Simple Summary
In this review, I discuss our recent finding that lysophospholipid metabolism is essential for the maintenance of chronic myelogenous leukemia (CML) stem cells. Lysophospholipids have only one fatty acid chain and so are more hydrophilic than phospholipids, allowing them to act as lipid second messengers. We demonstrated that the stem cell quiescence and TKI resistance displayed by CML stem cells in vivo are sustained by the Gdpd3 enzyme involved in lysophospholipid metabolism. At the mechanistic level, Gdpd3 function allows lysophospholipid metabolism to suppress the AKT/mTORC1-mediated cell growth pathway while activating the stemness factors FOXO and β-catenin. Our results thus link lysophospholipid metabolism to CML stemness, and may thereby open up new therapeutic avenues to overcome CML relapse post-TKI therapy.
Abstract
It is well known that mature chronic myelogenous leukemia (CML) cells proliferate in response to oncogenic BCR–ABL1-dependent signaling, but how CML stem cells are able to survive in an oncogene-independent manner and cause disease relapse has long been elusive. Here, I put into the context of the broader literature our recent finding that lysophospholipid metabolism is essential for the maintenance of CML stem cells. I describe the fundamentals of lysophospholipid metabolism and discuss how one of its key enzymes, Glycerophosphodiester Phosphodiesterase Domain Containing 3 (Gdpd3), is responsible for maintaining the unique characteristics of CML stem cells. I also explore how this knowledge may be exploited to devise novel therapies for CML patients.
1. Introduction
1.1. CML Stem Cells in CML Disease
Chronic myeloid leukemia (CML) arises when the BCR–ABL1 fusion oncogene forms and is activated in hematopoietic stem cells (HSCs) [1,2]. CML stem cells bearing this oncogene are generated and in turn give rise to most mature CML cells, which also bear this aberration. The pro-proliferative signaling triggered by BCR–ABL1 can be blocked by tyrosine kinase inhibitors (TKIs), which has dramatically improved the prognosis of many CML patients [2]. The first such TKI was imatinib mesylate (imatinib), and second-generation TKIs include dasatinib, nilotinib and bosutinib. Indeed, about 40–70% of chronic phase (CP)-CML patients who show a deep molecular response (DMR) to TKI therapy enjoy significant relapse-free survival without further TKI treatment [3,4,5]. However, the remaining 30–60% of CML patients who exhibit a DMR unfortunately suffer a relapse of CML disease after discontinuing TKI treatment. A major question in the field has thus been “What distinguishes CML patients who relapse from those who don’t?”.
It turns out that the CML stem cells that generate most mature CML cells are responsible for disease relapse post-TKI therapy [6,7,8]. While mature CML cells actively proliferate due to BCR–ABL1-dependent signaling, CML stem cells are able to avoid proliferation and maintain quiescence in an oncogene-independent fashion [9]. TKIs therefore eliminate most proliferating mature CML cells but have no effect on the quiescent CML stem cells [10,11]. After TKI therapy is stopped, the surviving CML stem cells emerge from quiescence and give rise to a new cadre of mature CML cells. To date, much research effort has been devoted to investigating the molecular mechanisms by which CML stem cells exploit stem cell quiescence, and searching for new modes of therapy that can be combined with TKI therapy to eradicate not only mature CML cells but also CML stem cells. Many researchers reported that numerous molecular mechanisms regulate the quiescence and TKI resistance in CML stem cells in vivo [6,7,8]. It is reportedly known that several factors within the bone marrow microenvironmental niche are also responsible for the maintenance of self-renewal capacity in CML stem cells in a non-cell autonomous manner [6,7,8].
1.2. Transcriptional Control in CML Stem Cells
To date, it is reportedly known that several signaling pathways maintain the self-renewal capacity in CML stem cells, such as JAK/STAT, Hedgehog, Wnt/β-catenin, PI3K–Akt and TGF-β-FOXO signaling within the bone marrow microenvironmental niche [6,7,8]. Pellicano et al. and our group reported that the forkhead O transcription factor FOXO plays an essential role in the maintenance of human and murine CML stem cells [12,13]. In mature CML cells, proliferation is promoted by activation of the PI3K–Akt signaling pathway that is triggered by BCR–ABL1. Activated Akt phosphorylates nuclear Foxo3a, which is then exported to the cytoplasm. This export suppresses Foxo3a’s transcriptional capacity in mature CML cells. In contrast, in quiescent CML stem cells, Akt is inactive despite the presence of BCR–ABL1 and Foxo3a remains within the nucleus and drives transcriptional activity [12,13]. The question then arises: What is the factor that overcomes BCR–ABL1 and suppresses Akt activation in CML stem cells such that Foxo3a is allowed to function? Our investigation revealed that lysophospholipid metabolism inhibits Akt, and that it is the lysophospholipase D enzyme Glycerophosphodiester Phosphodiesterase Domain Containing 3 (Gdpd3) that plays an essential role in maintaining stem cell quiescence and TKI resistance in CML stem cells [14,15]. This involvement of lysophospholipid metabolism in CML stemness opens up a new field of investigation in the realm of novel CML treatments. In this review, I attempt to familiarize the reader with the biological fundamentals of lysophospholipid metabolism, and to highlight ways in which these mechanisms might be targeted as fresh avenues of therapy for CML patients.
