymphoma in a patient diagnosed during pregnancy. Stem Cell Investig. 2019, 6, 12. [Google Scholar] [CrossRef]
- Czeh, M.; Rosenbauer, F. Uncovering a new cellular origin for acute myeloid leukemia with lineage plasticity. Mol. Cell. Oncol. 2017, 4, e1268241. [Google Scholar] [CrossRef] [Green Version]
- Pisco, A.O.; Huang, S. Non-genetic cancer cell plasticity and therapy-induced stemness in tumour relapse: ‘What does not kill me strengthens me’. Br. J. Cancer 2015, 112, 1725–1732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, S.; Jiang, J.; Lu, Y.; Nice, E.C.; Huang, C.; Zhang, J.; He, W. Emerging role of tumor cell plasticity in modifying therapeutic response. Signal Transduct. Target. Ther. 2020, 5, 228. [Google Scholar] [CrossRef]
- Boumahdi, S.; de Sauvage, F.J. The great escape: Tumour cell plasticity in resistance to targeted therapy. Nat. Rev. Drug Discov. 2020, 19, 39–56. [Google Scholar] [CrossRef] [PubMed]
- Zou, M.; Toivanen, R.; Mitrofanova, A.; Floch, N.; Hayati, S.; Sun, Y.; Le Magnen, C.; Chester, D.; Mostaghel, E.A.; Califano, A.; et al. Transdifferentiation as a Mechanism of Treatment Resistance in a Mouse Model of Castration-Resistant Prostate Cancer. Cancer Discov. 2017, 7, 736–749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaurova, T.; Zhang, L.; Goodrich, D.W.; Hershberger, P.A. Understanding Lineage Plasticity as a Path to Targeted Therapy Failure in EGFR-Mutant Non-small Cell Lung Cancer. Front. Genet. 2020, 11, 281. [Google Scholar] [CrossRef]
- Farrell, A.S.; Joly, M.M.; Allen-Petersen, B.L.; Worth, P.J.; Lanciault, C.; Sauer, D.; Link, J.; Pelz, C.; Heiser, L.M.; Morton, J.P.; et al. MYC regulates ductal-neuroendocrine lineage plasticity in pancreatic ductal adenocarcinoma associated with poor outcome and chemoresistance. Nat. Commun. 2017, 8, 1728. [Google Scholar] [CrossRef]
- Roubaud, G.; Liaw, B.C.; Oh, W.K.; Mulholland, D.J. Strategies to avoid treatment-induced lineage crisis in advanced prostate cancer. Nat. Rev. Clin. Oncol. 2017, 14, 269–283. [Google Scholar] [CrossRef] [Green Version]
- Parimi, V.; Goyal, R.; Poropatich, K.; Yang, X.J. Neuroendocrine differentiation of prostate cancer: A review. Am. J. Clin. Exp. Urol. 2014, 2, 273–285. [Google Scholar] [PubMed]
- Beltran, H.; Prandi, D.; Mosquera, J.M.; Benelli, M.; Puca, L.; Cyrta, J.; Marotz, C.; Giannopoulou, E.; Chakravarthi, B.V.; Varambally, S.; et al. Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. Nat. Med. 2016, 22, 298–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eriksson, B.; Arnberg, H.; Oberg, K.; Hellman, U.; Lundqvist, G.; Wernstedt, C.; Wilander, E. A polyclonal antiserum against chromogranin A and B--a new sensitive marker for neuroendocrine tumours. Acta Endocrinol. 1990, 122, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Davies, A.H.; Beltran, H.; Zoubeidi, A. Cellular plasticity and the neuroendocrine phenotype in prostate cancer. Nat. Rev. Urol. 2018, 15, 271–286. [Google Scholar] [CrossRef] [PubMed]
- Blee, A.M.; Huang, H. Lineage plasticity-mediated therapy resistance in prostate cancer. Asian J. Androl. 2019, 21, 241–248. [Google Scholar] [CrossRef] [PubMed]
- Dardenne, E.; Beltran, H.; Benelli, M.; Gayvert, K.; Berger, A.; Puca, L.; Cyrta, J.; Sboner, A.; Noorzad, Z.; MacDonald, T.; et al. N-Myc Induces an EZH2-Mediated Transcriptional Program Driving Neuroendocrine Prostate Cancer. Cancer Cell 2016, 30, 563–577. [Google Scholar] [CrossRef] [Green Version]
- Cyrta, J.; Augspach, A.; De Filippo, M.R.; Prandi, D.; Thienger, P.; Benelli, M.; Cooley, V.; Bareja, R.; Wilkes, D.; Chae, S.S.; et al. Role of specialized composition of SWI/SNF complexes in prostate cancer lineage plasticity. Nat. Commun. 2020, 11, 5549. [Google Scholar] [CrossRef]
- Mu, P.; Zhang, Z.; Benelli, M.; Karthaus, W.R.; Hoover, E.; Chen, C.C.; Wongvipat, J.; Ku, S.Y.; Gao, D.; Cao, Z.; et al. SOX2 promotes lineage plasticity and antiandrogen resistance in TP53- and RB1-deficient prostate cancer. Science 2017, 355, 84–88. [Google Scholar] [CrossRef] [Green Version]
- Tuerff, D.; Sissung, T.; Figg, W.D. Cellular identity crisis: Antiandrogen resistance by lineage plasticity. Cancer Biol. Ther. 2017, 18, 841–842. [Google Scholar] [CrossRef]
- Bishop, J.L.; Thaper, D.; Vahid, S.; Davies, A.; Ketola, K.; Kuruma, H.; Jama, R.; Nip, K.M.; Angeles, A.; Johnson, F.; et al. The Master Neural Transcription Factor BRN2 Is an Androgen Receptor-Suppressed Driver of Neuroendocrine Differentiation in Prostate Cancer. Cancer Discov. 2017, 7, 54–71. [Google Scholar] [CrossRef] [Green Version]
- Yasumizu, Y.; Rajabi, H.; Jin, C.; Hata, T.; Pitroda, S.; Long, M.D.; Hagiwara, M.; Li, W.; Hu, Q.; Liu, S.; et al. MUC1-C regulates lineage plasticity driving progression to neuroendocrine prostate cancer. Nat. Commun. 2020, 11, 338. [Google Scholar] [CrossRef] [Green Version]
- Monga, J.; Adrianto, I.; Rogers, C.; Gadgeel, S.; Chitale, D.; Alumkal, J.J.; Beltran, H.; Zoubeidi, A.; Ghosh, J. Tribbles 2 confers enzalutamide resistance in prostate cancer by promoting lineage plasticity. Biorxiv Prepr 2021. Available online: https://www.biorxiv.org/content/10.1101/2021.03.26.437250v1 (accessed on 9 April 2021).
