Simple Summary
The diagnosis of myeloproliferative neoplasms requires assessment of a combination of clinical, morphological, immunophenotypic and genetic features, and this integrated, multimodal approach forms the basis for precise classification. Evaluation includes cell counts and morphology in the peripheral blood, bone marrow aspiration and trephine biopsy, and may encompass flow cytometry for specific questions. Diagnosis nowadays is completed by targeted molecular analysis for the detection of recurrent driver and, optionally, disease-modifying mutations. According to the current World Health Organization classification, all myeloproliferative disorders require assessment of molecular features to support the diagnosis or confirm a molecularly defined entity. This requires a structured molecular analysis workflow tailored for a rapid and cost-effective diagnosis. The review focuses on the morphological and molecular features of Ph-negative myeloproliferative neoplasms and their differential diagnoses, addresses open questions of classification, and emphasizes the enduring role of histopathological assessment in the molecular era.
Abstract
The diagnosis of a myeloid neoplasm relies on a combination of clinical, morphological, immunophenotypic and genetic features, and an integrated, multimodality approach is needed for precise classification. The basic diagnostics of myeloid neoplasms still rely on cell counts and morphology of peripheral blood and bone marrow aspirate, flow cytometry, cytogenetics and bone marrow trephine biopsy, but particularly in the setting of Ph− myeloproliferative neoplasms (MPN), the trephine biopsy has a crucial role. Nowadays, molecular studies are of great importance in confirming or refining a diagnosis and providing prognostic information. All myeloid neoplasms of chronic evolution included in this review, nowadays feature the presence or absence of specific genetic markers in their diagnostic criteria according to the current WHO classification, underlining the importance of molecular studies. Crucial differential diagnoses of Ph− MPN are the category of myeloid/lymphoid neoplasms with eosinophilia and gene rearrangement of PDGFRA, PDGFRB or FGFR1, or with PCM1-JAK2, and myelodysplastic/myeloproliferative neoplasms (MDS/MPN). This review focuses on morphological, immunophenotypical and molecular features of BCR-ABL1-negative MPN and their differential diagnoses. Furthermore, areas of difficulties and open questions in their classification are addressed, and the persistent role of morphology in the area of molecular medicine is discussed.
1. Introduction
The current World Health Organization (WHO) classification of tumors of hematopoietic and lymphoid tissue from 2017 classifies myeloid neoplasms by a combination of clinical, morphological, immunophenotypic and genetic features. Thus, an integrated, multimodal approach is needed for precise classification. Although the basis of diagnosis of myeloid neoplasms is still the morphology of peripheral blood and bone marrow aspirate, complemented by cytogenetics, flow cytometry and histology on trephine biopsies, increasing knowledge about molecular alterations has led to a paradigm shift in the classification, with molecular features gaining importance and resulting in entities defined in part, or even exclusively, by recurrent genetic alterations. However, especially in the setting of myeloproliferative and myelodysplastic/myeloproliferative neoplasms, the bone marrow trephine biopsy still has a crucial role to provide additional information about cellularity, histotopography of hematopoietic cells and their maturation, bone marrow stroma and bone structure [1]. Morphological assessment is enhanced by immunohistochemical staining, e.g., for assessing blast counts or highlighting atypical micromegakaryocytes difficult to identify by routine stains. The basis of a good investigation is an adequate biopsy of sufficient length (≥1.0 cm evaluable hematopoietic marrow) with good fixation and decalcification, and without significant aspiration or crush artifacts [1,2,3,4]. Decalcification with EDTA provides an advantage over acid decalcification in better preservation of nucleic acids for molecular studies and antigens for immunohistochemistry.
For the morphological review of a bone marrow trephine with a differential diagnosis of myeloproliferative neoplasm (MPN), hematoxylin and eosin (H&E) and Giemsa stains of good quality, a silver stain optionally complemented by a trichrome stain to evaluate fibrosis, an iron stain and, optionally, periodic acid-Schiff (PAS) and naphthol AS-D chloroacetate esterase (CAE) histochemical stains, render an optimal overview of the different components of hematopoiesis and supporting structures [1]. A basic immunohistochemical panel for myeloid analysis should contain CD34 and CD117 for blast screening and detection of mast cells, CD61 or CD42b for megakaryopoiesis, and CD71 or analogous markers for erythropoiesis, complemented by CD3 and CD20 or equivalent markers for lymphoid cells. Further immunohistochemical markers can be added for specific questions, e.g., CD14 and/or lysozyme for monocytic and monoblastic differentiation [1]. In contrast to other myeloid neoplasms, especially AML and MDS, flow cytometry has less impact on diagnosis and classification of MPN in comparison to morphological and molecular evaluation. Nevertheless, flow cytometry can demonstrate aberrant expression of surface markers, may show evidence of progression, and can help to separate MPN from other chronic myeloid disorders [5,6].
This review focuses on the histology and immunophenotype of BCR-ABL1-negative myeloproliferative neoplasms on bone marrow biopsies, including their molecular features and discusses questions, which remain open with regard to the current classification.
2. BCR-ABL1-Negative Myeloproliferative Neoplasms
The current update to the 4th Edition of the WHO classification of MPN mentions seven entities, including chronic myeloid leukemia-BCR-ABL1-positive, chronic neutrophilic leukemia (CNL), polycythemia vera (PV), primary myelofibrosis (PMF), essential thrombocythemia (ET), chronic eosinophilic leukemia (CEL)-not otherwise specified (NOS), and myeloproliferative neoplasm–unclassifiable (MPN-U) [1].
MPNs are clonal hematopoietic stem cell disorders characterized by an increased proliferation of one or more myeloid lineages predominantly arising in adults in their fifties to seventies, but also occasionally occurring in younger age groups including children [1,7,8,9]. In the peripheral blood, the levels of granulocytes, red blood cells, and/or platelets are often elevated due to effective maturation lacking dysplasia, and frequently a hypercellular bone marrow is found. Clinically, a splenomegaly and a hepatomegaly due to extramedullary hematopoiesis are common [1]. MPNs in the chronic phase are unique among myeloid neoplasms in that clinical symptoms and complications are primarily due to the overproduction of mature and functional hematopoietic elements, e.g., causing circulatory problems or thrombotic events. All MPNs, though at very different frequencies, can progress to blast crisis, characterized by >20% blasts in the blood or bone marrow, and development of hematopoietic insufficiency.
Chronic myeloid leukemia (CML) is a molecularly defined disease and requires the detection of the t(9;22) translocation, resulting in the Philadelphia (Ph) chromosome and the BCR-ABL1 fusion gene [10,11]. By this criterion, CML is set apart from other MPNs, in which molecular features are important, but not sufficient on their own to define an entity; therefore, this review focuses on BCR-ABL1-negative MPN.
2.1. Chronic Neutrophilic Leukemia (CNL)
This rare MPN shows significant neutrophilia in the peripheral blood and is characterized by hypercellularity of the bone marrow and hepatosplenomegaly. By definition, CNL lacks a BCR-ABL1 fusion gene and reactive neutrophilia must be excluded [12]. Its true incidence meeting the current diagnostic criteria is unknown. Over 200 cases are published, but if the criteria are applied strictly, some of these cases likely would be excluded [12,13,14]. CNL occurs almost always in older adults with a median of 66 years and a slight male predominance [12,13,14,15]. By WHO definition the blood white blood cell count in peripheral blood is ≥25 × 109/L and segmented neutrophils plus banded neutrophils are ≥80% of the leukocytes [12].
In bone marrow biopsies, the cellularity is typically hypercellular with a dominance of neutrophilic proliferation and a myeloid-to-erythroid ratio of up to 20:1, but without any significant increase of neutrophilic precursors. Both erythropoiesis and megakaryopoiesis show a normal maturation without significant cytological dysplasia [12,13]. If significant dysplasia is found, a diagnosis of atypical CML (aCML) should be entertained [12]. Typical CNL shows no or only minimal fibrosis on reticulin staining [12].
An important differential diagnosis of CNL is a neutrophilic leukemoid reaction in the setting of a plasma cell neoplasia. The WHO classification recommends in patients with leukocytosis and multiple myeloma or monoclonal gammopathy of undetermined significance the demonstration of cytogenetic or molecular clonality in the neutrophil lineage before an additional diagnosis of CNL is made [12,13].
The molecular landscape of CNL is dominated by CSF3R mutations, which are found in the majority of CNL cases (64% to 100%) (Table 1) [16,17,18,19,20,21,22], first described in 2013 [23]. CSF3R mutations can be divided into different groups: the common membrane proximal mutations, primarily T618I and T615A, resulting in ligand-independent receptor activation, and truncating mutations in the cytoplasmic tail between amino acid 651 and 836, which almost always are compound mutations found in about 25% of patients with membrane proximal mutations, suggesting that the latter on their own lack sufficient oncogenicity [23,24,25]. Patients with T618I mutation show adverse clinical and laboratory features as well as lower overall survival [17]. In addition to CSF3R alterations, other genes are mutated, including ASXL1 (47% to 77%), SETBP1 (0% to 75%), SRSF2 (44%), TET2 (20.5% to 50%), CALR (5% to 12.5%), and JAK2 (8%) (Table 1) [16,17,18,19,20,21,22]. Some of these mutations, e.g., in epigenetic modifier and spliceosome genes, are classic CHIP (clonal hematopoiesis of indeterminate potential) mutations (DNMT3A, ASXL1, TET2, JAK2, PPMID1, SF3B1, SRSF2, TP53, CBL, and others). CHIP can be detected in about 10% of all healthy individuals over 65 years of age and is defined as the occurrence of a clonal mutation in at least 4% of hematopoietic cells without evidence of a hematological neoplasm. CHIP is an optional preneoplasia, and is correlated with the development of hematological neoplasms with a probability of 0.5 to 1% per year, especially myelodysplastic syndrome (MDS) and acute myeloid leukemia (AML), depending on the clone size, number of mutations and affected genes [26,27]. Based on the common occurrence of CHIP mutations in CNL, it has been suggested that both CNL and atypical chronic myeloid leukemia (aCML), discussed below, should be considered disorders of clonal hematopoiesis [16].

Table 1.
Main genetic alterations in myeloproliferative diseases.
As mentioned above, a main differential diagnosis of CNL is aCML. There are morphological and laboratory criteria separating the two disorders, including neutrophil precursors ≥ 10% of the leukocytes and the presence of dysgranulopoiesis, which may include abnormal chromatin clumping, as well as dysplasia in other cell lines in aCML. Although in the first study CSF3R mutations were observed in 44% of aCML [23], further studies revealed a much lower frequency between 0 and 11%, [19,46,47,48]. There are also other entities which can contain a CSF3R alteration. In adult and pediatric acute myeloid leukemia about 0.5 to 1% and about 2 % harbor a CSF3R mutation, respectively [23,49,50,51], and in chronic myelomonocytic leukemia (CMML) about 4% show such a mutation [52]. The mutational landscape of aCML is dominated by ASXL1 mutations (81% to 92%), followed by SRSF2 (37% to 48%), TET2 (37%) and EZH2 (30% to 32%) [16,46]. SETBP1 mutations have a frequency between 7% and 38% in aCML [16,46,53,54]. In CNL, SETBP1 is mostly associated with the CSF3R mutation, so that despite a high frequency in both entities, the sole occurrence of SETBP1 is more in line with aCML [13,55]. Due to the overlapping mutational and expression profile of CNL and aCML, and to a lesser extent chronic myelomonocytic leukemia (CMML) and MDS/MPN unclassifiable, and their association with CHIP, it has recently been suggested that these entities might represent a continuum of closely related hematological disorders rather than discrete entities [16].
2.2. Polycythemia Vera (PV)
This MPN is characterized by an increase of red blood cell volume and absence of physiological regulation of erythropoiesis as evidenced by low erythropoietin levels. According to current WHO criteria, PV shows elevated hemoglobin concentration (>16.5 g/dL in men, >16.0 g/dL in women), or elevated hematocrit (>49% in men, >48% in women), or increased red blood cell mass (>25% above mean normal predicted value) (Table 2) [56]. It is also a disease of elderly people with a median age of 60 years, and with a slight male predominance with a male-to-female ratio of 1–2:1 [28,56,57,58]. PV can be divided into two phases: the polycythemic phase and the post-polycythemic myelofibrosis phase. The second phase is characterized by cytopenia (including anemia), bone marrow fibrosis, extramedullary hematopoiesis, and hypersplenism [56]. In the initial phase, some patients show obvious thrombocytosis and no elevated hemoglobin/ hematocrit, mimicking ET and designated the pre-polycythemic phase, but subsequent development of raised red blood cell counts and typical bone marrow pathology unmask the PV diagnosis [56,59,60,61].
Table 2.
Diagnostic criteria for the main Ph−myeloproliferative neoplasms [56,62,63].
