
Chromatin and Epigenetic Dysregulation of Prostate Cancer Development, Progression, and Therapeutic Response

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Prostate Lineage Commitment and Prostate Cancer Plasticity
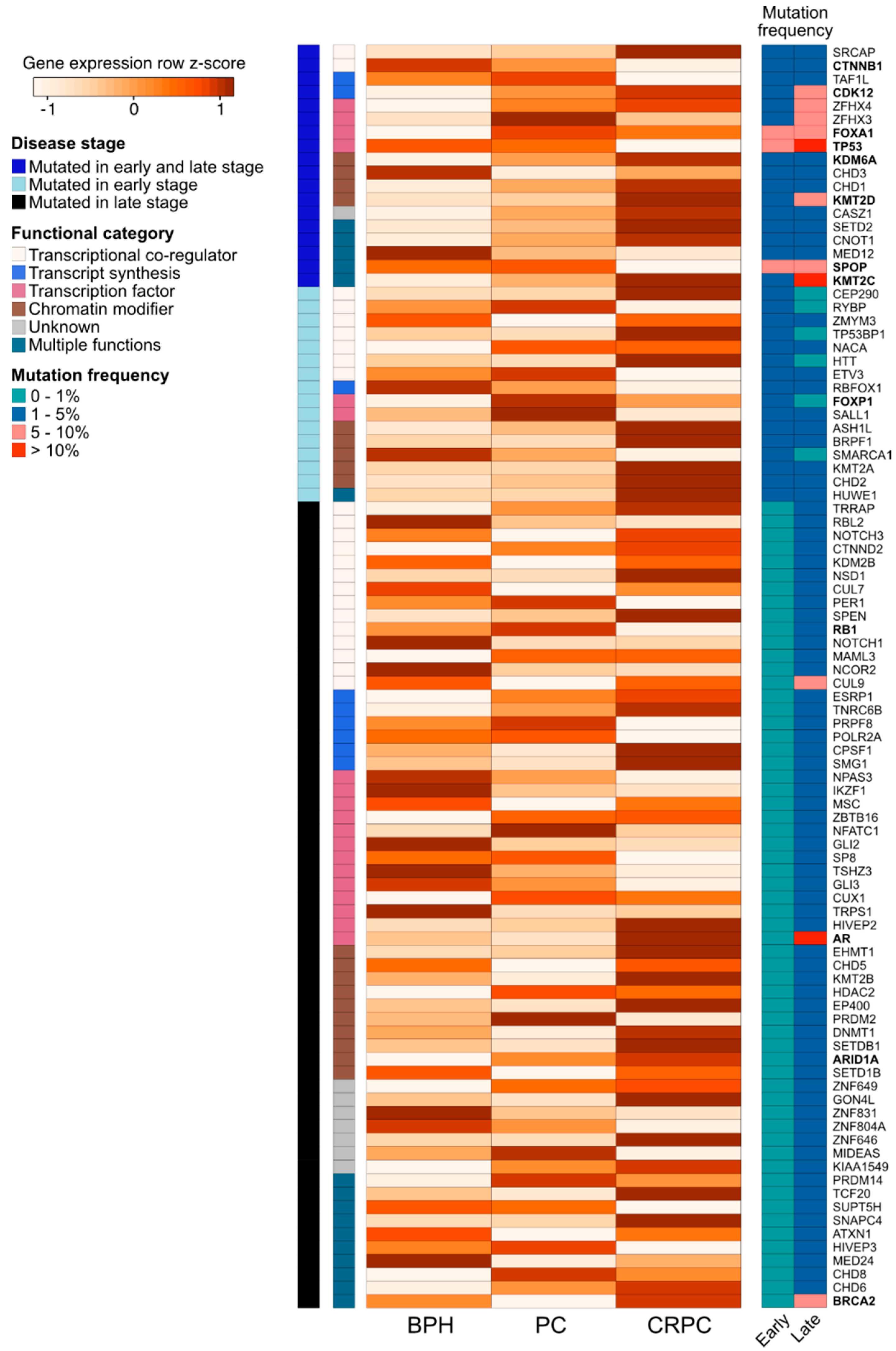
3. Mutations and Expression Dysregulation of Genes Coding for Chromatin-Associated Factors
4. The Role of DNA Methylation in Prostate Cancer
5. Dysregulation of Chromatin States through Histone Modification in Prostate Cancer
6. Dysregulation of Chromatin Accessibility
7. Dysregulation of Chromatin Accessibility and Regulation of Transcription Factors Binding to DNA
8. Chromatin Conformation and 3D Disorganization
9. Clinical Aspects and Future Directions
10. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef]
- Sathianathen, N.J.; Koschel, S.; Thangasamy, I.A.; Teh, J.; Alghazo, O.; Butcher, G.; Howard, H.; Kapoor, J.; Lawrentschuk, N.; Siva, S.; et al. Indirect Comparisons of Efficacy between Combination Approaches in Metastatic Hormone-Sensitive Prostate Cancer: A Systematic Review and Network Meta-Analysis. Eur. Urol. 2020, 77, 365–372. [Google Scholar] [CrossRef]
- Heidenreich, A.; Bastian, P.J.; Bellmunt, J.; Bolla, M.; Joniau, S.; van der Kwast, T.; Mason, M.; Matveev, V.; Wiegel, T.; Zattoni, F.; et al. EAU Guidelines on Prostate Cancer. Part II: Treatment of Advanced, Relapsing, and Castration-Resistant Prostate Cancer. Eur. Urol. 2014, 65, 467–479. [Google Scholar] [CrossRef]
- Tosco, L.; Briganti, A.; D’amico, A.V.; Eastham, J.; Eisenberger, M.; Gleave, M.; Haustermans, K.; Logothetis, C.J.; Saad, F.; Sweeney, C.; et al. Systematic Review of Systemic Therapies and Therapeutic Combinations with Local Treatments for High-Risk Localized Prostate Cancer. Eur. Urol. 2019, 75, 44–60. [Google Scholar] [CrossRef]
- Moris, L.; Cumberbatch, M.G.; Van den Broeck, T.; Gandaglia, G.; Fossati, N.; Kelly, B.; Pal, R.; Briers, E.; Cornford, P.; De Santis, M.; et al. Benefits and Risks of Primary Treatments for High-Risk Localized and Locally Advanced Prostate Cancer: An International Multidisciplinary Systematic Review. Eur. Urol. 2020, 77, 614–627. [Google Scholar] [CrossRef] [PubMed]
- Beltran, H.; Prandi, D.; Mosquera, J.M.; Benelli, M.; Puca, L.; Cyrta, J.; Marotz, C.; Giannopoulou, E.; Chakravarthi, B.V.S.K.; Varambally, S.; et al. Divergent Clonal Evolution of Castration-Resistant Neuroendocrine Prostate Cancer. Nat. Med. 2016, 22, 298–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Formaggio, N.; Rubin, M.A.; Theurillat, J.-P. Loss and Revival of Androgen Receptor Signaling in Advanced Prostate Cancer. Oncogene 2021, 40, 1205–1216. [Google Scholar] [CrossRef]
- Watson, P.A.; Arora, V.K.; Sawyers, C.L. Emerging Mechanisms of Resistance to Androgen Receptor Inhibitors in Prostate Cancer. Nat. Rev. Cancer 2015, 15, 701–711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beltran, H.; Tomlins, S.; Aparicio, A.; Arora, V.; Rickman, D.; Ayala, G.; Huang, J.; True, L.; Gleave, M.E.; Soule, H.; et al. Aggressive Variants of Castration-Resistant Prostate Cancer. Clin. Cancer Res. 2014, 20, 2846–2850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Handle, F.; Prekovic, S.; Helsen, C.; Van den Broeck, T.; Smeets, E.; Moris, L.; Eerlings, R.; El Kharraz, S.; Urbanucci, A.; Mills, I.G.; et al. Drivers of AR Indifferent Anti-Androgen Resistance in Prostate Cancer Cells. Sci. Rep. 2019, 9, 13786. [Google Scholar] [CrossRef] [Green Version]
- Bluemn, E.G.; Coleman, I.M.; Lucas, J.M.; Coleman, R.T.; Hernandez-Lopez, S.; Tharakan, R.; Bianchi-Frias, D.; Dumpit, R.F.; Kaipainen, A.; Corella, A.N.; et al. Androgen Receptor Pathway-Independent Prostate Cancer Is Sustained through FGF Signaling. Cancer Cell 2017, 32, 474–489.e6. [Google Scholar] [CrossRef] [Green Version]
- Crona, D.J.; Whang, Y.E. Androgen Receptor-Dependent and -Independent Mechanisms Involved in Prostate Cancer Therapy Resistance. Cancers 2017, 9, 67. [Google Scholar] [CrossRef]
- Isikbay, M.; Otto, K.; Kregel, S.; Kach, J.; Cai, Y.; Vander Griend, D.J.; Conzen, S.D.; Szmulewitz, R.Z. Glucocorticoid Receptor Activity Contributes to Resistance to Androgen-Targeted Therapy in Prostate Cancer. Horm. Cancer 2014, 5, 72–89. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Alyamani, M.; Zhang, A.; Chang, K.-H.; Berk, M.; Li, Z.; Zhu, Z.; Petro, M.; Magi-Galluzzi, C.; Taplin, M.-E.; et al. Aberrant Corticosteroid Metabolism in Tumor Cells Enables GR Takeover in Enzalutamide Resistant Prostate Cancer. eLife 2017, 6. [Google Scholar] [CrossRef] [Green Version]
- Mu, P.; Zhang, Z.; Benelli, M.; Karthaus, W.R.; Hoover, E.; Chen, C.-C.; Wongvipat, J.; Ku, S.-Y.; Gao, D.; Cao, Z.; et al. SOX2 Promotes Lineage Plasticity and Antiandrogen Resistance in TP53- and RB1-Deficient Prostate Cancer. Science 2017, 355, 84–88. [Google Scholar] [CrossRef] [Green Version]
- Braadland, P.R.; Ramberg, H.; Grytli, H.H.; Urbanucci, A.; Nielsen, H.K.; Guldvik, I.J.; Engedal, A.; Ketola, K.; Wang, W.; Svindland, A.; et al. The β2-Adrenergic Receptor Is a Molecular Switch for Neuroendocrine Transdifferentiation of Prostate Cancer Cells. Mol. Cancer Res. 2019, 17, 2154–2168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, L.; Shen, H.C.; Wantroba, M.; Khalid, O.; Liang, G.; Wang, Q.; Gentzschein, E.; Pinski, J.K.; Stanczyk, F.Z.; Jones, P.A.; et al. Locus-Wide Chromatin Remodeling and Enhanced Androgen Receptor-Mediated Transcription in Recurrent Prostate Tumor Cells. Mol. Cell. Biol. 2006, 26, 7331–7341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tewari, A.K.; Yardimci, G.G.; Shibata, Y.; Sheffield, N.C.; Song, L.; Taylor, B.S.; Georgiev, S.G.; Coetzee, G.A.; Ohler, U.; Furey, T.S.; et al. Chromatin Accessibility Reveals Insights into Androgen Receptor Activation and Transcriptional Specificity. Genome Biol. 2012, 13, R88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urbanucci, A.; Barfeld, S.J.; Kytölä, V.; Itkonen, H.M.; Coleman, I.M.; Vodák, D.; Sjöblom, L.; Sheng, X.; Tolonen, T.; Minner, S.; et al. Androgen Receptor Deregulation Drives Bromodomain-Mediated Chromatin Alterations in Prostate Cancer. Cell Rep. 2017, 19, 2045–2059. [Google Scholar] [CrossRef] [Green Version]
- Brücher, B.L.D.M.; Jamall, I.S. Somatic Mutation Theory—Why It’s Wrong for Most Cancers. Cell. Physiol. Biochem. 2016, 38, 1663–1680. [Google Scholar] [CrossRef] [PubMed]
- The ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Consortium. Pan-Cancer Analysis of Whole Genomes. Nature 2020, 578, 82–93. [Google Scholar] [CrossRef] [Green Version]
- Barbieri, C.E.; Baca, S.C.; Lawrence, M.S.; Demichelis, F.; Blattner, M.; Theurillat, J.-P.; White, T.A.; Stojanov, P.; Van Allen, E.; Stransky, N.; et al. Exome Sequencing Identifies Recurrent SPOP, FOXA1 and MED12 Mutations in Prostate Cancer. Nat. Genet. 2012, 44, 685–689. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. The Hallmarks of Cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Flavahan, W.A.; Gaskell, E.; Bernstein, B.E. Epigenetic Plasticity and the Hallmarks of Cancer. Science 2017, 357. [Google Scholar] [CrossRef] [Green Version]
- Darwiche, N. Epigenetic Mechanisms and the Hallmarks of Cancer: An Intimate Affair. Am. J. Cancer Res. 2020, 10, 1954–1978. [Google Scholar]
- Mikkelsen, T.S.; Ku, M.; Jaffe, D.B.; Issac, B.; Lieberman, E.; Giannoukos, G.; Alvarez, P.; Brockman, W.; Kim, T.-K.; Koche, R.P.; et al. Genome-Wide Maps of Chromatin State in Pluripotent and Lineage-Committed Cells. Nature 2007, 448, 553–560. [Google Scholar] [CrossRef]
- Rubin, M.A.; Bristow, R.G.; Thienger, P.D.; Dive, C.; Imielinski, M. Impact of Lineage Plasticity to and from a Neuroendocrine Phenotype on Progression and Response in Prostate and Lung Cancers. Mol. Cell 2020, 80, 562–577. [Google Scholar] [CrossRef] [PubMed]
- Brocks, D.; Assenov, Y.; Minner, S.; Bogatyrova, O.; Simon, R.; Koop, C.; Oakes, C.; Zucknick, M.; Lipka, D.B.; Weischenfeldt, J.; et al. Intratumor DNA Methylation Heterogeneity Reflects Clonal Evolution in Aggressive Prostate Cancer. Cell Rep. 2014, 8, 798–806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fontugne, J.; Davis, K.; Palanisamy, N.; Udager, A.; Mehra, R.; McDaniel, A.S.; Siddiqui, J.; Rubin, M.A.; Mosquera, J.M.; Tomlins, S.A. Clonal Evaluation of Prostate Cancer Foci in Biopsies with Discontinuous Tumor Involvement by Dual ERG/SPINK1 Immunohistochemistry. Mod. Pathol. 2016, 29, 157–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salami, S.S.; Hovelson, D.H.; Kaplan, J.B.; Mathieu, R.; Udager, A.M.; Curci, N.E.; Lee, M.; Plouffe, K.R.; de la Vega, L.L.; Susani, M.; et al. Transcriptomic Heterogeneity in Multifocal Prostate Cancer. JCI Insight 2018, 3. [Google Scholar] [CrossRef]