- Travis, W.D.; Brambilla, E.; Nicholson, A.G.; Yatabe, Y.; Austin, J.H.M.; Beasley, M.B.; Chirieac, L.R.; Dacic, S.; Duhig, E.; Flieder, D.B.; et al. The 2015 World Health Organization Classification of Lung Tumors: Impact of Genetic, Clinical and Radiologic Advances Since the 2004 Classification. J. Thorac. Oncol. Off. Publ. Int. Assoc. Study Lung Cancer 2015, 10, 1243–1260. [Google Scholar] [CrossRef] [Green Version]
- Park, K.S.; Liang, M.C.; Raiser, D.M.; Zamponi, R.; Roach, R.R.; Curtis, S.J.; Walton, Z.; Schaffer, B.E.; Roake, C.M.; Zmoos, A.F.; et al. Characterization of the cell of origin for small cell lung cancer. Cell Cycle 2011, 10, 2806–2815. [Google Scholar] [CrossRef] [Green Version]
- Yokomizo, A.; Tindall, D.J.; Drabkin, H.; Gemmill, R.; Franklin, W.; Yang, P.; Sugio, K.; Smith, D.I.; Liu, W. PTEN/MMAC1 mutations identified in small cell, but not in non-small cell lung cancers. Oncogene 1998, 17, 475–479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beasley, M.B.; Lantuejoul, S.; Abbondanzo, S.; Chu, W.S.; Hasleton, P.S.; Travis, W.D.; Brambilla, E. The P16/cyclin D1/Rb pathway in neuroendocrine tumors of the lung. Hum. Pathol. 2003, 34, 136–142. [Google Scholar] [CrossRef]
- Sutherland, K.D.; Proost, N.; Brouns, I.; Adriaensen, D.; Song, J.Y.; Berns, A. Cell of origin of small cell lung cancer: Inactivation of Trp53 and Rb1 in distinct cell types of adult mouse lung. Cancer Cell 2011, 19, 754–764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubin, M.A.; Bristow, R.G.; Thienger, P.D.; Dive, C.; Imielinski, M. Impact of Lineage Plasticity to and from a Neuroendocrine Phenotype on Progression and Response in Prostate and Lung Cancers. Mol. Cell 2020, 80, 562–577. [Google Scholar] [CrossRef] [PubMed]
- Shepherd, F.A.; Rodrigues Pereira, J.; Ciuleanu, T.; Tan, E.H.; Hirsh, V.; Thongprasert, S.; Campos, D.; Maoleekoonpiroj, S.; Smylie, M.; Martins, R.; et al. Erlotinib in previously treated non-small-cell lung cancer. N. Engl. J. Med. 2005, 353, 123–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sequist, L.V.; Waltman, B.A.; Dias-Santagata, D.; Digumarthy, S.; Turke, A.B.; Fidias, P.; Bergethon, K.; Shaw, A.T.; Gettinger, S.; Cosper, A.K.; et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci. Transl. Med. 2011, 3, 75ra26. [Google Scholar] [CrossRef] [Green Version]
- Clery, E.; Pisapia, P.; Feliciano, S.; Vigliar, E.; Marano, A.; De Luca, C.; Malapelle, U.; Troncone, G.; Bellevicine, C. There is still a role for cytology in the ‘liquid biopsy’ era. A lesson from a TKI-treated patient showing adenocarcinoma to squamous cell carcinoma transition during disease progression. J. Clin. Pathol. 2017, 70, 798–802. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Shim, J.H.; Lee, B.; Cho, I.; Park, W.Y.; Kim, Y.; Lee, S.H.; Choi, Y.; Han, J.; Ahn, J.S.; et al. Paired genomic analysis of squamous cell carcinoma transformed from EGFR-mutated lung adenocarcinoma. Lung Cancer 2019, 134, 7–15. [Google Scholar] [CrossRef]
- Ricciuti, B.; Metro, G.; Brambilla, M.; Ludovini, V.; Baglivo, S.; Siggillino, A.; Prosperi, E.; Chiari, R. Acquired Resistance to Afatinib Due to T790M-Positive Squamous Progression in EGFR-Mutant Adenosquamous Lung Carcinoma. J. Thorac. Oncol. Off. Publ. Int. Assoc. Study Lung Cancer 2018, 13, e9–e12. [Google Scholar] [CrossRef] [Green Version]
- Jakobsen, K.R.; Demuth, C.; Madsen, A.T.; Hussmann, D.; Vad-Nielsen, J.; Nielsen, A.L.; Sorensen, B.S. MET amplification and epithelial-to-mesenchymal transition exist as parallel resistance mechanisms in erlotinib-resistant, EGFR-mutated, NSCLC HCC827 cells. Oncogenesis 2017, 6, e307. [Google Scholar] [CrossRef]
- Uramoto, H.; Iwata, T.; Onitsuka, T.; Shimokawa, H.; Hanagiri, T.; Oyama, T. Epithelial-mesenchymal transition in EGFR-TKI acquired resistant lung adenocarcinoma. Anticancer Res. 2010, 30, 2513–2517. [Google Scholar]
