
Mitochondrial Metabolism in Carcinogenesis and Cancer Therapy


{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
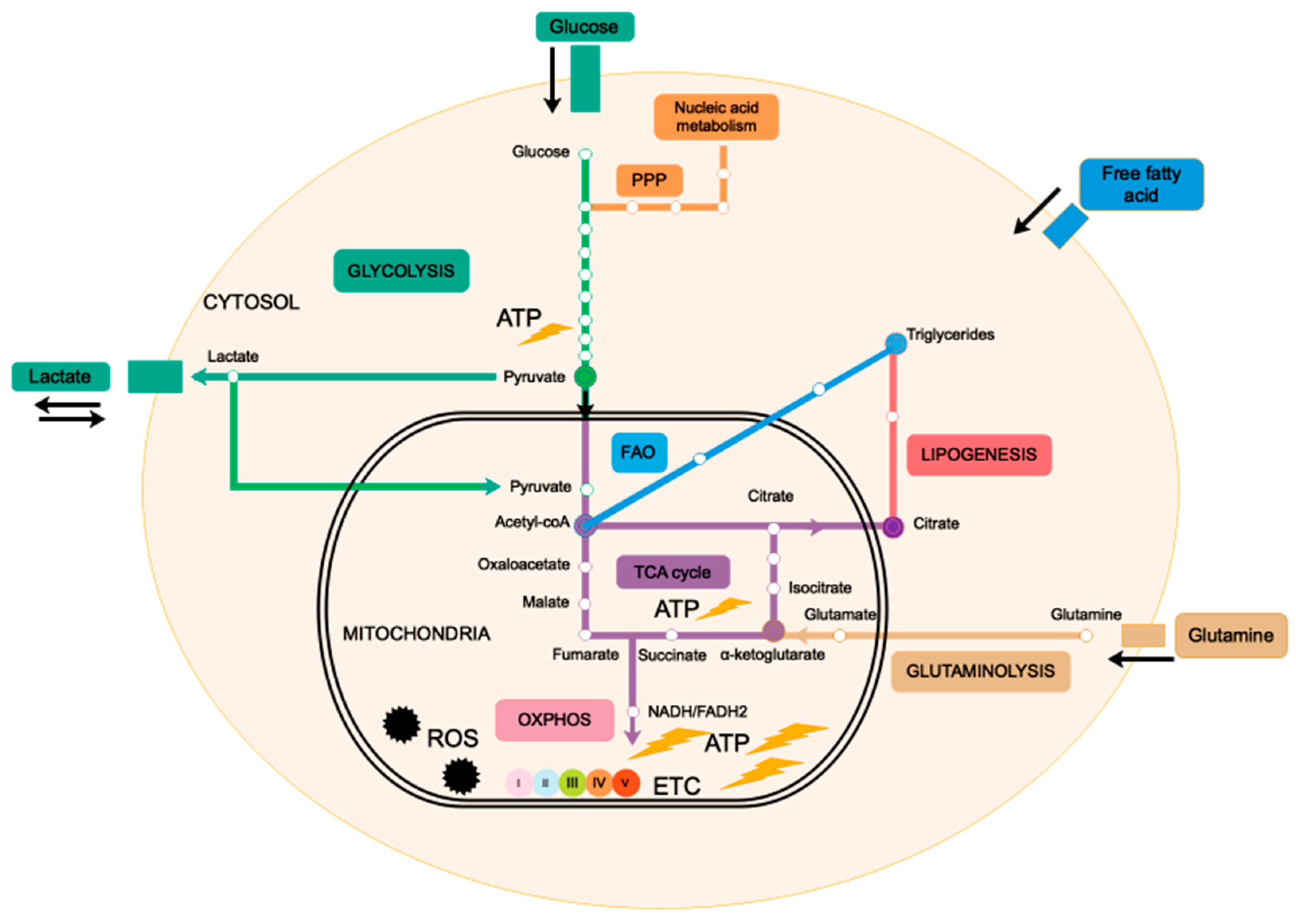
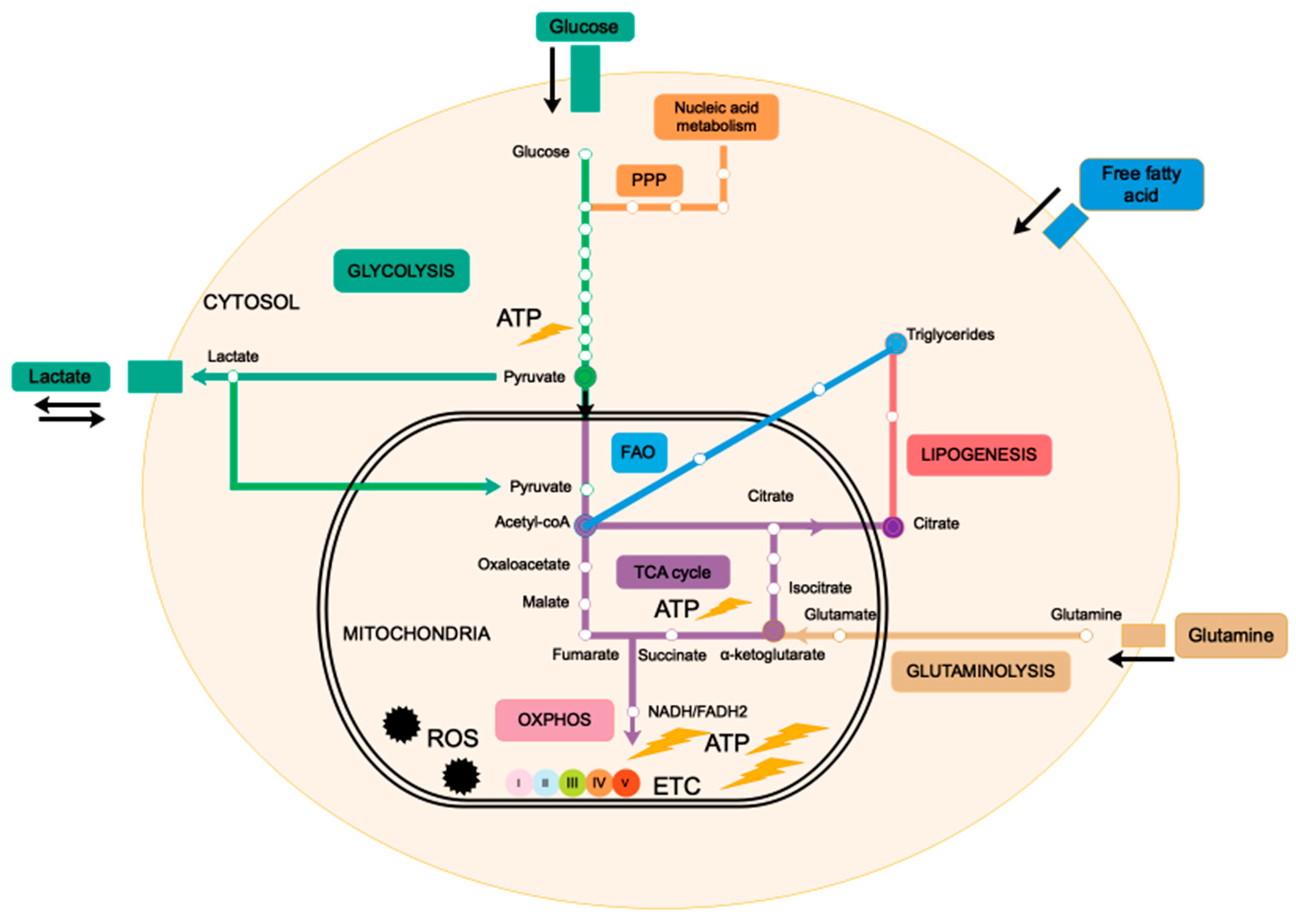
2. Mitochondria, the Powerhouse of the Cell
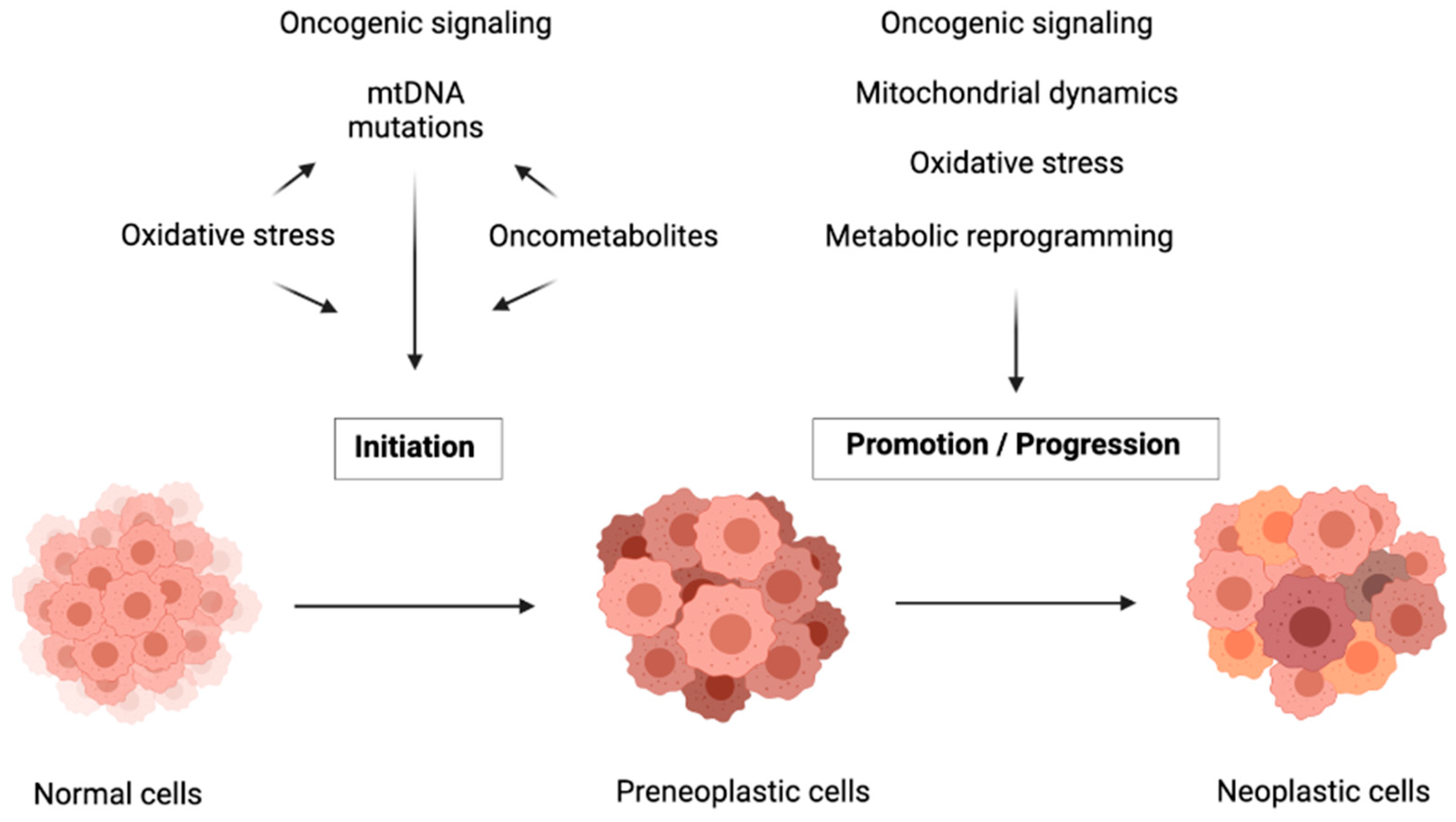
3. Mutations in Genes Involved in Mitochondrial Metabolism Drive Carcinogenesis Initiation
3.1. Mutations and Decreased Copy Number of mtDNA
3.2. Mutations in Nuclear-Encoded Mitochondrial Genes
3.2.1. Bioenergetic Metabolism Alteration
3.2.2. Oxidative Stress Promotion
3.2.3. Epigenetic Regulation