1.3. Biology of Lysophospholipids and Lysophosphatidic Acids
The lipid bilayer in the plasma membrane of most cells consists of the familiar glycerophospholipids (commonly referred to as “phospholipids”) that contain two fatty acid ester chains and one polar group (Figure 1). In contrast, lysophospholipids and lysophosphatidic acids (LPAs) have only one fatty acid ester chain and are more hydrophilic than phospholipids. Indeed, LPAs are not only intermediates in phospholipid biosynthesis but can also act as “lipid second messengers” themselves. With respect to phospholipid biosynthesis, glycerol 3-phosphate (G3P) is first converted into LPAs and then into various phospholipids via the Kennedy pathway (the so-called de novo pathway) [16] (Figure 1). The fatty acid ester chains and polar bases of phospholipids can then be chemically substituted via the Lands’ cycle (remodeling pathway) to sustain the production of a wide variety of phospholipid molecules [17,18]. Lysophospholipids are recycled back into LPAs by lysophospholipase D enzymes. To date, three lysophospholipase D enzymes, namely Autotaxin (ATX), GDPD3 (also termed GDE7) and GDPD1 (GDE4), have been shown to specifically hydrolyze the polar base of lysophospholipids, such as choline, ethanolamine, inositol and serine [19,20,21]. Recently, we reported that Gdpd3 plays an essential role in maintaining CML stemness in vivo [14].
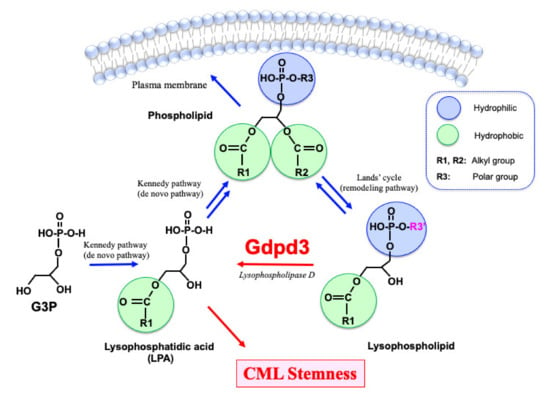
Figure 1.
Lipid metabolism in CML stem cells. Glycerophospholipids (phospholipids), which organize the lipid bilayer in a cell’s plasma membrane, are synthesized from lysophosphatidic acids (LPAs). LPAs originate from glycerol 3-phosphate (G3P) via the Kennedy pathway (de novo pathway). Phospholipids are converted to lysophospholipids via the Lands’ cycle (remodeling pathway), which can reverse to produce a wide variety of phospholipids. Lysophospholipids are recycled back into LPAs by lysophospholipase D enzymes such as Gdpd3. Whereas phospholipids have two hydrophobic fatty acid chains, lysophospholipids and LPAs have only one fatty acid chain. Thus, lysophospholipids and LPAs are more hydrophilic than phospholipids and can act as lipid second messengers. We recently demonstrated that CML stemness in vivo depends on Gdpd3 and its function in lysophospholipid metabolism [14].
2. Biological Significance of Gdpd3 in CML Stem Cells
2.1. Stem Cell Quiescence and TKI Resistance
To investigate gene expression changes specific to CML stem cells, we performed a comparative RNA-Seq analysis of murine normal HSCs and CML stem cells. We found that the most primitive long-term (LT) CML stem cells expressed the Gdpd3 gene more highly than normal LT-HSCs [14]. While lysophospholipase D enzymes were recycled back into LPA from lysophospholipids, a biological role of Gdpd3 was not identified. We thus established a Gdpd3-deficient mouse strain using genome-editing methodologies. Then, we established a CML-like disease model by retroviral BCR–ABL1 transduction into HSCs isolated from WT and Gdpd3-deficent mice, followed by bone marrow transplantation (BMT) into recipient mice. We isolated CML stem cells from these CML-affected mice and evaluated their self-renewal capacity in serial BMT experiments in vivo. After a first-round of BMT, mice that were transplanted with CML stem cells from Gdpd3-deficient mice (Gdpd3-deficient CML stem cells) and developed CML disease succumbed more rapidly than recipients transplanted with CML stem cells from WT mice (WT-CML stem cells). We then purified CML stem cells from these first-round CML-affected mice and transplanted them into a second set of recipients. To our surprise, Gdpd3-deficient CML stem cells showed a significant decrease in their ability to induce CML, whereas WT-CML stem cells maintained this capacity in a second-round of BMT. These results demonstrate that Gdpd3 is critical for the long-term maintenance of CML stem cells in vivo.