- Chung, J.H.; Rho, J.K.; Xu, X.; Lee, J.S.; Yoon, H.I.; Lee, C.T.; Choi, Y.J.; Kim, H.R.; Kim, C.H.; Lee, J.C. Clinical and molecular evidences of epithelial to mesenchymal transition in acquired resistance to EGFR-TKIs. Lung Cancer 2011, 73, 176–182. [Google Scholar] [CrossRef]
- Schulze, A.B.; Evers, G.; Kerkhoff, A.; Mohr, M.; Schliemann, C.; Berdel, W.E.; Schmidt, L.H. Future Options of Molecular-Targeted Therapy in Small Cell Lung Cancer. Cancers 2019, 11, 690. [Google Scholar] [CrossRef] [Green Version]
- Lupo, B.; Sassi, F.; Pinnelli, M.; Galimi, F.; Zanella, E.R.; Vurchio, V.; Migliardi, G.; Gagliardi, P.A.; Puliafito, A.; Manganaro, D.; et al. Colorectal cancer residual disease at maximal response to EGFR blockade displays a druggable Paneth cell-like phenotype. Sci. Transl. Med. 2020, 12. [Google Scholar] [CrossRef]
- Kong, F.E.; Li, G.M.; Tang, Y.Q.; Xi, S.Y.; Loong, J.H.C.; Li, M.M.; Li, H.L.; Cheng, W.; Zhu, W.J.; Mo, J.Q.; et al. Targeting tumor lineage plasticity in hepatocellular carcinoma using an anti-CLDN6 antibody-drug conjugate. Sci. Transl. Med. 2021, 13. [Google Scholar] [CrossRef] [PubMed]
- Jolly, M.K.; Kulkarni, P.; Weninger, K.; Orban, J.; Levine, H. Phenotypic Plasticity, Bet-Hedging, and Androgen Independence in Prostate Cancer: Role of Non-Genetic Heterogeneity. Front. Oncol. 2018, 8, 50. [Google Scholar] [CrossRef]
- Basak, O.; Beumer, J.; Wiebrands, K.; Seno, H.; van Oudenaarden, A.; Clevers, H. Induced Quiescence of Lgr5+ Stem Cells in Intestinal Organoids Enables Differentiation of Hormone-Producing Enteroendocrine Cells. Cell Stem Cell 2017, 20, 177–190.e4. [Google Scholar] [CrossRef] [Green Version]
- Buczacki, S.J.; Zecchini, H.I.; Nicholson, A.M.; Russell, R.; Vermeulen, L.; Kemp, R.; Winton, D.J. Intestinal label-retaining cells are secretory precursors expressing Lgr5. Nature 2013, 495, 65–69. [Google Scholar] [CrossRef]
- Barriga, F.M.; Montagni, E.; Mana, M.; Mendez-Lago, M.; Hernando-Momblona, X.; Sevillano, M.; Guillaumet-Adkins, A.; Rodriguez-Esteban, G.; Buczacki, S.J.A.; Gut, M.; et al. Mex3a Marks a Slowly Dividing Subpopulation of Lgr5+ Intestinal Stem Cells. Cell Stem Cell 2017, 20, 801–816.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.F.; de Oliveira, A.C.; Santoro, A.; Raoul, J.L.; Forner, A.; et al. Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef]
- Biehs, B.; Dijkgraaf, G.J.P.; Piskol, R.; Alicke, B.; Boumahdi, S.; Peale, F.; Gould, S.E.; de Sauvage, F.J. A cell identity switch allows residual BCC to survive Hedgehog pathway inhibition. Nature 2018, 562, 429–433. [Google Scholar] [CrossRef]
- Haddox, C.L.; Mangaonkar, A.A.; Chen, D.; Shi, M.; He, R.; Oliveira, J.L.; Litzow, M.R.; Al-Kali, A.; Hogan, W.J.; Elliott, M.A. Blinatumomab-induced lineage switch of B-ALL with t(4:11)(q21;q23) KMT2A/AFF1 into an aggressive AML: Pre- and post-switch phenotypic, cytogenetic and molecular analysis. Blood Cancer J. 2017, 7, e607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frauenfeld, L.; Bonzheim, I.; Wirths, S.; Steinhilber, J.; Horger, M.; Mankel, B.; Bethge, W.; Fend, F.; Federmann, B. Clonal evolution of chronic lymphocytic leukemia to Langerhans cell histiocytosis: A case report. Virchows Arch. Int. J. Pathol. 2019, 475, 795–798. [Google Scholar] [CrossRef]
- Zhang, Q.; Orlando, E.J.; Wang, H.Y.; Bogusz, A.M.; Liu, X.; Lacey, S.F.; Strauser, H.T.; Nunez-Cruz, S.; Nejati, R.; Zhang, P.; et al. Transdifferentiation of lymphoma into sarcoma associated with profound reprogramming of the epigenome. Blood 2020, 136, 1980–1983. [Google Scholar] [CrossRef] [PubMed]
