
The Genetic Analyses of French Canadians of Quebec Facilitate the Characterization of New Cancer Predisposing Genes Implicated in Hereditary Breast and/or Ovarian Cancer Syndrome Families

Simple Summary
Abstract
1. Introduction
2. Methods Applied in the Identification of HBC and/or HBOC Syndrome Predisposing Gene Candidates
3. Genetic Analyses of FC Cancer Cases Facilitate the Interpretation of Variants in BRCA1 and BRCA2
3.1. Haplotype Analyses Suggest Common Ancestors of Frequently Occurring BRCA1 and BRCA2 Variants in the FC Population
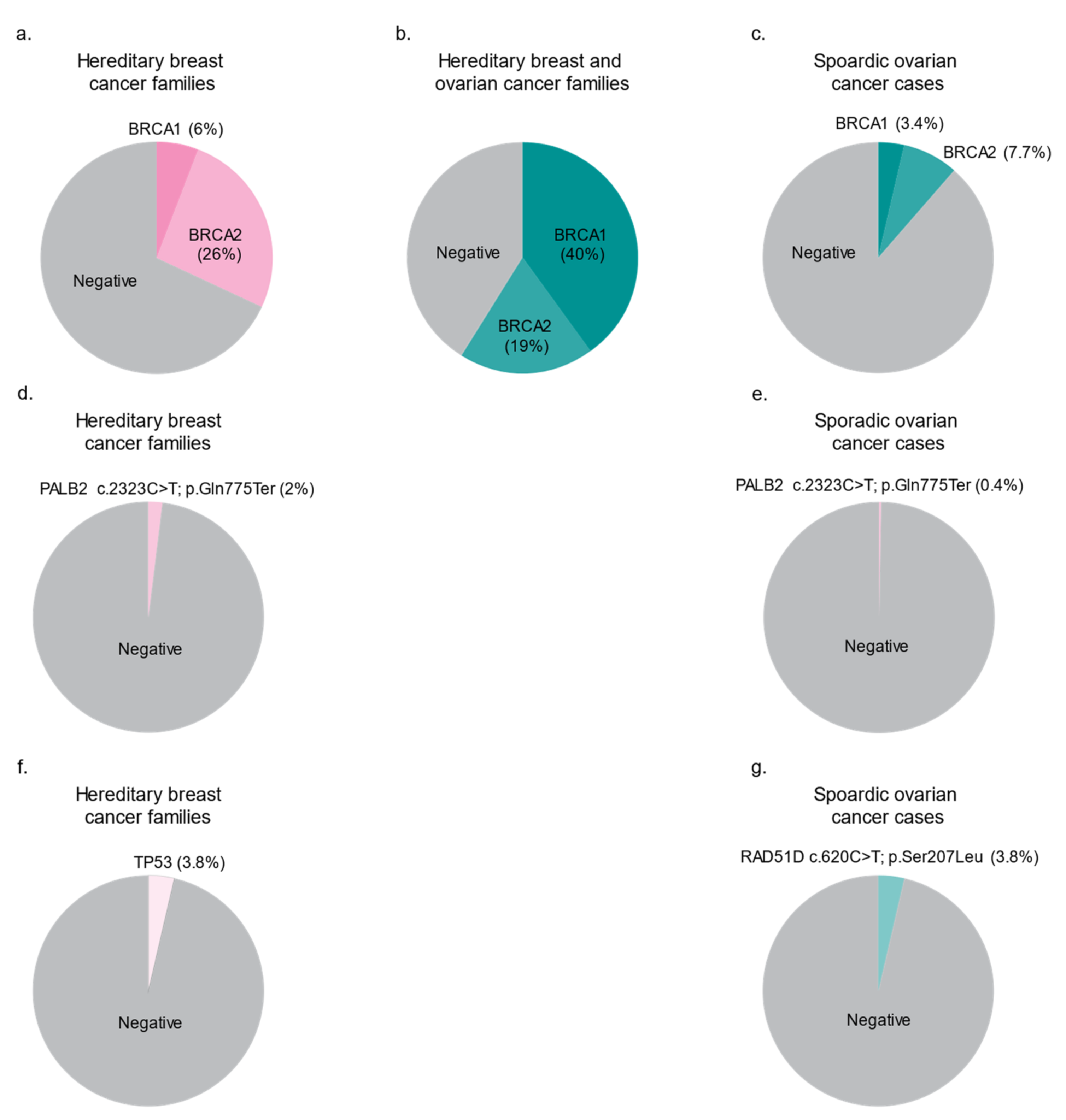
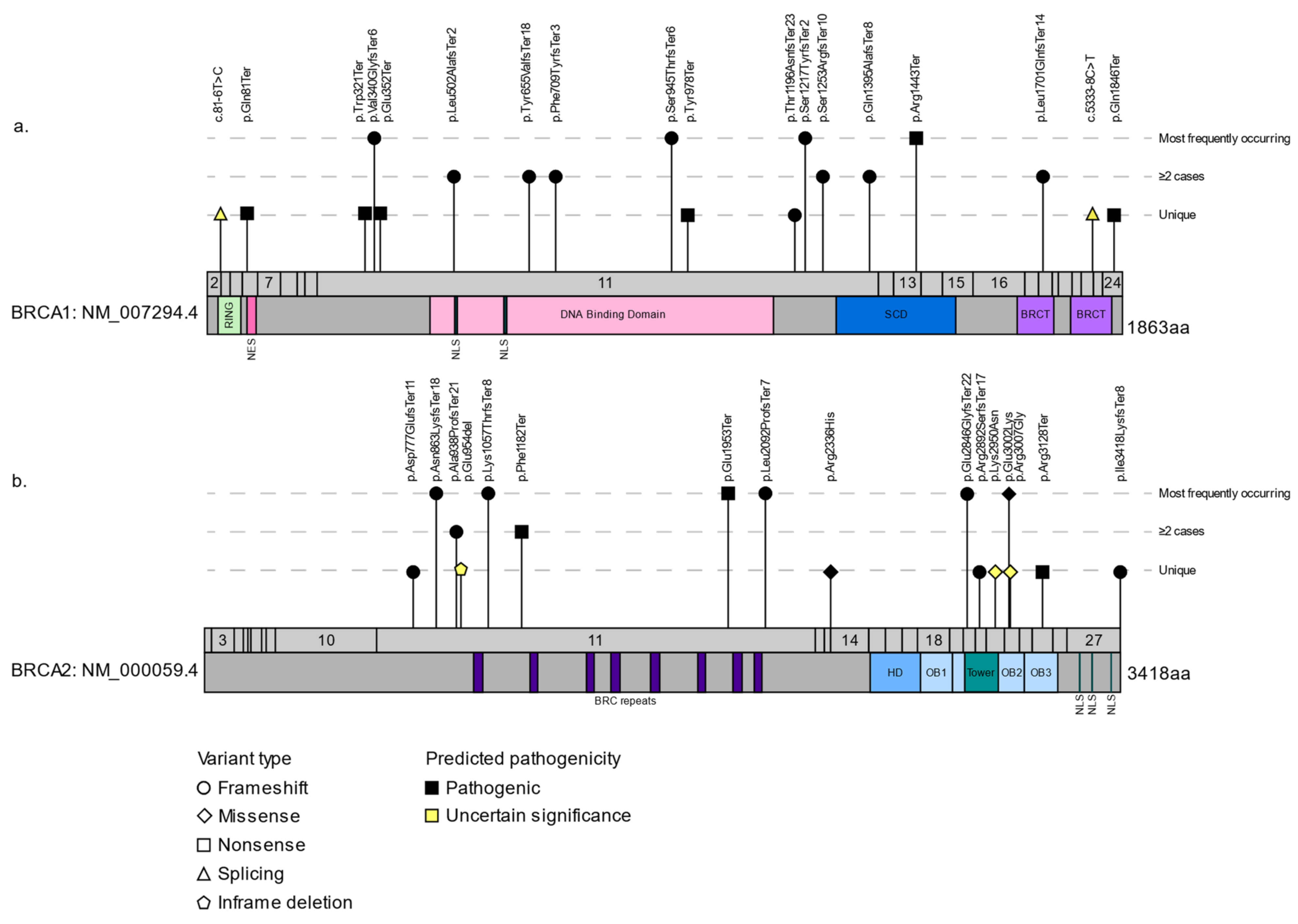
3.2. The Spectrum of BRCA1 and BRCA2 Variants in FCs
4. Genetic Analyses of FC Cancer Cases Helps Define the Role of New Candidate HBC/HBOC Predisposing Genes
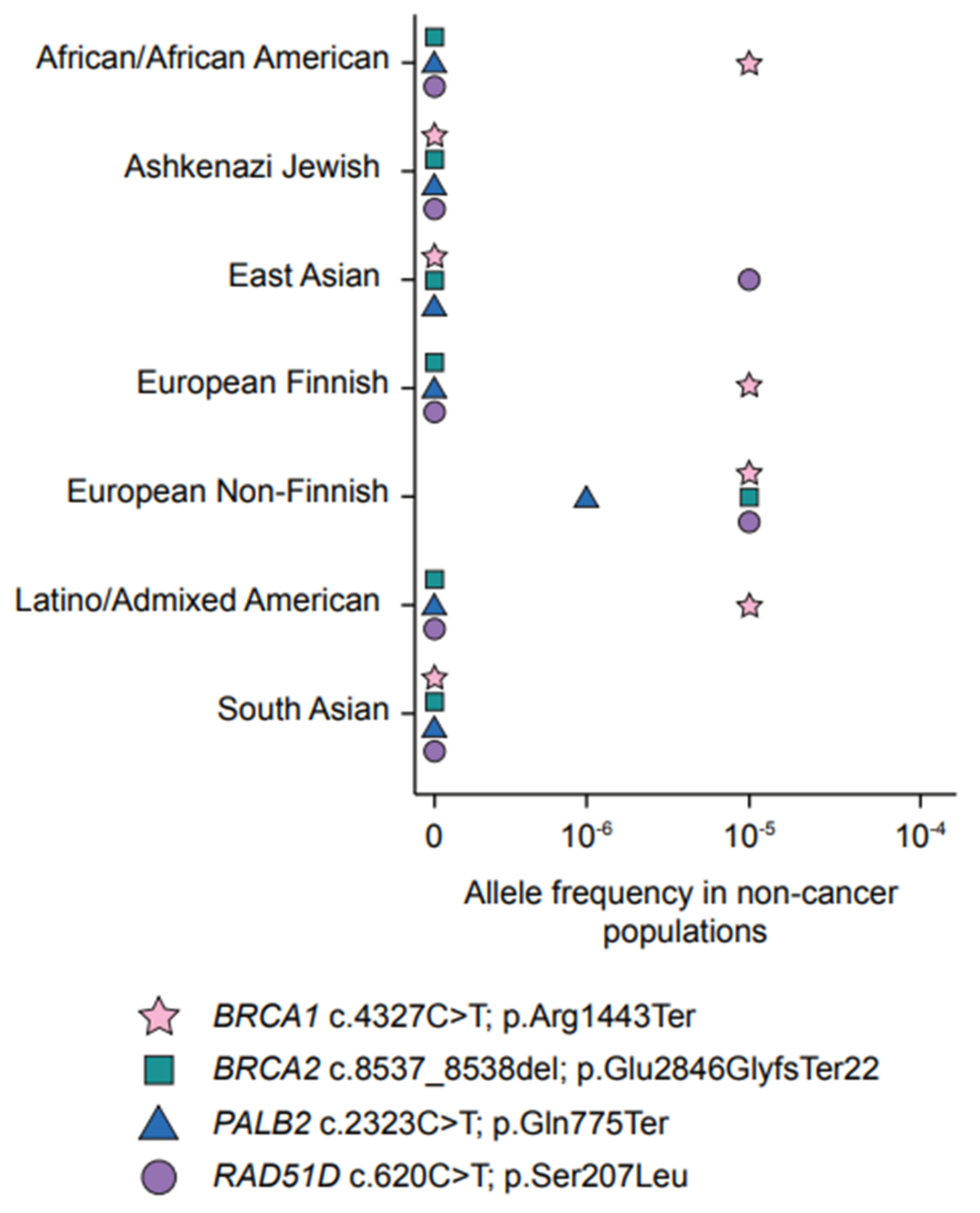
4.1. A Predominant PV in PALB2 Frequently Occurs in FC Hereditary BC Cases
4.2. A Frequently Occurring Missense Variant in FCs Supports a Role for RAD51D in Hereditary OC
4.3. BRIP1 and CHEK2 in FC BC and OC Cases
4.4. The Role of Proposed Cancer Predisposing Genes in FCs
5. Discovery of New Candidate HBC/HBOC Predisposing Genes Identified in the FC Population
6. Perspectives
7. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A. BRCA1 Discovery: A Paradigm for Genetic Linkage Analyses and Positional Cloning Approach for Identifying Cancer Predisposing Gene Candidates
Appendix B. The Unique Genetic Architecture of FCs of Quebec, Canada
References
- Mucci, L.A.; Hjelmborg, J.; Harris, J.R.; Czene, K.; Havelick, D.J.; Scheike, T.; Graff, R.E.; Holst, K.; Möller, S.; Unger, R.; et al. Familial Risk and Heritability of Cancer Among Twins in Nordic Countries. JAMA 2016, 315, 68–76. [Google Scholar] [CrossRef] [PubMed]
- Ziogas, A.; Gildea, M.; Cohen, P.; Bringman, D.; Taylor, T.H.; Seminara, D.; Barker, D.; Casey, G.; Haile, R.; Liao, S.Y.; et al. Cancer risk estimates for family members of a population-based family registry for breast and ovarian cancer. Cancer Epidemiol. Biomark. Prev. 2000, 9, 103–111. [Google Scholar]
- Miki, Y.; Swensen, J.; Shattuck-Eidens, D.; Futreal, P.A.; Harshman, K.; Tavtigian, S.; Liu, Q.; Cochran, C.; Bennett, L.M.; Ding, W.; et al. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science 1994, 266, 66–71. [Google Scholar] [CrossRef]
- Wooster, R.; Bignell, G.; Lancaster, J.; Swift, S.; Seal, S.; Mangion, J.; Collins, N.; Gregory, S.; Gumbs, C.; Micklem, G.; et al. Identification of the breast cancer susceptibility gene BRCA2. Nature 1995, 378, 789–792. [Google Scholar] [CrossRef] [PubMed]
- Rahman, N. Realizing the promise of cancer predisposition genes. Nat. Cell Biol. 2014, 505, 302–308. [Google Scholar] [CrossRef]
- Foulkes, W.D. Inherited Susceptibility to Common Cancers. N. Engl. J. Med. 2008, 359, 2143–2153. [Google Scholar] [CrossRef]
- Daly, M.B.; Pilarski, R.; Yurgelun, M.B.; Berry, M.P.; Buys, S.S.; Dickson, P.; Domchek, S.M.; Elkhanany, A.; Friedman, S.; Garber, J.E.; et al. NCCN Guidelines Insights: Genetic/Familial High-Risk Assessment: Breast, Ovarian, and Pancreatic, Version 1.2020. J. Natl. Compr. Cancer Netw. 2020, 18, 380–391. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Cline, M.S.; Liao, R.G.; Parsons, M.T.; Paten, B.; Alquaddoomi, F.; Antoniou, A.; Baxter, S.; Brody, L.; Cook-Deegan, R.; Coffin, A.; et al. BRCA Challenge: BRCA Exchange as a global resource for variants in BRCA1 and BRCA2. PLoS Genet. 2018, 14, e1007752. [Google Scholar] [CrossRef]
- Suszynska, M.; Klonowska, K.; Jasinska, A.J.; Kozlowski, P. Large-scale meta-analysis of mutations identified in panels of breast/ovarian cancer-related genes—Providing evidence of cancer predisposition genes. Gynecol. Oncol. 2019, 153, 452–462. [Google Scholar] [CrossRef]
- Yoshida, R. Hereditary breast and ovarian cancer (HBOC): Review of its molecular characteristics, screening, treatment, and prognosis. Breast Cancer 2020, 1–14. [Google Scholar] [CrossRef]
- Ford, D.; Easton, D.; Stratton, M.; Narod, S.; Goldgar, D.; Devilee, P.; Bishop, D.; Weber, B.; Lenoir, G.; Chang-Claude, J.; et al. Genetic Heterogeneity and Penetrance Analysis of the BRCA1 and BRCA2 Genes in Breast Cancer Families. Am. J. Hum. Genet. 1998, 62, 676–689. [Google Scholar] [CrossRef]
- Lynch, H.T.; Lynch, J.; Conway, T.; Watson, P.; Feunteun, J.; Lenoir, G.; Narod, S.; Fitzgibbons, R.; Feunteum, J. Hereditary breast cancer and family cancer syndromes. World J. Surg. 1994, 18, 21–31. [Google Scholar] [CrossRef]
- Narod, S.A.; Foulkes, W.D. BRCA1 and BRCA2: 1994 and beyond. Nat. Rev. Cancer 2004, 4, 665–676. [Google Scholar] [CrossRef]
- I H G S International Human Genome Sequencing Consortium. Finishing the euchromatic sequence of the human genome. Nat. Cell Biol. 2004, 431, 931–945. [Google Scholar] [CrossRef]
- Templeton, A.R. The Theory of Speciation via the Founder Principle. Genetics 1980, 94, 1011–1038. [Google Scholar] [CrossRef] [PubMed]
- Provine, W.B. Ernst Mayr: Genetics and speciation. Genetics 2004, 167, 1041–1046. [Google Scholar] [CrossRef] [PubMed]
- Gulcher, J.; Stefansson, K. Population Genomics: Laying the Groundwork for Genetic Disease Modeling and Targeting. Clin. Chem. Lab. Med. 1998, 36, 523–527. [Google Scholar] [CrossRef] [PubMed]
- Slatkin, M. A Population-Genetic Test of Founder Effects and Implications for Ashkenazi Jewish Diseases. Am. J. Hum. Genet. 2004, 75, 282–293. [Google Scholar] [CrossRef] [PubMed]
- Ebenesersdóttir, S.S.; Sandoval-Velasco, M.; Gunnarsdóttir, E.D.; Jagadeesan, A.; Guðmundsdóttir, V.B.; Thordardóttir, E.L.; Einarsdóttir, M.S.; Moore, K.H.S.; Sigurðsson, Á.; Magnúsdóttir, D.N.; et al. Ancient genomes from Iceland reveal the making of a human population. Science 2018, 360, 1028–1032. [Google Scholar] [CrossRef] [PubMed]
- Kääriäinen, H.; Muilu, J.; Perola, M.; Kristiansson, K. Genetics in an isolated population like Finland: A different basis for genomic medicine? J. Community Genet. 2017, 8, 319–326. [Google Scholar] [CrossRef] [PubMed]
- Scriver, C.R. Human genetics: Lessons from Quebec Populations. Annu. Rev. Genom. Hum. Genet. 2001, 2, 69–101. [Google Scholar] [CrossRef] [PubMed]