Given that stem cell quiescence is vital for the maintenance of CML stem cells, we used in vivo BrdU incorporation assays to evaluate the cell cycle distribution of Gdpd3-deficient and WT-CML stem cells in CML-affected mice after a first-round of BMT. Importantly, the frequency of S-phase cells was strikingly increased among Gdpd3-deficient CML stem cells compared to WT-CML stem cells, indicating that Gdpd3 loss activates the cell division of quiescent CML stem cells. Because sustained stem cell quiescence is the key to TKI resistance, we then investigated whether loss of Gdpd3 would affect TKI resistance in vivo. Strikingly, recipient mice that harbored Gdpd3-deficient CML stem cells and were treated with the TKI dasatinib showed a decrease in disease relapse even after a first-round of BMT. Thus, Gdpd3-mediated lysophospholipid metabolism in CML stem cells is critical for the maintenance of their quiescence and thus TKI resistance in vivo.
2.2. Lipidomics Analyses of the Gdpd3-Deficient CML Cells
Researchers have used numerous animal models and human patient samples to explore the roles of lysophospholipids in human diseases. For example, enforced transgenic expression of the Atx gene encoding the lysophospholipase D enzyme Autotaxin promoted tumor cell metastasis in a mouse model of breast cancer [22,23]. In ascites of human gastric cancer patients, lysophospholipids such as lysophosphatidyl serine (LPS) and lysophosphatidyl glycerol (LPG) were elevated [24]. Several LPAs were found to be increased in the plasma of acute coronary syndrome patients as well as in lumbar spinal cord stenoses in a rat model of cauda equina compression [25,26]. However, whether lysophospholipid metabolism is crucial in normal tissue stem cells and/or cancer stem cells has yet to be reported. Because Gdpd3 has lysophospholipase D activity that generates LPAs, we performed comparative lipidomics analyses of bone marrow CML stem cells from WT- and Gdpd3-deficient CML-affected mice. We observed that the mutant CML stem cells showed decreased LPAs compared to WT-CML stem cells [14]. These results suggest that lysophospholipid metabolism is indeed vital for CML stem cell functionality in vivo.
Lipid mediators and the signaling pathways they trigger play important roles in immune responses, inflammation and carcinogenesis [27,28,29]. Although phospholipids are known to be sources of several lipid mediators, including prostaglandins, leukotrienes and eicosanoids, it is not clear whether lysophospholipids and LPAs can also generate these lipid messengers. We conducted lipidomics analyses of 196 lipid mediators in total bone marrow CML cells and found that levels of prostaglandins, eicosanoids and a docosanoid were decreased in Gdpd3-deficient CML cells compared to WT-CML cells [14]. While it is still unknown exactly how Gdpd3 is involved in producing lipid mediators, these results suggest that at least some important messengers originate from lysophospholipid metabolism.
2.3. A Signaling Pathway That Regulates CML Stem Cell Quiescence
We have sought to define the underlying molecular mechanisms by which lysophospholipid metabolism affects stem cell quiescence and CML stemness. Because Gdpd3 loss activated the cell division of CML stem cells but attenuated their self-renewal capacity, we examined the phosphorylation of Akt and S6 ribosomal protein in WT- and Gdpd3-deficient LT-CML stem cells. Levels of phospho-Akt and phospho-S6 were increased in Gdpd3-deficient LT-CML stem cells compared to WT-LT-CML stem cells, indicating that Gdpd3 loss activates the Akt–mTORC1 signaling pathway in CML stem cells. Consistent with this finding, Foxo3a was exported to cytoplasm and inactivated in Gdpd3-deficient LT-CML stem cells, in contrast to its nuclear (activated) localization in WT-LT-CML stem cells [14]. Thus, Gdpd3-mediated lysophospholipid metabolism regulates CML stem cell quiescence by suppressing the Akt–mTORC1 pathway and promoting nuclear Foxo3a localization. Collectively, our results indicate that lysophospholipid metabolism governs CML stemness in vivo in a manner that is independent of oncogenic BCR–ABL1 signaling.