The typical bone marrow biopsy (Figure 1a–d) in the polycythemic phase is hypercellular with a generalized panmyelosis, including the subcortical, normally hypocellular, spaces [56,65,66,67]. Most prominent are the usually normoblastic erythroid precursors, which lie in enlarged islands, and the megakaryocytes, which often show hypersegmented or pleomorphic nuclei and significant size variability, and may form loose, but not tight clusters. Granulopoiesis is in general morphologically unremarkable but increased for age [65,66,68,69]. As mentioned above, the so-called masked/ prodromal PV can mimic ET at the beginning, especially due to increased megakaryopoiesis with often hypersegmented nuclei in the context of high platelet counts and no elevated hemoglobin/hematocrit [60,61,70]. PV has no significant increase in reticulin fiber network in up to 80% of cases, with the remainder showing mild to moderate fibrosis at initial diagnosis, a sign for early progression to post-polycythemic myelofibrosis [65,66,67,71,72,73,74,75]. In nearly all cases of PV no stainable iron can be found in bone marrow aspirate and biopsy. Reactive lymphoid aggregates exist in about one-fifth of cases [66,68,69,75,76]. Post-polycythemic myelofibrosis is characterized by an overt reticulin and collagen fibrosis of the bone marrow with increasing hypocellularity, decreased erythropoiesis resulting in anemia, more clustered megakaryocytes with often hyperchromatic and dysmorphic nuclei, and in the late stage decreased granulopoiesis. Post-PV myelofibrosis occurs in 4.9–6% of cases at 10 years and 6–14% of cases at 15 years [77]. Whether post-polycythemic myelofibrosis can be discerned from PMF remains controversial, but increased cellularity, less prominent atypia and less clustering of megakaryocytes have been identified as features of fibrotic progression of PV [66,68,75,78,79,80]. In addition to anemia, additional clinical criteria are increasing splenomegaly, leukoerythroblastosis, weight loss, night sweats and unexplained fever [78]. Neutrophilic leukocytosis (CNL-like) in this stage is linked to an adverse course of the disease (see below) [81].
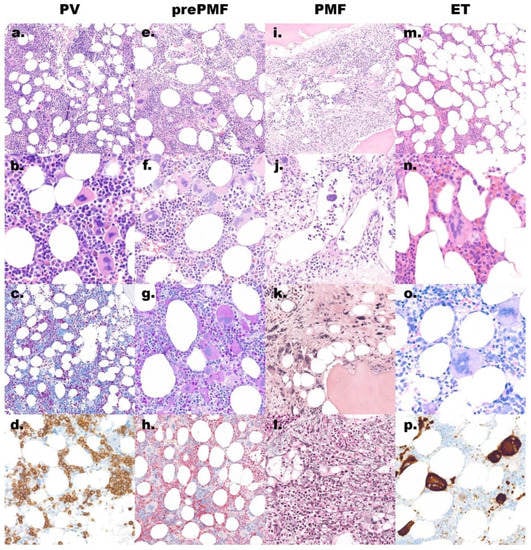
Figure 1.
Morphology overview of the main discussed entities. (a–d) Polycythemia vera (PV) shows a hypercellular bone marrow biopsy: (a) hematoxylin and eosin (H&E) stain, original magnification 200×; with increased, morphologically quite variable megakaryocytes: (b) H&E stain, original magnification 600×; increased erythropoiesis and reduced granulopoiesis: (c) naphthol AS-D chloracetate esterase stain, original magnification 200×; CD71 marks the increased and left shifted erythropoiesis: (d) immunoperoxidase, original magnification 400×; (e–h) Prefibrotic/early primary myelofibrosis (pre-PMF) shows a hypercellular bone marrow: (e) H&E stain, original magnification 200×; with increased megakaryocytes with tight clusters and large, atypical, often cloudy nuclei: (f,g) H&E and PAS stain, original magnification 400×; the granulopoiesis is often increased: (h) naphthol AS-D chloracetate esterase stain, original magnification 200×; (i–l) Overt primary myelofibrosis (PMF) shows a hypercellular bone marrow with typical paratrabecular fat tissue: (i) H&E stain, original magnification 200×; open sinus with hematopoiesis: (j) H&E stain, original magnification 400×; in late stage a reduced cellularity with irregular megakaryocytes lying in a streaming pattern with paratrabecular fat tissue: (k) H&E stain; original magnification 400× and increased reticulin fibers: (l) silver stain, original magnification 400×; (m–p) Essential thrombocythemia (ET) has usually a normocellular bone marrow: (m) H&E stain, original magnification 200×; with increased megakaryocytes with hypersegmented nuclei: (n) H&E stain and (o) Giemsa stain, original magnification 600×; the CD61 stain (p) marks also these large megakaryocytes, immunoperoxidase, original magnification 400×.
Nearly all patients with PV have a JAK2 (Janus kinase 2) mutation, of which 96% harbor the classic JAK2 V617F mutation in exon 14 and about 3% an exon 12 mutation (Table 1) [28,29,30,31,32,33]. These lead to constitutive activation of the JAK-STAT pathway and independent growth [82,83]. Both mutations are prognostically similar, but patients with an exon 12 mutation usually show prominent erythroid hematopoiesis and lower leukocytes and platelet counts, whereas a younger age at diagnosis and higher hemoglobin levels have been found in some, but not all studies of these patients [84,85]. In 2014, two cases of JAK2 V617F and JAK2 exon 12-negative PVs were described with a calreticulin (CALR) mutation (52 base pair deletion) in peripheral granulocytes, providing an alternative pathogenesis for JAK2-negative PV [64]. Recently, we observed a similar case with a predominance of erythropoiesis, enlarged and irregular megakaryocytes without clustering and a confirmed CALR mutation (type 1), which was classified in synopsis with the peripheral blood values (hemoglobin 17.5 g/dL, thrombocytes 565 × 109/L) as a JAK2WT/CALRmut PV.
Patients with PV tend to have higher allelic burden of the JAK2 mutation in comparison to patients with ET, with about 25 to 30% of PV and 2 to 4% of ET harboring a homozygous JAK2 mutation, usually due to uniparental disomy. Homozygosity is associated with a more symptomatic course and a higher progression to secondary myelofibrosis in both PV and ET, with a higher risk of thrombotic events in patients with ET [86]. Additionally, an allelic burden ≥ 75% in PV at the time of diagnosis was associated with high-risk disease [87]. In recent years, additional mutations to the driver mutation were detected, especially classic CHIP mutations. In PV, these include TET2 (10–20%), ASXL1 (up to 10%), DNMT3A (5%), and SF3B1 (5%), which affect DNA methylation, histone modification and mRNA splicing (Table 1) [34,35,36]. An interesting feature is the time point of acquisition of these secondary mutations and their correlation with the clinical phenotype. When JAK2 is the first mutation, the patient presents more commonly with PV, whereas when TET2 or DNMT3A occur prior to JAK2, the phenotype is more likely ET [35,88]. The additional mutations also have a prognostic impact, with ASXL1, SRSF2, and IDH2 mutated cases showing an adverse prognosis in PV [36].
2.3. Primary Myelofibrosis (PMF)
PMF is a myeloproliferative neoplasm with a dominance of the granulocytic lineage and atypical, large megakaryocytes in the bone marrow, progressive fibrosis and development of significant extramedullary hematopoiesis during the course of the disease. There are two stages of development: initially a prefibrotic/ early stage, followed by an overt fibrotic stage. It is mostly a diagnosis of the sixth and seventh decade of life and the gender distribution is roughly even [62]. About 15% of PV patients, and a small minority of patients with ET, show progression to a myelofibrotic phase, usually many years after primary diagnosis, called secondary myelofibrosis [78,89]. Whereas the prefibrotic stage of PMF frequently shows leuko- and thrombocytosis in the peripheral blood very similar to ET, the typical features of the overt fibrotic stage are increasing anemia, a blood smear with leukoerythroblastosis and teardrop-shaped red blood cells [62].
The molecular landscape of PMF shows one of the typical and disease-defining mutations in JAK2 (virtually always V617F), CALR encoding for calreticulin and MPL (myeloproliferative leukemia virus oncogene) encoding the thrombopoietin receptor. More than half of the cases (about 50–65%) show the JAK2 mutation in exon 14, followed by 25 to 30% with a CALR mutation and 8 to 10% with a MPL mutation (Table 1) [29,37,38]. Calreticulin is a molecular chaperone residing in the endoplasmatic reticulum, and a crucial protein in calcium homeostasis and protein folding [90]. About 80% of all CALR alterations are made up of two main mutations, both affecting exon 9: the first one (type 1) is a long deletion (c.1092_1143del, p.L367fs*46), and the second (type 2) is a short insertion (c.1154_1155insTTGTC, p.K385fs*47). The remaining mutations are grouped with one of the two main alterations based on their similarity [91,92]. In PMF, the type 1 alteration is more common than type 2 (70%/13%), whereas in ET the two types are more equally distributed (51%/39%) [93]. All CALR mutations result in a + 1 frameshift resulting in a new C-terminus lacking the KDEL motif required for retention in the endoplasmatic reticulum. Functional studies have shown that mutant CALR binds to MPL resulting in activation of MPL and thereby a downstream activation of the JAK-STAT pathway [94,95,96,97]. The constitutive MPL activation common to both MPL and CALR mutated MPN, induces megakaryopoiesis and platelet production and explains the thrombocytosis as a common laboratory feature. Patients with a type 1 CALR mutation have a significantly longer survival in comparison to JAK2, MPL, type 2 CALR and the rare triple-negative cases. The absence of a type 1 CALR alteration, or the presence of an additional ASXL1/SRSF2 mutation, were separately prognostic for lower survival. Moreover, patients with the type 1 CALR mutation and additional ASXL1/SRSF2 alterations have a significantly longer survival in comparison to patients with these additional mutations and a nontype 1 alteration, suggesting that type 1 mutations ameliorate the actual negative effect of these additional mutations [98]. The most common MPL mutations are point mutations in exon 10 at position 515 (mostly W515L and W515K) within a transmembrane domain of the protein resulting in a gain of function, but other alterations are also described like the S505N mutation [99,100,101,102]. As mentioned above, there are additional secondary mutations in PMF cases, including CHIP mutations such as ASXL1 (up to 35%), TET2 (20%), SRSF2 (up to 20%), U2AF1 (16%), ZRSR2 (10%), SF3B1 (10%), DNMT3A (5–15%), among others, which are less common (Table 1) [39,40,41,42]. These secondary mutations mainly affect genes involved in DNA methylation, mRNA splicing and histone modification and transcription factors. These secondary mutations, more typically found in CHIP and MDS, are more common in PMF than ET and PV, and might, in part, explain the worse prognosis of PMF. A small number of cases, up to 12% of PMF are so-called triple negative cases lacking the classic driver mutations [34,41]. Deep sequencing identified mutations outside the classic regions of MPL exon 10 in about 10% of these triple-negative cases [103]. Although PMF shows the worst prognosis of the three classical Ph− MPN, there is a wide range of clinical behavior, with some patients, especially those with pre-PMF, show a relatively stable course over many years, whereas others progress rapidly and die within a few years after diagnosis [37,89]. In order to stratify patients according to their risk for disease progression and selection of appropriate therapy, including allogeneic stem cell transplant, a variety of risk scores such as the DPISS, MIPSS70 and GIPSS, containing clinical, genetic or a combination of these parameters, have been developed over the years [37].
2.3.1. Prefibrotic/Early Primary Myelofibrosis
The diagnosis of a prefibrotic PMF is based on a set of clinical, laboratory, morphological and molecular criteria (Table 2) [62]. Clinically, these include anaemia (without any other cause), thrombocytosis, leukocytosis above 11 × 199 /L, splenomegaly or an increase of lactate dehydrogenase (LDH) [62,104]. Thrombocytosis can be quite high, and if the remaining blood values are normal or borderline the differentiation from ET is difficult and a bone marrow biopsy is mandatory [105,106,107]. About 30% to 50% of cases with PMF are diagnosed in this prefibrotic stage [73,108,109,110].
The bone marrow is hypercellular and shows a proliferation of megakaryopoiesis and granulopoiesis with usually decreased erythropoiesis (Figure 1e–h). Granulopoiesis can be left shifted, but myeloblasts are usually not increased [109,110,111]. The most prominent and distinctive feature of prefibrotic PMF is megakayopoiesis. Distribution and morphology are important criteria for the distinction from ET. The megakaryocytes often form dense clusters and are commonly found next to vessels and bone trabeculae. Megakaryocytes are usually enlarged but scattered small cells may also be present; they show an increased nuclear/cytoplasmic ratio, an abnormal density of the chromatin with cloud-like nuclei and in part also dispersed naked nuclei. In contrast to the other MPN, megakaryocytes are distinctly more atypical in pre-PMF [69,70,73,76,107,110,112]. For diagnosis of this stage, a maximum of grade 1 fibrosis may be present according to the semiquantitative bone marrow fibrosis grading system [67,113]. In up to 20% of BM, trephines reactive lymphoid aggregates can be found [107,110].
2.3.2. Overt Primary Myelofibrosis
More than 50% of patients with PMF are diagnosed in the overt fibrosis stage [73,109,110,114]. The main feature of this stage is the clear increase in reticulin or collagen fibers (grade 2 or 3), frequently associated with osteosclerosis (Table 2) [67,113]. The bone marrow itself (Figure 1i–k) is usually normo or hypocellular and only rarely hypercellular, with reduced hematopoiesis and areas of loose connective tissue. A characteristic feature of PMF is the redistribution of fat cells along the bone trabecules. Megakaryocytes are very conspicuous with numerous atypical shapes and frequent clustering. Granulopoiesis, and especially erythropoiesis, are often significantly reduced. Important stromal changes are a high density of vessels and dilated sinuses with characteristic intrasinusoidal hematopoiesis [110,115,116,117]. Late stage PMF cases may show almost complete absence of hematopoiesis with extensive fibrosis, including collagen and osteosclerosis with irregular apposition of osteoid. As the disease progresses, the occurrence of extramedullary hematopoiesis becomes more evident, mainly in the spleen and less frequently in the liver and other organs. In the spleen, there is hyperplasia of the red pulp, with cells of maturing trilineage hematopoiesis. The appearance of dense monomorphic round cell areas and nodules should suggest a differential diagnosis of transformation to an extramedullary manifestation of AML/myelosarcoma and trigger additional immunohistochemical staining for blasts (CD34, CD117) [118,119,120,121,122].