- Talos, F.; Mitrofanova, A.; Bergren, S.K.; Califano, A.; Shen, M.M. A Computational Systems Approach Identifies Synergistic Specification Genes That Facilitate Lineage Conversion to Prostate Tissue. Nat. Commun. 2017, 8, 14662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, Q.; Liu, Y.; Cai, T.; Horton, C.; Stefanson, J.; Wang, Z.A. Dissecting Cell-Type-Specific Roles of Androgen Receptor in Prostate Homeostasis and Regeneration through Lineage Tracing. Nat. Commun. 2017, 8, 14284. [Google Scholar] [CrossRef] [Green Version]
- Dutta, A.; Le Magnen, C.; Mitrofanova, A.; Ouyang, X.; Califano, A.; Abate-Shen, C. Identification of an NKX3.1-G9a-UTY Transcriptional Regulatory Network That Controls Prostate Differentiation. Science 2016, 352, 1576–1580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, L.; Pu, Y.; Hepps, D.; Danielpour, D.; Prins, G.S. Posterior Hox Gene Expression and Differential Androgen Regulation in the Developing and Adult Rat Prostate Lobes. Endocrinology 2007, 148, 1235–1245. [Google Scholar] [CrossRef] [PubMed]
- Javed, S.; Langley, S.E.M. Importance of HOX Genes in Normal Prostate Gland Formation, Prostate Cancer Development and Its Early Detection. BJU Int. 2014, 113, 535–540. [Google Scholar] [CrossRef]
- Guo, W.; Li, L.; He, J.; Liu, Z.; Han, M.; Li, F.; Xia, X.; Zhang, X.; Zhu, Y.; Wei, Y.; et al. Single-Cell Transcriptomics Identifies a Distinct Luminal Progenitor Cell Type in Distal Prostate Invagination Tips. Nat. Genet. 2020, 52, 908–918. [Google Scholar] [CrossRef]
- Isaacs, J.T. Control of cell proliferation and cell death in the normal and neoplastic prostate: A stem cell model. In Benign Prostatic Hyperplasia; Rogers, C.H., Coffey, D.S., Cunha, G., Grayhack, J.T., Hinman, F., Jr., Horton, R., Eds.; Report No. NIH 87-2881; US Department of Health and Human Services: Washington, DC, USA, 1987; pp. 85–94. [Google Scholar]
- Tsujimura, A.; Koikawa, Y.; Salm, S.; Takao, T.; Coetzee, S.; Moscatelli, D.; Shapiro, E.; Lepor, H.; Sun, T.-T.; Wilson, E.L. Proximal Location of Mouse Prostate Epithelial Stem Cells: A Model of Prostatic Homeostasis. J. Cell Biol. 2002, 157, 1257–1265. [Google Scholar] [CrossRef] [Green Version]
- Henry, G.H.; Malewska, A.; Joseph, D.B.; Malladi, V.S.; Lee, J.; Torrealba, J.; Mauck, R.J.; Gahan, J.C.; Raj, G.V.; Roehrborn, C.G.; et al. A Cellular Anatomy of the Normal Adult Human Prostate and Prostatic Urethra. Cell Rep. 2018, 25, 3530–3542.e5. [Google Scholar] [CrossRef] [Green Version]
- Karthaus, W.R.; Hofree, M.; Choi, D.; Linton, E.L.; Turkekul, M.; Bejnood, A.; Carver, B.; Gopalan, A.; Abida, W.; Laudone, V.; et al. Regenerative Potential of Prostate Luminal Cells Revealed by Single-Cell Analysis. Science 2020, 368, 497–505. [Google Scholar] [CrossRef]
- Montoro, D.T.; Haber, A.L.; Biton, M.; Vinarsky, V.; Lin, B.; Birket, S.E.; Yuan, F.; Chen, S.; Leung, H.M.; Villoria, J.; et al. A Revised Airway Epithelial Hierarchy Includes CFTR-Expressing Ionocytes. Nature 2018, 560, 319–324. [Google Scholar] [CrossRef]
- Maitland, N.J.; Collins, A.T. Prostate Cancer Stem Cells: A New Target for Therapy. J. Clin. Oncol. 2008, 26, 2862–2870. [Google Scholar] [CrossRef]
- Hudson, D.L.; Guy, A.T.; Fry, P.; O’Hare, M.J.; Watt, F.M.; Masters, J.R. Epithelial Cell Differentiation Pathways in the Human Prostate: Identification of Intermediate Phenotypes by Keratin Expression. J. Histochem. Cytochem. 2001, 49, 271–278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.A.; Mitrofanova, A.; Bergren, S.K.; Abate-Shen, C.; Cardiff, R.D.; Califano, A.; Shen, M.M. Lineage Analysis of Basal Epithelial Cells Reveals Their Unexpected Plasticity and Supports a Cell-of-Origin Model for Prostate Cancer Heterogeneity. Nat. Cell Biol. 2013, 15, 274–283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Julio, M.K.; Economides, K.D.; Walker, D.; Yu, H.; Vivienne Halili, M.; Hu, Y.-P.; Price, S.M.; Abate-Shen, C.; Shen, M.M. A Luminal Epithelial Stem Cell That Is a Cell of Origin for Prostate Cancer. Nature 2009, 461, 495–500. [Google Scholar] [CrossRef]
- Goldstein, A.S.; Huang, J.; Guo, C.; Garraway, I.P.; Witte, O.N. Identification of a Cell of Origin for Human Prostate Cancer. Science 2010, 329, 568–571. [Google Scholar] [CrossRef] [Green Version]
- Choi, N.; Zhang, B.; Zhang, L.; Ittmann, M.; Xin, L. Adult Murine Prostate Basal and Luminal Cells Are Self-Sustained Lineages That Can Both Serve as Targets for Prostate Cancer Initiation. Cancer Cell 2012, 21, 253–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.A.; Toivanen, R.; Bergren, S.K.; Chambon, P.; Shen, M.M. Luminal Cells Are Favored as the Cell of Origin for Prostate Cancer. Cell Rep. 2014, 8, 1339–1346. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.H.; Shen, M.M. Cell Types of Origin for Prostate Cancer. Curr. Opin. Cell Biol. 2015, 37, 35–41. [Google Scholar] [CrossRef]
- Sfanos, K.S.; Yegnasubramanian, S.; Nelson, W.G.; De Marzo, A.M. The Inflammatory Microenvironment and Microbiome in Prostate Cancer Development. Nat. Rev. Urol. 2018, 15, 11–24. [Google Scholar] [CrossRef]
- Guo, H.; Ci, X.; Ahmed, M.; Hua, J.T.; Soares, F.; Lin, D.; Puca, L.; Vosoughi, A.; Xue, H.; Li, E.; et al. ONECUT2 Is a Driver of Neuroendocrine Prostate Cancer. Nat. Commun. 2019, 10, 278. [Google Scholar] [CrossRef] [Green Version]
- Giunchi, F.; Fiorentino, M.; Loda, M. The Metabolic Landscape of Prostate Cancer. Eur. Urol. Oncol. 2019, 2, 28–36. [Google Scholar] [CrossRef]
- Corbin, J.M.; Ruiz-Echevarría, M.J. One-Carbon Metabolism in Prostate Cancer: The Role of Androgen Signaling. Int. J. Mol. Sci. 2016, 17, 1208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gurel, B.; Iwata, T.; Koh, C.M.; Jenkins, R.B.; Lan, F.; Van Dang, C.; Hicks, J.L.; Morgan, J.; Cornish, T.C.; Sutcliffe, S.; et al. Nuclear MYC Protein Overexpression Is an Early Alteration in Human Prostate Carcinogenesis. Mod. Pathol. 2008, 21, 1156–1167. [Google Scholar] [CrossRef] [PubMed]
- Civenni, G.; Malek, A.; Albino, D.; Garcia-Escudero, R.; Napoli, S.; Di Marco, S.; Pinton, S.; Sarti, M.; Carbone, G.M.; Catapano, C.V. RNAi-Mediated Silencing of Myc Transcription Inhibits Stem-like Cell Maintenance and Tumorigenicity in Prostate Cancer. Cancer Res. 2013, 73, 6816–6827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vander Griend, D.J.; Litvinov, I.V.; Isaacs, J.T. Conversion of Androgen Receptor Signaling from a Growth Suppressor in Normal Prostate Epithelial Cells to an Oncogene in Prostate Cancer Cells Involves a Gain of Function in c-Myc Regulation. Int. J. Biol. Sci. 2014, 10, 627–642. [Google Scholar] [CrossRef] [Green Version]
- Antony, L.; van der Schoor, F.; Dalrymple, S.L.; Isaacs, J.T. Androgen Receptor (AR) Suppresses Normal Human Prostate Epithelial Cell Proliferation via AR/β-catenin/TCF-4 Complex Inhibition of c-MYC Transcription. Prostate 2014, 74, 1118–1131. [Google Scholar] [CrossRef] [Green Version]
- Maitland, N.J.; Collins, A. A Tumour Stem Cell Hypothesis for the Origins of Prostate Cancer. BJU Int. 2005, 96, 1219–1223. [Google Scholar] [CrossRef]
- Song, H.; Weinstein, H.N.W.; Allegakoen, P.; Wadsworth, M.H.; Xie, J.; Yang, H.; Feng, F.Y.; Carroll, P.R.; Wang, B.; Cooperberg, M.R.; et al. Single-Cell Analysis of Human Primary Prostate Cancer Reveals the Heterogeneity of Tumor-Associated Epithelial Cell States. bioRxiv 2020. [Google Scholar] [CrossRef]
- Pomerantz, M.M.; Li, F.; Takeda, D.Y.; Lenci, R.; Chonkar, A.; Chabot, M.; Cejas, P.; Vazquez, F.; Cook, J.; Shivdasani, R.A.; et al. The Androgen Receptor Cistrome Is Extensively Reprogrammed in Human Prostate Tumorigenesis. Nat. Genet. 2015, 47, 1346–1351. [Google Scholar] [CrossRef]
- Grbesa, I.; Augello, M.A.; Liu, D.; McNally, D.R.; Gaffney, C.D. SPOP Mutation Confers Sensitivity to AR-Targeted Therapy in Prostate Cancer by Reshaping the Androgen-Driven Chromatin Landscape. bioRxiv 2021. [Google Scholar] [CrossRef]
- Sharma, N.L.; Massie, C.E.; Ramos-Montoya, A.; Zecchini, V.; Scott, H.E.; Lamb, A.D.; MacArthur, S.; Stark, R.; Warren, A.Y.; Mills, I.G.; et al. The Androgen Receptor Induces a Distinct Transcriptional Program in Castration-Resistant Prostate Cancer in Man. Cancer Cell 2013, 23, 35–47. [Google Scholar] [CrossRef] [Green Version]
- Pomerantz, M.M.; Qiu, X.; Zhu, Y.; Takeda, D.Y.; Pan, W.; Baca, S.C.; Gusev, A.; Korthauer, K.D.; Severson, T.M.; Ha, G.; et al. Prostate Cancer Reactivates Developmental Epigenomic Programs during Metastatic Progression. Nat. Genet. 2020, 52, 790–799. [Google Scholar] [CrossRef] [PubMed]