- Laberge, A.-M.; Michaud, J.; Richter, A.; Lemyre, E.; Lambert, M.; Brais, B.; Mitchell, G. Population history and its impact on medical genetics in Quebec. Clin. Genet. 2005, 68, 287–301. [Google Scholar] [CrossRef]
- Ferla, R.; Calò, V.; Cascio, S.; Rinaldi, G.; Badalamenti, G.; Carreca, I.; Surmacz, E.; Colucci, G.; Bazan, V.; Russo, A. Founder mutations in BRCA1 and BRCA2 genes. Ann. Oncol. 2007, 18, vi93–vi98. [Google Scholar] [CrossRef] [PubMed]
- Lea, C.S.; Gordon, N.P.; Prebil, L.A.; Ereman, R.; Uratsu, C.S.; Powell, M. Differences in reproductive risk factors for breast cancer in middle-aged women in Marin County, California and a sociodemographically similar area of Northern California. BMC Womens Health 2009, 9, 6. [Google Scholar] [CrossRef]
- Ewertz, M.; Duffy, S.W.; Adami, H.-O.; Kvåle, G.; Lund, E.; Meirik, O.; Mellemgaard, A.; Soini, I.; Tulinius, H. Age at first birth, parity and risk of breast cancer: A meta-analysis of 8 studies from the nordic countries. Int. J. Cancer 1990, 46, 597–603. [Google Scholar] [CrossRef]
- Friedman, L.S.; Szabo, C.I.; Ostermeyer, E.A.; Dowd, P.; Butler, L.; Park, T.; Lee, M.K.; Goode, E.L.; Rowell, S.E.; King, M.C. Novel inherited mutations and variable expressivity of BRCA1 alleles, including the founder mutation 185delAG in Ashkenazi Jewish families. Am. J. Hum. Genet. 1995, 57, 1284–1297. [Google Scholar]
- Struewing, J.; Abeliovich, D.; Peretz, T.; Avishai, N.; Kaback, M.M.; Collins, F.S.; Brody, L.C. The carrier frequency of the BRCA1 185delAG mutation is approximately 1 percent in Ashkenazi Jewish individuals. Nat. Genet. 1995, 11, 198–200. [Google Scholar] [CrossRef] [PubMed]
- Barkardottir, R.B.; Sarantaus, L.; Arason, A.; Vehmanen, P.; Bendahl, P.-O.; Kainu, T.; Syrjäkoski, K.; Krahe, R.; Huusko, P.; Pyrhönen, S.; et al. Haplotype analysis in Icelandic and Finnish BRCA2 999del5 breast cancer families. Eur. J. Hum. Genet. 2001, 9, 773–779. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, R.; Oak, N.; Plon, S.E. Evaluation of in silico algorithms for use with ACMG/AMP clinical variant interpretation guidelines. Genome Biol. 2017, 18, 1–12. [Google Scholar] [CrossRef]
- Alenezi, W.M.; Fierheller, C.T.; Recio, N.; Tonin, P.N. Literature Review of BARD1 as a Cancer Predisposing Gene with a Focus on Breast and Ovarian Cancers. Genes 2020, 11, 856. [Google Scholar] [CrossRef]
- Matulonis, U.A.; Sood, A.K.; Fallowfield, L.; Howitt, B.E.; Sehouli, J.; Karlan, B.Y. Ovarian cancer. Nat. Rev. Dis. Prim. 2016, 2, 16061. [Google Scholar] [CrossRef] [PubMed]
- Harbeck, N.; Penault-Llorca, F.; Cortes, J.; Gnant, M.; Houssami, N.; Poortmans, P.; Ruddy, K.; Tsang, J.; Cardoso, F. Breast cancer. Nat. Rev. Dis. Prim. 2019, 5, 1–31. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, F.C.; van Overeem Hansen, T.; Sørensen, C.S. Hereditary breast and ovarian cancer: New genes in confined pathways. Nat. Rev. Cancer 2016, 16, 599–612. [Google Scholar] [CrossRef] [PubMed]
- Oros, K.K.; Ghadirian, P.; Greenwood, C.M.; Perret, C.; Shen, Z.; Paredes, Y.; Arcand, S.L.; Mes-Masson, A.-M.; Narod, S.A.; Foulkes, W.D.; et al. Significant proportion of breast and/or ovarian cancer families of French Canadian descent harbor 1 of 5BRCA1 andBRCA2 mutations. Int. J. Cancer 2004, 112, 411–419. [Google Scholar] [CrossRef]
- Tonin, P.N.; Maugard, C.M.; Perret, C.; Mes-Masson, A.-M.; Provencher, D.M. A review of histopathological subtypes of ovarian cancer in BRCA-related French Canadian cancer families. Fam. Cancer 2007, 6, 491–497. [Google Scholar] [CrossRef]
- Arason, A.; Gunnarsson, H.; Johannesdottir, G.; Jonasson, K.; Bendahl, P.-O.; Gillanders, E.M.; Agnarsson, B.A.; Jönsson, G.; Pylkäs, K.; Mustonen, A.; et al. Genome-wide search for breast cancer linkage in large Icelandic non-BRCA1/2 families. Breast Cancer Res. 2010, 12, R50. [Google Scholar] [CrossRef]
- Marikkannu, R.; Aravidis, C.; Rantala, J.; Picelli, S.; Adamovic, T.; Keihas, M.; Liu, T.; Kontham, V.; Nilsson, D.; Lindblom, A. Whole-genome Linkage Analysis and Sequence Analysis of Candidate Loci in Familial Breast Cancer. Anticancer Res. 2015, 35, 3155–3165. [Google Scholar]
- Belanger, M.H.; Dolman, L.; Arcand, S.L.; Shen, Z.; Chong, G.; Mes-Masson, A.-M.; Provencher, D.; Tonin, P.N. A targeted analysis identifies a high frequency of BRCA1 and BRCA2 mutation carriers in women with ovarian cancer from a founder population. J. Ovarian Res. 2015, 8, 1–10. [Google Scholar] [CrossRef]
- Tischkowitz, M.; Sabbaghian, N.; Hamel, N.; Pouchet, C.; Foulkes, W.D.; Mes-Masson, A.-M.; Provencher, D.M.; Tonin, P.N. Contribution of the PALB2 c.2323C>T [p.Q775X] Founder mutation in well-defined breast and/or ovarian cancer families and unselected ovarian cancer cases of French Canadian descent. BMC Med. Genet. 2013, 14, 5. [Google Scholar] [CrossRef]
- Arcand, S.L.; Maugard, C.M.; Ghadirian, P.; Robidoux, A.; Perret, C.; Zhang, P.; Fafard, E.; Mes-Masson, A.-M.; Foulkes, W.D.; Provencher, D.; et al. Germline TP53 mutations in BRCA1 and BRCA2 mutation-negative French Canadian breast cancer families. Breast Cancer Res. Treat. 2008, 108, 399–408. [Google Scholar] [CrossRef]
- Rivera, B.; Di Iorio, M.; Frankum, J.; Nadaf, J.; Fahiminiya, S.; Arcand, S.L.; Burk, D.L.; Grapton, D.; Tomiak, E.; Hastings, V.; et al. Functionally Null RAD51D Missense Mutation Associates Strongly with Ovarian Carcinoma. Cancer Res. 2017, 77, 4517–4529. [Google Scholar] [CrossRef]
- Ramsey, S.D.; Yoon, P.; Moonesinghe, R.; Khoury, M.J. Population-based study of the prevalence of family history of cancer: Implications for cancer screening and prevention. Genet. Med. 2006, 8, 571–575. [Google Scholar] [CrossRef] [PubMed]
- Weber-Lassalle, N.; Borde, J.; Weber-Lassalle, K.; Horváth, J.; Niederacher, D.; Arnold, N.; Kaulfuß, S.; Ernst, C.; Paul, V.G.; Honisch, E.; et al. Germline loss-of-function variants in the BARD1 gene are associated with early-onset familial breast cancer but not ovarian cancer. Breast Cancer Res. 2019, 21, 1–6. [Google Scholar] [CrossRef]