2.4. Essential Thrombocythemia (ET)
ET is characterized by dominance of megakaryopoiesis with prominent thrombocytosis in the peripheral blood [63]. The WHO classification requires a platelet count ≥ 450 × 109 /L, a typical bone marrow morphology, the exclusion of other WHO-defined diseases such as CML, PV and PMF or other myeloid neoplasms, and one of the driver mutations in JAK2, CALR, or MPL. In their absence, the WHO classification calls for the presence of another clonal marker or the exclusion of reactive thrombocytosis (Table 2) [63]. Like the other classical MPNs, ET is also a disease of older people, with a peak between 50 and 60 years and a minimal predilection of women; especially in women there is a further peak around 30 years [38,105,123,124,125]. ET can also be observed in children. Like in PV, there is a small risk to develop a so-called post-ET myelofibrosis, ranging from <0.8–4.9% after 10 years [43,77]. The varying frequencies of post-ET myelofibrosis in part can be explained in the difficult separation of ET from pre-PMF.
A bone marrow biopsy (Figure 1m–p) is, in general, normocellular, and only few cases are hypercellular [61,67,76]. The most important and prominent finding is the increased megakaryopoiesis with many big, “aged” megakaryocytes, showing large cytoplasm and lobated and hypersegmented “staghorn” nuclei, which are mostly distributed singly throughout the bone marrow, sometimes in loose clusters. The erythropoiesis and granulopoiesis, in general, are normal, but due to hemorrhage, erythropoiesis may be left shifted [63,105,106,126,127,128]. By definition, significant fibrosis is absent, and only grade 1 fibrosis is allowed, which occurs in less than 5% of cases. [38,67,74,76,106,123,126,127,129].
The differential diagnoses are broad and the morphological findings in the bone marrow trephine are crucial. With increase in granulocytic and erythroid lineage and the JAK2 V617F mutation, a masked/prodromal PV should be considered [60,61,70]. In comparison to PMF, megakaryocyte clusters are rare and normally loose, and the megakaryocytes are not atypical, without clumped chromatin and cloud-like nuclei [107,112,126,127,130]. In the presence of anemia, an MDS/MPN with ring sideroblasts and thrombocytosis (RARS-T) needs to be considered.
A diagnosis of post-ET myelofibrosis requires a documented diagnosis of ET and a bone marrow fibrosis of grade 2 or 3, and additionally at least two more criteria of the following are needed: anaemia, leukoerythroblastosis, increasing splenomegaly, elevated LDH value, or two or three of the following symptoms: >10% weight loss within six months, night sweats or unexplained fever above 37.5°C [63,78].
The mutational landscape of ET is similar to PMF with regard to driver mutations: about 50–60% of cases harbor the JAK2 mutation V617F, but no JAK2 exon 12 mutations, up to 30% show CALR and 3–8% MPL mutations; up to 15% are “triple-negative” for the classic MPN driver mutations and are difficult to separate from reactive thrombocytosis (Table 1) [29,34,38,43,44]. As mentioned above, cases which acquire TET2 or DNMT3A mutations before the JAK2 mutation more often have an ET phenotype [35,88], and the two different types of CALR mutations are more balanced in ET vs. PMF [93]. The type 1 CALR mutation shows a significantly higher risk of transformation to myelofibrosis in ET, and patients with both CALR types show a higher platelet count, lower hemoglobin and leukocyte values in comparison to JAK2-positive ET [131,132]. Patients with CALR mutated ET are also younger, have a lower risk for thrombosis, and show no progression to PV (vs. 29% at 15 years), assuming that JAK2-positive ET and PV are different stages/phenotypes of a single disease, and CALR-positive ET is a different nosological entity [133]. Atypical JAK2 mutations are found in single, classic triple-negative ET cases including V625F and F556V [103]. Although at a lower frequency than in PMF, additional mutations including CHIP mutations, occur in ET, including TET2 (10–15%), ASXL1 (5–10%), DNMT3A (5%), SF3B1 (3%), and others with lower frequencies (Table 1) [34,36]. Some additional mutations show an adverse prognostic impact in ET, including SH2B3/LNK, SF3B1, U2AF1, TP53, IDH2, and EZH2 [36].
2.5. Chronic Eosinophilic Leukemia, Not Otherwise Specified (CEL, NOS)
CEL, NOS is still a diagnosis of exclusion and is characterized by an increase of eosinophils and their precursors in the bone marrow, the peripheral blood and tissue, and consecutive damage of organs [134]. The diagnostic criteria for CEL in the WHO classification are an eosinophil count ≥ 1.5 × 109 /L and evidence of a clonal cytogenetic/molecular genetic abnormality, or blast cell count of ≥2% of cells in the peripheral blood or ≥5% in the bone marrow. Additionally, other WHO-defined neoplasms, including all other MPNs, CMML, aCML, and especially myeloid/lymphoid neoplasms with eosinophilia and gene rearrangement of PDGFRA, PDGFRB or FGFR1, need to be excluded, and no PCM1-JAK2, ETV6-JAK2 or BCR-JAK2 fusion, blasts ≥ 20% or inv(16)(p13.1;q22), t(16;16)(p13.1;q22), or t(8;21)(q22;q22.1) are allowed for a diagnosis of CEL [134]. If eosinophilia persists for ≥6 months, and any other MN and reactive eosinophilia have been excluded, a diagnosis of idiopathic hypereosinophilic syndrome (HES) is made in the absence of both an increase in blasts and clonal cytogenetic/molecular genetic abnormality [134,135,136]. Without any evidence of organ damage or dysfunction relatable to tissue hyereosinophilia, the term “(idiopathic) hyereosinophilia” is suitable [134,135,136]. Due to the diagnostic difficulties in differentiating CEL, NOS from other related eosinophilic diseases, valid epidemiological data are rare, but the disease seems to be more common in men and occurs in median in the seventh decade [45,137].
The bone marrow biopsy in CEL NOS is hypercellular with a dominance of eosinophils with mostly normal maturation. The amount of myeloblasts can be increased above 5%, another defining criterion for CEL in the absence of cytogenetic or molecular genetic alterations. The red cell lineage and the megakaryocytes are typically normal, but some dysplastic changes can occur. Charcot-Leyden crystals can be present. Only rare cases show relevant fibrosis (grade 2/3) [45,134,137,138].
For a diagnosis of CEL, NOS extensive molecular analyses are needed, including mutational analysis, RNA-based fusion examination or fluorescence in situ hybridization (FISH) for rearrangement analysis, and cytogenetic studies. Typical nonspecific cytogenetic alterations in CEL, NOS are trisomy 8, loss of chromosome 7, or isochromosome 17 [134,139,140]. Next-generation sequencing identified recurrent mutations in primary idiopathic HES, which showed no significant difference in disease-specific survival to CEL, NOS and, therefore, could be reclassified as CEL, NOS. The identified mutations were ASXL1 (43%), TET2 (36%), EZH2 (29%), SETBP1 (22%), CBL (14%), and NOTCH1 (14%) [45]. A further study showed known or predicted pathogenic mutations in TET2, ASXL1, KIT, IDH2, JAK2, SF3B1 and TP53 [141]. In 2018, STAT5B N642H was identified as an additional hotspot mutation in the setting of eosinophilia. The study also showed additional mutations in most cases; patients with a solitary STAT5B or with a further SF3B1 mutation had a significantly better overall survival in comparison to patients with other additional alterations [142]. Since in elderly persons, many of these mutations with the exception of STAT5B can occur as CHIP mutations, it is recommended to eliminate all possible reasons of reactive eosinophilia before making a CEL, NOS diagnosis based on these alterations in an older person [134]. Currently, cases with rearrangements of ABL1, FLT3 and JAK2, with the exception of PCM1-JAK2, are also classified as CEL, NOS even though they show similarities and often indistinguishable clinical features to the category of “myeloid/lymphoid neoplasms with eosinophilia and fusion genes”, indicating that these cases might be classified differently in the future [135,143]. One additional interesting alteration is the ETV6-ABL1 fusion, which can occur both in children predominantly as acute lymphoblastic leukemia (ALL), and in adults mostly as myeloid neoplasm (MPN-U or AML) strongly reminiscent of BCR-ABL1 positive CML [135,144]. All MPN and AML cases showed eosinophilia, but only a minority of the ALL cases. Therefore, all cases of BCR-ABL1-negative MPN resembling CML should additionally be analyzed for this ETV6-ABL1 fusion [144]. Of note, the formation of this fusion gene needs a minimum of three chromosomal breaks, thus routine karyotyping is frequently noncontributory and detailed molecular analysis is needed [144,145]. In conclusion, the detection of one of the disease-specific genetic alterations described above leads to a specific diagnosis associated with eosinophilia instead of CEL, NOS.
2.6. Myloproliferative Neoplasm, Unclassifiable (MPN-U)
This category is a waste basket category of cases, which show clear features (clinical, laboratory, morphological, molecular) of MPN, but cannot be categorized to any specific WHO-defined entity, or show overlap between entities. Any disease-defining genetic alterations exclude the diagnosis of MPN-U, such as BCR-ABL1 and PCM1-JAK2, and rearrangements of PDGFRA, PDGFRB, or FGFR1. [146]. Larger series diagnosed 10–15% of all MPNs as unclassifiable [147,148,149], but accurate analysis including clinical, morphological and molecular characteristics should reduce the frequency to less than 5% [146,147,150,151]. Molecular analysis identifies the same driver mutations as in other MPNs. Gianelli et al. found a JAK2 mutation in 71.8%, in 11.3% a CALR alteration (type 1: 5.6%, type 2: 2.8%, others: 2.8%), and in 2.8% a MPL mutation [147]. As mentioned above, some cases of MPN-U may contain a t(9;12) with ETV6-ABL1 fusion. Altogether, the category of MPN-U is quite heterogeneous and reflects the biological continuum between the defined entities. Nevertheless, a careful examination is needed to diagnose or exclude a distinct entity before making an MPN-U diagnosis [146,147].
3. Special Issues and Still Open Questions
3.1. Early Stage Classic Ph− MPN–Clinico-Pathological Versus Molecular Classification
With increasing knowledge about the molecular profiles of classic Ph− MPN, it has become clear that the type of driver mutation, the presence of additional mutations, and their sequence, have a significant influence on prognosis. Furthermore, differentiation between early-stage MPN can be difficult on a clinical morphological basis. For example, a major morphological challenge is the differential diagnosis between essential thrombocythemia and prefibrotic PMF, which show significant differences in progression and survival in many, but not all published studies. The 15-year survival rate is 59% vs. 80% in prefibrotic PMF and ET, respectively; the leukemic transformation rate after 15 years is 11.7% vs. 2.1%, the progression to overt PMF is 16.9% to 9.3%, and the survival of ET patients was similar to the general European population [123]. However, studies from different groups have shown conflicting results concerning reproducibility of the WHO criteria for the separation of these entities and the prognostic impact [152]. Therefore, progression rates for ET and pre-PMF show significant variation in different studies, depending on diagnostic criteria (WHO versus polycythemia vera study group (PVSG)) and morphological evaluation. Therefore, changes in diagnostic criteria, mainly a stronger inclusion of driver mutation status and secondary mutations, have been advocated [37,153]. Given the importance of mutation order for the clinical phenotype of MPN, as mentioned above, examining the order of appearance of secondary mutations based on allelic frequencies might help to predict disease progression better [35,88]. However, despite the increasing weight of molecular markers, clinico-pathological classification remains important, e.g., as demonstrated for the triple negative MPN group. Whereas triple negative ET showed the best overall survival in a univariate, but not in multivariate analysis [154], triple negative PMF showed the worst median overall survival of all PMF [155]. In order to reduce the influence of interobserver variation, advanced image analysis tools have been used to address the morphological separation of Ph− MPN [156].
A related issue showing the importance of clinico-pathological correlation is the occurrence of JAK2 mutations in individuals without clinical or laboratory evidence of an MPN, which in this setting is considered a CHIP mutation [157]. It is currently unclear how many of these individuals will progress to develop manifest MPN, and what are the underlying mechanisms.
3.2. Myeloproliferative Neoplasms with Multiple Driver Mutations
Although the classical driver mutations including JAK2, CALR, MPL and BCR-ABL1 usually are mutually exclusive, rare cases of chronic myeloid neoplasms with multiple driver mutations have been observed. One group of cases shows, in addition to the classic BCR-ABL1 fusion, a JAK2 or CALR mutation. The CML-associated thrombocytosis and leukocytosis often obscure the Ph– MPN, which is only discovered after successful treatment of the CML with tyrosine kinase inhibitors [158,159]. The temporal sequence of these mutations is variable, with the BCR-ABL1 fusion preceding or following a CALR or JAK2 alteration. In the latter case, classic ET or pre-PMF features in the bone marrow are superseded by the classic CML morphology and leukocytosis, and reverse back to the old morphological picture upon successful treatment [160,161,162]. Although the two alterations potentially might reside in the same clone, the evidence of these cases indicates that BCR-ABL1 and JAK2 or CALR mutations reside in distinct clones. In addition, rare cases with combinations of JAK2, MPL or CALR mutations have been observed [159].