- Kwon, O.-J.; Zhang, L.; Jia, D.; Zhou, Z.; Li, Z.; Haffner, M.; Lee, J.K.; True, L.; Morrissey, C.; Xin, L. De Novo Induction of Lineage Plasticity from Human Prostate Luminal Epithelial Cells by Activated AKT1 and c-Myc. Oncogene 2020, 39, 7142–7151. [Google Scholar] [CrossRef] [PubMed]
- Carceles-Cordon, M.; Kelly, W.K.; Gomella, L.; Knudsen, K.E.; Rodriguez-Bravo, V.; Domingo-Domenech, J. Cellular Rewiring in Lethal Prostate Cancer: The Architect of Drug Resistance. Nat. Rev. Urol. 2020, 17, 292–307. [Google Scholar] [CrossRef]
- Linja, M.J.; Savinainen, K.J.; Saramäki, O.R.; Tammela, T.L.; Vessella, R.L.; Visakorpi, T. Amplification and Overexpression of Androgen Receptor Gene in Hormone-Refractory Prostate Cancer. Cancer Res. 2001, 61, 3550–3555. [Google Scholar] [PubMed]
- Zhang, Z.; Zhou, C.; Li, X.; Barnes, S.D.; Deng, S.; Hoover, E.; Chen, C.-C.; Lee, Y.S.; Zhang, Y.; Wang, C.; et al. Loss of CHD1 Promotes Heterogeneous Mechanisms of Resistance to AR-Targeted Therapy via Chromatin Dysregulation. Cancer Cell 2020, 37, 584–598.e11. [Google Scholar] [CrossRef] [PubMed]
- Brennen, W.N.; Nathaniel Brennen, W.; Zhu, Y.; Coleman, I.M.; Dalrymple, S.L.; Antony, L.; Patel, R.A.; Hanratty, B.; Chikarmane, R.; Meeker, A.K.; et al. Resistance to Androgen Receptor Signaling Inhibition Does Not Necessitate Development of Neuroendocrine Prostate Cancer. JCI Insight 2021, 6, e146827. [Google Scholar] [CrossRef]
- Nyquist, M.D.; Corella, A.; Coleman, I.; De Sarkar, N.; Kaipainen, A.; Ha, G.; Gulati, R.; Ang, L.; Chatterjee, P.; Lucas, J.; et al. Combined TP53 and RB1 Loss Promotes Prostate Cancer Resistance to a Spectrum of Therapeutics and Confers Vulnerability to Replication Stress. Cell Rep. 2020, 31, 107669. [Google Scholar] [CrossRef]
- He, M.X.; Cuoco, M.S.; Crowdis, J.; Bosma-Moody, A.; Zhang, Z.; Bi, K.; Kanodia, A.; Su, M.-J.; Ku, S.-Y.; Garcia, M.M.; et al. Transcriptional Mediators of Treatment Resistance in Lethal Prostate Cancer. Nat. Med. 2021, 27, 426–433. [Google Scholar] [CrossRef]
- Uusi-Mäkelä, J.; Afyounian, E.; Tabaro, F.; Häkkinen, T.; Lussana, A.; Shcherban, A.; Annala, M.; Nurminen, R.; Kivinummi, K.; Tammela, T.L.J.; et al. Chromatin Accessibility Analysis Uncovers Regulatory Element Landscape in Prostate Cancer Progression. bioRxiv 2020. [Google Scholar] [CrossRef]
- Ku, S.Y.; Rosario, S.; Wang, Y.; Mu, P.; Seshadri, M.; Goodrich, Z.W.; Goodrich, M.M.; Labbé, D.P.; Gomez, E.C.; Wang, J.; et al. Rb1 and Trp53 Cooperate to Suppress Prostate Cancer Lineage Plasticity, Metastasis, and Antiandrogen Resistance. Science 2017, 355, 78–83. [Google Scholar] [CrossRef] [Green Version]
- Mandigo, A.C.; Yuan, W.; Xu, K.; Gallagher, P.; Pang, A.; Guan, Y.F.; Shafi, A.A.; Thangavel, C.; Sheehan, B.; Bogdan, D.; et al. RB/E2F1 as a Master Regulator of Cancer Cell Metabolism in Advanced Disease. Cancer Discov. 2021. [Google Scholar] [CrossRef] [PubMed]
- Kaarijärvi, R.; Kaljunen, H.; Ketola, K. Molecular and Functional Links between Neurodevelopmental Processes and Treatment-Induced Neuroendocrine Plasticity in Prostate Cancer Progression. Cancers 2021, 13, 692. [Google Scholar] [CrossRef] [PubMed]
- Park, J.W.; Lee, J.K.; Sheu, K.M.; Wang, L.; Balanis, N.G.; Nguyen, K.; Smith, B.A.; Cheng, C.; Tsai, B.L.; Cheng, D.; et al. Reprogramming Normal Human Epithelial Tissues to a Common, Lethal Neuroendocrine Cancer Lineage. Science 2018, 362, 91–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baca, S.C.; Takeda, D.Y.; Seo, J.-H.; Hwang, J.; Ku, S.Y.; Arafeh, R.; Arnoff, T.; Agarwal, S.; Bell, C.; O’Connor, E.; et al. Reprogramming of the FOXA1 Cistrome in Treatment-Emergent Neuroendocrine Prostate Cancer. Nat. Commun. 2021, 12, 1979. [Google Scholar] [CrossRef] [PubMed]
- Flores-Morales, A.; Bergmann, T.B.; Lavallee, C.; Batth, T.S.; Lin, D.; Lerdrup, M.; Friis, S.; Bartels, A.; Kristensen, G.; Krzyzanowska, A.; et al. Proteogenomic Characterization of Patient-Derived Xenografts Highlights the Role of REST in Neuroendocrine Differentiation of Castration-Resistant Prostate Cancer. Clin. Cancer Res. 2019, 25, 595–608. [Google Scholar] [CrossRef] [Green Version]
- Svensson, C.; Ceder, J.; Iglesias-Gato, D.; Chuan, Y.-C.; Pang, S.T.; Bjartell, A.; Martinez, R.M.; Bott, L.; Helczynski, L.; Ulmert, D.; et al. REST Mediates Androgen Receptor Actions on Gene Repression and Predicts Early Recurrence of Prostate Cancer. Nucleic Acids Res. 2014, 42, 999–1015. [Google Scholar] [CrossRef]
- Prager, B.C.; Xie, Q.; Bao, S.; Rich, J.N. Cancer Stem Cells: The Architects of the Tumor Ecosystem. Cell Stem Cell 2019, 24, 41–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ge, R.; Wang, Z.; Montironi, R.; Jiang, Z.; Cheng, M.; Santoni, M.; Huang, K.; Massari, F.; Lu, X.; Cimadamore, A.; et al. Epigenetic Modulations and Lineage Plasticity in Advanced Prostate Cancer. Ann. Oncol. 2020, 31, 470–479. [Google Scholar] [CrossRef] [Green Version]
- Berger, A.; Brady, N.J.; Bareja, R.; Robinson, B.; Conteduca, V.; Augello, M.A.; Puca, L.; Ahmed, A.; Dardenne, E.; Lu, X.; et al. N-Myc–mediated Epigenetic Reprogramming Drives Lineage Plasticity in Advanced Prostate Cancer. J. Clin. Investig. 2019, 129, 3924–3940. [Google Scholar] [CrossRef]
- Chatterjee, A.; Rodger, E.J.; Eccles, M.R. Epigenetic Drivers of Tumourigenesis and Cancer Metastasis. Semin. Cancer Biol. 2018, 51, 149–159. [Google Scholar] [CrossRef] [PubMed]
- Ruggero, K.; Farran-Matas, S.; Martinez-Tebar, A.; Aytes, A. Epigenetic Regulation in Prostate Cancer Progression. Curr. Mol. Biol. Rep. 2018, 4, 101–115. [Google Scholar] [CrossRef]
- Heinäniemi, M.; Nykter, M.; Kramer, R.; Wienecke-Baldacchino, A.; Sinkkonen, L.; Zhou, J.X.; Kreisberg, R.; Kauffman, S.A.; Huang, S.; Shmulevich, I. Gene-Pair Expression Signatures Reveal Lineage Control. Nat. Methods 2013, 10, 577–583. [Google Scholar] [CrossRef] [Green Version]
- International Cancer Genome Consortium; Hudson, T.J.; Anderson, W.; Artez, A.; Barker, A.D.; Bell, C.; Bernabé, R.R.; Bhan, M.K.; Calvo, F.; Eerola, I.; et al. International Network of Cancer Genome Projects. Nature 2010, 464, 993–998. [Google Scholar]
- Robinson, D.; Van Allen, E.M.; Wu, Y.-M.; Schultz, N.; Lonigro, R.J.; Mosquera, J.-M.; Montgomery, B.; Taplin, M.-E.; Pritchard, C.C.; Attard, G.; et al. Integrative Clinical Genomics of Advanced Prostate Cancer. Cell 2015, 161, 1215–1228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grasso, C.S.; Wu, Y.-M.; Robinson, D.R.; Cao, X.; Dhanasekaran, S.M.; Khan, A.P.; Quist, M.J.; Jing, X.; Lonigro, R.J.; Brenner, J.C.; et al. The Mutational Landscape of Lethal Castration-Resistant Prostate Cancer. Nature 2012, 487, 239–243. [Google Scholar] [CrossRef] [Green Version]
- Ylipää, A.; Kivinummi, K.; Kohvakka, A.; Annala, M.; Latonen, L.; Scaravilli, M.; Kartasalo, K.; Leppänen, S.-P.; Karakurt, S.; Seppälä, J.; et al. Transcriptome Sequencing Reveals PCAT5 as a Novel ERG-Regulated Long Noncoding RNA in Prostate Cancer. Cancer Res. 2015, 75, 4026–4031. [Google Scholar] [CrossRef] [Green Version]
- Han, Y.-C.; Zheng, Z.-L.; Zuo, Z.-H.; Yu, Y.P.; Chen, R.; Tseng, G.C.; Nelson, J.B.; Luo, J.-H. Metallothionein 1 H Tumour Suppressor Activity in Prostate Cancer Is Mediated by Euchromatin Methyltransferase 1. J. Pathol. 2013, 230, 184–193. [Google Scholar] [CrossRef]
- Augello, M.A.; Hickey, T.E.; Knudsen, K.E. FOXA1: Master of Steroid Receptor Function in Cancer. EMBO J. 2011, 30, 3885–3894. [Google Scholar] [CrossRef] [Green Version]
- Teng, M.; Zhou, S.; Cai, C.; Lupien, M.; He, H.H. Pioneer of Prostate Cancer: Past, Present and the Future of FOXA1. Protein Cell 2021, 12, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Lupien, M.; Eeckhoute, J.; Meyer, C.A.; Wang, Q.; Zhang, Y.; Li, W.; Carroll, J.S.; Liu, X.S.; Brown, M. FoxA1 Translates Epigenetic Signatures into Enhancer-Driven Lineage-Specific Transcription. Cell 2008, 132, 958–970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heinlein, C.A.; Chang, C. Androgen Receptor in Prostate Cancer. Endocr. Rev. 2004, 25, 276–308. [Google Scholar] [CrossRef] [Green Version]
- Rao, R.C.; Dou, Y. Hijacked in Cancer: The KMT2 (MLL) Family of Methyltransferases. Nat. Rev. Cancer 2015, 15, 334–346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stokes, D.G.; Tartof, K.D.; Perry, R.P. CHD1 Is Concentrated in Interbands and Puffed Regions of Drosophila Polytene Chromosomes. Proc. Natl. Acad. Sci. USA 1996, 93, 7137–7142. [Google Scholar] [CrossRef] [Green Version]