2.4. Downstream Targets Underlying CML Stemness
To understand gene expression patterns related to lysophospholipid metabolism in CML stem cells, we conducted comparative RNA-Seq analysis of WT- and Gdpd3-deficient LT-CML stem cells. We observed that expression levels of several genes encoding prototypical seven-transmembrane G-protein-coupled receptors (GPCRs) were attenuated in Gdpd3-deficient CML stem cells (Figure 2). Among these GPCR genes, mRNA levels of the Lgr4/GPR48 gene, which encodes a leucine-rich repeat (LRR)-containing GPCR, were decreased in Gdpd3-deficient LT-CML stem cells compared to WT-LT-CML stem cells. Lgr4/GPR48 is known to be involved in the canonical Wnt/β-catenin signaling pathway [30,31,32], and β-catenin cooperates with active Foxo3a to regulate the metastasis of colon cancer cells [33]. β-catenin and Foxo3a are also known to be stemness factors for CML stem cells [12,34]. Thus, we investigated if active Foxo3a interacted with β-catenin in LT-CML stem cells. Such an interaction was readily detected in the nuclei of WT-LT-CML stem cells, but was dramatically reduced in LT-CML stem cells isolated from either our Gdpd3-deficient CML mouse model or an Lgr4/Gpr48-hypomorphic mutant CML mouse model [14]. We also determined if treatment in vitro with prostaglandin E2 (PGE2), which can activate β-catenin, could rescue the defective interaction between active Foxo3a and β-catenin in mutant LT-CML stem cells [35]. To our surprise, enforced treatment in vitro with PGE2 restored this interaction within the nuclei of Lgr4/Gpr48-hypomorphic mutant LT-CML stem cells, but not in Gdpd3-deficient LT-CML stem cells (where Foxo3a was inactivated in the cytoplasm). These results suggest that Gdpd3-mediated lysophospholipid metabolism may maintain the self-renewal capacity of CML stem cells by activating the stemness factors Foxo3a and β-catenin [14,15].
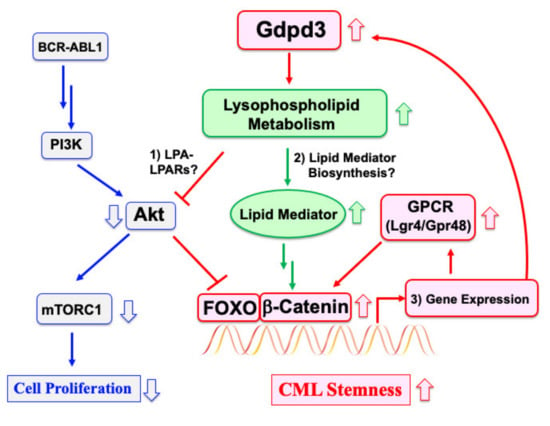
Figure 2.
Regulation of CML stemness is independent of BCR–ABL1 oncogenic signaling. In the vast majority of mature CML cells, cell proliferation is driven by BCR–ABL1-mediated activation of the PI3K–Akt–mTORC1 signaling pathway. CML stem cells are able to maintain stem cell quiescence despite possessing the oncogene and so are TKI resistant. We have shown that Gdpd3 and lysophospholipid metabolism are essential for maintaining CML stem cell functions in vivo. Elevated lysophospholipid metabolism contributes to CML stemness by regulating an interaction between active Foxo3a and β-catenin (although the exact mechanism remains unclear). It is possible that Gdpd3-mediated lysophospholipid metabolism: (1) suppresses Akt via an LPA–LPARs pathway; (2) contributes to the biosynthesis of lipid mediators; and/or (3) participates in a gene expression program involving Gdpd3 and GPCRs by FOXO/β-catenin. Targeting any one of these elements of lysophospholipid metabolism specific to CML stem cells might provide fresh therapies to overcome disease relapse in many CML patients.
3. Additional Perspectives
3.1. Suppression of Akt by an LPA–LPARs Pathway
How exactly does Gdpd3-mediated lysophospholipid metabolism suppress the Akt–mTORC1 pathway and thus activate Foxo3a, leading to the quiescence essential for self-renewal capacity? (Figure 2). Taniguchi et al. performed a crystal structure analysis demonstrating that LPAs transduce their signaling by binding to the appropriate LPA receptors (LPARs) [36]. In our study, we found that expression levels of the Lpar4/Gpr23 gene were decreased in Gdpd3-deficient LT-CML stem cells compared to WT-LT-CML stem cells. Igarashi et al. previously showed that bone marrow stromal cells isolated from Lpar4-deficient mice have a deficit in hematopoiesis-supporting capacity compared to those from WT mice [37]. It is therefore possible that LPA–LPAR4 binding may transduce signals between CML stem cells and stromal cells in their microenvironmental niche in a non-cell autonomous manner that preserves CML stemness. Further investigations are required to understand the precise mechanisms, but it is clear they must be independent of BCR–ABL1 signaling.