3.3. Genetic Overlaps with Other Chronic Myeloid Neoplasms
Similar to the phenomenon described above, MPN may show genetic alterations usually associated with other chronic myeloid neoplasms, and vice versa. Typical MPN driver mutations can occur in the setting of other disorders, such as in myelodysplastic syndromes. [159]. Classification in these unusual cases can be difficult but should rely on the aggregated clinical and pathological features. Examples described in the Workshop Report of the 2017 EAHP/SH meeting included cases of ET with del(5q) or PMF with SF3B1 p.K666N mutation and ring sideroblasts, in addition to a canonical JAK2 V617F mutation. Based on morphology, peripheral blood counts and other clinical features, these cases were classified as MPN. On the other hand, cases with classic MDS features may show additional JAK2 or MPL mutations. As long as the clinical and morphological criteria for an MDS are met, classic MPN mutations can occur without the diagnosis being changed, and may represent a background CHIP mutation.
3.4. Unusual Types of Progression of MPN
Increasing myelofibrosis, called secondary MF in ET and PV, or transformation to acute leukemia, sometimes in the form of extramedullary myelosarcoma or pure erythroid leukemia, are the two traditionally recognized forms of disease progression in MPN [163].
As briefly mentioned above, neutrophilic leukocytosis can occur in PV patients around the time point of post-polycythemic myelofibrosis [81]. These patients showed persistent absolute leukocytosis of ≥13 × 109 /L (median: 25.1 × 109 /L) with neutrophilic granulocytes usually ≥75% of all white blood cells. The bone marrow presented a prominently increased myeloid to erythroid ratio simulating CNL or CML, but BCR-ABL1 fusions or additional mutations in CSF3R, SETBP1, or SRSF2, potentially explaining the leukocytosis, could not be detected [78]. In comparison with a cohort without leukocytosis in the postpolycythemic myelofibrosis phase, the first group had a shorter overall survival (median overall survival 181 vs. 252 months).
Another unusual type of progression heralding worse prognosis is the development of absolute monocytosis (>1 × 109 /L) in patients with PMF [164]. Interestingly, bone marrow biopsies revealed a prominent myelomonocytic increase resembling CMML after the onset of monocytosis instead of the characteristic PMF morphology. Of note, cases of PMF with monocytosis at onset may be misdiagnosed as CMML if bone marrow biopsy and testing for MPN driver mutations are not performed [165].
3.5. How Much Molecular Testing Is Needed for a Diagnosis of MPN?
Recent technical advances have made high throughput molecular testing, including next generation sequencing, more accessible for many clinical laboratories, and have resulted in its widespread use for the diagnostic workup of MPN. Limited approaches such as single or few gene assays for the main drivers BCR-ABL1, JAK2, MPL and CALR can still serve as the method of choice in straightforward cases of MPN, but unusual clinico-pathological features, or absence of MPN driver mutations, should prompt more comprehensive testing. In addition, detection of the nonspecific disease-modifying genetic alterations described above may serve for improved prognostication, especially in PMF. Ultimately, more comprehensive genotyping will allow a personalized risk assessment for patients with MPN and better therapeutic stratification. For cases lacking typical driver mutations, translocations involving PDGFRA/B, FGFR1 and JAK2, and potentially AML-type alterations, should be investigated in addition to mutations found in myeloid neoplasms (MN) in general. In addition to BM aspirates and peripheral blood leukocytes, EDTA-decalcified bone marrow biopsies are also suitable for extensive testing, including RNA analysis for translocation detection.
Cases requiring more extensive analyses are, for example: MN with clinical/morphological/phenotypical overlaps, such as MPN-U and MDS/MPN-U; MDS with fibrosis or systemic mastocytosis with associated hematological neoplasm; MN presenting as suspected accelerated phase or blast crisis to identify an underlying/antecendent chronic MPN, in the setting of a relapse or progression/transformation to identify the clonal evolution and resistance mutations, and after allogeneic stem cell transplantation to separate a relapse or a second neoplasm from reactive changes.
An example for the value of comprehensive testing is shown in Figure 2. A 71-year-old man showed leukocytosis with left shift, eosinophilia and an elevated LDH of 500, raising a differential diagnosis of MPN versus a myeloid neoplasm with eosinophilia. The bone marrow biopsy was hypercellular with eosinophilia, marked left shifted erythropoiesis, fibrosis grade 1, and mild increase in mast cells, but no blast increase. The initial molecular analyses showed no evidence for BCL-ABL1, PDGFRA or PDGFRB alterations or the JAK2 mutation. Additional extended molecular analysis, including NGS-based search for fusions using RNA from a formalin-fixed, paraffin-embedded bone marrow biopsy, identified a PCM1(36)-JAK2(11) rearrangement, subsequently confirmed by FISH and the presence of a t(8;9)(p22;p24) in conventional cytogenetics, resulting in a diagnosis of a myeloid/lymphoid neoplasm with PCM1-JAK2 rearrangement, a provisional entity newly incorporated into the update of the 4th Edition of the WHO classification.
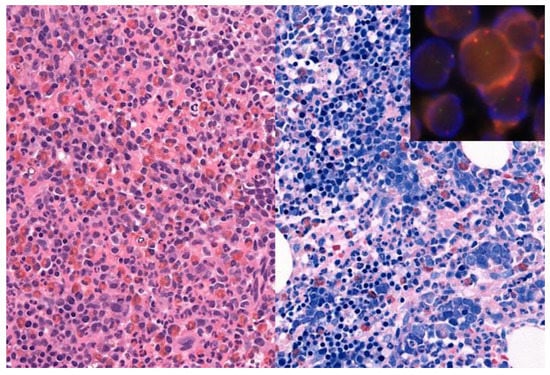
Figure 2.
71-year-old man with a hypercellular bone marrow biopsy with eosinophilia (left part: hematoxylin and eosin staining, original magnification 400×) and prominent left shifted erythropoiesis (right part: Giemsa staining, original magnification 400×). Insert: Fluorescence in situ hybridization showed a JAK2 rearrangement with a JAK2 break apart probe. Next generation sequencing identified a PCM1(36)-JAK2(11) rearrangement, in addition to DNMT3A and a NRAS mutations, resulting in a diagnosis of a myeloid/lymphoid neoplasm with PCM1-JAK2 rearrangement.
4. Conclusions—The Superior Value of a Synoptic Diagnosis
Myeloid neoplasms are diagnosed by a combination of clinical, morphological, immunophenotypic and genetic features in the current WHO classification, and an integrated, multimodal approach is needed for a precise and clinically applicable diagnosis. Although genetic features will gain further importance, and more MN likely will primarily be genetically defined in the future, clinical features and morphology on the bone marrow trephine biopsy, especially in the setting of MPN and MDS/MPN, still have a crucial role to provide additional information about cellularity, histotopography and distribution of hematopoietic cells, with bone marrow stroma and bone structure remaining important for classification [1]. Molecular features, on the other hand, require integration with clinical and laboratory features, as well as morphology and immunophenotyping, in order to avoid misclassification of MN. Many genetic alterations lack specificity for a certain entity, and background CHIP mutations, or unusual mutational combinations, present diagnostic pitfalls best avoided by a synoptic approach. Novel approaches currently tested in the research setting, such as integrating multiomics and deep learning algorithms for disease classification, will provide new insights for classification and therapy. This review summarizes the salient clinical, morphological and genetic features of Ph− MPN and their differential diagnosis from a practical standpoint, and highlights open questions to be addressed in the future evolution of MPN classification.
Author Contributions
Conceptualization and data collection, D.N. and F.F.; writing—original draft preparation, D.N.; writing—review and editing, F.F.; visualization, D.N.; supervision, F.F.; project administration, F.F. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Acknowledgments
The authors thank the team of the immunohistochemistry and molecular pathology labs for their support.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Arber, D.A.; Orazi, A.; Hasserjian, R.P.; Brunning, R.D.; Le Beau, M.M.; Porwit, A.; Tefferi, A.; Levine, R.; Bloomfield, C.D.; Cazzola, M.; et al. Introduction and overview of the classification of myeloid neoplasms. In WHO Classification of Tumours of the Haematopoietic and Lymphoid Tissues, revised, 4th ed.; Swerdlow, S.H., Campo, E., Harris, N.L., Jaffe, E.S., Pileri, S.A., Stein, H., Thiele, J., Eds.; IARC Press: Lyon, France, 2017; pp. 16–27. [Google Scholar]
- Wilkins, B.S. Pitfalls in bone marrow pathology: Avoiding errors in bone marrow trephine biopsy diagnosis. J. Clin. Pathol. 2011, 64, 380–386. [Google Scholar] [CrossRef]
- Torlakovic, E.E.; Brynes, R.K.; Hyjek, E.; Lee, S.H.; Kreipe, H.; Kremer, M.; McKenna, R.; Sadahira, Y.; Tzankov, A.; Reis, M.; et al. ICSH guidelines for the standardization of bone marrow immunohistochemistry. Int. J. Lab. Hematol. 2015, 37, 431–449. [Google Scholar] [CrossRef]
- Lee, S.H.; Erber, W.N.; Porwit, A.; Tomonaga, M.; Peterson, L.C.; International Council for Standardization in Hematology. ICSH guidelines for the standardization of bone marrow specimens and reports. Int. J. Lab. Hematol. 2008, 30, 349–364. [Google Scholar] [CrossRef] [PubMed]
- Feng, B.; Verstovsek, S.; Jorgensen, J.L.; Lin, P. Aberrant myeloid maturation identified by flow cytometry in primary myelofibrosis. Am. J. Clin. Pathol. 2010, 133, 314–320. [Google Scholar] [CrossRef]
- Ouyang, J.; Zheng, W.; Shen, Q.; Goswami, M.; Jorgensen, J.L.; Medeiros, L.J.; Wang, S.A. Flow cytometry immunophenotypic analysis of Philadelphia-negative myeloproliferative neoplasms: Correlation with histopathologic features. Cytom. B Clin. Cytom. 2014, 11, 21215. [Google Scholar] [CrossRef] [PubMed]
- Titmarsh, G.J.; Duncombe, A.S.; McMullin, M.F.; O’Rorke, M.; Mesa, R.; De Vocht, F.; Horan, S.; Fritschi, L.; Clarke, M.; Anderson, L.A. How common are myeloproliferative neoplasms? A systematic review and meta-analysis. Am. J. Hematol. 2014, 89, 581–587. [Google Scholar] [CrossRef] [PubMed]
- Moulard, O.; Mehta, J.; Fryzek, J.; Olivares, R.; Iqbal, U.; Mesa, R.A. Epidemiology of myelofibrosis, essential thrombocythemia, and polycythemia vera in the European Union. Eur. J. Haematol. 2014, 92, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Johansson, P. Epidemiology of the myeloproliferative disorders polycythemia vera and essential thrombocythemia. Semin. Thromb. Hemost. 2006, 32, 171–173. [Google Scholar] [CrossRef]