- Boysen, G.; Rodrigues, D.N.; Rescigno, P.; Seed, G.; Dolling, D.; Riisnaes, R.; Crespo, M.; Zafeiriou, Z.; Sumanasuriya, S.; Bianchini, D.; et al. SPOP-Mutated/CHD1-Deleted Lethal Prostate Cancer and Abiraterone Sensitivity. Clin. Cancer Res. 2018, 24, 5585–5593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shenoy, T.R.; Boysen, G.; Wang, M.Y.; Xu, Q.Z.; Guo, W.; Koh, F.M.; Wang, C.; Zhang, L.Z.; Wang, Y.; Gil, V.; et al. CHD1 Loss Sensitizes Prostate Cancer to DNA Damaging Therapy by Promoting Error-Prone Double-Strand Break Repair. Ann. Oncol. 2017, 28, 1495–1507. [Google Scholar] [CrossRef]
- Aparicio, A.M.; Shen, L.; Tapia, E.L.N.; Lu, J.-F.; Chen, H.-C.; Zhang, J.; Wu, G.; Wang, X.; Troncoso, P.; Corn, P.; et al. Combined Tumor Suppressor Defects Characterize Clinically Defined Aggressive Variant Prostate Cancers. Clin. Cancer Res. 2016, 22, 1520–1530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazrooei, P.; Kron, K.J.; Zhu, Y.; Zhou, S.; Grillo, G.; Mehdi, T.; Ahmed, M.; Severson, T.M.; Guilhamon, P.; Armstrong, N.S.; et al. Cistrome Partitioning Reveals Convergence of Somatic Mutations and Risk Variants on Master Transcription Regulators in Primary Prostate Tumors. Cancer Cell 2019, 36, 674–689.e6. [Google Scholar] [CrossRef]
- Takeda, D.Y.; Spisák, S.; Seo, J.-H.; Bell, C.; O’Connor, E.; Korthauer, K.; Ribli, D.; Csabai, I.; Solymosi, N.; Szállási, Z.; et al. A Somatically Acquired Enhancer of the Androgen Receptor Is a Noncoding Driver in Advanced Prostate Cancer. Cell 2018, 174, 422–432.e13. [Google Scholar] [CrossRef] [Green Version]
- Lee, W.H.; Morton, R.A.; Epstein, J.I.; Brooks, J.D.; Campbell, P.A.; Bova, G.S.; Hsieh, W.S.; Isaacs, W.B.; Nelson, W.G. Cytidine Methylation of Regulatory Sequences near the Pi-Class Glutathione S-Transferase Gene Accompanies Human Prostatic Carcinogenesis. Proc. Natl. Acad. Sci. USA 1994, 91, 11733–11737. [Google Scholar] [CrossRef] [Green Version]
- Lee, W.H.; Isaacs, W.B.; Bova, G.S.; Nelson, W.G. CG Island Methylation Changes near the GSTP1 Gene in Prostatic Carcinoma Cells Detected Using the Polymerase Chain Reaction: A New Prostate Cancer Biomarker. Cancer Epidemiol. Biomark. Prev. 1997, 6, 443–450. [Google Scholar]
- Martignano, F.; Gurioli, G.; Salvi, S.; Calistri, D.; Costantini, M.; Gunelli, R.; De Giorgi, U.; Foca, F.; Casadio, V. GSTP1 Methylation and Protein Expression in Prostate Cancer: Diagnostic Implications. Dis. Markers 2016, 2016, 4358292. [Google Scholar] [CrossRef] [Green Version]
- Mahapatra, S.; Klee, E.W.; Young, C.Y.F.; Sun, Z.; Jimenez, R.E.; Klee, G.G.; Tindall, D.J.; Donkena, K.V. Global Methylation Profiling for Risk Prediction of Prostate Cancer. Clin. Cancer Res. 2012, 18, 2882–2895. [Google Scholar] [CrossRef] [Green Version]
- Börno, S.T.; Fischer, A.; Kerick, M.; Fälth, M.; Laible, M. Genome-Wide DNA Methylation Events in TMPRSS2–ERG Fusion-Negative Prostate Cancers Implicate an EZH2-Dependent Mechanism with miR-26a Hypermethylation. Cancer Discov. 2012. [Google Scholar] [CrossRef] [Green Version]
- Friedlander, T.W.; Roy, R.; Tomlins, S.A.; Ngo, V.T.; Kobayashi, Y.; Azameera, A.; Rubin, M.A.; Pienta, K.J.; Chinnaiyan, A.; Ittmann, M.M.; et al. Common Structural and Epigenetic Changes in the Genome of Castration-Resistant Prostate Cancer. Cancer Res. 2012, 72, 616–625. [Google Scholar] [CrossRef] [Green Version]
- Zhao, S.G.; Chen, W.S.; Li, H.; Foye, A.; Zhang, M.; Sjöström, M.; Aggarwal, R.; Playdle, D.; Liao, A.; Alumkal, J.J.; et al. The DNA Methylation Landscape of Advanced Prostate Cancer. Nat. Genet. 2020, 52, 778–789. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Dhanasekaran, S.M.; Prensner, J.R.; Cao, X.; Robinson, D.; Kalyana-Sundaram, S.; Huang, C.; Shankar, S.; Jing, X.; Iyer, M.; et al. Deep Sequencing Reveals Distinct Patterns of DNA Methylation in Prostate Cancer. Genome Res. 2011, 21, 1028–1041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kron, K.; Pethe, V.; Briollais, L.; Sadikovic, B.; Ozcelik, H.; Sunderji, A.; Venkateswaran, V.; Pinthus, J.; Fleshner, N.; van der Kwast, T.; et al. Discovery of Novel Hypermethylated Genes in Prostate Cancer Using Genomic CpG Island Microarrays. PLoS ONE 2009, 4, e4830. [Google Scholar] [CrossRef] [PubMed]
- Kron, K.; Trudel, D.; Pethe, V.; Briollais, L.; Fleshner, N.; van der Kwast, T.; Bapat, B. Altered DNA Methylation Landscapes of Polycomb-Repressed Loci Are Associated with Prostate Cancer Progression and ERG Oncogene Expression in Prostate Cancer. Clin. Cancer Res. 2013, 19, 3450–3461. [Google Scholar] [CrossRef] [Green Version]
- Jones, P.A.; Baylin, S.B. The Fundamental Role of Epigenetic Events in Cancer. Nat. Rev. Genet. 2002, 3, 415–428. [Google Scholar] [CrossRef]
- Smith, Z.D.; Meissner, A. DNA Methylation: Roles in Mammalian Development. Nat. Rev. Genet. 2013, 14, 204–220. [Google Scholar] [CrossRef]
- Parry, A.; Rulands, S.; Reik, W. Active Turnover of DNA Methylation during Cell Fate Decisions. Nat. Rev. Genet. 2021, 22, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, M.V.C.; Bourc’his, D. The Diverse Roles of DNA Methylation in Mammalian Development and Disease. Nat. Rev. Mol. Cell Biol. 2019, 20, 590–607. [Google Scholar] [CrossRef]
- Massie, C.E.; Mills, I.G.; Lynch, A.G. The Importance of DNA Methylation in Prostate Cancer Development. J. Steroid Biochem. Mol. Biol. 2017, 166, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alvarez, S.; Germain, P.; Alvarez, R.; Rodríguez-Barrios, F.; Gronemeyer, H.; de Lera, A.R. Structure, Function and Modulation of Retinoic Acid Receptor Beta, a Tumor Suppressor. Int. J. Biochem. Cell Biol. 2007, 39, 1406–1415. [Google Scholar] [CrossRef]
- Wang, W.; Liu, S.; Jiang, C.; Wang, Y.; Zhu, H.; Wang, X. High Expression of RARβ Is a Favorable Factor in Colorectal Cancer. Dis. Markers 2019, 2019, 7138754. [Google Scholar] [CrossRef] [Green Version]
- Buijs, J.T.; Rentsch, C.A.; van der Horst, G.; van Overveld, P.G.M.; Wetterwald, A.; Schwaninger, R.; Henriquez, N.V.; Ten Dijke, P.; Borovecki, F.; Markwalder, R.; et al. BMP7, a Putative Regulator of Epithelial Homeostasis in the Human Prostate, Is a Potent Inhibitor of Prostate Cancer Bone Metastasis in Vivo. Am. J. Pathol. 2007, 171, 1047–1057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panja, S.; Hayati, S.; Epsi, N.J.; Parrott, J.S.; Mitrofanova, A. Integrative (epi) genomic analysis to predict response to androgen-deprivation therapy in prostate cancer. EBioMedicine 2018, 31, 110–121. [Google Scholar] [CrossRef] [Green Version]
- Fiano, V.; Zugna, D.; Grasso, C.; Trevisan, M.; Delsedime, L.; Molinaro, L.; Gillio-Tos, A.; Merletti, F.; Richiardi, L. LINE-1 Methylation Status in Prostate Cancer and Non-Neoplastic Tissue Adjacent to Tumor in Association with Mortality. Epigenetics 2017, 12, 11–18. [Google Scholar] [CrossRef] [Green Version]
- Storebjerg, T.M.; Strand, S.H.; Høyer, S.; Lynnerup, A.-S.; Borre, M.; Ørntoft, T.F.; Sørensen, K.D. Dysregulation and Prognostic Potential of 5-Methylcytosine (5mC), 5-Hydroxymethylcytosine (5hmC), 5-Formylcytosine (5fC), and 5-Carboxylcytosine (5caC) Levels in Prostate Cancer. Clin. Epigenet. 2018, 10, 105. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas Research Network. The Molecular Taxonomy of Primary Prostate Cancer. Cell 2015, 163, 1011–1025. [Google Scholar] [CrossRef] [Green Version]
- Noushmehr, H.; Weisenberger, D.J.; Diefes, K.; Phillips, H.S.; Pujara, K.; Berman, B.P.; Pan, F.; Pelloski, C.E.; Sulman, E.P.; Bhat, K.P.; et al. Identification of a CpG Island Methylator Phenotype That Defines a Distinct Subgroup of Glioma. Cancer Cell 2010, 17, 510–522. [Google Scholar] [CrossRef] [Green Version]
- Gerhauser, C.; Favero, F.; Risch, T.; Simon, R.; Feuerbach, L.; Assenov, Y.; Heckmann, D.; Sidiropoulos, N.; Waszak, S.M.; Hübschmann, D.; et al. Molecular Evolution of Early-Onset Prostate Cancer Identifies Molecular Risk Markers and Clinical Trajectories. Cancer Cell 2018, 34, 996–1011.e8. [Google Scholar] [CrossRef] [Green Version]
- Brunskill, E.W.; Sequeira-Lopez, M.L.S.; Pentz, E.S.; Lin, E.; Yu, J.; Aronow, B.J.; Potter, S.S.; Gomez, R.A. Genes That Confer the Identity of the Renin Cell. J. Am. Soc. Nephrol. 2011, 22, 2213–2225. [Google Scholar] [CrossRef] [PubMed]
- Ebihara, T.; Song, C.; Ryu, S.H.; Plougastel-Douglas, B.; Yang, L.; Levanon, D.; Groner, Y.; Bern, M.D.; Stappenbeck, T.S.; Colonna, M.; et al. Runx3 Specifies Lineage Commitment of Innate Lymphoid Cells. Nat. Immunol. 2015, 16, 1124–1133. [Google Scholar] [CrossRef] [Green Version]