- Jacoby, E.; Nguyen, S.M.; Fountaine, T.J.; Welp, K.; Gryder, B.; Qin, H.; Yang, Y.; Chien, C.D.; Seif, A.E.; Lei, H.; et al. CD19 CAR immune pressure induces B-precursor acute lymphoblastic leukaemia lineage switch exposing inherent leukaemic plasticity. Nat. Commun. 2016, 7, 12320. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhou, Y.; Yuan, Q.; Xiao, M. Myeloid Blast Crisis of Chronic Myeloid Leukemia Followed by Lineage Switch to B-Lymphoblastic Leukemia: A Case Report. Oncotargets Ther. 2020, 13, 3259–3264. [Google Scholar] [CrossRef] [Green Version]
- Novakova, M.; Zaliova, M.; Fiser, K.; Vakrmanova, B.; Slamova, L.; Musilova, A.; Bruggemann, M.; Ritgen, M.; Fronkova, E.; Kalina, T.; et al. DUX4r, ZNF384r and PAX5-P80R mutated B-cell precursor acute lymphoblastic leukemia frequently undergo monocytic switch. Haematologica 2020. [Google Scholar] [CrossRef]
- Mo, G.; Wang, H.W.; Talleur, A.C.; Shahani, S.A.; Yates, B.; Shalabi, H.; Douvas, M.G.; Calvo, K.R.; Shern, J.F.; Chaganti, S.; et al. Diagnostic approach to the evaluation of myeloid malignancies following CAR T-cell therapy in B-cell acute lymphoblastic leukemia. J. Immunother. Cancer 2020, 8. [Google Scholar] [CrossRef] [PubMed]
- Mundada, M.C.; Ahmed, F.; Pasam, M.K.; Murthy, S.; Santa, A.; Patil, V. The intriguing occurrence of Acute Myeloid Leukemia in a case of Acute Lymphoblastic leukemia:Report of two cases. J. Appl. Hematol. 2020, 11, 21–24. [Google Scholar] [CrossRef]
- Quintanal-Villalonga, A.; Chan, J.M.; Yu, H.A.; Pe’er, D.; Sawyers, C.L.; Sen, T.; Rudin, C.M. Lineage plasticity in cancer: A shared pathway of therapeutic resistance. Nat. Rev. Clin. Oncol. 2020, 17, 360–371. [Google Scholar] [CrossRef] [PubMed]
- Ku, S.Y.; Rosario, S.; Wang, Y.; Mu, P.; Seshadri, M.; Goodrich, Z.W.; Goodrich, M.M.; Labbe, D.P.; Gomez, E.C.; Wang, J.; et al. Rb1 and Trp53 cooperate to suppress prostate cancer lineage plasticity, metastasis, and antiandrogen resistance. Science 2017, 355, 78–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meuwissen, R.; Linn, S.C.; Linnoila, R.I.; Zevenhoven, J.; Mooi, W.J.; Berns, A. Induction of small cell lung cancer by somatic inactivation of both Trp53 and Rb1 in a conditional mouse model. Cancer Cell 2003, 4, 181–189. [Google Scholar] [CrossRef] [Green Version]
- Ghanei, Z.; Jamshidizad, A.; Joupari, M.D.; Shamsara, M. Isolation and characterization of breast cancer stem cell-like phenotype by Oct4 promoter-mediated activity. J. Cell. Physiol. 2020, 235, 7840–7848. [Google Scholar] [CrossRef]
- Koo, B.S.; Lee, S.H.; Kim, J.M.; Huang, S.; Kim, S.H.; Rho, Y.S.; Bae, W.J.; Kang, H.J.; Kim, Y.S.; Moon, J.H.; et al. Oct4 is a critical regulator of stemness in head and neck squamous carcinoma cells. Oncogene 2015, 34, 2317–2324. [Google Scholar] [CrossRef]
- Liu, H.L.; Tang, H.T.; Yang, H.L.; Deng, T.T.; Xu, Y.P.; Xu, S.Q.; Peng, L.; Wang, Z.; Fang, Q.; Kuang, X.Y.; et al. Oct4 Regulates the Transition of Cancer Stem-Like Cells to Tumor Endothelial-Like Cells in Human Liver Cancer. Front. Cell Dev. Biol. 2020, 8, 563316. [Google Scholar] [CrossRef]
- Lim, E.; Vaillant, F.; Wu, D.; Forrest, N.C.; Pal, B.; Hart, A.H.; Asselin-Labat, M.L.; Gyorki, D.E.; Ward, T.; Partanen, A.; et al. Aberrant luminal progenitors as the candidate target population for basal tumor development in BRCA1 mutation carriers. Nat. Med. 2009, 15, 907–913. [Google Scholar] [CrossRef] [PubMed]
- Molyneux, G.; Geyer, F.C.; Magnay, F.A.; McCarthy, A.; Kendrick, H.; Natrajan, R.; Mackay, A.; Grigoriadis, A.; Tutt, A.; Ashworth, A.; et al. BRCA1 basal-like breast cancers originate from luminal epithelial progenitors and not from basal stem cells. Cell Stem Cell 2010, 7, 403–417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keller, P.J.; Arendt, L.M.; Skibinski, A.; Logvinenko, T.; Klebba, I.; Dong, S.; Smith, A.E.; Prat, A.; Perou, C.M.; Gilmore, H.; et al. Defining the cellular precursors to human breast cancer. Proc. Natl. Acad. Sci. USA 2012, 109, 2772–2777. [Google Scholar] [CrossRef] [Green Version]