- Vardiman, J.W.; Melo, J.V.; Baccarani, M.; Radich, J.P.; Kvasnicka, H.M. Chronic myeloid leukaemia, BCR-ABL1-positive. In WHO Classification of Tumours of the Haematopoietic and Lymphoid Tissues, revised 4th ed.; Swerdlow, S.H., Campo, E., Harris, N.L., Jaffe, E.S., Pileri, S.A., Stein, H., Thiele, J., Eds.; IARC Press: Lyon, France, 2017; pp. 30–36. [Google Scholar]
- Bartram, C.R.; de Klein, A.; Hagemeijer, A.; van Agthoven, T.; Geurts van Kessel, A.; Bootsma, D.; Grosveld, G.; Ferguson-Smith, M.A.; Davies, T.; Stone, M.; et al. Translocation of c-ab1 oncogene correlates with the presence of a Philadelphia chromosome in chronic myelocytic leukaemia. Nature 1983, 306, 277–280. [Google Scholar] [CrossRef]
- Bain, B.J.; Brunning, R.D.; Orazi, A.; Thiele, J. Chronic neutrophilic leukaemia. In WHO Classification of Tumours of the Haematopoietic and Lymphoid Tissues, revised 4th ed.; Swerdlow, S.H., Campo, E., Harris, N.L., Jaffe, E.S., Pileri, S.A., Stein, H., Thiele, J., Eds.; IARC Press: Lyon, France, 2017; pp. 37–38. [Google Scholar]
- Szuber, N.; Elliott, M.; Tefferi, A. Chronic neutrophilic leukemia: 2020 update on diagnosis, molecular genetics, prognosis, and management. Am. J. Hematol. 2020, 95, 212–224. [Google Scholar] [CrossRef]
- Bain, B.J.; Ahmad, S. Chronic neutrophilic leukaemia and plasma cell-related neutrophilic leukaemoid reactions. Br. J. Haematol. 2015, 171, 400–410. [Google Scholar] [CrossRef] [PubMed]
- Hasle, H.; Olesen, G.; Kerndrup, G.; Philip, P.; Jacobsen, N. Chronic neutrophil leukaemia in adolescence and young adulthood. Br. J. Haematol. 1996, 94, 628–630. [Google Scholar] [CrossRef]
- Zhang, H.; Wilmot, B.; Bottomly, D.; Dao, K.T.; Stevens, E.; Eide, C.A.; Khanna, V.; Rofelty, A.; Savage, S.; Reister Schultz, A.; et al. Genomic landscape of neutrophilic leukemias of ambiguous diagnosis. Blood 2019, 134, 867–879. [Google Scholar] [CrossRef]
- Szuber, N.; Finke, C.M.; Lasho, T.L.; Elliott, M.A.; Hanson, C.A.; Pardanani, A.; Tefferi, A. CSF3R-mutated chronic neutrophilic leukemia: Long-term outcome in 19 consecutive patients and risk model for survival. Blood Cancer J. 2018, 8, 1–5. [Google Scholar] [CrossRef]
- Ouyang, Y.; Qiao, C.; Chen, Y.; Zhang, S.J. Clinical significance of CSF3R, SRSF2 and SETBP1 mutations in chronic neutrophilic leukemia and chronic myelomonocytic leukemia. Oncotarget 2017, 8, 20834–20841. [Google Scholar] [CrossRef]
- Meggendorfer, M.; Haferlach, T.; Alpermann, T.; Jeromin, S.; Haferlach, C.; Kern, W.; Schnittger, S. Specific molecular mutation patterns delineate chronic neutrophilic leukemia, atypical chronic myeloid leukemia, and chronic myelomonocytic leukemia. Haematologica 2014, 99, e244–e246. [Google Scholar] [CrossRef]
- Langabeer, S.E.; Haslam, K.; Kelly, J.; Quinn, J.; Morrell, R.; Conneally, E. Targeted next-generation sequencing identifies clinically relevant mutations in patients with chronic neutrophilic leukemia at diagnosis and blast crisis. Clin. Transl. Oncol. 2018, 20, 420–423. [Google Scholar] [CrossRef]
- Elliott, M.A.; Pardanani, A.; Hanson, C.A.; Lasho, T.L.; Finke, C.M.; Belachew, A.A.; Tefferi, A. ASXL1 mutations are frequent and prognostically detrimental in CSF3R-mutated chronic neutrophilic leukemia. Am. J. Hematol. 2015, 90, 653–656. [Google Scholar] [CrossRef]
- Cui, Y.; Li, B.; Gale, R.P.; Jiang, Q.; Xu, Z.; Qin, T.; Zhang, P.; Zhang, Y.; Xiao, Z. CSF3R, SETBP1 and CALR mutations in chronic neutrophilic leukemia. J. Hematol. Oncol. 2014, 7, 77. [Google Scholar] [CrossRef]
- Maxson, J.E.; Gotlib, J.; Pollyea, D.A.; Fleischman, A.G.; Agarwal, A.; Eide, C.A.; Bottomly, D.; Wilmot, B.; McWeeney, S.K.; Tognon, C.E.; et al. Oncogenic CSF3R mutations in chronic neutrophilic leukemia and atypical CML. N. Engl. J. Med. 2013, 368, 1781–1790. [Google Scholar] [CrossRef]
- Zhang, H.; Reister Schultz, A.; Luty, S.; Rofelty, A.; Su, Y.; Means, S.; Bottomly, D.; Wilmot, B.; McWeeney, S.K.; Tyner, J.W. Characterization of the leukemogenic potential of distal cytoplasmic CSF3R truncation and missense mutations. Leukemia 2017, 31, 2752–2760. [Google Scholar] [CrossRef]
- Gotlib, J.; Maxson, J.E.; George, T.I.; Tyner, J.W. The new genetics of chronic neutrophilic leukemia and atypical CML: Implications for diagnosis and treatment. Blood 2013, 122, 1707–1711. [Google Scholar] [CrossRef]
- Genovese, G.; Kahler, A.K.; Handsaker, R.E.; Lindberg, J.; Rose, S.A.; Bakhoum, S.F.; Chambert, K.; Mick, E.; Neale, B.M.; Fromer, M.; et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N. Engl. J. Med. 2014, 371, 2477–2487. [Google Scholar] [CrossRef]
- Jaiswal, S.; Ebert, B.L. Clonal hematopoiesis in human aging and disease. Science 2019, 366, eaan4673. [Google Scholar] [CrossRef]
- Tefferi, A.; Rumi, E.; Finazzi, G.; Gisslinger, H.; Vannucchi, A.M.; Rodeghiero, F.; Randi, M.L.; Vaidya, R.; Cazzola, M.; Rambaldi, A.; et al. Survival and prognosis among 1545 patients with contemporary polycythemia vera: An international study. Leukemia 2013, 27, 1874–1881. [Google Scholar] [CrossRef]
- Tefferi, A. Novel mutations and their functional and clinical relevance in myeloproliferative neoplasms: JAK2, MPL, TET2, ASXL1, CBL, IDH and IKZF1. Leukemia 2010, 24, 1128–1138. [Google Scholar] [CrossRef]
- Scott, L.M.; Tong, W.; Levine, R.L.; Scott, M.A.; Beer, P.A.; Stratton, M.R.; Futreal, P.A.; Erber, W.N.; McMullin, M.F.; Harrison, C.N.; et al. JAK2 exon 12 mutations in polycythemia vera and idiopathic erythrocytosis. N. Engl. J. Med. 2007, 356, 459–468. [Google Scholar] [CrossRef]
- Pardanani, A.; Lasho, T.L.; Finke, C.; Hanson, C.A.; Tefferi, A. Prevalence and clinicopathologic correlates of JAK2 exon 12 mutations in JAK2V617F-negative polycythemia vera. Leukemia 2007, 21, 1960–1963. [Google Scholar] [CrossRef] [PubMed]
- Lakey, M.A.; Pardanani, A.; Hoyer, J.D.; Nguyen, P.L.; Lasho, T.L.; Tefferi, A.; Hanson, C.A. Bone marrow morphologic features in polycythemia vera with JAK2 exon 12 mutations. Am. J. Clin. Pathol. 2010, 133, 942–948. [Google Scholar] [CrossRef]
- Kralovics, R.; Passamonti, F.; Buser, A.S.; Teo, S.S.; Tiedt, R.; Passweg, J.R.; Tichelli, A.; Cazzola, M.; Skoda, R.C. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N. Engl. J. Med. 2005, 352, 1779–1790. [Google Scholar] [CrossRef]
- Loscocco, G.G.; Guglielmelli, P.; Vannucchi, A.M. Impact of Mutational Profile on the Management of Myeloproliferative Neoplasms: A Short Review of the Emerging Data. OncoTargets Ther. 2020, 13, 12367–12382. [Google Scholar] [CrossRef]
- Ortmann, C.A.; Kent, D.G.; Nangalia, J.; Silber, Y.; Wedge, D.C.; Grinfeld, J.; Baxter, E.J.; Massie, C.E.; Papaemmanuil, E.; Menon, S.; et al. Effect of mutation order on myeloproliferative neoplasms. N. Engl. J. Med. 2015, 372, 601–612. [Google Scholar] [CrossRef]
- Tefferi, A.; Lasho, T.L.; Guglielmelli, P.; Finke, C.M.; Rotunno, G.; Elala, Y.; Pacilli, A.; Hanson, C.A.; Pancrazzi, A.; Ketterling, R.P.; et al. Targeted deep sequencing in polycythemia vera and essential thrombocythemia. Blood Adv. 2016, 1, 21–30. [Google Scholar] [CrossRef]
- Tefferi, A. Primary myelofibrosis: 2021 update on diagnosis, risk-stratification and management. Am. J. Hematol. 2021, 96, 145–162. [Google Scholar] [CrossRef]
- Tefferi, A.; Vainchenker, W. Myeloproliferative neoplasms: Molecular pathophysiology, essential clinical understanding, and treatment strategies. J. Clin. Oncol. 2011, 29, 573–582. [Google Scholar] [CrossRef]
- Tefferi, A.; Finke, C.M.; Lasho, T.L.; Hanson, C.A.; Ketterling, R.P.; Gangat, N.; Pardanani, A. U2AF1 mutation types in primary myelofibrosis: Phenotypic and prognostic distinctions. Leukemia 2018, 32, 2274–2278. [Google Scholar] [CrossRef]
- Tefferi, A.; Guglielmelli, P.; Lasho, T.L.; Rotunno, G.; Finke, C.; Mannarelli, C.; Belachew, A.A.; Pancrazzi, A.; Wassie, E.A.; Ketterling, R.P.; et al. CALR and ASXL1 mutations-based molecular prognostication in primary myelofibrosis: An international study of 570 patients. Leukemia 2014, 28, 1494–1500. [Google Scholar] [CrossRef] [PubMed]
- Tefferi, A.; Lasho, T.L.; Finke, C.M.; Elala, Y.; Hanson, C.A.; Ketterling, R.P.; Gangat, N.; Pardanani, A. Targeted deep sequencing in primary myelofibrosis. Blood Adv. 2016, 1, 105–111. [Google Scholar] [CrossRef]
- Vannucchi, A.M.; Lasho, T.L.; Guglielmelli, P.; Biamonte, F.; Pardanani, A.; Pereira, A.; Finke, C.; Score, J.; Gangat, N.; Mannarelli, C.; et al. Mutations and prognosis in primary myelofibrosis. Leukemia 2013, 27, 1861–1869. [Google Scholar] [CrossRef] [PubMed]
- Tefferi, A.; Guglielmelli, P.; Larson, D.R.; Finke, C.; Wassie, E.A.; Pieri, L.; Gangat, N.; Fjerza, R.; Belachew, A.A.; Lasho, T.L.; et al. Long-term survival and blast transformation in molecularly annotated essential thrombocythemia, polycythemia vera, and myelofibrosis. Blood 2014, 124, 2507–2513. [Google Scholar] [CrossRef]
- Tefferi, A.; Wassie, E.A.; Lasho, T.L.; Finke, C.; Belachew, A.A.; Ketterling, R.P.; Hanson, C.A.; Pardanani, A.; Gangat, N.; Wolanskyj, A.P. Calreticulin mutations and long-term survival in essential thrombocythemia. Leukemia 2014, 28, 2300–2303. [Google Scholar] [CrossRef]
- Wang, S.A.; Tam, W.; Tsai, A.G.; Arber, D.A.; Hasserjian, R.P.; Geyer, J.T.; George, T.I.; Czuchlewski, D.R.; Foucar, K.; Rogers, H.J.; et al. Targeted next-generation sequencing identifies a subset of idiopathic hypereosinophilic syndrome with features similar to chronic eosinophilic leukemia, not otherwise specified. Mod. Pathol. 2016, 29, 854–864. [Google Scholar] [CrossRef] [PubMed]
- Palomo, L.; Meggendorfer, M.; Hutter, S.; Twardziok, S.; Adema, V.; Fuhrmann, I.; Fuster-Tormo, F.; Xicoy, B.; Zamora, L.; Acha, P.; et al. Molecular landscape and clonal architecture of adult myelodysplastic/myeloproliferative neoplasms. Blood 2020, 136, 1851–1862. [Google Scholar] [CrossRef]
- Pardanani, A.; Lasho, T.L.; Laborde, R.R.; Elliott, M.; Hanson, C.A.; Knudson, R.A.; Ketterling, R.P.; Maxson, J.E.; Tyner, J.W.; Tefferi, A. CSF3R T618I is a highly prevalent and specific mutation in chronic neutrophilic leukemia. Leukemia 2013, 27, 1870–1873. [Google Scholar] [CrossRef]
- Wang, S.A.; Hasserjian, R.P.; Fox, P.S.; Rogers, H.J.; Geyer, J.T.; Chabot-Richards, D.; Weinzierl, E.; Hatem, J.; Jaso, J.; Kanagal-Shamanna, R.; et al. Atypical chronic myeloid leukemia is clinically distinct from unclassifiable myelodysplastic/myeloproliferative neoplasms. Blood 2014, 123, 2645–2651. [Google Scholar] [CrossRef]
- Beekman, R.; Valkhof, M.; van Strien, P.; Valk, P.J.; Touw, I.P. Prevalence of a new auto-activating colony stimulating factor 3 receptor mutation (CSF3R-T595I) in acute myeloid leukemia and severe congenital neutropenia. Haematologica 2013, 98, e62–e63. [Google Scholar] [CrossRef]