- Meijers-Heijboer, H.; Van den Ouweland, A.; Klijn, J.; Wasielewski, M.; De Snoo, A.; Oldenburg, R.; Hollestelle, A.; Houben, M.; Crepin, E.; Van Veghel-Plandsoen, M.; et al. Low-penetrance susceptibility to breast cancer due to CHEK2*1100delC in noncarriers of BRCA1 or BRCA2 mutations. Nat. Genet. 2002, 31, 55–59. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, S.; Kwong, A.; Kim, S.-W.; Iau, P.; Patmasiriwat, P.; Dofitas, R.; Aryandono, T.; Hu, Z.; Huang, C.-S.; Ginsburg, O.; et al. Current Status of the Management of Hereditary Breast and Ovarian Cancer in Asia: First Report by the Asian BRCA Consortium. Public Health Genom. 2015, 19, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Okano, M.; Nomizu, T.; Tachibana, K.; Nagatsuka, M.; Matsuzaki, M.; Katagata, N.; Ohtake, T.; Yokoyama, S.; Arai, M.; Nakamura, S. The relationship between BRCA-associated breast cancer and age factors: An analysis of the Japanese HBOC consortium database. J. Hum. Genet. 2021, 66, 307–314. [Google Scholar] [CrossRef] [PubMed]
- Oliver, J.; Urban, R.Q.; Cortés, C.A.F.; Velásquez, C.E.D.; Paez, A.L.M.; Pacheco-Orozco, R.A.; Rojas, C.C.; García-Robles, R.; Rivera, J.J.L.; Chaparro, S.G.; et al. Latin American Study of Hereditary Breast and Ovarian Cancer LACAM: A Genomic Epidemiology Approach. Front. Oncol. 2019, 9, 1429. [Google Scholar] [CrossRef] [PubMed]
- Pharoah, P.D.P.; Dunning, A.M.; Ponder, B.A.J.; Easton, D.F. Association studies for finding cancer-susceptibility genetic variants. Nat. Rev. Cancer 2004, 4, 850–860. [Google Scholar] [CrossRef] [PubMed]
- Renwick, A.; Thompson, D.; Seal, S.; Kelly, P.; Chagtai, T.; Ahmed, M.; North, B.; Jayatilake, H.; Barfoot, R.; Spanova, K.; et al. ATM mutations that cause ataxia-telangiectasia are breast cancer susceptibility alleles. Nat. Genet. 2006, 38, 873–875. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Hart, S.N.; Gnanaolivu, R.; Huang, H.; Lee, K.Y.; Na, J.; Gao, C.; Lilyquist, J.; Yadav, S.; Boddicker, N.J.; et al. A Population-Based Study of Genes Previously Implicated in Breast Cancer. N. Engl. J. Med. 2021, 384, 440–451. [Google Scholar] [CrossRef]
- Dorling, L.; Carvalho, S.; Allen, J.; González-Neira, A.; Luccarini, C.; Wahlström, C.; Pooley, K.A.; Parsons, M.T.; Fortuno, C.; Wang, Q.; et al. Breast Cancer Risk Genes—Association Analysis in More than 113,000 Women. N. Engl. J. Med. 2021, 384, 428–439. [Google Scholar] [CrossRef]
- Seal, S.; Thompson, D.; Renwick, A.; Elliott, A.; Kelly, P.; Barfoot, R.; Chagtai, T.; Jayatilake, H.; Ahmed, M.; Spanova, K.; et al. Truncating mutations in the Fanconi anemia J gene BRIP1 are low-penetrance breast cancer susceptibility alleles. Nat. Genet. 2006, 38, 1239–1241. [Google Scholar] [CrossRef]
- Erkko, H.; Xia, B.; Nikkilä, J.; Schleutker, J.; Syrjäkoski, K.; Mannermaa, A.; Kallioniemi, A.; Pylkäs, K.; Karppinen, S.-M.; Rapakko, K.; et al. A recurrent mutation in PALB2 in Finnish cancer families. Nat. Cell Biol. 2007, 446, 316–319. [Google Scholar] [CrossRef] [PubMed]
- Meindl, A.; Hellebrand, H.; Wiek, C.; Erven, V.; Wappenschmidt, B.; Niederacher, D.; Freund, M.; Lichtner, P.; Hartmann, L.; Schaal, H.; et al. Germline mutations in breast and ovarian cancer pedigrees establish RAD51C as a human cancer susceptibility gene. Nat. Genet. 2010, 42, 410–414. [Google Scholar] [CrossRef] [PubMed]
- Loveday, C.; Turnbull, C.; Ramsay, E.; Hughes, D.; Ruark, E.; Frankum, J.R.; Bowden, G.; Kalmyrzaev, B.; Warren-perry, M.; Snape, K.; et al. Germline mutations in RAD51D confer susceptibility to ovarian cancer. Nat. Genet. 2011, 43, 879–882. [Google Scholar] [CrossRef] [PubMed]
- Cybulski, C.; Carrot-Zhang, J.; Kluźniak, W.; Rivera, B.; Kashyap, A.; Wokołorczyk, D.; Giroux, S.; Nadaf, J.; Hamel, N.; Zhang, S.; et al. Germline RECQL mutations are associated with breast cancer susceptibility. Nat. Genet. 2015, 47, 643–646. [Google Scholar] [CrossRef] [PubMed]
- Chandler, M.R.; Bilgili, E.P.; Merner, N. A Review of Whole-Exome Sequencing Efforts Toward Hereditary Breast Cancer Susceptibility Gene Discovery. Hum. Mutat. 2016, 37, 835–846. [Google Scholar] [CrossRef] [PubMed]
- Szabo, C.; Masiello, A.; Ryan, J.; Brody, L.C. The BIC Consortium the Breast Cancer Information Core: Database design, structure, and scope. Hum. Mutat. 2000, 16, 123–131. [Google Scholar] [CrossRef]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.R.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W.; et al. ClinVar: Improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018, 46, D1062–D1067. [Google Scholar] [CrossRef]
- Simard, J.; Tonin, P.; Durocher, F.; Morgan, K.; Rommens, J.; Gingras, S.; Samson, C.; Leblanc, J.-F.; Bélanger, C.; Dion, F.; et al. Common origins of BRCA1 mutations in Canadian breast and ovarian cancer families. Nat. Genet. 1994, 8, 392–398. [Google Scholar] [CrossRef] [PubMed]
- Tonin, P.; Ghadirian, P.; Phelan, C.; Lenoir, G.M.; Lynch, H.T.; Letendre, F.; Belanger, D.; Monte, M.; Narod, S.A. A large multisite cancer family is linked to BRCA2. J. Med. Genet. 1995, 32, 982–984. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Tonin, P.N.; Mes-Masson, A.-M.; Futreal, P.A.; Morgan, K.; Mahon, M.; Foulkes, W.D.; Cole, D.E.; Provencher, D.; Ghadirian, P.; Narod, S.A. Founder BRCA1 and BRCA2 Mutations in French Canadian Breast and Ovarian Cancer Families. Am. J. Hum. Genet. 1998, 63, 1341–1351. [Google Scholar] [CrossRef] [PubMed]
- Gauvin, H.; Moreau, C.; Lefebvre, J.-F.; Laprise, C.; Vézina, H.; Labuda, D.; Roy-Gagnon, M.-H. Genome-wide patterns of identity-by-descent sharing in the French Canadian founder population. Eur. J. Hum. Genet. 2014, 22, 814–821. [Google Scholar] [CrossRef]
- Charbonneau, H.; Desjardins, B.; Légaré, J.; Denis, H.; Haines, M.R. The Population of the St-Lawrence Valley 1608–1760; Steckel, R.H., Ed.; Cambridge University Press: Cambridge, UK, 2000. [Google Scholar]
- Vézina, H.; Durocher, F.; Dumont, M.; Houde, L.; Szabo, C.; Tranchant, M.; Chiquette, J.; Plante, M.; Laframboise, R.; Lépine, J.; et al. Molecular and genealogical characterization of the R1443X BRCA1 mutation in high-risk French-Canadian breast/ovarian cancer families. Qual. Life Res. 2005, 117, 119–132. [Google Scholar] [CrossRef]
- Palomba, G.; Cossu, A.; Friedman, E.; Budroni, M.; Farris, A.; Contu, A.; Pisano, M.; Baldinu, P.; Sini, M.C.; Tanda, F.; et al. Origin and distribution of the BRCA2-8765delAG mutation in breast cancer. BMC Cancer 2007, 7, 132. [Google Scholar] [CrossRef] [PubMed]