- Ruiz, S.; Santos, E.; Bustelo, X.R. RasGRF2, a Guanosine Nucleotide Exchange Factor for Ras GTPases, Participates in T-Cell Signaling Responses. Mol. Cell. Biol. 2007, 27, 8127–8142. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Deng, T.; Long, X.; Lin, X.; Wu, S.; Wang, H.; Ge, R.; Zhang, Z.; Wu, C.-L.; Taplin, M.-E.; et al. Methylation of SRD5A2 Promoter Predicts a Better Outcome for Castration-Resistant Prostate Cancer Patients Undergoing Androgen Deprivation Therapy. PLoS ONE 2020, 15, e0229754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peter, M.R.; Bilenky, M.; Davies, A.; Isserlin, R.; Bader, G.D.; Fleshner, N.E.; Hirst, M.; Zoubeidi, A.; Bapat, B. Distinct DNA Methylation Patterns Associated with Treatment Resistance in Metastatic Castration Resistant Prostate Cancer. Sci. Rep. 2021, 11, 6630. [Google Scholar] [CrossRef] [PubMed]
- Hyun, K.; Jeon, J.; Park, K.; Kim, J. Writing, Erasing and Reading Histone Lysine Methylations. Exp. Mol. Med. 2017, 49, e324. [Google Scholar] [CrossRef] [Green Version]
- Bannister, A.J.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395. [Google Scholar] [CrossRef] [PubMed]
- Ernst, J.; Kellis, M. ChromHMM: Automating Chromatin-State Discovery and Characterization. Nat. Methods 2012, 9, 215–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ernst, J.; Kellis, M. Chromatin-State Discovery and Genome Annotation with ChromHMM. Nat. Protoc. 2017, 12, 2478–2492. [Google Scholar] [CrossRef]
- Roadmap Epigenomics Consortium; Kundaje, A.; Meuleman, W.; Ernst, J.; Bilenky, M.; Yen, A.; Heravi-Moussavi, A.; Kheradpour, P.; Zhang, Z.; Wang, J.; et al. Integrative Analysis of 111 Reference Human Epigenomes. Nature 2015, 518, 317–330. [Google Scholar]
- Chen, Z.; Wang, L.; Wang, Q.; Li, W. Histone Modifications and Chromatin Organization in Prostate Cancer. Epigenomics 2010, 2, 551–560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, Z.; Jänne, O.A.; Palvimo, J.J. Coregulator Recruitment and Histone Modifications in Transcriptional Regulation by the Androgen Receptor. Mol. Endocrinol. 2004, 18, 2633–2648. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Carroll, J.S.; Brown, M. Spatial and Temporal Recruitment of Androgen Receptor and Its Coactivators Involves Chromosomal Looping and Polymerase Tracking. Mol. Cell 2005, 19, 631–642. [Google Scholar] [CrossRef]
- Shang, Y.; Myers, M.; Brown, M. Formation of the Androgen Receptor Transcription Complex. Mol. Cell 2002, 9, 601–610. [Google Scholar] [CrossRef]
- Zhong, J.; Ding, L.; Bohrer, L.R.; Pan, Y.; Liu, P.; Zhang, J.; Sebo, T.J.; Karnes, R.J.; Tindall, D.J.; van Deursen, J.; et al. p300 Acetyltransferase Regulates Androgen Receptor Degradation and PTEN-Deficient Prostate Tumorigenesis. Cancer Res. 2014, 74, 1870–1880. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; He, D.; Cheng, L.; Huang, C.; Zhang, Y.; Rao, X.; Kong, Y.; Li, C.; Zhang, Z.; Liu, J.; et al. p300/CBP Inhibition Enhances the Efficacy of Programmed Death-Ligand 1 Blockade Treatment in Prostate Cancer. Oncogene 2020, 39, 3939–3951. [Google Scholar] [CrossRef]
- Yamane, K.; Toumazou, C.; Tsukada, Y.-I.; Erdjument-Bromage, H.; Tempst, P.; Wong, J.; Zhang, Y. JHDM2A, a JmjC-Containing H3K9 Demethylase, Facilitates Transcription Activation by Androgen Receptor. Cell 2006, 125, 483–495. [Google Scholar] [CrossRef] [Green Version]
- Wissmann, M.; Yin, N.; Müller, J.M.; Greschik, H.; Fodor, B.D.; Jenuwein, T.; Vogler, C.; Schneider, R.; Günther, T.; Buettner, R.; et al. Cooperative Demethylation by JMJD2C and LSD1 Promotes Androgen Receptor-Dependent Gene Expression. Nat. Cell Biol. 2007, 9, 347–353. [Google Scholar] [CrossRef]
- Metzger, E.; Wissmann, M.; Yin, N.; Müller, J.M.; Schneider, R.; Peters, A.H.F.M.; Günther, T.; Buettner, R.; Schüle, R. LSD1 Demethylates Repressive Histone Marks to Promote Androgen-Receptor-Dependent Transcription. Nature 2005, 437, 436–439. [Google Scholar] [CrossRef]
- Seligson, D.B.; Horvath, S.; McBrian, M.A.; Mah, V.; Yu, H.; Tze, S.; Wang, Q.; Chia, D.; Goodglick, L.; Kurdistani, S.K. Global Levels of Histone Modifications Predict Prognosis in Different Cancers. Am. J. Pathol. 2009, 174, 1619–1628. [Google Scholar] [CrossRef] [Green Version]
- Ellinger, J.; Kahl, P.; von der Gathen, J.; Rogenhofer, S.; Heukamp, L.C.; Gütgemann, I.; Walter, B.; Hofstädter, F.; Büttner, R.; Müller, S.C.; et al. Global Levels of Histone Modifications Predict Prostate Cancer Recurrence. Prostate 2010, 70, 61–69. [Google Scholar] [CrossRef]
- Bianco-Miotto, T.; Chiam, K.; Buchanan, G.; Jindal, S.; Day, T.K.; Thomas, M.; Pickering, M.A.; O’Loughlin, M.A.; Ryan, N.K.; Raymond, W.A.; et al. Global Levels of Specific Histone Modifications and an Epigenetic Gene Signature Predict Prostate Cancer Progression and Development. Cancer Epidemiol. Biomark. Prev. 2010, 19, 2611–2622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ke, X.-S.; Qu, Y.; Rostad, K.; Li, W.-C.; Lin, B.; Halvorsen, O.J.; Haukaas, S.A.; Jonassen, I.; Petersen, K.; Goldfinger, N.; et al. Genome-Wide Profiling of Histone h3 Lysine 4 and Lysine 27 Trimethylation Reveals an Epigenetic Signature in Prostate Carcinogenesis. PLoS ONE 2009, 4, e4687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gal-Yam, E.N.; Egger, G.; Iniguez, L.; Holster, H.; Einarsson, S.; Zhang, X.; Lin, J.C.; Liang, G.; Jones, P.A.; Tanay, A. Frequent Switching of Polycomb Repressive Marks and DNA Hypermethylation in the PC3 Prostate Cancer Cell Line. Proc. Natl. Acad. Sci. USA 2008, 105, 12979–12984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Welti, J.; Sharp, A.; Brooks, N.; Yuan, W.; McNair, C.; Chand, S.N.; Pal, A.; Figueiredo, I.; Riisnaes, R.; Gurel, B.; et al. Targeting the p300/CBP Axis in Lethal Prostate Cancer. Cancer Discov. 2021. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Fan, L.; Jeon, H.-Y.; Zhang, F.; Cui, X.; Mickle, M.B.; Peng, G.; Hussain, A.; Fazli, L.; Gleave, M.E.; et al. p300-Mediated Acetylation of Histone Demethylase JMJD1A Prevents Its Degradation by Ubiquitin Ligase STUB1 and Enhances Its Activity in Prostate Cancer. Cancer Res. 2020, 80, 3074–3087. [Google Scholar] [CrossRef] [PubMed]
- Sahu, B.; Laakso, M.; Ovaska, K.; Mirtti, T.; Lundin, J.; Rannikko, A.; Sankila, A.; Turunen, J.-P.; Lundin, M.; Konsti, J.; et al. Dual Role of FoxA1 in Androgen Receptor Binding to Chromatin, Androgen Signalling and Prostate Cancer. EMBO J. 2011, 30, 3962–3976. [Google Scholar] [CrossRef]
- Wang, Q.; Li, W.; Zhang, Y.; Yuan, X.; Xu, K.; Yu, J.; Chen, Z.; Beroukhim, R.; Wang, H.; Lupien, M.; et al. Androgen Receptor Regulates a Distinct Transcription Program in Androgen-Independent Prostate Cancer. Cell 2009, 138, 245–256. [Google Scholar] [CrossRef] [Green Version]
- Pellakuru, L.G.; Iwata, T.; Gurel, B.; Schultz, D.; Hicks, J.; Bethel, C.; Yegnasubramanian, S.; De Marzo, A.M. Global Levels of H3K27me3 Track with Differentiation in Vivo and Are Deregulated by MYC in Prostate Cancer. Am. J. Pathol. 2012, 181, 560–569. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.; Cao, Q.; Mehra, R.; Laxman, B.; Yu, J.; Tomlins, S.A.; Creighton, C.J.; Dhanasekaran, S.M.; Shen, R.; Chen, G.; et al. Integrative Genomics Analysis Reveals Silencing of Beta-Adrenergic Signaling by Polycomb in Prostate Cancer. Cancer Cell 2007, 12, 419–431. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.; Yu, J.; Rhodes, D.R.; Tomlins, S.A.; Cao, X.; Chen, G.; Mehra, R.; Wang, X.; Ghosh, D.; Shah, R.B.; et al. A Polycomb Repression Signature in Metastatic Prostate Cancer Predicts Cancer Outcome. Cancer Res. 2007, 67, 10657–10663. [Google Scholar] [CrossRef] [Green Version]
- Bryant, R.J.; Cross, N.A.; Eaton, C.L.; Hamdy, F.C.; Cunliffe, V.T. EZH2 Promotes Proliferation and Invasiveness of Prostate Cancer Cells. Prostate 2007, 67, 547–556. [Google Scholar] [CrossRef] [PubMed]
- Dundr, P.; Bártů, M.; Hojný, J.; Michálková, R.; Hájková, N.; Stružinská, I.; Krkavcová, E.; Hadravský, L.; Kleissnerová, L.; Kopejsková, J.; et al. HNF1B, EZH2 and ECI2 in Prostate Carcinoma. Molecular, Immunohistochemical and Clinico-Pathological Study. Sci. Rep. 2020, 10, 14365. [Google Scholar] [CrossRef]
- Varambally, S.; Dhanasekaran, S.M.; Zhou, M.; Barrette, T.R.; Kumar-Sinha, C.; Sanda, M.G.; Ghosh, D.; Pienta, K.J.; Sewalt, R.G.A.B.; Otte, A.P.; et al. The Polycomb Group Protein EZH2 Is Involved in Progression of Prostate Cancer. Nature 2002, 419, 624–629. [Google Scholar] [CrossRef] [PubMed]
- Melling, N.; Thomsen, E.; Tsourlakis, M.C.; Kluth, M.; Hube-Magg, C.; Minner, S.; Koop, C.; Graefen, M.; Heinzer, H.; Wittmer, C.; et al. Overexpression of Enhancer of Zeste Homolog 2 (EZH2) Characterizes an Aggressive Subset of Prostate Cancers and Predicts Patient Prognosis Independently from Pre- and Postoperatively Assessed Clinicopathological Parameters. Carcinogenesis 2015, 36, 1333–1340. [Google Scholar] [CrossRef] [Green Version]