- Maxson, J.E.; Ries, R.E.; Wang, Y.C.; Gerbing, R.B.; Kolb, E.A.; Thompson, S.L.; Guidry Auvil, J.M.; Marra, M.A.; Ma, Y.; Zong, Z.; et al. CSF3R mutations have a high degree of overlap with CEBPA mutations in pediatric AML. Blood 2016, 127, 3094–3098. [Google Scholar] [CrossRef]
- Sano, H.; Ohki, K.; Park, M.J.; Shiba, N.; Hara, Y.; Sotomatsu, M.; Tomizawa, D.; Taga, T.; Kiyokawa, N.; Tawa, A.; et al. CSF3R and CALR mutations in paediatric myeloid disorders and the association of CSF3R mutations with translocations, including t(8; 21). Br. J. Haematol. 2015, 170, 391–397. [Google Scholar] [CrossRef]
- Kosmider, O.; Itzykson, R.; Chesnais, V.; Lasho, T.; Laborde, R.; Knudson, R.; Gauthier, A.; Merlevede, J.; Ades, L.; Morabito, M.; et al. Mutation of the colony-stimulating factor-3 receptor gene is a rare event with poor prognosis in chronic myelomonocytic leukemia. Leukemia 2013, 27, 1946–1949. [Google Scholar] [CrossRef]
- Meggendorfer, M.; Bacher, U.; Alpermann, T.; Haferlach, C.; Kern, W.; Gambacorti-Passerini, C.; Haferlach, T.; Schnittger, S. SETBP1 mutations occur in 9% of MDS/MPN and in 4% of MPN cases and are strongly associated with atypical CML, monosomy 7, isochromosome i (17) (q10), ASXL1 and CBL mutations. Leukemia 2013, 27, 1852–1860. [Google Scholar] [CrossRef]
- Piazza, R.; Valletta, S.; Winkelmann, N.; Redaelli, S.; Spinelli, R.; Pirola, A.; Antolini, L.; Mologni, L.; Donadoni, C.; Papaemmanuil, E.; et al. Recurrent SETBP1 mutations in atypical chronic myeloid leukemia. Nat. Genet. 2013, 45, 18–24. [Google Scholar] [CrossRef]
- Maxson, J.E.; Tyner, J.W. Genomics of chronic neutrophilic leukemia. Blood 2017, 129, 715–722. [Google Scholar] [CrossRef] [PubMed]
- Thiele, J.; Kvasnicka, H.M.; Orazi, A.; Tefferi, A.; Birgegard, G.; Barbui, T. Polycythaemia vera. In WHO Classification of Tumours of the Haematopoietic and Lymphoid Tissues, revised, 4th ed.; Swerdlow, S.H., Campo, E., Harris, N.L., Jaffe, E.S., Pileri, S.A., Stein, H., Thiele, J., Eds.; IARC Press: Lyon, France, 2017; pp. 39–43. [Google Scholar]
- Gruppo Italiano Studio Policitemia. Polycythemia vera: The natural history of 1213 patients followed for 20 years. Ann. Intern. Med. 1995, 123, 656–664. [Google Scholar] [CrossRef] [PubMed]
- Marchioli, R.; Finazzi, G.; Landolfi, R.; Kutti, J.; Gisslinger, H.; Patrono, C.; Marilus, R.; Villegas, A.; Tognoni, G.; Barbui, T. Vascular and neoplastic risk in a large cohort of patients with polycythemia vera. J. Clin. Oncol. 2005, 23, 2224–2232. [Google Scholar] [CrossRef] [PubMed]
- Barbui, T.; Thiele, J.; Carobbio, A.; Guglielmelli, P.; Rambaldi, A.; Vannucchi, A.M.; Tefferi, A. Discriminating between essential thrombocythemia and masked polycythemia vera in JAK2 mutated patients. Am. J. Hematol. 2014, 89, 588–590. [Google Scholar] [CrossRef]
- Gianelli, U.; Iurlo, A.; Vener, C.; Moro, A.; Fermo, E.; Bianchi, P.; Graziani, D.; Radaelli, F.; Coggi, G.; Bosari, S.; et al. The significance of bone marrow biopsy and JAK2V617F mutation in the differential diagnosis between the “early” prepolycythemic phase of polycythemia vera and essential thrombocythemia. Am. J. Clin. Pathol. 2008, 130, 336–342. [Google Scholar] [CrossRef]
- Thiele, J.; Kvasnicka, H.M.; Diehl, V. Initial (latent) polycythemia vera with thrombocytosis mimicking essential thrombocythemia. Acta Haematol. 2005, 113, 213–219. [Google Scholar] [CrossRef]
- Thiele, J.; Kvasnicka, H.M.; Orazi, A.; Gianelli, U.; Barbui, T.; Barosi, G.; Tefferi, A. Primary myelofibrosis. In WHO Classification of Tumours of the Haematopoietic and Lymphoid Tissues, revised, 4th ed.; Swerdlow, S.H., Campo, E., Harris, N.L., Jaffe, E.S., Pileri, S.A., Stein, H., Thiele, J., Eds.; IARC Press: Lyon, France, 2017; pp. 44–50. [Google Scholar]
- Thiele, J.; Kvasnicka, H.M.; Orazi, A.; Gianelli, U.; Tefferi, A.; Gisslinger, H.; Barbui, T. Essential thrombocythaemia. In WHO Classification of Tumours of the Haematopoietic and Lymphoid Tissues, revised, 4th ed.; Swerdlow, S.H., Campo, E., Harris, N.L., Jaffe, E.S., Pileri, S.A., Stein, H., Thiele, J., Eds.; IARC Press: Lyon, France, 2017; pp. 50–53. [Google Scholar]
- Broseus, J.; Park, J.H.; Carillo, S.; Hermouet, S.; Girodon, F. Presence of calreticulin mutations in JAK2-negative polycythemia vera. Blood 2014, 124, 3964–3966. [Google Scholar] [CrossRef]
- Georgii, A.; Buesche, G.; Kreft, A. The histopathology of chronic myeloproliferative diseases. Baillière’s Clin. Haematol. 1998, 11, 721–749. [Google Scholar] [CrossRef]
- Thiele, J.; Kvasnicka, H.M. Diagnostic impact of bone marrow histopathology in polycythemia vera (PV). Histol. Histopathol. 2005, 20, 317–328. [Google Scholar] [CrossRef]
- Thiele, J.; Kvasnicka, H.M.; Facchetti, F.; Franco, V.; van der Walt, J.; Orazi, A. European consensus on grading bone marrow fibrosis and assessment of cellularity. Haematologica 2005, 90, 1128–1132. [Google Scholar]
- Ellis, J.T.; Peterson, P.; Geller, S.A.; Rappaport, H. Studies of the bone marrow in polycythemia vera and the evolution of myelofibrosis and second hematologic malignancies. Semin. Hematol. 1986, 23, 144–155. [Google Scholar]
- Thiele, J.; Kvasnicka, H.M.; Orazi, A. Bone marrow histopathology in myeloproliferative disorders—current diagnostic approach. Semin. Hematol. 2005, 42, 184–195. [Google Scholar] [CrossRef]
- Kvasnicka, H.M.; Thiele, J. Prodromal myeloproliferative neoplasms: The 2008 WHO classification. Am. J. Hematol. 2010, 85, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Abdulkarim, K.; Ridell, B.; Johansson, P.; Kutti, J.; Safai-Kutti, S.; Andreasson, B. The impact of peripheral blood values and bone marrow findings on prognosis for patients with essential thrombocythemia and polycythemia vera. Eur. J. Haematol. 2011, 86, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Barbui, T.; Thiele, J.; Passamonti, F.; Rumi, E.; Boveri, E.; Randi, M.L.; Bertozzi, I.; Marino, F.; Vannucchi, A.M.; Pieri, L.; et al. Initial bone marrow reticulin fibrosis in polycythemia vera exerts an impact on clinical outcome. Blood 2012, 119, 2239–2241. [Google Scholar] [CrossRef] [PubMed]
- Buhr, T.; Georgii, A.; Choritz, H. Myelofibrosis in chronic myeloproliferative disorders. Incidence among subtypes according to the Hannover Classification. Pathol. Res. Pract. 1993, 189, 121–132. [Google Scholar] [CrossRef]
- Kreft, A.; Buche, G.; Ghalibafian, M.; Buhr, T.; Fischer, T.; Kirkpatrick, C.J. The incidence of myelofibrosis in essential thrombocythaemia, polycythaemia vera and chronic idiopathic myelofibrosis: A retrospective evaluation of sequential bone marrow biopsies. Acta Haematol. 2005, 113, 137–143. [Google Scholar] [CrossRef]
- Thiele, J.; Kvasnicka, H.M.; Vardiman, J. Bone marrow histopathology in the diagnosis of chronic myeloproliferative disorders: A forgotten pearl. Best Pract. Res. Clin. Haematol. 2006, 19, 413–437. [Google Scholar] [CrossRef]
- Thiele, J.; Kvasnicka, H.M. The 2008 WHO diagnostic criteria for polycythemia vera, essential thrombocythemia, and primary myelofibrosis. Curr. Hematol. Malig. Rep. 2009, 4, 33–40. [Google Scholar] [CrossRef]
- Cerquozzi, S.; Tefferi, A. Blast transformation and fibrotic progression in polycythemia vera and essential thrombocythemia: A literature review of incidence and risk factors. Blood Cancer J. 2015, 5, e366. [Google Scholar] [CrossRef]
- Barosi, G.; Mesa, R.A.; Thiele, J.; Cervantes, F.; Campbell, P.J.; Verstovsek, S.; Dupriez, B.; Levine, R.L.; Passamonti, F.; Gotlib, J.; et al. Proposed criteria for the diagnosis of post-polycythemia vera and post-essential thrombocythemia myelofibrosis: A consensus statement from the International Working Group for Myelofibrosis Research and Treatment. Leukemia 2008, 22, 437–438. [Google Scholar] [CrossRef]
- Boiocchi, L.; Mathew, S.; Gianelli, U.; Iurlo, A.; Radice, T.; Barouk-Fox, S.; Knowles, D.M.; Orazi, A. Morphologic and cytogenetic differences between post-polycythemic myelofibrosis and primary myelofibrosis in fibrotic stage. Mod. Pathol. 2013, 26, 1577–1585. [Google Scholar] [CrossRef]
- Sangle, N.; Cook, J.; Perkins, S.; Teman, C.J.; Bahler, D.; Hickman, K.; Wilson, A.; Prchal, J.; Salama, M.E. Myelofibrotic transformations of polycythemia vera and essential thrombocythemia are morphologically, biologically, and prognostically indistinguishable from primary myelofibrosis. Appl. Immunohistochem. Mol. Morphol. AIMM/Off. Publ. Soc. Appl. Immunohistochem. 2014, 22, 663–668. [Google Scholar] [CrossRef] [PubMed]
- Boiocchi, L.; Gianelli, U.; Iurlo, A.; Fend, F.; Bonzheim, I.; Cattaneo, D.; Knowles, D.M.; Orazi, A. Neutrophilic leukocytosis in advanced stage polycythemia vera: Hematopathologic features and prognostic implications. Mod. Pathol. 2015, 28, 1448–1457. [Google Scholar] [CrossRef]
- Miklossy, G.; Hilliard, T.S.; Turkson, J. Therapeutic modulators of STAT signalling for human diseases. Nat. Rev. Drug Discov. 2013, 12, 611–629. [Google Scholar] [CrossRef] [PubMed]
- Levine, R.L.; Wadleigh, M.; Cools, J.; Ebert, B.L.; Wernig, G.; Huntly, B.J.; Boggon, T.J.; Wlodarska, I.; Clark, J.J.; Moore, S.; et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell 2005, 7, 387–397. [Google Scholar] [CrossRef]
- Tefferi, A.; Lavu, S.; Mudireddy, M.; Lasho, T.L.; Finke, C.M.; Gangat, N.; Pardanani, A.; Hanson, C.A.; Mannarelli, C.; Guglielmelli, P.; et al. JAK2 exon 12 mutated polycythemia vera: Mayo-Careggi MPN Alliance study of 33 consecutive cases and comparison with JAK2V617F mutated disease. Am. J. Hematol. 2018, 93, E93–E96. [Google Scholar] [CrossRef]
- Passamonti, F.; Elena, C.; Schnittger, S.; Skoda, R.C.; Green, A.R.; Girodon, F.; Kiladjian, J.J.; McMullin, M.F.; Ruggeri, M.; Besses, C.; et al. Molecular and clinical features of the myeloproliferative neoplasm associated with JAK2 exon 12 mutations. Blood 2011, 117, 2813–2816. [Google Scholar] [CrossRef] [PubMed]
- Vannucchi, A.M.; Antonioli, E.; Guglielmelli, P.; Rambaldi, A.; Barosi, G.; Marchioli, R.; Marfisi, R.M.; Finazzi, G.; Guerini, V.; Fabris, F.; et al. Clinical profile of homozygous JAK2 617V > F mutation in patients with polycythemia vera or essential thrombocythemia. Blood 2007, 110, 840–846. [Google Scholar] [CrossRef]
- Vannucchi, A.M.; Antonioli, E.; Guglielmelli, P.; Longo, G.; Pancrazzi, A.; Ponziani, V.; Bogani, C.; Ferrini, P.R.; Rambaldi, A.; Guerini, V.; et al. Prospective identification of high-risk polycythemia vera patients based on JAK2(V617F) allele burden. Leukemia 2007, 21, 1952–1959. [Google Scholar] [CrossRef] [PubMed]
- Nangalia, J.; Nice, F.L.; Wedge, D.C.; Godfrey, A.L.; Grinfeld, J.; Thakker, C.; Massie, C.E.; Baxter, J.; Sewell, D.; Silber, Y.; et al. DNMT3A mutations occur early or late in patients with myeloproliferative neoplasms and mutation order influences phenotype. Haematologica 2015, 100, e438–e442. [Google Scholar] [CrossRef] [PubMed]