- Lin, S.C.; Chou, Y.T.; Jiang, S.S.; Chang, J.L.; Chung, C.H.; Kao, Y.R.; Chang, I.S.; Wu, C.W. Epigenetic Switch between SOX2 and SOX9 Regulates Cancer Cell Plasticity. Cancer Res. 2016, 76, 7036–7048. [Google Scholar] [CrossRef] [Green Version]
- Dang, C.V. MYC on the path to cancer. Cell 2012, 149, 22–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Kim, J.; Roh, M.; Franco, O.E.; Hayward, S.W.; Wills, M.L.; Abdulkadir, S.A. Pim1 kinase synergizes with c-MYC to induce advanced prostate carcinoma. Oncogene 2010, 29, 2477–2487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruneel, K.; Verstappe, J.; Vandamme, N.; Berx, G. Intrinsic Balance between ZEB Family Members Is Important for Melanocyte Homeostasis and Melanoma Progression. Cancers 2020, 12, 2248. [Google Scholar] [CrossRef] [PubMed]
- De Craene, B.; Berx, G. Regulatory networks defining EMT during cancer initiation and progression. Nat. Rev. Cancer 2013, 13, 97–110. [Google Scholar] [CrossRef] [PubMed]
- Denecker, G.; Vandamme, N.; Akay, O.; Koludrovic, D.; Taminau, J.; Lemeire, K.; Gheldof, A.; De Craene, B.; Van Gele, M.; Brochez, L.; et al. Identification of a ZEB2-MITF-ZEB1 transcriptional network that controls melanogenesis and melanoma progression. Cell Death Differ. 2014, 21, 1250–1261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoek, K.S.; Eichhoff, O.M.; Schlegel, N.C.; Dobbeling, U.; Kobert, N.; Schaerer, L.; Hemmi, S.; Dummer, R. In vivo switching of human melanoma cells between proliferative and invasive states. Cancer Res. 2008, 68, 650–656. [Google Scholar] [CrossRef] [Green Version]
- Konieczkowski, D.J.; Johannessen, C.M.; Abudayyeh, O.; Kim, J.W.; Cooper, Z.A.; Piris, A.; Frederick, D.T.; Barzily-Rokni, M.; Straussman, R.; Haq, R.; et al. A melanoma cell state distinction influences sensitivity to MAPK pathway inhibitors. Cancer Discov. 2014, 4, 816–827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fagnocchi, L.; Poli, V.; Zippo, A. Enhancer reprogramming in tumor progression: A new route towards cancer cell plasticity. Cell. Mol. Life Sci. Cmls 2018, 75, 2537–2555. [Google Scholar] [CrossRef]
- Suva, M.L.; Riggi, N.; Bernstein, B.E. Epigenetic reprogramming in cancer. Science 2013, 339, 1567–1570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scaffidi, P.; Misteli, T. Cancer epigenetics: From disruption of differentiation programs to the emergence of cancer stem cells. Cold Spring Harb. Symp. Quant. Biol. 2010, 75, 251–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Easwaran, H.; Tsai, H.C.; Baylin, S.B. Cancer epigenetics: Tumor heterogeneity, plasticity of stem-like states, and drug resistance. Mol. Cell 2014, 54, 716–727. [Google Scholar] [CrossRef] [Green Version]
- Bueno-Costa, A.; Pineyro, D.; Soler, M.; Javierre, B.M.; Raurell-Vila, H.; Subirana-Granes, M.; Pasquali, L.; Martinez-Climent, J.A.; Esteller, M. B-cell leukemia transdifferentiation to macrophage involves reconfiguration of DNA methylation for long-range regulation. Leukemia 2020, 34, 1158–1162. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.G.; Chen, W.S.; Li, H.; Foye, A.; Zhang, M.; Sjostrom, M.; Aggarwal, R.; Playdle, D.; Liao, A.; Alumkal, J.J.; et al. The DNA methylation landscape of advanced prostate cancer. Nat. Genet. 2020, 52, 778–789. [Google Scholar] [CrossRef]
- Gan, L.; Yang, Y.; Li, Q.; Feng, Y.; Liu, T.; Guo, W. Epigenetic regulation of cancer progression by EZH2: From biological insights to therapeutic potential. Biomark. Res. 2018, 6, 10. [Google Scholar] [CrossRef]
- Patil, S.; Steuber, B.; Kopp, W.; Kari, V.; Urbach, L.; Wang, X.; Kuffer, S.; Bohnenberger, H.; Spyropoulou, D.; Zhang, Z.; et al. EZH2 Regulates Pancreatic Cancer Subtype Identity and Tumor Progression via Transcriptional Repression of GATA6. Cancer Res. 2020, 80, 4620–4632. [Google Scholar] [CrossRef]
- Sharma, S.V.; Lee, D.Y.; Li, B.; Quinlan, M.P.; Takahashi, F.; Maheswaran, S.; McDermott, U.; Azizian, N.; Zou, L.; Fischbach, M.A.; et al. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell 2010, 141, 69–80. [Google Scholar] [CrossRef] [Green Version]