3.2. Functional Links between Lysophospholipid Metabolism and Lipid Mediator Biosynthesis
Although it has yet to be clarified how lysophospholipid metabolism is involved in the biosynthesis of downstream lipid mediators, several previous reports have established that certain lipid mediators and their downstream signaling targets are required for CML stemness. For example: (1) Murine CML stem cells isolated from mice lacking either arachidonate 5-oxygenase (Alox5) or Alox15 display decreased self-renewal capacity in vivo [38,39]. (2) The administration of the anti-diabetic drug pioglitazone induces DMR in TKI-insensitive CML patients by activating the proliferator-activated receptor γ (PPARγ) signaling pathway and thereby eradicating CML stem cells [40]. (3) Treatment of CML-affected mice with PGE1 plus the TKI imatinib increases therapeutic benefit over treatment with imatinib alone. Indeed, treatment of recipient mice transplanted with CML stem cells with misoprostol, an agonist of the E-type proteinoid receptor-4 (EP4), has therapeutic effects [35]. (4) Lpar3-deficient female mice show decreased Cox2 mRNA and defects in embryo implantation, a phenotype similar to that of Cox2-deficient mice lacking the Cox2 enzyme essential for generating prostaglandins [41,42]. Treatment of Lpar3-deficient female mice with PGE2 or cPGI (a stable PGI2 analogue) partially rescued these defects [41]. (5) In our study, Gdpd3-deficient CML stem cells showed decreased PGE2 levels, and in vitro treatment of Lgr4-hypomorphic mutant CML stem cells with PGE2 restored the interaction between active Foxo3a and β-catenin [14]. Collectively, these reports reinforce our contention that stimulation of lipid mediators underlying lysophospholipid metabolism promotes CML stemness.
Our knowledge is still limited as to precisely how lysophospholipids are involved in the production of the lipid mediators needed to maintain CML stem cells. It is possible that lysophospholipids are direct sources of such lipid mediators, and/or that lysophospholipids regulate the expression of genes critical for lipid mediator biosynthesis. To distinguish between these possibilities, we should determine the substrates and products of the metabolic reaction governed by Gdpd3 by using 13C-stable isotopic metabolite tracing experiments. The results of these studies will shed much-needed additional light on how lysophospholipid metabolism is linked to vital lipid mediators in CML stem cells.
3.3. A Gene Expression Program Involving Gdpd3 and GPCRs by FOXO/β-Catenin
Another unanswered question in the field is how lysophospholipid metabolism regulates the transcription of GPCR mRNAs. We observed that Gdpd3-deficient LT-CML stem cells showed decreased expression of GPCR genes, including Lgr4/GPR48, compared to WT-LT-CML stem cells [14]. Interestingly, Lgr4/GPR48 is involved in canonical Wnt/β-catenin signaling as a receptor for R-Spondins [30,31,32]. Recently, Salik et al. reported that R-Spondin-3 and Lgr4/GPR48 regulate self-renewal capacity in acute myelogenous leukemia (AML) stem cells in vivo [43], paralleling our finding that Lgr4/GPR48 is essential for the maintenance of CML stem cells in mice [14]. We also showed that LT-CML stem cells from Lgr4/GPR48-hypomorphic mutant mice had a defect in active Foxo3a/β-catenin interaction that could be rescued in vitro by PGE2. However, this treatment could not restore Foxo3a/β-catenin interaction lost due to lack of Gdpd3. Notably, Beulac et al. reported that Foxo3 induces Gdpd3 expression in a mouse model of noise-induced hearing loss [44]. These data suggest the existence of a gene expression program involving Gdpd3 and GPCRs by Foxo3a/β-catenin (Figure 2). Future work should clarify the transcriptional targets of the putative Foxo3a/β-catenin complex that governs gene expression patterns required for CML stem cell functions in vivo.
4. Conclusions
In this review, I have attempted to put into a broader biological context our recent findings on the role of lysophospholipid metabolism in general, and Gdpd3 in particular, in CML stem cells in vivo. Importantly, healthy Gdpd3-deficient mice show no obvious defects [14,45], suggesting that specific inhibition of Gdpd3-mediated lysophospholipid metabolism may be a viable means of therapeutically targeting CML stem cells without generating harmful side effects in normal tissue stem cells. Future work should determine if lysophospholipid metabolism is critical in other types of hematological cancer stem cells and/or in solid tumors. Regulating pathways within the lysophospholipid metabolome may open up new avenues for maintaining the quiescence of cancer stem cells, thereby potentially preventing disease relapse in cancer patients.
Funding
The author was supported by grants-in-aid for Scientific Research (B) (KAKENHI Grant Numbers 20H0351700) from the Ministry of Education, Culture, Sports, Science and Technology, Japan, and by the Takashi Ogimura Special Award from the Friends of Leukemia Research Fund, Japan.
Conflicts of Interest
The author was supported by a grant-in-aid from Carna Biosciences Inc., Japan.