- Xu, K.; Wu, Z.J.; Groner, A.C.; He, H.H.; Cai, C.; Lis, R.T.; Wu, X.; Stack, E.C.; Loda, M.; Liu, T.; et al. EZH2 Oncogenic Activity in Castration-Resistant Prostate Cancer Cells Is Polycomb-Independent. Science 2012, 338, 1465–1469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clapier, C.R.; Iwasa, J.; Cairns, B.R.; Peterson, C.L. Mechanisms of Action and Regulation of ATP-Dependent Chromatin-Remodelling Complexes. Nat. Rev. Mol. Cell Biol. 2017, 18, 407–422. [Google Scholar] [CrossRef]
- Hargreaves, D.C. Chromatin Openness Requires Continuous SWI/SNF Activity. Nat. Genet. 2021, 53, 263–264. [Google Scholar] [CrossRef]
- Cyrta, J.; Augspach, A.; De Filippo, M.R.; Prandi, D.; Thienger, P.; Benelli, M.; Cooley, V.; Bareja, R.; Wilkes, D.; Chae, S.-S.; et al. Role of Specialized Composition of SWI/SNF Complexes in Prostate Cancer Lineage Plasticity. Nat. Commun. 2020, 11, 5549. [Google Scholar] [CrossRef]
- Giles, K.A.; Gould, C.M.; Achinger-Kawecka, J.; Page, S.G.; Kafer, G.R.; Rogers, S.; Luu, P.-L.; Cesare, A.J.; Clark, S.J.; Taberlay, P.C. BRG1 Knockdown Inhibits Proliferation through Multiple Cellular Pathways in Prostate Cancer. Clin. Epigenet. 2021, 13, 37. [Google Scholar] [CrossRef]
- Sun, A.; Tawfik, O.; Gayed, B.; Thrasher, J.B.; Hoestje, S.; Li, C.; Li, B. Aberrant Expression of SWI/SNF Catalytic Subunits BRG1/BRM Is Associated with Tumor Development and Increased Invasiveness in Prostate Cancers. Prostate 2007, 67, 203–213. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Li, N.; Dong, B.; Guo, W.; Wei, H.; Chen, Q.; Yuan, H.; Han, Y.; Chang, H.; Kan, S.; et al. Chromatin Remodeling ATPase BRG1 and PTEN Are Synthetic Lethal in Prostate Cancer. J. Clin. Investig. 2019, 129, 759–773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muthuswami, R.; Bailey, L.; Rakesh, R.; Imbalzano, A.N.; Nickerson, J.A.; Hockensmith, J.W. BRG1 Is a Prognostic Indicator and a Potential Therapeutic Target for Prostate Cancer. J. Cell. Physiol. 2019. [Google Scholar] [CrossRef] [Green Version]
- Jamaspishvili, T.; Berman, D.M.; Ross, A.E.; Scher, H.I.; De Marzo, A.M.; Squire, J.A.; Lotan, T.L. Clinical Implications of PTEN Loss in Prostate Cancer. Nat. Rev. Urol. 2018, 15, 222–234. [Google Scholar] [CrossRef] [PubMed]
- Balasubramaniam, S.; Comstock, C.E.S.; Ertel, A.; Jeong, K.W.; Stallcup, M.R.; Addya, S.; McCue, P.A.; Ostrander, W.F., Jr.; Augello, M.A.; Knudsen, K.E. Aberrant BAF57 Signaling Facilitates Prometastatic Phenotypes. Clin. Cancer Res. 2013, 19, 2657–2667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heebøll, S.; Borre, M.; Ottosen, P.D.; Andersen, C.L.; Mansilla, F.; Dyrskjøt, L.; Orntoft, T.F.; Tørring, N. SMARCC1 Expression Is Upregulated in Prostate Cancer and Positively Correlated with Tumour Recurrence and Dedifferentiation. Histol. Histopathol. 2008, 23, 1069–1076. [Google Scholar] [PubMed]
- Link, K.A.; Balasubramaniam, S.; Sharma, A.; Comstock, C.E.S.; Godoy-Tundidor, S.; Powers, N.; Cao, K.H.; Haelens, A.; Claessens, F.; Revelo, M.P.; et al. Targeting the BAF57 SWI/SNF Subunit in Prostate Cancer: A Novel Platform to Control Androgen Receptor Activity. Cancer Res. 2008, 68, 4551–4558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stelloo, S.; Nevedomskaya, E.; van der Poel, H.G.; de Jong, J.; van Leenders, G.J.L.H.; Jenster, G.; Wessels, L.F.A.; Bergman, A.M.; Zwart, W. Androgen Receptor Profiling Predicts Prostate Cancer Outcome. EMBO Mol. Med. 2015, 7, 1450–1464. [Google Scholar] [CrossRef]
- Xu, Q.; Liu, X.; Zhu, S.; Hu, X.; Niu, H.; Zhang, X.; Zhu, D.; Nesa, E.U.; Tian, K.; Yuan, H. Hyper-Acetylation Contributes to the Sensitivity of Chemo-Resistant Prostate Cancer Cells to Histone Deacetylase Inhibitor Trichostatin A. J. Cell. Mol. Med. 2018, 22, 1909–1922. [Google Scholar] [CrossRef] [Green Version]
- Seligson, D.B.; Horvath, S.; Shi, T.; Yu, H.; Tze, S.; Grunstein, M.; Kurdistani, S.K. Global Histone Modification Patterns Predict Risk of Prostate Cancer Recurrence. Nature 2005, 435, 1262–1266. [Google Scholar] [CrossRef]
- Zhou, L.-X.; Li, T.; Huang, Y.-R.; Sha, J.-J.; Sun, P.; Li, D. Application of Histone Modification in the Risk Prediction of the Biochemical Recurrence after Radical Prostatectomy. Asian J. Androl. 2010, 12, 171–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Devaiah, B.N.; Case-Borden, C.; Gegonne, A.; Hsu, C.H.; Chen, Q.; Meerzaman, D.; Dey, A.; Ozato, K.; Singer, D.S. BRD4 Is a Histone Acetyltransferase That Evicts Nucleosomes from Chromatin. Nat. Struct. Mol. Biol. 2016, 23, 540–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Surface, L.E.; Fields, P.A.; Subramanian, V.; Behmer, R.; Udeshi, N.; Peach, S.E.; Carr, S.A.; Jaffe, J.D.; Boyer, L.A. H2A.Z.1 Monoubiquitylation Antagonizes BRD2 to Maintain Poised Chromatin in ESCs. Cell Rep. 2016, 14, 1142–1155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stathis, A.; Bertoni, F. BET Proteins as Targets for Anticancer Treatment. Cancer Discov. 2018, 8, 24–36. [Google Scholar] [CrossRef] [Green Version]
- Asangani, I.A.; Dommeti, V.L.; Wang, X.; Malik, R.; Cieslik, M.; Yang, R.; Escara-Wilke, J.; Wilder-Romans, K.; Dhanireddy, S.; Engelke, C.; et al. Therapeutic Targeting of BET Bromodomain Proteins in Castration-Resistant Prostate Cancer. Nature 2014, 510, 278–282. [Google Scholar] [CrossRef] [PubMed]
- Asangani, I.A.; Wilder-Romans, K.; Dommeti, V.L.; Krishnamurthy, P.M.; Apel, I.J.; Escara-Wilke, J.; Plymate, S.R.; Navone, N.M.; Wang, S.; Feng, F.Y.; et al. BET Bromodomain Inhibitors Enhance Efficacy and Disrupt Resistance to AR Antagonists in the Treatment of Prostate Cancer. Mol. Cancer Res. 2016, 14, 324–331. [Google Scholar] [CrossRef] [Green Version]
- Raina, K.; Lu, J.; Qian, Y.; Altieri, M.; Gordon, D.; Rossi, A.M.K.; Wang, J.; Chen, X.; Dong, H.; Siu, K.; et al. PROTAC-Induced BET Protein Degradation as a Therapy for Castration-Resistant Prostate Cancer. Proc. Natl. Acad. Sci. USA 2016, 113, 7124–7129. [Google Scholar] [CrossRef] [Green Version]
- Wyce, A.; Degenhardt, Y.; Bai, Y.; Le, B.C.; Korenchuk, S. Inhibition of BET Bromodomain Proteins as a Therapeutic Approach in Prostate Cancer. Oncotarget 2013, 4, 2419–2429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blee, A.M.; Liu, S.; Wang, L.; Huang, H. BET Bromodomain-Mediated Interaction between ERG and BRD4 Promotes Prostate Cancer Cell Invasion. Oncotarget 2016, 7, 38319–38332. [Google Scholar] [CrossRef] [Green Version]
- Shafran, J.S.; Andrieu, G.P.; Györffy, B.; Denis, G.V. BRD4 Regulates Metastatic Potential of Castration-Resistant Prostate Cancer through AHNAK. Mol. Cancer Res. 2019, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faivre, E.J.; McDaniel, K.F.; Albert, D.H.; Mantena, S.R. Selective Inhibition of the BD2 Bromodomain of BET Proteins in Prostate Cancer. Nature 2020, 578, 306–310. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.-Q.; Li, J.; Sachs, L.M.; Cole, P.A.; Wong, J. A Role for Cofactor-Cofactor and Cofactor-Histone Interactions in Targeting p300, SWI/SNF and Mediator for Transcription. EMBO J. 2003, 22, 2146–2155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, M.L.; Kim, Y.W.; Jeong, K.W. BAF53A Regulates Androgen Receptor-Mediated Gene Expression and Proliferation in LNCaP Cells. Biochem. Biophys. Res. Commun. 2018, 505, 618–623. [Google Scholar] [CrossRef]
- Link, K.A.; Burd, C.J.; Williams, E.; Marshall, T.; Rosson, G.; Henry, E.; Weissman, B.; Knudsen, K.E. BAF57 Governs Androgen Receptor Action and Androgen-Dependent Proliferation through SWI/SNF. Mol. Cell. Biol. 2005, 25, 2200–2215. [Google Scholar] [CrossRef] [Green Version]
- Marshall, T.W.; Link, K.A.; Petre-Draviam, C.E.; Knudsen, K.E. Differential Requirement of SWI/SNF for Androgen Receptor Activity. J. Biol. Chem. 2003, 278, 30605–30613. [Google Scholar] [CrossRef] [Green Version]
- Urbanucci, A.; Mills, I.G. Bromodomain-Containing Proteins in Prostate Cancer. Mol. Cell. Endocrinol. 2018, 462, 31–40. [Google Scholar] [CrossRef] [Green Version]
- Klemm, S.L.; Shipony, Z.; Greenleaf, W.J. Chromatin accessibility and the regulatory epigenome. Nat. Rev. Genet. 2019, 20, 207–220. [Google Scholar] [CrossRef] [PubMed]
- Corces, M.R.; Granja, J.M.; Shams, S.; Louie, B.H.; Seoane, J.A.; Zhou, W.; Silva, T.C.; Groeneveld, C.; Wong, C.K.; Cho, S.W.; et al. The Chromatin Accessibility Landscape of Primary Human Cancers. Science 2018, 362. [Google Scholar] [CrossRef] [Green Version]
- Gasperini, M.; Tome, J.M.; Shendure, J. Towards a Comprehensive Catalogue of Validated and Target-Linked Human Enhancers. Nat. Rev. Genet. 2020, 21, 292–310. [Google Scholar] [CrossRef]