- Liau, B.B.; Sievers, C.; Donohue, L.K.; Gillespie, S.M.; Flavahan, W.A.; Miller, T.E.; Venteicher, A.S.; Hebert, C.H.; Carey, C.D.; Rodig, S.J.; et al. Adaptive Chromatin Remodeling Drives Glioblastoma Stem Cell Plasticity and Drug Tolerance. Cell Stem Cell 2017, 20, 233–246.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medina, P.P.; Sanchez-Cespedes, M. Involvement of the chromatin-remodeling factor BRG1/SMARCA4 in human cancer. Epigenetics 2008, 3, 64–68. [Google Scholar] [CrossRef] [Green Version]
- Xu, G.; Chhangawala, S.; Cocco, E.; Razavi, P.; Cai, Y.; Otto, J.E.; Ferrando, L.; Selenica, P.; Ladewig, E.; Chan, C.; et al. ARID1A determines luminal identity and therapeutic response in estrogen-receptor-positive breast cancer. Nat. Genet. 2020, 52, 198–207. [Google Scholar] [CrossRef]
- Liu, T.; Han, C.; Fang, P.; Zhu, H.; Wang, S.; Ma, Z.; Zhang, Q.; Xia, W.; Wang, J.; Xu, L.; et al. Long non-coding RNAs in lung cancer: Implications for lineage plasticity-mediated TKI resistance. Cell. Mol. Life Sci. Cmls 2021, 78, 1983–2000. [Google Scholar] [CrossRef] [PubMed]
- Crea, F.; Venalainen, E.; Ci, X.; Cheng, H.; Pikor, L.; Parolia, A.; Xue, H.; Nur Saidy, N.R.; Lin, D.; Lam, W.; et al. The role of epigenetics and long noncoding RNA MIAT in neuroendocrine prostate cancer. Epigenomics 2016, 8, 721–731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramnarine, V.R.; Alshalalfa, M.; Mo, F.; Nabavi, N.; Erho, N.; Takhar, M.; Shukin, R.; Brahmbhatt, S.; Gawronski, A.; Kobelev, M.; et al. The long noncoding RNA landscape of neuroendocrine prostate cancer and its clinical implications. GigaScience 2018, 7. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.H.; Zhou, L.Y.; Xu, S.; Zheng, Y.L.; Wan, Y.F.; Hu, C.P. Overexpression of lncRNA HOXA11-AS promotes cell epithelial-mesenchymal transition by repressing miR-200b in non-small cell lung cancer. Cancer Cell Int. 2017, 17, 64. [Google Scholar] [CrossRef]
- Di Croce, L.; Helin, K. Transcriptional regulation by Polycomb group proteins. Nat. Struct. Mol. Biol. 2013, 20, 1147–1155. [Google Scholar] [CrossRef]
- Colotta, F.; Allavena, P.; Sica, A.; Garlanda, C.; Mantovani, A. Cancer-related inflammation, the seventh hallmark of cancer: Links to genetic instability. Carcinogenesis 2009, 30, 1073–1081. [Google Scholar] [CrossRef] [Green Version]
- Giroux, V.; Rustgi, A.K. Metaplasia: Tissue injury adaptation and a precursor to the dysplasia-cancer sequence. Nat. Rev. Cancer 2017, 17, 594–604. [Google Scholar] [CrossRef]
- Yuan, S.; Norgard, R.J.; Stanger, B.Z. Cellular Plasticity in Cancer. Cancer Discov. 2019, 9, 837–851. [Google Scholar] [CrossRef] [Green Version]
- Meyer, A.R.; Goldenring, J.R. Injury, repair, inflammation and metaplasia in the stomach. J. Physiol. 2018, 596, 3861–3867. [Google Scholar] [CrossRef] [Green Version]
- Le Magnen, C.; Virk, R.K.; Dutta, A.; Kim, J.Y.; Panja, S.; Lopez-Bujanda, Z.A.; Califano, A.; Drake, C.G.; Mitrofanova, A.; Abate-Shen, C. Cooperation of loss of NKX3.1 and inflammation in prostate cancer initiation. Dis. Models Mech. 2018, 11. [Google Scholar] [CrossRef] [Green Version]
- Baram, T.; Rubinstein-Achiasaf, L.; Ben-Yaakov, H.; Ben-Baruch, A. Inflammation-Driven Breast Tumor Cell Plasticity: Stemness/EMT, Therapy Resistance and Dormancy. Front. Oncol. 2020, 10, 614468. [Google Scholar] [CrossRef]
- Lu, H.; Clauser, K.R.; Tam, W.L.; Frose, J.; Ye, X.; Eaton, E.N.; Reinhardt, F.; Donnenberg, V.S.; Bhargava, R.; Carr, S.A.; et al. A breast cancer stem cell niche supported by juxtacrine signalling from monocytes and macrophages. Nat. Cell Biol. 2014, 16, 1105–1117. [Google Scholar] [CrossRef] [Green Version]
- Tiwari, R.; Manzar, N.; Ateeq, B. Dynamics of Cellular Plasticity in Prostate Cancer Progression. Front. Mol. Biosci. 2020, 7, 130. [Google Scholar] [CrossRef]