- Manning, A.P.; Abelovich, D.; Ghadirian, P.; Lambert, J.A.; Frappier, D.; Provencher, D.; Robidoux, A.; Peretz, T.; Narod, S.A.; Mes-Masson, A.-M.; et al. Haplotype analysis of BRCA2 8765delAG mutation carriers in French Canadian and Yemenite Jewish hereditary breast cancer families. Hum. Hered. 2001, 52, 116–120. [Google Scholar] [CrossRef]
- Oros, K.K.; Leblanc, G.; Arcand, S.L.; Shen, Z.; Perret, C.; Mes-Masson, A.-M.; Foulkes, W.D.; Ghadirian, P.; Provencher, D.; Tonin, P.N. Haplotype analysis suggest common founders in carriers of the recurrent BRCA2mutation, 3398delAAAAG, in French Canadian hereditary breast and/ovarian cancer families. BMC Med. Genet. 2006, 7, 23. [Google Scholar] [CrossRef]
- Biswas, K.; Das, R.; Eggington, J.M.; Qiao, H.; North, S.L.; Stauffer, S.; Burkett, S.S.; Martin, B.K.; Southon, E.; Sizemore, S.C.; et al. Functional evaluation of BRCA2 variants mapping to the PALB2-binding and C-terminal DNA-binding domains using a mouse ES cell-based assay. Hum. Mol. Genet. 2012, 21, 3993–4006. [Google Scholar] [CrossRef]
- Cote, S.; Arcand, S.L.; Royer, R.; Nolet, S.; Mes-Masson, A.-M.; Ghadirian, P.; Foulkes, W.D.; Tischkowitz, M.; Narod, S.A.; Provencher, D.; et al. The BRCA2 c.9004G>A (E2002K) [corrected] variant is likely pathogenic and recurs in breast and/or ovarian cancer families of French Canadian descent. Breast Cancer Res. Treat. 2012, 131, 333–340. [Google Scholar] [CrossRef]
- Mesman, R.L.S.; Calléja, F.M.G.R.; Hendriks, G.; Morolli, B.; Misovic, B.; Devilee, P.; van Asperen, C.J.; Vrieling, H.; Vreeswijk, M.P.G. The functional impact of variants of uncertain significance in BRCA2. Genet. Med. 2019, 21, 293–302. [Google Scholar] [CrossRef]
- Lohmann, D.R.; Gallie, B.L. Retinoblastoma; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Stephens, K., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- D’Andrea, E.; Marzuillo, C.; De Vito, C.; Di Marco, M.; Pitini, E.; Vacchio, M.R.; Villari, P. Which BRCA genetic testing programs are ready for implementation in health care? A systematic review of economic evaluations. Genet. Med. 2016, 18, 1171–1180. [Google Scholar] [CrossRef] [PubMed]
- Gabai-Kapara, E.; Lahad, A.; Kaufman, B.; Friedman, E.; Segev, S.; Renbaum, P.; Beeri, R.; Gal, M.; Grinshpun-Cohen, J.; Djemal, K.; et al. Population-based screening for breast and ovarian cancer risk due toBRCA1andBRCA2. Proc. Natl. Acad. Sci. USA 2014, 111, 14205–14210. [Google Scholar] [CrossRef] [PubMed]
- Struewing, J.; Hartge, P.; Wacholder, S.; Baker, S.M.; Berlin, M.; McAdams, M.; Timmerman, M.M.; Brody, L.C.; Tucker, M.A. The Risk of Cancer Associated with Specific Mutations ofBRCA1andBRCA2among Ashkenazi Jews. N. Engl. J. Med. 1997, 336, 1401–1408. [Google Scholar] [CrossRef] [PubMed]
- Metcalfe, K.A.; Poll, A.; Royer, R.; Llacuachaqui, M.; Tulman, A.; Sun, P.; Narod, S.A. Screening for Founder Mutations in BRCA1 and BRCA2 in Unselected Jewish Women. J. Clin. Oncol. 2010, 28, 387–391. [Google Scholar] [CrossRef] [PubMed]
- Whittemore, A.S.; Gong, G.; John, E.M.; McGuire, V.; Li, F.P.; Ostrow, K.L.; DiCioccio, R.; Felberg, A.; West, D.W. Prevalence of BRCA1 mutation carriers among U.S. non-Hispanic Whites. Cancer Epidemiol. Biomark. Prev. 2004, 13, 2078–2084. [Google Scholar]
- Risch, H.A.; McLaughlin, J.R.; Cole, D.E.; Rosen, B.; Bradley, L.; Kwan, E.; Jack, E.; Vesprini, D.J.; Kuperstein, G.; Abrahamson, J.L.; et al. Prevalence and Penetrance of Germline BRCA1 and BRCA2 Mutations in a Population Series of 649 Women with Ovarian Cancer. Am. J. Hum. Genet. 2001, 68, 700–710. [Google Scholar] [CrossRef]
- Behl, S.; Hamel, N.; De Ladurantaye, M.; Lepage, S.; Lapointe, R.; Mes-Masson, A.-M.; Foulkes, W.D. Founder BRCA1/BRCA2/PALB2 pathogenic variants in French-Canadian breast cancer cases and controls. Sci. Rep. 2020, 10, 1–7. [Google Scholar] [CrossRef]
- Tonin, P.N.; Perret, C.; Lambert, J.A.; Kantemiroff, T.; Martin, G.; Foulkes, W.D.; Ghadirian, P. Founder BRCA1 and BRCA2 mutations in early-onset french Canadian breast cancer cases unselected for family history. Int. J. Cancer 2001, 95, 189–193. [Google Scholar] [CrossRef]
- Chappuis, P.O.; Hamel, N.; Paradis, A.-J.; Deschênes, J.; Robidoux, A.; Potvin, C.; Cantin, J.; Tonin, P.; Ghadirian, P.; Foulkes, W. Prevalence of founder BRCA1 and BRCA2 mutations in unselected French Canadian women with breast cancer. Clin. Genet. 2001, 59, 418–423. [Google Scholar] [CrossRef]
- Tonin, P.N.; Mes-Masson, A.-M.; Narod, S.A.; Ghadirian, P.; Provencher, D. Founder BRCA1 and BRCA2 mutations in French Canadian ovarian cancer cases unselected for family history. Clin. Genet. 1999, 55, 318–324. [Google Scholar] [CrossRef]
- Kurian, A.W.; Ward, K.C.; Howlader, N.; Deapen, D.; Hamilton, A.S.; Mariotto, A.; Miller, D.; Penberthy, L.S.; Katz, S.J. Genetic Testing and Results in a Population-Based Cohort of Breast Cancer Patients and Ovarian Cancer Patients. J. Clin. Oncol. 2019, 37, 1305–1315. [Google Scholar] [CrossRef]
- Phelan, C.M.; Lancaster, J.M.; Tonin, P.N.; Gumbs, C.; Cochran, C.; Carter, R.L.; Ghadirian, P.; Perret, C.; Moslehi, R.; Dion, F.; et al. Mutation analysis of the BRCA2 gene in 49 site–specific breast cancer families. Nat. Genet. 1996, 13, 120–122. [Google Scholar] [CrossRef] [PubMed]
- Rudkin, T.M.; Hamel, N.; Galvez, M.; Hogervorst, F.; Gille, J.J.P.; Møller, P.; Apold, J.; Foulkes, W.D. The frequent BRCA1 mutation 1135insA has multiple origins: A haplotype study in different populations. BMC Med. Genet. 2006, 7, 15. [Google Scholar] [CrossRef] [PubMed]
- Simard, J.-C.; Dumont, M.; Moisan, A.-M.; Gaborieau, V.; Vézina, H.; Durocher, F.; Chiquette, J.; Plante, M.; Avard, D.; Bessette, P.; et al. Evaluation of BRCA1 and BRCA2 mutation prevalence, risk prediction models and a multistep testing approach in French-Canadian families with high risk of breast and ovarian cancer. J. Med. Genet. 2007, 44, 107–121. [Google Scholar] [CrossRef] [PubMed]
- Ghadirian, P.; Robidoux, A.; Zhang, P.; Royer, R.; Akbari, M.; Zhang, S.; Fafard, E.; Costa, M.; Martin, G.; Potvin, C.; et al. The contribution of founder mutations to early-onset breast cancer in French-Canadian women. Clin. Genet. 2009, 76, 421–426. [Google Scholar] [CrossRef] [PubMed]