References
- Ren, R. Mechanisms of BCR-ABL in the pathogenesis of chronic myelogenous leukaemia. Nat. Rev. Cancer 2005, 5, 172–183. [Google Scholar] [CrossRef]
- O’Hare, T.; Zabriskie, M.S.; Eiring, A.M.; Deininger, M.W. Pushing the limits of targeted therapy in chronic myeloid leukaemia. Nat. Rev. Cancer 2012, 12, 513–526. [Google Scholar] [CrossRef]
- Mahon, F.X.; Rea, D.; Guilhot, J.; Guilhot, F.; Huguet, F.; Nicolini, F.; Legros, L.; Charbonnier, A.; Guerci, A.; Varet, B.; et al. Discontinuation of imatinib in patients with chronic myeloid leukaemia who have maintained complete molecular remission for at least 2 years: The prospective, multicentre Stop Imatinib (STIM) trial. Lancet Oncol. 2010, 11, 1029–1035. [Google Scholar] [CrossRef]
- Saußele, S.; Richter, J.; Hochhaus, A.; Mahon, F.X. The concept of treatment-free remission in chronic myeloid leukemia. Leukemia 2016, 30, 1638–1647. [Google Scholar] [CrossRef]
- Ureshino, H. Treatment-free remission and immunity in chronic myeloid leukemia. Int. J. Hematol. 2021, 113, 642–647. [Google Scholar] [CrossRef]
- Holyoake, T.L.; Vetrie, D. The chronic myeloid leukemia stem cell: Stemming the tide of persistence. Blood 2017, 129, 1595–1606. [Google Scholar] [CrossRef]
- Houshmand, M.; Simonetti, G.; Circosta, P.; Gaidano, V.; Cignetti, A.; Martinelli, G.; Saglio, G.; Gale, R.P. Chronic myeloid leukemia stem cells. Leukemia 2019, 33, 1543–1556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minciacchi, V.R.; Kumar, R.; Krause, D.S. Chronic Myeloid Leukemia: A Model Disease of the Past, Present and Future. Cells 2021, 10, 117. [Google Scholar] [CrossRef]
- Corbin, A.S.; Agarwal, A.; Loriaux, M.; Cortes, J.; Deininger, M.W.; Druker, B.J. Human chronic myeloid leukemia stem cells are insensitive to imatinib despite inhibition of BCR-ABL activity. J. Clin. Investig. 2011, 121, 396–409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graham, S.M.; Jorgensen, H.G.; Allan, E.; Pearson, C.; Alcorn, M.J.; Richmond, L.; Holyoake, T.L. Primitive, quiescent, Philadelphia-positive stem cells from patients with chronic myeloid leukemia are insensitive to STI571 in vitro. Blood 2002, 99, 319–325. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, R.; Holtz, M.; Niu, N.; Gray, R.; Snyder, D.S.; Sawyers, C.L.; Arber, D.A.; Slovak, M.L.; Forman, S.J. Persistence of malignant hematopoietic progenitors in chronic myelogenous leukemia patients in complete cytogenetic remission following imatinib mesylate treatment. Blood 2003, 101, 4701–4707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naka, K.; Hoshii, T.; Muraguchi, T.; Tadokoro, Y.; Ooshio, T.; Kondo, Y.; Nakao, S.; Motoyama, N.; Hirao, A. TGF-β-FOXO signalling maintains leukaemia-initiating cells in chronic myeloid leukaemia. Nature 2010, 463, 676–680. [Google Scholar] [CrossRef] [PubMed]
- Pellicano, F.; Scott, M.T.; Helgason, G.V.; Hopcroft, L.E.; Allan, E.K.; Aspinall-O’Dea, M.; Copland, M.; Pierce, A.; Huntly, B.J.; Whetton, A.D.; et al. The anti-proliferative activity of kinase inhibitors in chronic myeloid leukaemia cells is mediated by FOXO transcription factors. Stem Cells 2014, 32, 2324–2337. [Google Scholar] [CrossRef] [Green Version]
- Naka, K.; Ochiai, R.; Matsubara, E.; Kondo, C.; Yang, K.M.; Hoshii, T.; Araki, M.; Araki, K.; Sotomaru, Y.; Sasaki, K.; et al. The lysophospholipase D enzyme Gdpd3 is required to maintain chronic myelogenous leukaemia stem cells. Nat. Commun. 2020, 11, 4681. [Google Scholar] [CrossRef] [PubMed]
- Naka, K. New routes to eradicating chronic myelogenous leukemia stem cells by targeting metabolism. Int. J. Hematol. 2021, 113, 648–655. [Google Scholar] [CrossRef]
- Kennedy, E.P.; Weiss, S.B. The function of cytidine coenzymes in the biosynthesis of phospholipides. J. Biol. Chem. 1956, 222, 193–214. [Google Scholar] [CrossRef]
