
Differential but Concerted Expression of HSD17B2, HSD17B3, SHBG and SRD5A1 Testosterone Tetrad Modulate Therapy Response and Susceptibility to Disease Relapse in Patients with Prostate Cancer

 ,
,
Abstract
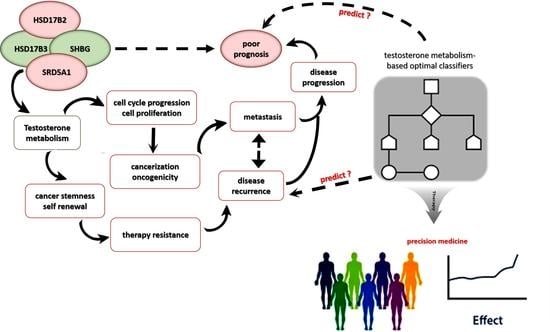
Simple Summary
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
1.1. Clinical and Molecular Characteristics of Prostate Cancer
1.2. Testosterone Metabolism, Therapy Response, and Susceptibility to Disease Relapse in Patients with PCa
1.3. The Clinical Implication of Dysregulated Testosterone Metabolism in Therapy Response and Susceptibility to Disease Relapse in Patients with PCa
1.4. The Complicity of Testosterone-Addiction and Testosterone Metabolic Reprogramming in the Response of PCa Cells to Therapy, and Propensity for Recurrence
1.5. Translational Relevance of Present Study
2. Material and Methods
2.1. Prostate Cancer Tissue Samples
2.2. Cell Culture and Chemicals
2.3. Antibodies and Reagents
2.4. Cell Viability and Proliferation Colorimetric Assay
2.5. Drug Combination Assay
2.6. Knockout of SRD5A1 by CRISPR Interference
2.7. Construction and Transfection of Plasmids Expressing SHBG
2.8. Immunohistochemical (IHC) and Immunofluorescence (IFC) Staining Assays
2.9. Western Blotting Assay
2.10. Tumorsphere Formation and Self-Renewal Assay
2.11. Scratch-Wound Healing Migration Assay
2.12. Invasion Assay
2.13. Tumor Xenograft In Vivo Studies
2.14. Statistical Analysis
3. Results
3.1. The Aberrant Expression of the Testosterone Metabolic Tetrad HSD17B2, HSD17B3, SHBG, and SRD5A1 Characterize Androgen-Addicted PCa and Is Associated with Disease Progression
3.2. Variation in HSD17B2, HSD17B3, SHBG, and SRD5A1 Expression Co-Occur and Concertedly Bear Significant Prognostic Relevance in Patients with PCa
3.3. The Differential but Concerted Expression of HSD17B2, HSD17B3, SHBG, and SRD5A1 Is Associated with the Metastatic and Recurrent Phenotype of Patients with PCa
3.4. Distinct Interaction between the Testosterone Tetrad Elicits Androgenic Signals That Drive Cell Cycle Progression, Enhanced Motility, Cancer Stemness, and Resistance to Therapy in Patients with PCa
3.5. Molecular Fine-Tuning of Components of the 4-Gene PCa Signature Modulate the Highly Proliferative, Metastatic and Cancer Stem-Cell-Like (Cum Disease Recurrent) Phenotypes of PCa Cells
3.6. Compared with PSA, the HSD17B2/HSD17B3/SHBG/SRD5A1 4-Gene Signature Is Capable of Differentiating Recurrent/Nonresponsive from Nonrecurrent/Responsive PCa
3.7. Inhibition of SRD5A1 with Dutasteride Synergistically Enhances the Anticancer Potential of Low Dose Docetaxel, Reduced Metastatic Burden, and Confer Survival Advantage, In Vivo
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ferlay, J.; Ervik, M.; Lam, F.; Colombet, M.; Mery, L.; Piñeros, M.; Znaor, A.; Soerjomataram, I.; Bray, F. Global Cancer Observatory: Cancer Tomorrow; International Agency for Research on Cancer: Lyon, France, 2018; Available online: https://gco.iarc.fr/tomorrow (accessed on 14 December 2020).