- Tefferi, A.; Saeed, L.; Hanson, C.A.; Ketterling, R.P.; Pardanani, A.; Gangat, N. Application of current prognostic models for primary myelofibrosis in the setting of post-polycythemia vera or post-essential thrombocythemia myelofibrosis. Leukemia 2017, 31, 2851–2852. [Google Scholar] [CrossRef] [PubMed]
- Gold, L.I.; Eggleton, P.; Sweetwyne, M.T.; Van Duyn, L.B.; Greives, M.R.; Naylor, S.M.; Michalak, M.; Murphy-Ullrich, J.E. Calreticulin: Non-endoplasmic reticulum functions in physiology and disease. FASEB J. 2010, 24, 665–683. [Google Scholar] [CrossRef] [PubMed]
- Klampfl, T.; Gisslinger, H.; Harutyunyan, A.S.; Nivarthi, H.; Rumi, E.; Milosevic, J.D.; Them, N.C.; Berg, T.; Gisslinger, B.; Pietra, D.; et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. N. Engl. J. Med. 2013, 369, 2379–2390. [Google Scholar] [CrossRef]
- Nangalia, J.; Massie, C.E.; Baxter, E.J.; Nice, F.L.; Gundem, G.; Wedge, D.C.; Avezov, E.; Li, J.; Kollmann, K.; Kent, D.G.; et al. Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. N. Engl. J. Med. 2013, 369, 2391–2405. [Google Scholar] [CrossRef]
- Cabagnols, X.; Defour, J.P.; Ugo, V.; Ianotto, J.C.; Mossuz, P.; Mondet, J.; Girodon, F.; Alexandre, J.H.; Mansier, O.; Viallard, J.F.; et al. Differential association of calreticulin type 1 and type 2 mutations with myelofibrosis and essential thrombocytemia: Relevance for disease evolution. Leukemia 2015, 29, 249–252. [Google Scholar] [CrossRef]
- Balligand, T.; Achouri, Y.; Pecquet, C.; Chachoua, I.; Nivarthi, H.; Marty, C.; Vainchenker, W.; Plo, I.; Kralovics, R.; Constantinescu, S.N. Pathologic activation of thrombopoietin receptor and JAK2-STAT5 pathway by frameshift mutants of mouse calreticulin. Leukemia 2016, 30, 1775–1778. [Google Scholar] [CrossRef]
- Chachoua, I.; Pecquet, C.; El-Khoury, M.; Nivarthi, H.; Albu, R.I.; Marty, C.; Gryshkova, V.; Defour, J.P.; Vertenoeil, G.; Ngo, A.; et al. Thrombopoietin receptor activation by myeloproliferative neoplasm associated calreticulin mutants. Blood 2016, 127, 1325–1335. [Google Scholar] [CrossRef]
- Elf, S.; Abdelfattah, N.S.; Chen, E.; Perales-Paton, J.; Rosen, E.A.; Ko, A.; Peisker, F.; Florescu, N.; Giannini, S.; Wolach, O.; et al. Mutant Calreticulin Requires Both Its Mutant C-terminus and the Thrombopoietin Receptor for Oncogenic Transformation. Cancer Discov. 2016, 6, 368–381. [Google Scholar] [CrossRef]
- Nivarthi, H.; Chen, D.; Cleary, C.; Kubesova, B.; Jager, R.; Bogner, E.; Marty, C.; Pecquet, C.; Vainchenker, W.; Constantinescu, S.N.; et al. Thrombopoietin receptor is required for the oncogenic function of CALR mutants. Leukemia 2016, 30, 1759–1763. [Google Scholar] [CrossRef]
- Tefferi, A.; Nicolosi, M.; Mudireddy, M.; Szuber, N.; Finke, C.M.; Lasho, T.L.; Hanson, C.A.; Ketterling, R.P.; Pardanani, A.; Gangat, N.; et al. Driver mutations and prognosis in primary myelofibrosis: Mayo-Careggi MPN alliance study of 1095 patients. Am. J. Hematol. 2018, 93, 348–355. [Google Scholar] [CrossRef] [PubMed]
- Bridgford, J.L.; Lee, S.M.; Lee, C.M.M.; Guglielmelli, P.; Rumi, E.; Pietra, D.; Wilcox, S.; Chhabra, Y.; Rubin, A.F.; Cazzola, M.; et al. Novel drivers and modifiers of MPL-dependent oncogenic transformation identified by deep mutational scanning. Blood 2020, 135, 287–292. [Google Scholar] [CrossRef]
- Defour, J.P.; Chachoua, I.; Pecquet, C.; Constantinescu, S.N. Oncogenic activation of MPL/thrombopoietin receptor by 17 mutations at W515: Implications for myeloproliferative neoplasms. Leukemia 2016, 30, 1214–1216. [Google Scholar] [CrossRef]
- Pikman, Y.; Lee, B.H.; Mercher, T.; McDowell, E.; Ebert, B.L.; Gozo, M.; Cuker, A.; Wernig, G.; Moore, S.; Galinsky, I.; et al. MPLW515L is a novel somatic activating mutation in myelofibrosis with myeloid metaplasia. PLoS Med. 2006, 3, e270. [Google Scholar] [CrossRef]
- Staerk, J.; Lacout, C.; Sato, T.; Smith, S.O.; Vainchenker, W.; Constantinescu, S.N. An amphipathic motif at the transmembrane-cytoplasmic junction prevents autonomous activation of the thrombopoietin receptor. Blood 2006, 107, 1864–1871. [Google Scholar] [CrossRef]
- Milosevic Feenstra, J.D.; Nivarthi, H.; Gisslinger, H.; Leroy, E.; Rumi, E.; Chachoua, I.; Bagienski, K.; Kubesova, B.; Pietra, D.; Gisslinger, B.; et al. Whole-exome sequencing identifies novel MPL and JAK2 mutations in triple-negative myeloproliferative neoplasms. Blood 2016, 127, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Tefferi, A. Myelofibrosis with myeloid metaplasia. N. Engl. J. Med. 2000, 342, 1255–1265. [Google Scholar] [CrossRef] [PubMed]
- Thiele, J.; Kvasnicka, H.M.; Mullauer, L.; Buxhofer-Ausch, V.; Gisslinger, B.; Gisslinger, H. Essential thrombocythemia versus early primary myelofibrosis: A multicenter study to validate the WHO classification. Blood 2011, 117, 5710–5718. [Google Scholar] [CrossRef]
- Thiele, J.; Kvasnicka, H.M. Clinicopathological criteria for differential diagnosis of thrombocythemias in various myeloproliferative disorders. Semin. Thromb. Hemost. 2006, 32, 219–230. [Google Scholar] [CrossRef]
- Thiele, J.; Kvasnicka, H.M. Diagnostic differentiation of essential thrombocythaemia from thrombocythaemias associated with chronic idiopathic myelofibrosis by discriminate analysis of bone marrow features—A clinicopathological study on 272 patients. Histol. Histopathol. 2003, 18, 93–102. [Google Scholar] [CrossRef]
- Barosi, G. Essential thrombocythemia vs. early/prefibrotic myelofibrosis: Why does it matter. Best Pract. Res. Clin. Haematol. 2014, 27, 129–140. [Google Scholar] [CrossRef] [PubMed]
- Thiele, J.; Kvasnicka, H.M. Prefibrotic chronic idiopathic myelofibrosis—A diagnostic enigma? Acta Haematol. 2004, 111, 155–159. [Google Scholar] [CrossRef] [PubMed]
- Thiele, J.; Kvasnicka, H.M. Hematopathologic findings in chronic idiopathic myelofibrosis. Semin. Oncol. 2005, 32, 380–394. [Google Scholar] [CrossRef] [PubMed]
- Thiele, J.; Kvasnicka, H.M.; Diehl, V. Bone marrow CD34+ progenitor cells in Philadelphia chromosome-negative chronic myeloproliferative disorders—A clinicopathological study on 575 patients. Leuk. Lymphoma 2005, 46, 709–715. [Google Scholar] [CrossRef] [PubMed]
- Kvasnicka, H.M. WHO classification of myeloproliferative neoplasms (MPN): A critical update. Curr. Hematol. Malig. Rep. 2013, 8, 333–341. [Google Scholar] [CrossRef]
- Kvasnicka, H.M.; Beham-Schmid, C.; Bob, R.; Dirnhofer, S.; Hussein, K.; Kreipe, H.; Kremer, M.; Schmitt-Graeff, A.; Schwarz, S.; Thiele, J.; et al. Problems and pitfalls in grading of bone marrow fibrosis, collagen deposition and osteosclerosis—A consensus-based study. Histopathology 2016, 68, 905–915. [Google Scholar] [CrossRef]
- Barosi, G.; Hoffman, R. Idiopathic myelofibrosis. Semin. Hematol. 2005, 42, 248–258. [Google Scholar] [CrossRef]
- Boveri, E.; Passamonti, F.; Rumi, E.; Pietra, D.; Elena, C.; Arcaini, L.; Pascutto, C.; Castello, A.; Cazzola, M.; Magrini, U.; et al. Bone marrow microvessel density in chronic myeloproliferative disorders: A study of 115 patients with clinicopathological and molecular correlations. Br. J. Haematol. 2008, 140, 162–168. [Google Scholar] [CrossRef] [PubMed]
- Buhr, T.; Busche, G.; Choritz, H.; Langer, F.; Kreipe, H. Evolution of myelofibrosis in chronic idiopathic myelofibrosis as evidenced in sequential bone marrow biopsy specimens. Am. J. Clin. Pathol. 2003, 119, 152–158. [Google Scholar] [CrossRef] [PubMed]
- Kvasnicka, H.M.; Thiele, J. Bone marrow angiogenesis: Methods of quantification and changes evolving in chronic myeloproliferative disorders. Histol. Histopathol. 2004, 19, 1245–1260. [Google Scholar] [CrossRef] [PubMed]
- Thiele, J.; Kvasnicka, H.M.; Czieslick, C. CD34+ progenitor cells in idiopathic (primary) myelofibrosis: A comparative quantification between spleen and bone marrow tissue. Ann. Hematol. 2002, 81, 86–89. [Google Scholar] [CrossRef]
- O’Malley, D.P.; Kim, Y.S.; Perkins, S.L.; Baldridge, L.; Juliar, B.E.; Orazi, A. Morphologic and immunohistochemical evaluation of splenic hematopoietic proliferations in neoplastic and benign disorders. Mod. Pathol. 2005, 18, 1550–1561. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Prakash, S.; Lu, M.; Tripodi, J.; Ye, F.; Najfeld, V.; Li, Y.; Schwartz, M.; Weinberg, R.; Roda, P.; et al. Spleens of myelofibrosis patients contain malignant hematopoietic stem cells. J. Clin. Investig. 2012, 122, 3888–3899. [Google Scholar] [CrossRef]
- Prakash, S.; Hoffman, R.; Barouk, S.; Wang, Y.L.; Knowles, D.M.; Orazi, A. Splenic extramedullary hematopoietic proliferation in Philadelphia chromosome-negative myeloproliferative neoplasms: Heterogeneous morphology and cytological composition. Mod. Pathol. 2012, 25, 815–827. [Google Scholar] [CrossRef] [PubMed]
- Thiele, J.; Bennewitz, F.G.; Bertsch, H.P.; Falk, S.; Fischer, R.; Stutte, H.J. Splenic haematopoiesis in primary (idiopathic) osteomyelofibrosis: Immunohistochemical and morphometric evaluation of proliferative activity of erytro- and endoreduplicative capacity of megakaryopoiesis (PCNA- and Ki-67 staining). Virchows Arch. B Cell Pathol. Incl. Mol. Pathol. 1993, 64, 281–286. [Google Scholar] [CrossRef]
- Barbui, T.; Thiele, J.; Passamonti, F.; Rumi, E.; Boveri, E.; Ruggeri, M.; Rodeghiero, F.; d’Amore, E.S.; Randi, M.L.; Bertozzi, I.; et al. Survival and disease progression in essential thrombocythemia are significantly influenced by accurate morphologic diagnosis: An international study. J. Clin. Oncol. 2011, 29, 3179–3184. [Google Scholar] [CrossRef]
- Finazzi, G.; Harrison, C. Essential thrombocythemia. Semin. Hematol. 2005, 42, 230–238. [Google Scholar] [CrossRef] [PubMed]
- Harrison, C.N.; Green, A.R. Essential thrombocythemia. Essential thrombocythemia. Hematol./ Oncol. Clin. 2003, 17, 1175–1190. [Google Scholar] [CrossRef]
- Florena, A.M.; Tripodo, C.; Iannitto, E.; Porcasi, R.; Ingrao, S.; Franco, V. Value of bone marrow biopsy in the diagnosis of essential thrombocythemia. Haematologica 2004, 89, 911–919. [Google Scholar]
- Gianelli, U.; Vener, C.; Raviele, P.R.; Moro, A.; Savi, F.; Annaloro, C.; Somalvico, F.; Radaelli, F.; Franco, V.; Deliliers, G.L. Essential thrombocythemia or chronic idiopathic myelofibrosis? A single-center study based on hematopoietic bone marrow histology. Leuk. Lymphoma 2006, 47, 1774–1781. [Google Scholar] [CrossRef] [PubMed]
- Thiele, J.; Kvasnicka, H.M. Chronic myeloproliferative disorders with thrombocythemia: A comparative study of two classification systems (PVSG, WHO) on 839 patients. Ann. Hematol. 2003, 82, 148–152. [Google Scholar] [CrossRef] [PubMed]
- Thiele, J.; Kvasnicka, H.M.; Schmitt-Graeff, A.; Zankovich, R.; Diehl, V. Follow-up examinations including sequential bone marrow biopsies in essential thrombocythemia (ET): A retrospective clinicopathological study of 120 patients. Am. J. Hematol. 2002, 70, 283–291. [Google Scholar] [CrossRef] [PubMed]