- Lands, W.E. Metabolism of glycerolipides; a comparison of lecithin and triglyceride synthesis. J. Biol. Chem. 1958, 231, 883–888. [Google Scholar] [CrossRef]
- Shindou, H.; Shimizu, T. Acyl-CoA: Lysophospholipid acyltransferases. J. Biol. Chem. 2009, 284, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Tokumura, A.; Majima, E.; Kariya, Y.; Tominaga, K.; Kogure, K.; Yasuda, K.; Fukuzawa, K. Identification of human plasma lysophospholipase D, a lysophosphatidic acid-producing enzyme, as autotaxin, a multifunctional phosphodiesterase. J. Biol. Chem. 2002, 277, 39436–39442. [Google Scholar] [CrossRef] [Green Version]
- Ohshima, N.; Kudo, T.; Yamashita, Y.; Mariggio, S.; Araki, M.; Honda, A.; Nagano, T.; Isaji, C.; Kato, N.; Corda, D.; et al. New members of the mammalian glycerophosphodiester phosphodiesterase family: GDE4 and GDE7 produce lysophosphatidic acid by lysophospholipase D activity. J. Biol. Chem. 2015, 290, 4260–4271. [Google Scholar] [CrossRef] [Green Version]
- Rahman, I.A.; Tsuboi, K.; Hussain, Z.; Yamashita, R.; Okamoto, Y.; Uyama, T.; Yamazaki, N.; Tanaka, T.; Tokumura, A.; Ueda, N. Calcium-dependent generation of N-acylethanolamines and lysophosphatidic acids by glycerophosphodiesterase GDE7. Biochim. Biophys. Acta 2016, 1861, 1881–1892. [Google Scholar] [CrossRef]
- Liu, S.; Umezu-Goto, M.; Murph, M.; Lu, Y.; Liu, W.; Zhang, F.; Yu, S.; Stephens, L.C.; Cui, X.; Murrow, G.; et al. Expression of autotaxin and lysophosphatidic acid receptors increases mammary tumorigenesis, invasion, and metastases. Cancer Cell 2009, 15, 539–550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moolenaar, W.H.; Perrakis, A. Insights into autotaxin: How to produce and present a lipid mediator. Nat. Rev. Mol. Cell Biol. 2011, 12, 674–679. [Google Scholar] [CrossRef] [PubMed]
- Emoto, S.; Kurano, M.; Kano, K.; Matsusaki, K.; Yamashita, H.; Nishikawa, M.; Igarashi, K.; Ikeda, H.; Aoki, J.; Kitayama, J.; et al. Analysis of glycero-lysophospholipids in gastric cancerous ascites. J. Lipid Res. 2017, 58, 763–771. [Google Scholar] [CrossRef] [Green Version]
- Kurano, M.; Suzuki, A.; Inoue, A.; Tokuhara, Y.; Kano, K.; Matsumoto, H.; Igarashi, K.; Ohkawa, R.; Nakamura, K.; Dohi, T.; et al. Possible involvement of minor lysophospholipids in the increase in plasma lysophosphatidic acid in acute coronary syndrome. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 463–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uranbileg, B.; Ito, N.; Kurano, M.; Saigusa, D.; Saito, R.; Uruno, A.; Kano, K.; Ikeda, H.; Yamada, Y.; Sumitani, M.; et al. Alteration of the lysophosphatidic acid and its precursor lysophosphatidylcholine levels in spinal cord stenosis: A study using a rat cauda equina compression model. Sci. Rep. 2019, 9, 16578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Meer, G.; Voelker, D.R.; Feigenson, G.W. Membrane lipids: Where they are and how they behave. Nat. Rev. Mol. Cell Biol. 2008, 9, 112–124. [Google Scholar] [CrossRef]
- Serhan, C.N. Pro-resolving lipid mediators are leads for resolution physiology. Nature 2014, 510, 92–101. [Google Scholar] [CrossRef] [Green Version]
- Röhrig, F.; Schulze, A. The multifaceted roles of fatty acid synthesis in cancer. Nat. Rev. Cancer 2016, 16, 732–749. [Google Scholar] [CrossRef]
- Carmon, K.S.; Gong, X.; Lin, Q.; Thomas, A.; Liu, Q. R-spondins function as ligands of the orphan receptors LGR4 and LGR5 to regulate Wnt/b-catenin signaling. Proc. Natl. Acad. Sci. USA 2011, 108, 11452–11457. [Google Scholar] [CrossRef] [Green Version]
- De Lau, W.; Barker, N.; Low, T.Y.; Koo, B.K.; Li, V.S.; Teunissen, H.; Kujala, P.; Haegebarth, A.; Peters, P.J.; van de Wetering, M.; et al. Lgr5 homologues associate with Wnt receptors and mediate R-spondin signalling. Nature 2011, 476, 293–297. [Google Scholar] [CrossRef] [PubMed]