- Prostate Cancer Recurrence: Genomic Clues. Cancer Discov. 2017, 7, 240. [CrossRef][Green Version]
- Mansinho, A.; Macedo, D.; Fernandes, I.; Costa, L. Castration-Resistant Prostate Cancer: Mechanisms, Targets and Treatment. Adv. Exp. Med. Biol. 2018, 1096, 117–133. [Google Scholar] [CrossRef]
- Corona, G.; Baldi, E.; Maggi, M. Androgen regulation of prostate cancer: Where are we now? J. Endocrinol. Investig. 2011, 34, 232–243. [Google Scholar] [CrossRef]
- Audet-Walsh, É.; Yee, T.; Tam, I.S.; Giguère, V. Inverse Regulation of DHT Synthesis Enzymes 5α-Reductase Types 1 and 2 by the Androgen Receptor in Prostate Cancer. Endocrinology 2017, 158, 1015–1021. [Google Scholar] [CrossRef] [PubMed]
- Michaud, J.E.; Billups, K.L.; Partin, A.W. Testosterone and prostate cancer: An evidence-based review of pathogenesis and oncologic risk. Ther. Adv. Urol. 2015, 7, 378–387. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Pham, T.; McWhinney, B.C.; Ungerer, J.P.; Pretorius, C.J.; Richard, D.J.; Mortimer, R.H.; d’Emden, M.C.; Richard, K. Sex Hormone Binding Globulin Modifies Testosterone Action and Metabolism in Prostate Cancer Cells. Int. J. Endocrinol. 2016, 2016, 6437585. [Google Scholar] [CrossRef]
- Mostaghel, E.A.; Zhang, A.; Hernandez, S.; Marck, B.T.; Zhang, X.; Tamae, D.; Biehl, H.E.; Tretiakova, M.; Bartlett, J.; Burns, J.; et al. Contribution of Adrenal Glands to Intratumor Androgens and Growth of Castration-Resistant Prostate Cancer. Clin. Cancer Res. 2019, 25, 426–439. [Google Scholar] [CrossRef]
- Gao, X.; Dai, C.; Huang, S.; Tang, J.; Chen, G.; Li, J.; Zhu, Z.; Zhu, X.; Zhou, S.; Gao, Y.; et al. Functional Silencing of HSD17B2 in Prostate Cancer Promotes Disease Progression. Clin. Cancer Res. 2019, 25, 1291–1301. [Google Scholar] [CrossRef] [PubMed]
- Danila, D.C.; Fleisher, M.; Scher, H.I. Circulating tumor cells as biomarkers in prostate cancer. Clin. Cancer Res. 2011, 17, 3903–3912. [Google Scholar] [CrossRef]
- Torti, D.; Trusolino, L. Oncogene addiction as a foundational rationale for targeted anti-cancer therapy: Promises and perils. EMBO Mol. Med. 2011, 3, 623–636. [Google Scholar] [CrossRef] [PubMed]
- May, E.J.; Viers, L.D.; Viers, B.R.; Kawashima, A.; Kwon, E.D.; Karnes, R.J.; Froemming, A.T. Prostate cancer post-treatment follow-up and recurrence evaluation. Abdom. Radiol. 2016, 41, 862–876. [Google Scholar] [CrossRef]
- Zoni, E.; Minoli, M.; Bovet, C.; Wehrhan, A.; Piscuoglio, S.; Ng, C.K.Y.; Gray, P.C.; Spahn, M.; Thalmann, G.N.; Kruithof-de Julio, M. Preoperative plasma fatty acid metabolites inform risk of prostate cancer progression and may be used for personalized patient stratification. BMC Cancer 2019, 19, 1216. [Google Scholar] [CrossRef]
- Chuang, H.Y.; Lee, Y.P.; Lin, W.C.; Lin, Y.H.; Hwang, J.J. Fatty Acid Inhibition Sensitizes Androgen-Dependent and -Independent Prostate Cancer to Radiotherapy via FASN/NF-κB Pathway. Sci. Rep. 2019, 9, 13284. [Google Scholar] [CrossRef] [PubMed]
- Madigan, A.A.; Rycyna, K.J.; Parwani, A.V.; Datiri, Y.J.; Basudan, A.M.; Sobek, K.M.; Cummings, J.L.; Basse, P.H.; Bacich, D.J.; O’Keefe, D.S. Novel nuclear localization of fatty acid synthase correlates with prostate cancer aggressiveness. Am. J. Pathol. 2014, 184, 2156–2162. [Google Scholar] [CrossRef] [PubMed]
- Auchus, R.J.; Sharifi, N. Sex Hormones and Prostate Cancer. Annu. Rev. Med. 2020, 71, 33–45. [Google Scholar] [CrossRef]
- Boroughs, L.K.; DeBerardinis, R.J. Metabolic pathways promoting cancer cell survival and growth. Nat. Cell Biol. 2015, 17, 351–359. [Google Scholar] [CrossRef]
- Jia, D.; Lu, M.; Jung, K.H.; Park, J.H.; Yu, L.; Onuchic, J.N.; Kaipparettu, B.A.; Levine, H. Elucidating cancer metabolic plasticity by coupling gene regulation with metabolic pathways. Proc. Natl. Acad. Sci. USA 2019, 116, 3909–3918. [Google Scholar] [CrossRef]