- Baek, S.; Goldstein, I.; Hager, G.L. Bivariate Genomic Footprinting Detects Changes in Transcription Factor Activity. Cell Rep. 2017, 19, 1710–1722. [Google Scholar] [CrossRef] [Green Version]
- Mills, I.G. Maintaining and Reprogramming Genomic Androgen Receptor Activity in Prostate Cancer. Nat. Rev. Cancer 2014, 14, 187–198. [Google Scholar] [CrossRef]
- Braadland, P.R.; Urbanucci, A. Chromatin Reprogramming as an Adaptation Mechanism in Advanced Prostate Cancer. Endocr. Relat. Cancer 2019, 26, R211–R235. [Google Scholar] [CrossRef] [Green Version]
- Urbanucci, A.; Sahu, B.; Seppälä, J.; Larjo, A.; Latonen, L.M.; Waltering, K.K.; Tammela, T.L.J.; Vessella, R.L.; Lähdesmäki, H.; Jänne, O.A.; et al. Overexpression of Androgen Receptor Enhances the Binding of the Receptor to the Chromatin in Prostate Cancer. Oncogene 2012, 31, 2153–2163. [Google Scholar] [CrossRef] [Green Version]
- Senapati, D.; Kumari, S.; Heemers, H.V. Androgen Receptor Co-Regulation in Prostate Cancer. Asian J. Urol. 2020, 7, 219–232. [Google Scholar] [CrossRef]
- Tomlins, S.A.; Rhodes, D.R.; Perner, S.; Dhanasekaran, S.M.; Mehra, R.; Sun, X.-W.; Varambally, S.; Cao, X.; Tchinda, J.; Kuefer, R.; et al. Recurrent Fusion of TMPRSS2 and ETS Transcription Factor Genes in Prostate Cancer. Science 2005, 310, 644–648. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Yu, J.; Mani, R.-S.; Cao, Q.; Brenner, C.J.; Cao, X.; Wang, X.; Wu, L.; Li, J.; Hu, M.; et al. An Integrated Network of Androgen Receptor, Polycomb, and TMPRSS2-ERG Gene Fusions in Prostate Cancer Progression. Cancer Cell 2010, 17, 443–454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chng, K.R.; Chang, C.W.; Tan, S.K.; Yang, C.; Hong, S.Z.; Sng, N.Y.W.; Cheung, E. A Transcriptional Repressor Co-Regulatory Network Governing Androgen Response in Prostate Cancers: Corepressor Regulation of AR Signalling. EMBO J. 2012, 31, 2810–2823. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Chi, P.; Rockowitz, S.; Iaquinta, P.J.; Shamu, T.; Shukla, S.; Gao, D.; Sirota, I.; Carver, B.S.; Wongvipat, J.; et al. ETS Factors Reprogram the Androgen Receptor Cistrome and Prime Prostate Tumorigenesis in Response to PTEN Loss. Nat. Med. 2013, 19, 1023–1029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kron, K.J.; Murison, A.; Zhou, S.; Huang, V.; Yamaguchi, T.N.; Shiah, Y.-J.; Fraser, M.; van der Kwast, T.; Boutros, P.C.; Bristow, R.G.; et al. TMPRSS2--ERG Fusion Co-Opts Master Transcription Factors and Activates NOTCH Signaling in Primary Prostate Cancer. Nat. Genet. 2017, 49, 1336. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Yuan, Q.; Di, W.; Xia, X.; Liu, Z.; Mao, N.; Li, L.; Li, C.; He, J.; Li, Y.; et al. ERG Orchestrates Chromatin Interactions to Drive Prostate Cell Fate Reprogramming. J. Clin. Investig. 2020, 130, 5924–5941. [Google Scholar] [CrossRef] [PubMed]
- Weischenfeldt, J.; Simon, R.; Feuerbach, L.; Schlangen, K.; Weichenhan, D.; Minner, S.; Wuttig, D.; Warnatz, H.-J.; Stehr, H.; Rausch, T.; et al. Integrative Genomic Analyses Reveal an Androgen-Driven Somatic Alteration Landscape in Early-Onset Prostate Cancer. Cancer Cell 2013, 23, 159–170. [Google Scholar] [CrossRef] [Green Version]
- Koh, C.M.; Bieberich, C.J.; Dang, C.V.; Nelson, W.G.; Yegnasubramanian, S.; De Marzo, A.M. MYC and Prostate Cancer. Genes Cancer 2010, 1, 617–628. [Google Scholar] [CrossRef] [Green Version]
- Barfeld, S.J.; Urbanucci, A.; Itkonen, H.M.; Fazli, L.; Hicks, J.L.; Thiede, B.; Rennie, P.S.; Yegnasubramanian, S.; DeMarzo, A.M.; Mills, I.G. C-Myc Antagonises the Transcriptional Activity of the Androgen Receptor in Prostate Cancer Affecting Key Gene Networks. EBioMedicine 2017, 18, 83–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiu, X.; Boufaied, N.; Hallal, T.; Feit, A.; de Polo, A.; Luoma, A.M.; Larocque, J.; Zadra, G.; Xie, Y.; Gu, S.; et al. MYC Drives Aggressive Prostate Cancer by Disrupting Transcriptional Pause Release at Androgen Receptor Targets. bioRxiv 2021. [Google Scholar] [CrossRef]
- He, Y.; Wei, T.; Ye, Z.; Orme, J.J.; Lin, D.; Sheng, H.; Fazli, L.; Karnes, R.J.; Jimenez, R.; Wang, L.; et al. A Noncanonical AR Addiction Drives Enzalutamide Resistance in Prostate Cancer. Nat. Commun. 2021, 12, 1–14. [Google Scholar] [CrossRef]
- Taavitsainen, S.; Engedal, N.; Cao, S.; Handle, F.; Prekovic, S.; Wetterskog, D.; Vuorinen, E.M.; Kiviaho, A.; Nätkin, R.; Devlies, W.; et al. Single-Cell ATAC and RNA Sequencing Reveal Pre-Existing and Persistent Subpopulations of Cells Associated with Relapse of Prostate Cancer. bioRxiv 2021. [Google Scholar] [CrossRef]
- Ayaz, G.; Razizadeh, N.; Yaşar, P.; Kars, G.; Kahraman, D.C.; Saatci, Ö.; Şahin, Ö.; Çetin-Atalay, R.; Muyan, M. CXXC5 as an Unmethylated CpG Dinucleotide Binding Protein Contributes to Estrogen-Mediated Cellular Proliferation. Sci. Rep. 2020, 10, 5971, Correction in 2020, 10, 9943. [Google Scholar] [CrossRef] [Green Version]
- Solary, E.; Bernard, O.A.; Tefferi, A.; Fuks, F.; Vainchenker, W. The Ten-Eleven Translocation-2 (TET2) Gene in Hematopoiesis and Hematopoietic Diseases. Leukemia 2014, 28, 485–496. [Google Scholar] [CrossRef] [PubMed]
- McAuley, E.; Moline, D.; VanOpstall, C.; Lamperis, S.; Brown, R.; Vander Griend, D.J. Sox2 Expression Marks Castration-Resistant Progenitor Cells in the Adult Murine Prostate. Stem Cells 2019, 37, 690–700. [Google Scholar] [CrossRef]
- Mevel, R.; Steiner, I.; Mason, S.; Galbraith, L.C.A.; Patel, R.; Fadlullah, M.Z.H.; Ahmad, I.; Leung, H.Y.; Oliveira, P.; Blyth, K.; et al. RUNX1 Marks a Luminal Castration-Resistant Lineage Established at the Onset of Prostate Development. eLife 2020, 9, e60225. [Google Scholar] [CrossRef]
- Shah, N.; Wang, P.; Wongvipat, J.; Karthaus, W.R.; Abida, W.; Armenia, J.; Rockowitz, S.; Drier, Y.; Bernstein, B.E.; Long, H.W.; et al. Regulation of the Glucocorticoid Receptor via a BET-Dependent Enhancer Drives Antiandrogen Resistance in Prostate Cancer. eLife 2017, 6, e27861. [Google Scholar] [CrossRef]
- Hepburn, A.C.; Steele, R.E.; Veeratterapillay, R.; Wilson, L.; Kounatidou, E.E.; Barnard, A.; Berry, P.; Cassidy, J.R.; Moad, M.; El-Sherif, A.; et al. The Induction of Core Pluripotency Master Regulators in Cancers Defines Poor Clinical Outcomes and Treatment Resistance. Oncogene 2019, 38, 4412–4424. [Google Scholar] [CrossRef]
- Dixon, J.R.; Gorkin, D.U.; Ren, B. Chromatin Domains: The Unit of Chromosome Organization. Mol. Cell 2016, 62, 668–680. [Google Scholar] [CrossRef] [Green Version]
- Misteli, T.; Finn, E.H. Chromatin Architecture Is a Flexible Foundation for Gene Expression. Nat. Genet. 2021, 53, 426–427. [Google Scholar] [CrossRef] [PubMed]
- Achinger-Kawecka, J.; Taberlay, P.C.; Clark, S.J. Alterations in Three-Dimensional Organization of the Cancer Genome and Epigenome. Cold Spring Harb. Symp. Quant. Biol. 2016, 81, 41–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taberlay, P.C.; Achinger-Kawecka, J.; Lun, A.T.L.; Buske, F.A.; Sabir, K.; Gould, C.M.; Zotenko, E.; Bert, S.A.; Giles, K.A.; Bauer, D.C.; et al. Three-Dimensional Disorganization of the Cancer Genome Occurs Coincident with Long-Range Genetic and Epigenetic Alterations. Genome Res. 2016, 26, 719–731. [Google Scholar] [CrossRef] [Green Version]
- Rhie, S.K.; Perez, A.A.; Lay, F.D.; Schreiner, S.; Shi, J.; Polin, J.; Farnham, P.J. A High-Resolution 3D Epigenomic Map Reveals Insights into the Creation of the Prostate Cancer Transcriptome. Nat. Commun. 2019, 10, 4154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taslim, C.; Chen, Z.; Huang, K.; Huang, T.H.-M.; Wang, Q.; Lin, S. Integrated Analysis Identifies a Class of Androgen-Responsive Genes Regulated by Short Combinatorial Long-Range Mechanism Facilitated by CTCF. Nucleic Acids Res. 2012, 40, 4754–4764. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Zhang, C.; Wu, D.; Chen, H.; Rorick, A.; Zhang, X.; Wang, Q. Phospho-MED1-Enhanced UBE2C Locus Looping Drives Castration-Resistant Prostate Cancer Growth: MED1 Phosphorylation Enhances DNA Looping. EMBO J. 2011, 30, 2405–2419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.H.; Ku, J.Y.; Ha, J.M.; Bae, S.S.; Lee, J.Z.; Kim, C.-S.; Ha, H.K. Transcript Levels of Androgen Receptor Variant 7 and Ubiquitin-Conjugating Enzyme 2C in Hormone Sensitive Prostate Cancer and Castration-Resistant Prostate Cancer. Prostate 2017, 77, 60–71. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, J.; Tang, Q.; Ren, G. Identification of UBE2C as Hub Gene in Driving Prostate Cancer by Integrated Bioinformatics Analysis. PLoS ONE 2021, 16, e0247827. [Google Scholar]