- Cavallone, L.; Arcand, S.L.; Maugard, C.M.; Nolet, S.; Gaboury, L.A.; Mes-Masson, A.-M.; Ghadirian, P.; Provencher, D.; Tonin, P.N. Comprehensive BRCA1 and BRCA2 mutation analyses and review of French Canadian families with at least three cases of breast cancer. Fam. Cancer 2010, 9, 507–517. [Google Scholar] [CrossRef]
- Durocher, F.; Tonin, P.; Shattuck-Eidens, D.; Skolnick, M.; Narod, S.A.; Simard, J. Mutation analysis of the BRCA1 gene in 23 families with cases of cancer of the breast, ovary, and multiple other sites. J. Med. Genet. 1996, 33, 814–819. [Google Scholar] [CrossRef][Green Version]
- Ghadirian, P.; Robidoux, A.; Nassif, E.; Martin, G.; Potvin, C.; Patocskai, E.; Younan, R.; Larouche, N.; Venne, A.; Zhang, S.; et al. Screening for BRCA1 and BRCA2 mutations among French-Canadian breast cancer cases attending an outpatient clinic in Montreal. Clin. Genet. 2014, 85, 31–35. [Google Scholar] [CrossRef]
- Chen, C.-F.; Li, S.; Chen, Y.; Chen, P.-L.; Sharp, Z.D.; Lee, W.-H. The Nuclear Localization Sequences of the BRCA1 Protein Interact with the Importin-α Subunit of the Nuclear Transport Signal Receptor. J. Biol. Chem. 1996, 271, 32863–32868. [Google Scholar] [CrossRef] [PubMed]
- Spain, B.H.; Larson, C.J.; Shihabuddin, L.S.; Gage, F.H.; Verma, I.M. Truncated BRCA2 is cytoplasmic: Implications for cancer-linked mutations. Proc. Natl. Acad. Sci. USA 1999, 96, 13920–13925. [Google Scholar] [CrossRef] [PubMed]
- Tibbetts, R.; Cortez, D.; Brumbaugh, K.M.; Scully, R.; Livingston, D.; Elledge, S.J.; Abraham, R.T. Functional interactions between BRCA1 and the checkpoint kinase ATR during genotoxic stress. Genes Dev. 2000, 14, 2989–3002. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Jeffrey, P.D.; Miller, J.; Kinnucan, E.; Sun, Y.; Thomä, N.H.; Zheng, N.; Chen, P.-L.; Lee, W.-H.; Pavletich, N.P. BRCA2 Function in DNA Binding and Recombination from a BRCA2-DSS1-ssDNA Structure. Science 2002, 297, 1837–1848. [Google Scholar] [CrossRef] [PubMed]
- Pavlicek, A.; Noskov, V.N.; Kouprina, N.; Barrett, J.C.; Jurka, J.; Larionov, V. Evolution of the tumor suppressor BRCA1 locus in primates: Implications for cancer predisposition. Hum. Mol. Genet. 2004, 13, 2737–2751. [Google Scholar] [CrossRef] [PubMed]
- Teng, D.H.-R.; Bogden, R.; Mitchell, J.; Baumgard, M.; Bell, R.; Berry, S.; Davis, T.; Ha, P.C.; Kehrer, R.; Jammulapati, S.; et al. Low incidence of BRCA2 mutations in breast carcinoma and other cancers. Nat. Genet. 1996, 13, 241–244. [Google Scholar] [CrossRef] [PubMed]
- Paull, T.T.; Cortez, D.; Bowers, B.; Elledge, S.J.; Gellert, M. Direct DNA binding by Brca1. Proc. Natl. Acad. Sci. USA 2001, 98, 6086–6091. [Google Scholar] [CrossRef]
- Quintana-Murci, L.; Gal, I.; Bakhan, T.; Quach, H.; Sayar, S.H.; Shiri-Sverdlov, R.; Baruch, R.G.; McElreavey, K.; Dagan, E.; Narod, S.; et al. The Tyr978X BRCA1 mutation: Occurrence in non-Jewish Iranians and haplotype in French-Canadian and non-Ashkenazi Jews. Fam. Cancer 2005, 4, 85–88. [Google Scholar] [CrossRef]
- Vreeswijk, M.P.; Kraan, J.N.; Van Der Klift, H.M.; Vink, G.R.; Cornelisse, C.J.; Wijnen, J.T.; Bakker, E.; Van Asperen, C.J.; Devilee, P. Intronic variants inBRCA1andBRCA2that affect RNA splicing can be reliably selected by splice-site prediction programs. Hum. Mutat. 2009, 30, 107–114. [Google Scholar] [CrossRef]
- Anantha, R.W.; Simhadri, S.; Foo, T.K.; Miao, S.; Liu, J.; Shen, Z.; Ganesan, S.; Xia, B. Functional and mutational landscapes of BRCA1 for homology-directed repair and therapy resistance. eLife 2017, 6, e21350. [Google Scholar] [CrossRef]
- Guidugli, L.; Shimelis, H.; Masica, D.L.; Pankratz, V.S.; Lipton, G.B.; Singh, N.; Hu, C.; Monteiro, A.N.A.; Lindor, N.M.; Goldgar, D.E.; et al. Assessment of the clinical relevance of BRCA2 missense variants by functional and computational approaches. Am. J. Hum. Genet. 2018, 102, 233–248. [Google Scholar] [CrossRef]
- Baughan, S.; Tainsky, M. K3326X and Other C-Terminal BRCA2 Variants Implicated in Hereditary Cancer Syndromes: A Review. Cancers 2021, 13, 447. [Google Scholar] [CrossRef]
- Butz, H.; Papp, J.; Bozsik, A.; Krokker, L.; Pócza, T.; Oláh, E.; Patócs, A. Application of Multilayer Evidence for Annotation of C-Terminal BRCA2 Variants. Cancers 2021, 13, 881. [Google Scholar] [CrossRef]
- Thompson, E.R.; Gorringe, K.L.; Rowley, S.M.; Li, N.; McInerny, S.; Wong-Brown, M.W.; Devereux, L.; Li, J.; Trainer, A.H.; Mitchell, G.; et al. Reevaluation of the BRCA2 truncating allele c.9976A > T (p.Lys3326Ter) in a familial breast cancer context. Sci. Rep. 2015, 5, 14800. [Google Scholar] [CrossRef] [PubMed]
- Meeks, H.D.; Song, H.; Michailidou, K.; Bolla, M.K.; Dennis, J.; Wang, Q.; Barrowdale, D.; Frost, D.; McGuffog, L.; Ellis, S.; et al. BRCA2 Polymorphic Stop Codon K3326X and the Risk of Breast, Prostate, and Ovarian Cancers. J. Natl. Cancer Inst. 2015, 108, djv315. [Google Scholar] [CrossRef] [PubMed]
- Moisan, A.-M.; Fortin, J.; Moisan, M.D.; Samson, C.; Bessette, P.; Chiquette, J.; LaFramboise, R.; Lépine, J.; Lespérance, B.; Pichette, R.; et al. No Evidence of BRCA1/2 Genomic Rearrangements in High-Risk French-Canadian Breast/Ovarian Cancer Families. Genet. Test. 2006, 10, 104–115. [Google Scholar] [CrossRef] [PubMed]
- Rebbeck, T.R.; Mitra, N.; Wan, F.; Sinilnikova, O.M.; Healey, S.; McGuffog, L.; Mazoyer, S.; Chenevix-Trench, G.; Easton, D.F.; Antoniou, A.C.; et al. Association of Type and Location of BRCA1 and BRCA2 Mutations with Risk of Breast and Ovarian Cancer. JAMA 2015, 313, 1347–1361. [Google Scholar] [CrossRef] [PubMed]
- Malkin, D.; Li, F.P.; Strong, L.C.; Fraumeni, J.F.; Nelson, C.E.; Kim, D.H.; Kassel, J.; Gryka, M.A.; Bischoff, F.Z.; Tainsky, M.; et al. Germ line p53 mutations in a familial syndrome of breast cancer, sarcomas, and other neoplasms. Science 1990, 250, 1233–1238. [Google Scholar] [CrossRef] [PubMed]
- Jenne, D.E.; Reomann, H.; Nezu, J.-I.; Friedel, W.; Loff, S.; Jeschke, R.; Müller, O.; Back, W.; Zimmer, M. Peutz-Jeghers syndrome is caused by mutations in a novel serine threoninekinase. Nat. Genet. 1998, 18, 38–43. [Google Scholar] [CrossRef]