- Roswall, P.; Bocci, M.; Bartoschek, M.; Li, H.; Kristiansen, G.; Jansson, S.; Lehn, S.; Sjolund, J.; Reid, S.; Larsson, C.; et al. Microenvironmental control of breast cancer subtype elicited through paracrine platelet-derived growth factor-CC signaling. Nat. Med. 2018, 24, 463–473. [Google Scholar] [CrossRef]
- Lin, Y.; Xu, J.; Lan, H. Tumor-associated macrophages in tumor metastasis: Biological roles and clinical therapeutic applications. J. Hematol. Oncol. 2019, 12, 76. [Google Scholar] [CrossRef]
- Huang, R.; Wang, S.; Wang, N.; Zheng, Y.; Zhou, J.; Yang, B.; Wang, X.; Zhang, J.; Guo, L.; Wang, S.; et al. CCL5 derived from tumor-associated macrophages promotes prostate cancer stem cells and metastasis via activating beta-catenin/STAT3 signaling. Cell Death Dis. 2020, 11, 234. [Google Scholar] [CrossRef]
- Ding, H.; Zhao, L.; Dai, S.; Li, L.; Wang, F.; Shan, B. CCL5 secreted by tumor associated macrophages may be a new target in treatment of gastric cancer. Biomed. Pharmacother. Biomed. Pharmacother. 2016, 77, 142–149. [Google Scholar] [CrossRef]
- Poltavets, V.; Kochetkova, M.; Pitson, S.M.; Samuel, M.S. The Role of the Extracellular Matrix and Its Molecular and Cellular Regulators in Cancer Cell Plasticity. Front. Oncol. 2018, 8, 431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, Y.; Chakraborty, P.; Jolly, M.K.; Levine, H. A Theoretical Approach to Coupling the Epithelial-Mesenchymal Transition (EMT) to Extracellular Matrix (ECM) Stiffness via LOXL2. Cancers 2021, 13, 1609. [Google Scholar] [CrossRef]
- Tian, B.; Luo, Q.; Ju, Y.; Song, G. A Soft Matrix Enhances the Cancer Stem Cell Phenotype of HCC Cells. Int. J. Mol. Sci. 2019, 20, 2831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pang, M.F.; Siedlik, M.J.; Han, S.; Stallings-Mann, M.; Radisky, D.C.; Nelson, C.M. Tissue Stiffness and Hypoxia Modulate the Integrin-Linked Kinase ILK to Control Breast Cancer Stem-like Cells. Cancer Res. 2016, 76, 5277–5287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peris-Torres, C.; Plaza-Calonge, M.D.C.; Lopez-Dominguez, R.; Dominguez-Garcia, S.; Barrientos-Duran, A.; Carmona-Saez, P.; Rodriguez-Manzaneque, J.C. Extracellular Protease ADAMTS1 Is Required at Early Stages of Human Uveal Melanoma Development by Inducing Stemness and Endothelial-Like Features on Tumor Cells. Cancers 2020, 12, 801. [Google Scholar] [CrossRef] [Green Version]
- Casal, C.; Torres-Collado, A.X.; Plaza-Calonge Mdel, C.; Martino-Echarri, E.; Ramon, Y.C.S.; Rojo, F.; Griffioen, A.W.; Rodriguez-Manzaneque, J.C. ADAMTS1 contributes to the acquisition of an endothelial-like phenotype in plastic tumor cells. Cancer Res. 2010, 70, 4676–4686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serrano-Garrido, O.; Peris-Torres, C.; Redondo-Garcia, S.; Asenjo, H.G.; Plaza-Calonge, M.D.C.; Fernandez-Luna, J.L.; Rodriguez-Manzaneque, J.C. ADAMTS1 Supports Endothelial Plasticity of Glioblastoma Cells with Relevance for Glioma Progression. Biomolecules 2020, 11, 44. [Google Scholar] [CrossRef] [PubMed]
- Ruan, L.; Wang, L.; Wang, X.; He, M.; Yao, X. SIRT1 contributes to neuroendocrine differentiation of prostate cancer. Oncotarget 2018, 9, 2002–2016. [Google Scholar] [CrossRef]
- Smith, P.C.; Keller, E.T. Anti-interleukin-6 monoclonal antibody induces regression of human prostate cancer xenografts in nude mice. Prostate 2001, 48, 47–53. [Google Scholar] [CrossRef]
- Wallner, L.; Dai, J.; Escara-Wilke, J.; Zhang, J.; Yao, Z.; Lu, Y.; Trikha, M.; Nemeth, J.A.; Zaki, M.H.; Keller, E.T. Inhibition of interleukin-6 with CNTO328, an anti-interleukin-6 monoclonal antibody, inhibits conversion of androgen-dependent prostate cancer to an androgen-independent phenotype in orchiectomized mice. Cancer Res. 2006, 66, 3087–3095. [Google Scholar] [CrossRef] [Green Version]