- Moilanen, A.M.; Riikonen, R.; Oksala, R.; Ravanti, L.; Aho, E.; Wohlfahrt, G.; Nykänen, P.S.; Törmäkangas, O.P.; Palvimo, J.J.; Kallio, P.J. Discovery of ODM-201, a new-generation androgen receptor inhibitor targeting resistance mechanisms to androgen signaling-directed prostate cancer therapies. Sci. Rep. 2015, 5, 12007. [Google Scholar] [CrossRef]
- Santer, F.R.; Erb, H.H.; McNeill, R.V. Therapy escape mechanisms in the malignant prostate. Semin. Cancer Biol. 2015, 35, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Bianchini, D.; de Bono, J.S. Continued targeting of androgen receptor signalling: A rational and efficacious therapeutic strategy in metastatic castration-resistant prostate cancer. Eur. J. Cancer 2011, 47 (Suppl. S3), S189–S194. [Google Scholar] [CrossRef]
- Lévesque, É.; Huang, S.P.; Audet-Walsh, É.; Lacombe, L.; Bao, B.Y.; Fradet, Y.; Laverdière, I.; Rouleau, M.; Huang, C.Y.; Yu, C.C.; et al. Molecular markers in key steroidogenic pathways, circulating steroid levels, and prostate cancer progression. Clin. Cancer Res. 2013, 19, 699–709. [Google Scholar] [CrossRef]
- Khvostova, E.P.; Otpuschennikov, A.A.; Pustylnyak, V.O.; Gulyaeva, L.F. Gene expression of androgen metabolising enzymes in benign and malignant prostatic tissues. Horm. Metab. Res. 2015, 47, 119–124. [Google Scholar] [CrossRef][Green Version]
- Li, J.; Ding, Z.; Wang, Z.; Lu, J.; Maity, S.; Navone, N.; Logothetis, C.; Mills, G. Androgen regulation of 5α-reductase isoenzymes in prostate cancer: Implications for prostate cancer prevention. PLoS ONE 2011, 6, e28840. [Google Scholar] [CrossRef]
- Imamoto, T.; Suzuki, H.; Yano, M.; Kawamura, K.; Kamiya, N.; Araki, K.; Komiya, A.; Nihei, N.; Naya, Y.; Ichikawa, T. The role of testosterone in the pathogenesis of prostate cancer. Int. J. Urol. 2008, 15, 472–480. [Google Scholar] [CrossRef] [PubMed]
- Shiota, M.; Endo, S.; Fujimoto, N.; Tsukahara, S.; Ushijima, M.; Kashiwagi, E.; Takeuchi, A.; Inokuchi, J.; Uchiumi, T.; Eto, M. Polymorphisms in androgen metabolism genes with serum testosterone levels and prognosis in androgen-deprivation therapy. Urol. Oncol. 2020, 38, e11–e849. [Google Scholar] [CrossRef]
- Chandrasekar, T.; Yang, J.C.; Gao, A.C.; Evans, C.P. Mechanisms of resistance in castration-resistant prostate cancer (CRPC). Transl. Androl. Urol. 2015, 4, 365–380. [Google Scholar]
- O’Neill, A.J.; Prencipe, M.; Dowling, C.; Fan, Y.; Mulrane, L.; Gallagher, W.M.; O’Connor, D.; O’Connor, R.; Devey, A.; Corcoran, C.; et al. Characterisation and manipulation of docetaxel resistant prostate cancer cell lines. Mol. Cancer. 2011, 10, 126. [Google Scholar] [CrossRef] [PubMed]
- Otto, T.; Sicinski, P. Cell cycle proteins as promising targets in cancer therapy. Nat. Rev. Cancer 2017, 17, 93–115. [Google Scholar] [CrossRef]
- Otto, T.; Horn, S.; Brockmann, M.; Eilers, U.; Schüttrumpf, L.; Popov, N.; Kenney, A.M.; Schulte, J.H.; Beijersbergen, R.; Christiansen, H.; et al. Stabilization of N-Myc is a critical function of Aurora A in human neuroblastoma. Cancer Cell 2009, 15, 67–78. [Google Scholar] [CrossRef]
- Huang, T.; Song, X.; Xu, D.; Tiek, D.; Goenka, A.; Wu, B.; Sastry, N.; Hu, B.; Cheng, S.Y. Stem cell programs in cancer initiation, progression, and therapy resistance. Theranostics 2020, 10, 8721–8743. [Google Scholar] [CrossRef] [PubMed]
- Orlando, E.; Aebersold, D.M.; Medová, M.; Zimmer, Y. Oncogene addiction as a foundation of targeted cancer therapy: The paradigm of the MET receptor tyrosine kinase. Cancer Lett. 2019, 443, 189–202. [Google Scholar] [CrossRef] [PubMed]