- Hemminki, A.; Markie, D.; Tomlinson, I.; Avizienyte, E.; Roth, S.; Loukola, A.; Bignell, G.; Warren, W.; Aminoff, M.; Höglund, P.; et al. A serine/threonine kinase gene defective in Peutz–Jeghers syndrome. Nature 1998, 391, 184–187. [Google Scholar] [CrossRef]
- Arcand, S.L.; Akbari, M.R.; Mes-Masson, A.-M.; Provencher, D.; Foulkes, W.D.; Narod, S.A.; Tonin, P.N. Germline TP53 mutational spectrum in French Canadians with breast cancer. BMC Med. Genet. 2015, 16, 24. [Google Scholar] [CrossRef]
- Olivier, M.; Hollstein, M.; Hainaut, P. TP53 Mutations in Human Cancers: Origins, Consequences, and Clinical Use. Cold Spring Harb. Perspect. Biol. 2009, 2, a001008. [Google Scholar] [CrossRef]
- Guénard, F.; Pedneault, C.S.-L.; Ouellette, G.; Labrie, Y.; Simard, J.; Durocher, F. Evaluation of the Contribution of the Three Breast Cancer Susceptibility Genes CHEK2, STK11, and PALB2 in Non-BRCA1/2 French Canadian Families with High Risk of Breast Cancer. Genet. Test. Mol. Biomarkers 2010, 14, 515–526. [Google Scholar] [CrossRef]
- Xia, B.; Sheng, Q.; Nakanishi, K.; Ohashi, A.; Wu, J.; Christ, N.; Liu, X.; Jasin, M.; Couch, F.J.; Livingston, D.M. Control of BRCA2 Cellular and Clinical Functions by a Nuclear Partner, PALB2. Mol. Cell 2006, 22, 719–729. [Google Scholar] [CrossRef]
- Foulkes, W.D.; Ghadirian, P.; Akbari, M.R.; Hamel, N.; Giroux, S.; Sabbaghian, N.; Darnel, A.; Royer, R.; Poll, A.; Fafard, E.; et al. Identification of a novel truncating PALB2mutation and analysis of its contribution to early-onset breast cancer in French-Canadian women. Breast Cancer Res. 2007, 9, R83. [Google Scholar] [CrossRef]
- Yang, X.; Song, H.; Leslie, G.; Engel, C.; Hahnen, E.; Auber, B.; Horváth, J.; Kast, K.; Niederacher, D.; Turnbull, C.; et al. Ovarian and Breast Cancer Risks Associated With Pathogenic Variants in RAD51C and RAD51D. J. Natl. Cancer Inst. 2020, 112, 1242–1250. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Leslie, G.; Doroszuk, A.; Schneider, S.; Allen, J.; Decker, B.; Dunning, A.M.; Redman, J.; Scarth, J.; Plaskocinska, I.; et al. Cancer Risks Associated With Germline PALB2 Pathogenic Variants: An International Study of 524 Families. J. Clin. Oncol. 2020, 38, 674–685. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Dicks, E.M.; Tyrer, J.; Intermaggio, M.; Chenevix-Trench, G.; Bowtell, D.D.; Traficante, N.; AOCS Group; Brenton, J.; Goranova, T.; et al. Population-based targeted sequencing of 54 candidate genes identifies PALB2 as a susceptibility gene for high-grade serous ovarian cancer. J. Med. Genet. 2021, 58, 305–313. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Arnold, A.G.; Trottier, M.; Sonoda, Y.; Abu-Rustum, N.R.; Zivanovic, O.; Robson, M.E.; Stadler, Z.K.; Walsh, M.F.; Hyman, D.M.; et al. Characterization of a novel germline PALB2 duplication in a hereditary breast and ovarian cancer family. Breast Cancer Res. Treat. 2016, 160, 447–456. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Blanco, A.; De La Hoya, M.; Balmaña, J.; Cajal, T.R.Y.; Teulé, A.; Miramar, M.-D.; Esteban, E.; Infante, M.; Benítez, J.; Torres, A.; et al. Detection of a large rearrangement in PALB2 in Spanish breast cancer families with male breast cancer. Breast Cancer Res. Treat. 2012, 132, 307–315. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Song, H.; Dicks, E.; Ramus, S.J.; Tyrer, J.P.; Intermaggio, M.P.; Hayward, J.; Edlund, C.K.; Conti, D.; Harrington, P.; Fraser, L.; et al. Contribution of Germline Mutations in the RAD51B, RAD51C, and RAD51D Genes to Ovarian Cancer in the Population. J. Clin. Oncol. 2015, 33, 2901–2907. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.-M.; Li, S.; Black, M.H.; Lee, S.; Hoiness, R.; Wu, S.; Mu, W.; Huether, R.; Chen, J.; Sridhar, S.; et al. Association of Breast and Ovarian Cancers With Predisposition Genes Identified by Large-Scale Sequencing. JAMA Oncol. 2019, 5, 51–57. [Google Scholar] [CrossRef]
- Wickramanyake, A.; Bernier, G.; Pennil, C.; Casadei, S.; Agnew, K.J.; Stray, S.M.; Mandell, J.; Garcia, R.L.; Walsh, T.; King, M.-C.; et al. Loss of function germline mutations in RAD51D in women with ovarian carcinoma. Gynecol. Oncol. 2012, 127, 552–555. [Google Scholar] [CrossRef]
- Tung, N.; Lin, N.U.; Kidd, J.; Allen, B.A.; Singh, N.; Wenstrup, R.J.; Hartman, A.-R.; Winer, E.P.; Garber, J.E. Frequency of Germline Mutations in 25 Cancer Susceptibility Genes in a Sequential Series of Patients With Breast Cancer. J. Clin. Oncol. 2016, 34, 1460–1468. [Google Scholar] [CrossRef]
- Osher, D.J.; De Leeneer, K.; Michils, G.; Hamel, N.; Tomiak, E.; Poppe, B.; Leunen, K.; Legius, E.; Shuen, A.; Smith, E.; et al. Mutation analysis of RAD51D in non-BRCA1/2 ovarian and breast cancer families. Br. J. Cancer 2012, 106, 1460–1463. [Google Scholar] [CrossRef][Green Version]
- Akbari, M.R.; Tonin, P.; Foulkes, W.D.; Ghadirian, P.; Tischkowitz, M.; A Narod, S. RAD51C germline mutations in breast and ovarian cancer patients. Breast Cancer Res. 2010, 12, 404. [Google Scholar] [CrossRef] [PubMed]
- Pavanello, M.; Chan, I.H.; Ariff, A.; Pharoah, P.D.; Gayther, S.A.; Ramus, S.J. Rare Germline Genetic Variants and the Risks of Epithelial Ovarian Cancer. Cancers 2020, 12, 3046. [Google Scholar] [CrossRef] [PubMed]
- Cantor, S.B.; Bell, D.W.; Ganesan, S.; Kass, E.M.; Drapkin, R.; Grossman, S.; Wahrer, D.C.; Sgroi, D.C.; Lane, W.S.; Haber, D.A.; et al. BACH1, a Novel Helicase-like Protein, Interacts Directly with BRCA1 and Contributes to Its DNA Repair Function. Cell 2001, 105, 149–160. [Google Scholar] [CrossRef]
- Khanna, K.K.; Lavin, M.F.; Jackson, S.P.; Mulhern, T. ATM, a central controller of cellular responses to DNA damage. Cell Death Differ. 2001, 8, 1052–1065. [Google Scholar] [CrossRef]
- Antoni, L.; Sodha, N.; Collins, I.; Garrett, M.D. CHK2 kinase: Cancer susceptibility and cancer therapy—Two sides of the same coin? Nat. Rev. Cancer 2007, 7, 925–936. [Google Scholar] [CrossRef]
- Guénard, F.; Brcas, I.; Labrie, Y.; Ouellette, G.; Beauparlant, C.J.; Simard, J.; Durocher, F. Mutational analysis of the breast cancer susceptibility gene BRIP1 /BACH1/FANCJ in high-risk non-BRCA1/BRCA2 breast cancer families. J. Hum. Genet. 2008, 53, 579–591. [Google Scholar] [CrossRef][Green Version]