- Glinka, A.; Dolde, C.; Kirsch, N.; Huang, Y.L.; Kazanskaya, O.; Ingelfinger, D.; Boutros, M.; Cruciat, C.M.; Niehrs, C. LGR4 and LGR5 are R-spondin receptors mediating Wnt/b-catenin and Wnt/PCP signalling. EMBO Rep. 2011, 12, 1055–1061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tenbaum, S.P.; Ordonez-Moran, P.; Puig, I.; Chicote, I.; Arques, O.; Landolfi, S.; Fernandez, Y.; Herance, J.R.; Gispert, J.D.; Mendizabal, L.; et al. β-catenin confers resistance to PI3K and AKT inhibitors and subverts FOXO3a to promote metastasis in colon cancer. Nat. Med. 2012, 12, 892–901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heidel, F.H.; Bullinger, L.; Feng, Z.; Wang, Z.; Neff, T.A.; Stein, L.; Kalaitzidis, D.; Lane, S.W.; Armstrong, S.A. Genetic and pharmacologic inhibition of b-catenin targets imatinib-resistant leukemia stem cells in CML. Cell Stem Cell 2012, 10, 412–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, F.; He, B.; Ma, X.; Yu, S.; Bhave, R.R.; Lentz, S.R.; Tan, K.; Guzman, M.L.; Zhao, C.; Xue, H.H. Prostaglandin E1 and its analog Misoprostol inhibit human CML stem cell self-renewal via EP4 receptor activation and repression of AP-1. Cell Stem Cell 2017, 21, 359–373. [Google Scholar] [CrossRef] [Green Version]
- Taniguchi, R.; Inoue, A.; Sayama, M.; Uwamizu, A.; Yamashita, K.; Hirata, K.; Yoshida, M.; Tanaka, Y.; Kato, H.E.; Nakada-Nakura, Y.; et al. Structural insights into ligand recognition by the lysophosphatidic acid receptor LPA6. Nature 2017, 548, 356–360. [Google Scholar] [CrossRef]
- Igarashi, H.; Akahoshi, N.; Ohto-Nakanishi, T.; Yasuda, D.; Ishii, S. The lysophosphatidic acid receptor LPA4 regulates hematopoiesis-supporting activity of bone marrow stromal cells. Sci. Rep. 2015, 5, 11410. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Hu, Y.; Zhang, H.; Peng, C.; Li, S. Loss of the Alox5 gene impairs leukemia stem cells and prevents chronic myeloid leukemia. Nat. Genet. 2009, 41, 783–792. [Google Scholar] [CrossRef]
- Chen, Y.; Peng, C.; Abraham, S.A.; Shan, Y.; Guo, Z.; Desouza, N.; Cheloni, G.; Li, D.; Holyoake, T.L.; Li, S. Arachidonate 15-lipoxygenase is required for chronic myeloid leukemia stem cell survival. J. Clin. Investig. 2014, 124, 3847–3862. [Google Scholar] [CrossRef] [Green Version]
- Prost, S.; Relouzat, F.; Spentchian, M.; Ouzegdouh, Y.; Saliba, J.; Massonnet, G.; Beressi, J.P.; Verhoeyen, E.; Raggueneau, V.; Maneglier, B.; et al. Erosion of the chronic myeloid leukaemia stem cell pool by PPARg agonists. Nature 2015, 525, 380–383. [Google Scholar] [CrossRef]
- Ye, X.; Hama, K.; Contos, J.J.; Anliker, B.; Inoue, A.; Skinner, M.K.; Suzuki, H.; Amano, T.; Kennedy, G.; Arai, H.; et al. LPA3-mediated lysophosphatidic acid signalling in embryo implantation and spacing. Nature 2005, 435, 104–108. [Google Scholar] [CrossRef] [PubMed]
- Lim, H.; Paria, B.C.; Das, S.K.; Dinchuk, J.E.; Langenbach, R.; Trzaskos, J.M.; Dey, S.K. Multiple female reproductive failures in cyclooxygenase 2-deficient mice. Cell 1997, 91, 197–208. [Google Scholar] [CrossRef] [Green Version]
- Salik, B.; Yi, H.; Hassan, N.; Santiappillai, N.; Vick, B.; Connerty, P.; Duly, A.; Trahair, T.; Woo, A.J.; Beck, D.; et al. Targeting RSPO3-LGR4 Signaling for Leukemia Stem Cell Eradication in Acute Myeloid Leukemia. Cancer Cell 2020, 38, 263–278. [Google Scholar] [CrossRef] [PubMed]
- Beaulac, H.J.; Gilels, F.; Zhang, J.Y.; Jeoung, S.; White, P.A. Primed to Die: An Investigation of the Genetic Mechanisms Underlying Noise-Induced Hearing Loss and Cochlear Damage in Homozygous Foxo3-knockout Mice. Cell Death Dis. 2021, 12, 682. [Google Scholar] [CrossRef]
- Wijayatunge, R.; Holmstrom, S.R.; Foley, S.B.; Mgbemena, V.E.; Bhargava, V.; Perez, G.L.; McCrum, K.; Ross, T.S. Deficiency of the endocytic protein Hip1 leads to decreased Gdpd3 expression, low phosphocholine, and kypholordosis. Mol. Cell Biol. 2018, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).