- Thiele, J.; Kvasnicka, H.M.; Zankovich, R.; Diehl, V. Relevance of bone marrow features in the differential diagnosis between essential thrombocythemia and early stage idiopathic myelofibrosis. Haematologica 2000, 85, 1126–1134. [Google Scholar] [PubMed]
- Pietra, D.; Rumi, E.; Ferretti, V.V.; Di Buduo, C.A.; Milanesi, C.; Cavalloni, C.; Sant’Antonio, E.; Abbonante, V.; Moccia, F.; Casetti, I.C.; et al. Differential clinical effects of different mutation subtypes in CALR-mutant myeloproliferative neoplasms. Leukemia 2016, 30, 431–438. [Google Scholar] [CrossRef]
- Tefferi, A.; Wassie, E.A.; Guglielmelli, P.; Gangat, N.; Belachew, A.A.; Lasho, T.L.; Finke, C.; Ketterling, R.P.; Hanson, C.A.; Pardanani, A.; et al. Type 1 versus Type 2 calreticulin mutations in essential thrombocythemia: A collaborative study of 1027 patients. Am. J. Hematol. 2014, 89, E121–E124. [Google Scholar] [CrossRef]
- Rumi, E.; Pietra, D.; Ferretti, V.; Klampfl, T.; Harutyunyan, A.S.; Milosevic, J.D.; Them, N.C.; Berg, T.; Elena, C.; Casetti, I.C.; et al. JAK2 or CALR mutation status defines subtypes of essential thrombocythemia with substantially different clinical course and outcomes. Blood 2014, 123, 1544–1551. [Google Scholar] [CrossRef]
- Bain, B.J.; Horny, H.P.; Hasserjian, R.P.; Orazi, A. Chronic eosinophilic leukaemia, NOS. In WHO Classification of Tumours of the Haematopoietic and Lymphoid Tissues, revised, 4th ed.; Swerdlow, S.H., Campo, E., Harris, N.L., Jaffe, E.S., Pileri, S.A., Stein, H., Thiele, J., Eds.; IARC Press: Lyon, France, 2017; pp. 54–56. [Google Scholar]
- Reiter, A.; Gotlib, J. Myeloid neoplasms with eosinophilia. Blood 2017, 129, 704–714. [Google Scholar] [CrossRef]
- Valent, P.; Klion, A.D.; Horny, H.P.; Roufosse, F.; Gotlib, J.; Weller, P.F.; Hellmann, A.; Metzgeroth, G.; Leiferman, K.M.; Arock, M.; et al. Contemporary consensus proposal on criteria and classification of eosinophilic disorders and related syndromes. J. Allergy Clin. Immunol. 2012, 130, 607–612. [Google Scholar] [CrossRef]
- Helbig, G.; Soja, A.; Bartkowska-Chrobok, A.; Kyrcz-Krzemien, S. Chronic eosinophilic leukemia-not otherwise specified has a poor prognosis with unresponsiveness to conventional treatment and high risk of acute transformation. Am. J. Hematol. 2012, 87, 643–645. [Google Scholar] [CrossRef]
- Foucar, K.; McKenna, R.; Peterson, L.C.; Kroft, S.H. Chronic eosinophilic leukemia, NOS and hypereosinophilic syndrome. In Tumors of the Bone Marrow, 4th series ed.; Silverberg, S.G., DeLellis, R.A., Sobin, L.H., Eds.; AFIP atlas of the tumor pathology Series 4; ARP Press: Washington, DC, USA, 2016; pp. 297–300. [Google Scholar]
- Bain, B.J. Eosinophilic leukaemias and the idiopathic hypereosinophilic syndrome. Br. J. Haematol. 1996, 95, 2–9. [Google Scholar] [PubMed]
- Parreira, L.; Tavares de Castro, J.; Hibbin, J.A.; Marsh, J.C.; Marcus, R.E.; Babapulle, V.B.; Spry, C.J.; Goldman, J.M.; Catovsky, D. Chromosome and cell culture studies in eosinophilic leukaemia. Br. J. Haematol. 1986, 62, 659–669. [Google Scholar] [CrossRef]
- Pardanani, A.; Lasho, T.; Wassie, E.; Finke, C.; Zblewski, D.; Hanson, C.A.; Ketterling, R.P.; Gangat, N.; Tefferi, A. Predictors of survival in WHO-defined hypereosinophilic syndrome and idiopathic hypereosinophilia and the role of next-generation sequencing. Leukemia 2016, 30, 1924–1926. [Google Scholar] [CrossRef] [PubMed]
- Cross, N.C.P.; Hoade, Y.; Tapper, W.J.; Carreno-Tarragona, G.; Fanelli, T.; Jawhar, M.; Naumann, N.; Pieniak, I.; Lubke, J.; Ali, S.; et al. Recurrent activating STAT5B N642H mutation in myeloid neoplasms with eosinophilia. Leukemia 2019, 33, 415–425. [Google Scholar] [CrossRef]
- Bain, B.J.; Ahmad, S. Should myeloid and lymphoid neoplasms with PCM1-JAK2 and other rearrangements of JAK2 be recognized as specific entities? Br. J. Haematol. 2014, 166, 809–817. [Google Scholar] [CrossRef] [PubMed]
- Zaliova, M.; Moorman, A.V.; Cazzaniga, G.; Stanulla, M.; Harvey, R.C.; Roberts, K.G.; Heatley, S.L.; Loh, M.L.; Konopleva, M.; Chen, I.M.; et al. Characterization of leukemias with ETV6-ABL1 fusion. Haematologica 2016, 101, 1082–1093. [Google Scholar] [CrossRef]
- Tirado, C.A.; Siangchin, K.; Shabsovich, D.S.; Sharifian, M.; Schiller, G. A novel three-way rearrangement involving ETV6 (12p13) and ABL1 (9q34) with an unknown partner on 3p25 resulting in a possible ETV6-ABL1 fusion in a patient with acute myeloid leukemia: A case report and a review of the literature. Biomark. Res. 2016, 4, 16. [Google Scholar] [CrossRef]
- Kvasnicka, H.M.; Thiele, J.; Orazi, A.; Horny, H.P.; Bain, B.J. Myeloproliferative neoplasm, unclassifiable. In WHO Classification of Tumours of the Haematopoietic and Lymphoid Tissues, revised, 4th ed.; Swerdlow, S.H., Campo, E., Harris, N.L., Jaffe, E.S., Pileri, S.A., Stein, H., Thiele, J., Eds.; IARC Press: Lyon, France, 2017; pp. 57–59. [Google Scholar]
- Gianelli, U.; Cattaneo, D.; Bossi, A.; Cortinovis, I.; Boiocchi, L.; Liu, Y.C.; Augello, C.; Bonometti, A.; Fiori, S.; Orofino, N.; et al. The myeloproliferative neoplasms, unclassifiable: Clinical and pathological considerations. Mod. Pathol. 2017, 30, 1043. [Google Scholar] [CrossRef]
- Lin, Y.; Liu, E.; Sun, Q.; Ma, J.; Li, Q.; Cao, Z.; Wang, J.; Jia, Y.; Zhang, H.; Song, Z.; et al. The Prevalence of JAK2, MPL, and CALR Mutations in Chinese Patients With BCR-ABL1-Negative Myeloproliferative Neoplasms. Am. J. Clin. Pathol. 2015, 144, 165–171. [Google Scholar] [CrossRef]
- Srour, S.A.; Devesa, S.S.; Morton, L.M.; Check, D.P.; Curtis, R.E.; Linet, M.S.; Dores, G.M. Incidence and patient survival of myeloproliferative neoplasms and myelodysplastic/myeloproliferative neoplasms in the United States, 2001–2012. Br. J. Haematol. 2016, 174, 382–396. [Google Scholar] [CrossRef]
- Gisslinger, H.; Jeryczynski, G.; Gisslinger, B.; Wolfler, A.; Burgstaller, S.; Buxhofer-Ausch, V.; Schalling, M.; Krauth, M.T.; Schiefer, A.I.; Kornauth, C.; et al. Clinical impact of bone marrow morphology for the diagnosis of essential thrombocythemia: Comparison between the BCSH and the WHO criteria. Leukemia 2016, 30, 1126–1132. [Google Scholar] [CrossRef]
- Iurlo, A.; Gianelli, U.; Cattaneo, D.; Thiele, J.; Orazi, A. Impact of the 2016 revised WHO criteria for myeloproliferative neoplasms, unclassifiable: Comparison with the 2008 version. Am. J. Hematol. 2017, 92, E48–E51. [Google Scholar] [CrossRef]
- Wilkins, B.S.; Erber, W.N.; Bareford, D.; Buck, G.; Wheatley, K.; East, C.L.; Paul, B.; Harrison, C.N.; Green, A.R.; Campbell, P.J. Bone marrow pathology in essential thrombocythemia: Interobserver reliability and utility for identifying disease subtypes. Blood 2008, 111, 60–70. [Google Scholar] [CrossRef]
- Barbui, T.; Thiele, J.; Ferrari, A.; Vannucchi, A.M.; Tefferi, A. The new WHO classification for essential thrombocythemia calls for revision of available evidences. Blood Cancer J. 2020, 10, 22. [Google Scholar] [CrossRef]
- Elala, Y.C.; Lasho, T.L.; Gangat, N.; Finke, C.; Barraco, D.; Haider, M.; Abou Hussein, A.K.; Hanson, C.A.; Ketterling, R.P.; Pardanani, A.; et al. Calreticulin variant stratified driver mutational status and prognosis in essential thrombocythemia. Am. J. Hematol. 2016, 91, 503–506. [Google Scholar] [CrossRef]
- Rumi, E.; Pietra, D.; Pascutto, C.; Guglielmelli, P.; Martinez-Trillos, A.; Casetti, I.; Colomer, D.; Pieri, L.; Pratcorona, M.; Rotunno, G.; et al. Clinical effect of driver mutations of JAK2, CALR, or MPL in primary myelofibrosis. Blood 2014, 124, 1062–1069. [Google Scholar] [CrossRef] [PubMed]
- Sirinukunwattana, K.; Aberdeen, A.; Theissen, H.; Sousos, N.; Psaila, B.; Mead, A.J.; Turner, G.D.H.; Rees, G.; Rittscher, J.; Royston, D. Artificial intelligence-based morphological fingerprinting of megakaryocytes: A new tool for assessing disease in MPN patients. Blood Adv. 2020, 4, 3284–3294. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, S.; Fontanillas, P.; Flannick, J.; Manning, A.; Grauman, P.V.; Mar, B.G.; Lindsley, R.C.; Mermel, C.H.; Burtt, N.; Chavez, A.; et al. Age-related clonal hematopoiesis associated with adverse outcomes. N. Engl. J. Med. 2014, 371, 2488–2498. [Google Scholar] [CrossRef]
- Soderquist, C.R.; Ewalt, M.D.; Czuchlewski, D.R.; Geyer, J.T.; Rogers, H.J.; Hsi, E.D.; Wang, S.A.; Bueso-Ramos, C.E.; Orazi, A.; Arber, D.A.; et al. Myeloproliferative neoplasms with concurrent BCR-ABL1 translocation and JAK2 V617F mutation: A multi-institutional study from the bone marrow pathology group. Mod. Pathol. 2018, 31, 690–704. [Google Scholar] [CrossRef]
- Hasserjian, R.P.; Kelley, T.W.; Weinberg, O.K.; Morgan, E.A.; Fend, F. Genetic Testing in the Diagnosis and Biology of Myeloid Neoplasms (Excluding Acute Leukemias). Am. J. Clin. Pathol. 2019, 152, 302–321. [Google Scholar] [CrossRef] [PubMed]
- Bonzheim, I.; Mankel, B.; Klapthor, P.; Schmidt, J.; Hinrichsen, T.; Wachter, O.; Fend, F.; Quintanilla-Martinez, L. CALR-mutated essential thrombocythemia evolving to chronic myeloid leukemia with coexistent CALR mutation and BCR-ABL translocation. Blood 2015, 125, 2309–2311. [Google Scholar] [CrossRef]
- da Costa, V.E.F.; de Oliveira, R.D.; Traina, F.; Chahud, F.; Palma, L.C.; de Figueiredo-Pontes, L.L. Co-occurrence of BCR-ABL1-positive chronic myeloid leukaemia and CALR-mutated essential thrombocythaemia. Br. J. Haematol. 2020, 188, e21–e23. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Hu, R.; Du, Z.; Abecasis, M.; Wang, C. Atypical myeloproliferative neoplasm with concurrent BCR-ABL1 fusion and CALR mutation: A case report and literature review. Medicine (Baltimore) 2020, 99, e18811. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Hasserjian, R.P.; Kroft, S.H.; Harrington, A.M.; Wheaton, S.E.; Pildain, A.; Ewalt, M.D.; Gratzinger, D.; Hosking, P.; Olteanu, H. Pure Erythroid Leukemia and Erythroblastic Sarcoma Evolving From Chronic Myeloid Neoplasms. Am. J. Clin. Pathol. 2016, 145, 538–551. [Google Scholar] [CrossRef] [PubMed]
- Boiocchi, L.; Espinal-Witter, R.; Geyer, J.T.; Steinhilber, J.; Bonzheim, I.; Knowles, D.M.; Fend, F.; Orazi, A. Development of monocytosis in patients with primary myelofibrosis indicates an accelerated phase of the disease. Mod. Pathol. 2013, 26, 204–212. [Google Scholar] [CrossRef]
- Federmann, B.; Abele, M.; Rosero Cuesta, D.S.; Vogel, W.; Boiocchi, L.; Kanz, L.; Quintanilla-Martinez, L.; Orazi, A.; Bonzheim, I.; Fend, F. The detection of SRSF2 mutations in routinely processed bone marrow biopsies is useful in the diagnosis of chronic myelomonocytic leukemia. Hum. Pathol. 2014, 45, 2471–2479. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).