- Hu, Y.; Gu, Y.; Wang, H.; Huang, Y.; Zou, Y.M. Integrated Network Model Provides New Insights into Castration-Resistant Prostate Cancer. Sci. Rep. 2015, 5, 17280. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Zhang, C.; Rorick, A.; Wu, D.; Chiu, M. CCI-779 Inhibits Cell-Cycle G2–M Progression and Invasion of Castration-Resistant Prostate Cancer via Attenuation of UBE2C Transcription and mRNA Stability. Cancer Res. 2011. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.; Sprenger, C.; Wu, P.-J.; Sun, S.; Uo, T.; Haugk, K.; Epilepsia, K.S.; Plymate, S. MED1 Mediates Androgen Receptor Splice Variant Induced Gene Expression in the Absence of Ligand. Oncotarget 2015, 6, 288–304. [Google Scholar] [CrossRef] [Green Version]
- Du, M.; Tillmans, L.; Gao, J.; Gao, P.; Yuan, T.; Dittmar, R.L.; Song, W.; Yang, Y.; Sahr, N.; Wang, T.; et al. Chromatin Interactions and Candidate Genes at Ten Prostate Cancer Risk Loci. Sci. Rep. 2016, 6, 23202. [Google Scholar] [CrossRef] [Green Version]
- Cai, M.; Kim, S.; Wang, K.; Farnham, P.J.; Coetzee, G.A.; Lu, W. 4C-Seq Revealed Long-Range Interactions of a Functional Enhancer at the 8q24 Prostate Cancer Risk Locus. Sci. Rep. 2016, 6, 22462. [Google Scholar] [CrossRef] [Green Version]
- Luo, Z.; Rhie, S.K.; Lay, F.D.; Farnham, P.J. A Prostate Cancer Risk Element Functions as a Repressive Loop That Regulates HOXA13. Cell Rep. 2017, 21, 1411–1417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freedman, M.L.; Monteiro, A.N.A.; Gayther, S.A.; Coetzee, G.A.; Risch, A.; Plass, C.; Casey, G.; De Biasi, M.; Carlson, C.; Duggan, D.; et al. Principles for the Post-GWAS Functional Characterization of Cancer Risk Loci. Nat. Genet. 2011, 43, 513–518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Chng, K.R.; Lingadahalli, S.; Chen, Z.; Liu, M.H.; Do, H.H.; Cai, S.; Rinaldi, N.; Poh, H.M.; Li, G.; et al. An AR-ERG Transcriptional Signature Defined by Long-Range Chromatin Interactomes in Prostate Cancer Cells. Genome Res. 2019, 29, 223–235. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Yang, S.; Chen, X.; Stauffer, S.; Yu, F.; Lele, S.M.; Fu, K.; Datta, K.; Palermo, N.; Chen, Y.; et al. The Hippo Pathway Effector YAP Regulates Motility, Invasion, and Castration-Resistant Growth of Prostate Cancer Cells. Mol. Cell. Biol. 2015, 35, 1350–1362. [Google Scholar] [CrossRef] [Green Version]
- Coffey, K. Targeting the Hippo Pathway in Prostate Cancer: What’s New? Cancers 2021, 13, 611. [Google Scholar] [CrossRef]
- Yuan, G.; Flores, N.M.; Hausmann, S.; Lofgren, S.M.; Kharchenko, V.; Angulo-Ibanez, M.; Sengupta, D.; Lu, X.; Czaban, I.; Azhibek, D.; et al. Elevated NSD3 Histone Methylation Activity Drives Squamous Cell Lung Cancer. Nature 2021, 590, 504–508. [Google Scholar] [CrossRef]
- Li, W.; Tian, W.; Yuan, G.; Deng, P.; Sengupta, D.; Cheng, Z.; Cao, Y.; Ren, J.; Qin, Y.; Zhou, Y.; et al. Molecular Basis of Nucleosomal H3K36 Methylation by NSD Methyltransferases. Nature 2021, 590, 498–503. [Google Scholar] [CrossRef] [PubMed]
- Cabrera-Licona, A.; Pérez-Añorve, I.X.; Flores-Fortis, M.; Moral-Hernández, O.D.; González-de la Rosa, C.H.; Suárez-Sánchez, R.; Chávez-Saldaña, M.; Aréchaga-Ocampo, E. Deciphering the Epigenetic Network in Cancer Radioresistance. Radiother. Oncol. 2021, 159, 48–59. [Google Scholar] [CrossRef]
- Kvon, E.Z.; Waymack, R.; Gad, M.; Wunderlich, Z. Enhancer Redundancy in Development and Disease. Nat. Rev. Genet. 2021, 22, 324–336. [Google Scholar] [CrossRef]
- Ma, X.-X.; Cao, Z.-G.; Zhao, S.-L. m6A Methyltransferase METTL3 Promotes the Progression of Prostate Cancer via m6A-Modified LEF1. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 3565–3571. [Google Scholar]
- Yuan, Y.; Du, Y.; Wang, L.; Liu, X. The M6A Methyltransferase METTL3 Promotes the Development and Progression of Prostate Carcinoma via Mediating MYC Methylation. J. Cancer 2020, 11, 3588–3595. [Google Scholar] [CrossRef]
- Li, E.; Wei, B.; Wang, X.; Kang, R. METTL3 Enhances Cell Adhesion through Stabilizing Integrin β1 mRNA via an m6A-HuR-Dependent Mechanism in Prostatic Carcinoma. Am. J. Cancer Res. 2020, 10, 1012–1025. [Google Scholar] [PubMed]
- Itkonen, H.M.; Minner, S.; Guldvik, I.J.; Sandmann, M.J.; Tsourlakis, M.C.; Berge, V.; Svindland, A.; Schlomm, T.; Mills, I.G. O-GlcNAc Transferase Integrates Metabolic Pathways to Regulate the Stability of c-MYC in Human Prostate Cancer Cells. Cancer Res. 2013, 73, 5277–5287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itkonen, H.M.; Poulose, N.; Steele, R.E.; Martin, S.E.S.; Levine, Z.G.; Duveau, D.Y.; Carelli, R.; Singh, R.; Urbanucci, A.; Loda, M.; et al. Inhibition of O-GlcNAc Transferase Renders Prostate Cancer Cells Dependent on CDK9. Mol. Cancer Res. 2020, 18, 1512–1521. [Google Scholar] [CrossRef] [PubMed]
- Itkonen, H.M.; Urbanucci, A.; Martin, S.E.; Khan, A.; Mathelier, A.; Thiede, B.; Walker, S.; Mills, I.G. High OGT Activity Is Essential for MYC-Driven Proliferation of Prostate Cancer Cells. Theranostics 2019, 9, 2183–2197. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhu, G.; Yang, Y.; Wang, F.; Xiao, Y.-T.; Zhang, N.; Bian, X.; Zhu, Y.; Yu, Y.; Liu, F.; et al. Single-Cell Analysis Reveals Transcriptomic Remodellings in Distinct Cell Types That Contribute to Human Prostate Cancer Progression. Nat. Cell Biol. 2021, 23, 87–98. [Google Scholar] [CrossRef]
- Zhang, Z.; Karthaus, W.R.; Lee, Y.S.; Gao, V.R.; Wu, C.; Russo, J.W.; Liu, M.; Mota, J.M.; Abida, W.; Linton, E.; et al. Tumor Microenvironment-Derived NRG1 Promotes Antiandrogen Resistance in Prostate Cancer. Cancer Cell 2020, 38, 279–296.e9. [Google Scholar] [CrossRef]
- Drilon, A.; Somwar, R.; Mangatt, B.P.; Edgren, H.; Desmeules, P.; Ruusulehto, A.; Smith, R.S.; Delasos, L.; Vojnic, M.; Plodkowski, A.J.; et al. Response to ERBB3-Directed Targeted Therapy in NRG1-Rearranged Cancers. Cancer Discov. 2018, 8, 686–695. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Campisi, J.; Higano, C.; Beer, T.M.; Porter, P.; Coleman, I.; True, L.; Nelson, P.S. Treatment-Induced Damage to the Tumor Microenvironment Promotes Prostate Cancer Therapy Resistance through WNT16B. Nat. Med. 2012, 18, 1359–1368. [Google Scholar] [CrossRef]
- Zadra, G.; Loda, M. When Fat Goes Down, Prostate Cancer Is on the Ropes. Mol. Cell. Oncol. 2019, 6, 1595308. [Google Scholar] [CrossRef] [Green Version]
- Butler, L.M.; Perone, Y.; Dehairs, J.; Lupien, L.E.; de Laat, V.; Talebi, A.; Loda, M.; Kinlaw, W.B.; Swinnen, J.V. Lipids and Cancer: Emerging Roles in Pathogenesis, Diagnosis and Therapeutic Intervention. Adv. Drug Deliv. Rev. 2020, 159, 245–293. [Google Scholar] [CrossRef] [PubMed]
- Kumaraswamy, A.; Welker Leng, K.R.; Westbrook, T.C.; Yates, J.A.; Zhao, S.G.; Evans, C.P.; Feng, F.Y.; Morgan, T.M.; Alumkal, J.J. Recent Advances in Epigenetic Biomarkers and Epigenetic Targeting in Prostate Cancer. Eur. Urol. 2021. [Google Scholar] [CrossRef]
- Doultsinos, D.; Mills, I.G. Derivation and Application of Molecular Signatures to Prostate Cancer: Opportunities and Challenges. Cancers 2021, 13, 495. [Google Scholar] [CrossRef] [PubMed]
- Sjöström, M.; Zhao, S.; Small, E.J.; Ning, Y.; Maurice-Dror, C.; Foye, A.; Hua, J.J.T.; Li, H.; Beer, T.M.; Evans, C.P.; et al. 5-Hydroxymethylcytosine as a Liquid Biopsy Biomarker in mCRPC. J. Clin. Oncol. 2021, 39, 148. [Google Scholar] [CrossRef]
- Nakken, S.; Lilleby, W.; Switlyk, M.D.; Knudsen, K.E.; Lilleby, O.; Zhao, S.; Kaveh, F.; Ekstrøm, P.O.; Urbanucci, A.; Hovig, E. The Quandary of DNA-Based Treatment Assessment in De Novo Metastatic Prostate Cancer in the Era of Precision Oncology. J. Pers. Med. 2021, 11, 330. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kukkonen, K.; Taavitsainen, S.; Huhtala, L.; Uusi-Makela, J.; Granberg, K.J.; Nykter, M.; Urbanucci, A. Chromatin and Epigenetic Dysregulation of Prostate Cancer Development, Progression, and Therapeutic Response. Cancers 2021, 13, 3325. https://doi.org/10.3390/cancers13133325
Kukkonen K, Taavitsainen S, Huhtala L, Uusi-Makela J, Granberg KJ, Nykter M, Urbanucci A. Chromatin and Epigenetic Dysregulation of Prostate Cancer Development, Progression, and Therapeutic Response. Cancers. 2021; 13(13):3325. https://doi.org/10.3390/cancers13133325
Chicago/Turabian StyleKukkonen, Konsta, Sinja Taavitsainen, Laura Huhtala, Joonas Uusi-Makela, Kirsi J. Granberg, Matti Nykter, and Alfonso Urbanucci. 2021. "Chromatin and Epigenetic Dysregulation of Prostate Cancer Development, Progression, and Therapeutic Response" Cancers 13, no. 13: 3325. https://doi.org/10.3390/cancers13133325
APA StyleKukkonen, K., Taavitsainen, S., Huhtala, L., Uusi-Makela, J., Granberg, K. J., Nykter, M., & Urbanucci, A. (2021). Chromatin and Epigenetic Dysregulation of Prostate Cancer Development, Progression, and Therapeutic Response. Cancers, 13(13), 3325. https://doi.org/10.3390/cancers13133325