- Ramus, S.J.; Song, H.; Dicks, E.; Tyrer, J.P.; Rosenthal, A.N.; Intermaggio, M.P.; Fraser, L.; Gentry-Maharaj, A.; Hayward, J.; Philpott, S.; et al. Germline Mutations in the BRIP1, BARD1, PALB2, and NBN Genes in Women With Ovarian Cancer. J. Natl. Cancer Inst. 2015, 107, djv214. [Google Scholar] [CrossRef]
- Maxwell, K.N.; Ms, B.W.; Bs, K.D.; Bs, B.G.; Long, J.M.; Powers, J.; Bs, K.R.; Stopfer, J.E.; Zhu, J.; Bradbury, A.R.; et al. Prevalence of mutations in a panel of breast cancer susceptibility genes in BRCA1/2-negative patients with early-onset breast cancer. Genet. Med. 2015, 17, 630–638. [Google Scholar] [CrossRef] [PubMed]
- Novak, D.J.; Chen, L.Q.; Ghadirian, P.; Hamel, N.; Zhang, P.; Rossiny, V.; Cardinal, G.; Robidoux, A.; Tonin, P.N.; Rousseau, F.; et al. Identification of a novel CHEK2variant and assessment of its contribution to the risk of breast cancer in French Canadian women. BMC Cancer 2008, 8, 239. [Google Scholar] [CrossRef] [PubMed]
- Thai, T.H.; Du, F.; Tsan, J.T.; Jin, Y.; Phung, A.; Spillman, M.A.; Massa, H.F.; Muller, C.Y.; Ashfaq, R.; Mathis, J.M.; et al. Mutations in the BRCA1-associated RING domain (BARD1) gene in primary breast, ovarian and uterine cancers. Hum. Mol. Genet. 1998, 7, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.C.; Wang, Z.W.; Tsan, J.T.; Spillman, M.A.; Phung, A.; Xu, X.L.; Yang, M.-C.W.; Hwang, L.-Y.; Bowcock, A.; Baer, R. Identification of a RING protein that can interact in vivo with the BRCA1 gene product. Nat. Genet. 1996, 14, 430–440. [Google Scholar] [CrossRef] [PubMed]
- Bartkova, J.; Tommiska, J.; Oplustilova, L.; Aaltonen, K.; Tamminen, A.; Heikkinen, T.; Mistrik, M.; Aittomäki, K.; Blomqvist, C.; Heikkilä, P.; et al. Aberrations of the MRE11-RAD50-NBS1 DNA damage sensor complex in human breast cancer: MRE11 as a candidate familial cancer-predisposing gene. Mol. Oncol. 2008, 2, 296–316. [Google Scholar] [CrossRef]
- Damiola, F.; Pertesi, M.; Oliver, J.; Le Calvez-Kelm, F.; Voegele, C.; Young, E.L.; Robinot, N.; Forey, N.; Durand, G.; Vallée, M.P.; et al. Rare key functional domain missense substitutions in MRE11A, RAD50, and NBNcontribute to breast cancer susceptibility: Results from a Breast Cancer Family Registry case-control mutation-screening study. Breast Cancer Res. 2014, 16, R58. [Google Scholar] [CrossRef]
- Guénard, F.; Brcas, I.; Labrie, Y.; Ouellette, G.; Beauparlant, C.J.; Durocher, F. Genetic sequence variations of BRCA1-interacting genes AURKA, BAP1, BARD1 and DHX9 in French Canadian Families with high risk of breast cancer. J. Hum. Genet. 2009, 54, 152–161. [Google Scholar] [CrossRef]
- Desjardins, S.; Beauparlant, J.C.; Labrie, Y.; Ouellette, G.; Durocher, F. Variations in the NBN/NBS1 gene and the risk of breast cancer in non-BRCA1/2French Canadian families with high risk of breast cancer. BMC Cancer 2009, 9, 181. [Google Scholar] [CrossRef] [PubMed]
- Elkholi, I.E.; Di Iorio, M.; Fahiminiya, S.; Arcand, S.L.; Han, H.; Nogué, C.; Behl, S.; Hamel, N.; Giroux, S.; de Ladurantaye, M.; et al. Investigating the causal role of MRE11A p.E506* in breast and ovarian cancer. Sci. Rep. 2021, 11, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Wang, Y.; Xia, Y.; Xu, Y.; Ouyang, T.; Li, J.; Wang, T.; Fan, Z.; Fan, T.; Lin, B.; et al. Mutations in RECQL Gene Are Associated with Predisposition to Breast Cancer. PLoS Genet. 2015, 11, e1005228. [Google Scholar] [CrossRef]
- Bowden, A.R.; Tischkowitz, M. Clinical implications of germline mutations in breast cancer genes: RECQL. Breast Cancer Res. Treat. 2019, 174, 553–560. [Google Scholar] [CrossRef] [PubMed]
- Casals, F.; Hodgkinson, A.; Hussin, J.; Idaghdour, Y.; Bruat, V.; De Maillard, T.; Grenier, J.-C.; Gbeha, E.; Hamdan, F.F.; Girard, S.; et al. Whole-Exome Sequencing Reveals a Rapid Change in the Frequency of Rare Functional Variants in a Founding Population of Humans. PLoS Genet. 2013, 9, e1003815. [Google Scholar] [CrossRef]
- Thompson, E.R.; Doyle, M.A.; Ryland, G.L.; Rowley, S.M.; Choong, D.Y.H.; Tothill, R.W.; Thorne, H.; Barnes, D.R.; Li, J.; Ellul, J.; et al. Exome Sequencing Identifies Rare Deleterious Mutations in DNA Repair Genes FANCC and BLM as Potential Breast Cancer Susceptibility Alleles. PLoS Genet. 2012, 8, e1002894. [Google Scholar] [CrossRef]
- Dicks, E.; Song, H.; Ramus, S.J.; Van Oudenhove, E.; Tyrer, J.P.; Intermaggio, M.P.; Kar, S.P.; Harrington, P.; Bowtell, D.; Cicek, M.S.; et al. Germline whole exome sequencing and large-scale replication identifies FANCM as a likely high grade serous ovarian cancer susceptibility gene. Oncotarget 2017, 8, 50930–50940. [Google Scholar] [CrossRef]
- Kiiski, J.I.; Pelttari, L.M.; Khan, S.; Freysteinsdottir, E.S.; Reynisdottir, I.; Hart, S.N.; Shimelis, H.; Vilske, S.; Kallioniemi, A.; Schleutker, J.; et al. Exome sequencing identifies FANCM as a susceptibility gene for triple-negative breast cancer. Proc. Natl. Acad. Sci. USA 2014, 111, 15172–15177. [Google Scholar] [CrossRef]
- Giroux, S.; Dubé-Linteau, A.; Cardinal, G.; Labelle, Y.; Laflamme, N.; Giguère, Y.; Rousseau, F. Assessment of the prevalence of the 985A>G MCAD mutation in the French-Canadian population using allele-specific PCR. Clin. Genet. 2007, 71, 569–575. [Google Scholar] [CrossRef] [PubMed]
- Gagnon, A.; Heyer, E. Fragmentation of the Québec population genetic pool (Canada): Evidence from the genetic contribution of founders per region in the 17th and 18th centuries. Am. J. Phys. Anthropol. 2001, 114, 30–41. [Google Scholar] [CrossRef]
- Moreau, C.; Vézina, H.; Labuda, D. Founder effects and genetic variability in Quebec. Med. Sci. 2007, 23, 1008–1013. [Google Scholar] [CrossRef]
- Awadalla, P.; Boileau, C.; Payette, Y.; Idaghdour, Y.; Goulet, J.-P.; Knoppers, B.; Hamet, P.; Laberge, C. Cohort profile of the CARTaGENE study: Quebec’s population-based biobank for public health and personalized genomics. Int. J. Epidemiol. 2013, 42, 1285–1299. [Google Scholar] [CrossRef] [PubMed]
- Borugian, M.J.; Robson, P.; Fortier, I.; Parker, L.; McLaughlin, J.; Knoppers, B.M.; Bédard, K.; Gallagher, R.P.; Sinclair, S.; Ferretti, V.; et al. The Canadian Partnership for Tomorrow Project: Building a pan-Canadian research platform for disease prevention. Can. Med. Assoc. J. 2010, 182, 1197–1201. [Google Scholar] [CrossRef]