- Settleman, J. Oncogene addiction. Curr. Biol. 2012, 22, R43–R44. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Xu, H.; Cheng, C.; Ji, Z.; Zhao, H.; Sheng, Y.; Li, X.; Wang, J.; Shu, Y.; He, Y.; et al. Identification of a Zeb1 expressing basal stem cell subpopulation in the prostate. Nat. Commun. 2020, 11, 706. [Google Scholar] [CrossRef] [PubMed]
- Taylor, R.A.; Toivanen, R.; Frydenberg, M.; Pedersen, J.; Harewood, L. Australian Prostate Cancer Bioresource, Collins, A.T.; Maitland, N.J.; Risbridger, G.P. Human epithelial basal cells are cells of origin of prostate cancer, independent of CD133 status. Stem Cells 2012, 30, 1087–1096, Erratum in: Stem Cells 2012, 30, 1786. [Google Scholar] [CrossRef] [PubMed]
- Kong, D.; Banerjee, S.; Ahmad, A.; Li, Y.; Wang, Z.; Sethi, S.; Sarkar, F.H. Epithelial to mesenchymal transition is mechanistically linked with stem cell signatures in prostate cancer cells. PLoS ONE 2010, 27, e12445. [Google Scholar] [CrossRef] [PubMed]
- Pezaro, C.; Woo, H.H.; Davis, I.D. Prostate cancer: Measuring PSA. Intern. Med. J. 2014, 44, 433–440. [Google Scholar] [CrossRef]
- Hitzeman, N.; Molina, M. Screening for prostate cancer: Prostate-specific antigen testing is not effective. Am. Fam. Physician 2011, 83, 802–804. [Google Scholar]
- Van den Broeck, T.; van den Bergh, R.C.N.; Briers, E.; Cornford, P.; Cumberbatch, M.; Tilki, D.; De Santis, M.; Fanti, S.; Fossati, N.; Gillessen, S.; et al. Biochemical Recurrence in Prostate Cancer: The European Association of Urology Prostate Cancer Guidelines Panel Recommendations. Eur. Urol. Focus. 2020, 6, 231–234. [Google Scholar] [CrossRef]
- Rizzo, M. Mechanisms of docetaxel resistance in prostate cancer: The key role played by miRNAs. Biochim. Biophys. Acta Rev. Cancer 2021, 1875, 188481. [Google Scholar] [CrossRef]
- Azuma, T.; Matayoshi, Y.; Sato, Y.; Nagase, Y. Effect of dutasteride on castration-resistant prostate cancer. Mol. Clin. Oncol. 2018, 8, 133–136. [Google Scholar] [CrossRef]
- Fleshner, N.E.; Lucia, M.S.; Egerdie, B.; Aaron, L.; Eure, G.; Nandy, I.; Black, L.; Rittmaster, R.S. Dutasteride in localised prostate cancer management: The REDEEM randomised, double-blind, placebo-controlled trial. Lancet 2012, 379, 1103–1111. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bamodu, O.A.; Tzou, K.-Y.; Lin, C.-D.; Hu, S.-W.; Wang, Y.-H.; Wu, W.-L.; Chen, K.-C.; Wu, C.-C. Differential but Concerted Expression of HSD17B2, HSD17B3, SHBG and SRD5A1 Testosterone Tetrad Modulate Therapy Response and Susceptibility to Disease Relapse in Patients with Prostate Cancer. Cancers 2021, 13, 3478. https://doi.org/10.3390/cancers13143478
Bamodu OA, Tzou K-Y, Lin C-D, Hu S-W, Wang Y-H, Wu W-L, Chen K-C, Wu C-C. Differential but Concerted Expression of HSD17B2, HSD17B3, SHBG and SRD5A1 Testosterone Tetrad Modulate Therapy Response and Susceptibility to Disease Relapse in Patients with Prostate Cancer. Cancers. 2021; 13(14):3478. https://doi.org/10.3390/cancers13143478
Chicago/Turabian StyleBamodu, Oluwaseun Adebayo, Kai-Yi Tzou, Chia-Da Lin, Su-Wei Hu, Yuan-Hung Wang, Wen-Ling Wu, Kuan-Chou Chen, and Chia-Chang Wu. 2021. "Differential but Concerted Expression of HSD17B2, HSD17B3, SHBG and SRD5A1 Testosterone Tetrad Modulate Therapy Response and Susceptibility to Disease Relapse in Patients with Prostate Cancer" Cancers 13, no. 14: 3478. https://doi.org/10.3390/cancers13143478
APA StyleBamodu, O. A., Tzou, K.-Y., Lin, C.-D., Hu, S.-W., Wang, Y.-H., Wu, W.-L., Chen, K.-C., & Wu, C.-C. (2021). Differential but Concerted Expression of HSD17B2, HSD17B3, SHBG and SRD5A1 Testosterone Tetrad Modulate Therapy Response and Susceptibility to Disease Relapse in Patients with Prostate Cancer. Cancers, 13(14), 3478. https://doi.org/10.3390/cancers13143478