- Shen, S.; Clairambault, J. Cell plasticity in cancer cell populations. F1000Research 2020, 9. [Google Scholar] [CrossRef]
- Straussman, R.; Morikawa, T.; Shee, K.; Barzily-Rokni, M.; Qian, Z.R.; Du, J.; Davis, A.; Mongare, M.M.; Gould, J.; Frederick, D.T.; et al. Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature 2012, 487, 500–504. [Google Scholar] [CrossRef] [Green Version]
- Pattabiraman, D.R.; Bierie, B.; Kober, K.I.; Thiru, P.; Krall, J.A.; Zill, C.; Reinhardt, F.; Tam, W.L.; Weinberg, R.A. Activation of PKA leads to mesenchymal-to-epithelial transition and loss of tumor-initiating ability. Science 2016, 351, aad3680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Degos, L.; Dombret, H.; Chomienne, C.; Daniel, M.T.; Miclea, J.M.; Chastang, C.; Castaigne, S.; Fenaux, P. All-trans-retinoic acid as a differentiating agent in the treatment of acute promyelocytic leukemia. Blood 1995, 85, 2643–2653. [Google Scholar] [CrossRef]
- Turcan, S.; Fabius, A.W.; Borodovsky, A.; Pedraza, A.; Brennan, C.; Huse, J.; Viale, A.; Riggins, G.J.; Chan, T.A. Efficient induction of differentiation and growth inhibition in IDH1 mutant glioma cells by the DNMT Inhibitor Decitabine. Oncotarget 2013, 4, 1729–1736. [Google Scholar] [CrossRef] [Green Version]
- Rohle, D.; Popovici-Muller, J.; Palaskas, N.; Turcan, S.; Grommes, C.; Campos, C.; Tsoi, J.; Clark, O.; Oldrini, B.; Komisopoulou, E.; et al. An inhibitor of mutant IDH1 delays growth and promotes differentiation of glioma cells. Science 2013, 340, 626–630. [Google Scholar] [CrossRef] [Green Version]
- Ishay-Ronen, D.; Diepenbruck, M.; Kalathur, R.K.R.; Sugiyama, N.; Tiede, S.; Ivanek, R.; Bantug, G.; Morini, M.F.; Wang, J.; Hess, C.; et al. Gain Fat-Lose Metastasis: Converting Invasive Breast Cancer Cells into Adipocytes Inhibits Cancer Metastasis. Cancer Cell 2019, 35, 17–32. [Google Scholar] [CrossRef] [Green Version]
- Ishay-Ronen, D.; Christofori, G. Targeting Cancer Cell Metastasis by Converting Cancer Cells into Fat. Cancer Res. 2019, 79, 5471–5475. [Google Scholar] [CrossRef] [PubMed]
- Drapkin, B.J.; Minna, J.D. Studying Lineage Plasticity One Cell at a Time. Cancer Cell 2020, 38, 150–152. [Google Scholar] [CrossRef] [PubMed]
- LaFave, L.M.; Kartha, V.K.; Ma, S.; Meli, K.; Del Priore, I.; Lareau, C.; Naranjo, S.; Westcott, P.M.K.; Duarte, F.M.; Sankar, V.; et al. Epigenomic State Transitions Characterize Tumor Progression in Mouse Lung Adenocarcinoma. Cancer Cell 2020, 38, 212–228.e13. [Google Scholar] [CrossRef]
- Marjanovic, N.D.; Hofree, M.; Chan, J.E.; Canner, D.; Wu, K.; Trakala, M.; Hartmann, G.G.; Smith, O.C.; Kim, J.Y.; Evans, K.V.; et al. Emergence of a High-Plasticity Cell State during Lung Cancer Evolution. Cancer Cell 2020, 38, 229–246.e13. [Google Scholar] [CrossRef] [PubMed]
- Sahoo, S.; Mishra, A.; Kaur, H.; Hari, K.; Muralidharan, S.; Mandal, S.; Jolly, M.K. A mechanistic model captures the emergence and implications of non-genetic heterogeneity and reversible drug resistance in ER+ breast cancer cells. NAR Cancer 2021, 3, zcab027. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thankamony, A.P.; Subbalakshmi, A.R.; Jolly, M.K.; Nair, R. Lineage Plasticity in Cancer: The Tale of a Skin-Walker. Cancers 2021, 13, 3602. https://doi.org/10.3390/cancers13143602
Thankamony AP, Subbalakshmi AR, Jolly MK, Nair R. Lineage Plasticity in Cancer: The Tale of a Skin-Walker. Cancers. 2021; 13(14):3602. https://doi.org/10.3390/cancers13143602
Chicago/Turabian StyleThankamony, Archana P., Ayalur Raghu Subbalakshmi, Mohit Kumar Jolly, and Radhika Nair. 2021. "Lineage Plasticity in Cancer: The Tale of a Skin-Walker" Cancers 13, no. 14: 3602. https://doi.org/10.3390/cancers13143602
APA StyleThankamony, A. P., Subbalakshmi, A. R., Jolly, M. K., & Nair, R. (2021). Lineage Plasticity in Cancer: The Tale of a Skin-Walker. Cancers, 13(14), 3602. https://doi.org/10.3390/cancers13143602