- Winters, S.; Martin, C.; Murphy, D.; Shokar, N.K. Breast Cancer Epidemiology, Prevention, and Screening. Prog. Mol. Biol. Transl. Sci. 2017, 151, 1–32. [Google Scholar] [CrossRef]
- Hall, J.; Lee, M.; Newman, B.; Morrow, J.; Anderson, L.; Huey, B.; King, M. Linkage of early-onset familial breast cancer to chromosome 17q21. Science 1990, 250, 1684–1689. [Google Scholar] [CrossRef]
- Claus, E.B.; Risch, N.J.; Thompson, W.D. Age at onset as an indicator of familial risk of breast cancer. Am. J. Epidemiol. 1990, 131, 961–972. [Google Scholar] [CrossRef]
- Colditz, G.; Willett, W.C.; Hunter, D.J.; Stampfer, M.J.; Manson, J.E.; Hennekens, C.H.; Rosner, B.A.; Speizer, F.E. Family history, age, and risk of breast cancer. Prospective data from the Nurses’ Health Study. JAMA 1993, 270, 338–343. [Google Scholar] [CrossRef] [PubMed]
- Ott, J.; Wang, J.; Leal, S.M. Genetic linkage analysis in the age of whole-genome sequencing. Nat. Rev. Genet. 2015, 16, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Narod, S.; Feunteun, J.; Lynch, H.T.; Watson, P.; Conway, T.; Lynch, J.; Lenoir, G.M. Familial breast-ovarian cancer locus on chromosome 17q12-q23. Lancet 1991, 338, 82–83. [Google Scholar] [CrossRef]
- King, M.-C. “The Race” to Clone BRCA1. Science 2014, 343, 1462–1465. [Google Scholar] [CrossRef] [PubMed]
- Wooster, R.; Neuhausen, S.L.; Mangion, J.; Quirk, Y.; Ford, D.; Collins, N.; Nguyen, K.; Seal, S.; Tran, T.; Averill, D.; et al. Localization of a breast cancer susceptibility gene, BRCA2, to chromosome 13q12-13. Science 1994, 265, 2088–2090. [Google Scholar] [CrossRef]
- Bherer, C.; Labuda, D.; Roy-Gagnon, M.; Houde, L.; Tremblay, M.; Vézina, H. Admixed ancestry and stratification of Quebec regional populations. Am. J. Phys. Anthr. 2010, 144, 432–441. [Google Scholar] [CrossRef] [PubMed]
- Bray, S.M.; Mulle, J.G.; Dodd, A.F.; Pulver, A.E.; Wooding, S.; Warren, S.T. Signatures of founder effects, admixture, and selection in the Ashkenazi Jewish population. Proc. Natl. Acad. Sci. USA 2010, 107, 16222–16227. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Gene | Coding Change 2 | Protein Change 2 | Historical Nomenclature | Shared Haplotype in Carriers | Source(s) |
|---|---|---|---|---|---|
| BRCA1 | c.962G > A | p.Trp321Ter | 1081G > A | - | 35, 63, 85 |
| c.1054G > T | p.Glu352Ter | E352X | - | 63, 83 | |
| c.1961dup | p.Tyr655ValfsTer18 | 2080insA | - | 83 | |
| c.2125_2126insA | p.Phe709TyrfsTer3 | 2244insA | - | 83, 83, 84, 86 | |
| c.2834_2836delinsC | p.Ser945ThrfsTer6 | 2953del3 + C | Yes | 35, 59, 63,83, 85, 87, 88 | |
| c.3649_3650insA | p.Ser1217TyrfsTer2 | 3768insA | Yes | 35, 59, 86 | |
| c.3756_3759del | p.Ser1253ArgfsTer10 | 3875delGTCT | - | 35, 85, 88 | |
| c.4041_4042del | p.Gly1348AsnfsTer7 | 4160delAG | - | 35, 83 | |
| c.4327C > T | p.Arg1443Ter | C4446T | Yes | 35, 59, 63, 77, 79, 77, 83–86, 88 | |
| c.5102_5103del | p.Leu1701GlnfsTer14 | 5221delTG | - | 35, 77, 83 | |
| BRCA2 | c.2588dup | p.Asn863LysfsTer18 | 2816insA | Yes | 35, 59, 83, 86 |
| c.2808_2811del | p.Ala938ProfsTer21 | 3034del4 | Yes | 35, 83, 86 | |
| c.3170_3174del | p.Lys1057ThrfsTer8 | 3398del5 | Yes | 35, 84–86, 88 | |
| c.3545_3546del | p.Phe1182Ter | 3773delTT | - | 35, 63, 84, 86 | |
| c.5857G > T | p.Glu1953Ter | G6085T | Yes | 35, 59, 63, 78–79, 84–86, 88 | |
| c.6275_6276del | p.Leu2092ProfsTer7 | 6503delTT | Yes | 35, 59, 78, 83, 86 | |
| c.8537_8538del | p.Glu2846GlyfsTer22 | 8765delAG | Yes | 35, 59, 63, 77–79, 81, 83–86, 88 | |
| c.9004G > A | p.Glu3002Lys | E3002K | - | 63, 68, 86 |
| Gene | Canonical Transcript | Coding Change 2 | Protein Change 2 | Shared Haplotype in Carriers | Source(s) |
|---|---|---|---|---|---|
| BARD1 | NM_000465.4 | c.1075_1095dup | p.Leu359_Pro365dup | - | 134 |
| c.1930G > A | p.Val644Ile | - | 134 | ||
| c.2212A > G | p.Ile738Val | - | 134 | ||
| BRIP1 | NM_032043.3 | c.577G > A | p.Val193Ile | - | 126 |
| c.2097 + 7G > A | - | - | 126 | ||
| CHEK2 | NM_007194.4 | c.1100del | p.Thr367MetfsTer15 | - | 85, 105 |
| c.1217G > A | p.Arg406His | - | 129 | ||
| MRE11 | NM_005590.4 | c.1516G > T | p.Glu506Ter | - | 136 |
| PALB2 | NM_001005735.2 | c.226A > G | p.Ile76Val | - | 108 |
| c.1273G > A | p.Val425Met | - | 105 | ||
| c.1676A > G | p.Gln559Arg | - | 105 | ||
| c.2323C > T | p.Gln775Ter | Yes | 84, 85, 107, 108, 107 | ||
| c.2590C > T | p.Pro864Ser | - | 105 | ||
| RAD51D | NM_002878.4 | c.620C > T | p.Ser207Leu | Yes | 119 |
| RECQL | NM_032941.2 | c.643C > T | p.Arg215Ter | - | 53 |
| TP53 | NM_000546.5 | c.638G > A | p.Arg213Gln | - | 102, 103 |
| NM_000546.5 | c.703A > G | p.Asn235Asp | - | 103 | |
| NM_000546.5 | c.730G > A | p.Gly244Ser | - | 103 | |
| NM_000546.5 | c.742C > T | p.Arg248Trp | - | 103 | |
| NM_000546.5 | c.844C > T | p.Arg282Trp | - | 103 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fierheller, C.T.; Alenezi, W.M.; Tonin, P.N. The Genetic Analyses of French Canadians of Quebec Facilitate the Characterization of New Cancer Predisposing Genes Implicated in Hereditary Breast and/or Ovarian Cancer Syndrome Families. Cancers 2021, 13, 3406. https://doi.org/10.3390/cancers13143406
Fierheller CT, Alenezi WM, Tonin PN. The Genetic Analyses of French Canadians of Quebec Facilitate the Characterization of New Cancer Predisposing Genes Implicated in Hereditary Breast and/or Ovarian Cancer Syndrome Families. Cancers. 2021; 13(14):3406. https://doi.org/10.3390/cancers13143406
Chicago/Turabian StyleFierheller, Caitlin T., Wejdan M. Alenezi, and Patricia N. Tonin. 2021. "The Genetic Analyses of French Canadians of Quebec Facilitate the Characterization of New Cancer Predisposing Genes Implicated in Hereditary Breast and/or Ovarian Cancer Syndrome Families" Cancers 13, no. 14: 3406. https://doi.org/10.3390/cancers13143406
APA StyleFierheller, C. T., Alenezi, W. M., & Tonin, P. N. (2021). The Genetic Analyses of French Canadians of Quebec Facilitate the Characterization of New Cancer Predisposing Genes Implicated in Hereditary Breast and/or Ovarian Cancer Syndrome Families. Cancers, 13(14), 3406. https://doi.org/10.3390/cancers13143406