4. Mitochondrial Metabolic Reprogramming by Oncogenes
5. Mitochondrial Metabolic Reprogramming in the Progression of Carcinogenesis
5.1. Mitochondrial Dynamics
5.2. Mitophagy
5.3. Mitochondrial Retrograde Response
6. Mitochondria as Promising Targets in Cancer Therapies
6.1. Targeting mtDNA Transcription and Translation
6.2. Targeting ETC
6.3. Targeting the TCA Cycle
6.4. Targeting Redox Homeostasis
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pitot, H.C. The Molecular Biology of Carcinogenesis. Cancer 1993, 72, 962–970. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Rodrigues-Ferreira, S.; Moindjie, H.; Haykal, M.M.; Nahmias, C. Predicting and Overcoming Taxane Chemoresistance. Trends Mol. Med. 2021, 27, 138–151. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Ibekwe, E.; Chornenkyy, Y. Metabolic Alterations in Cancer Cells and the Emerging Role of Oncometabolites as Drivers of Neoplastic Change. Antioxidants 2018, 7, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corbet, C.; Feron, O. Cancer Cell Metabolism and Mitochondria: Nutrient Plasticity for TCA Cycle Fueling. Biochim. Biophys. Acta Rev. Cancer 2017, 1868, 7–15. [Google Scholar] [CrossRef]
- Spinelli, J.B.; Haigis, M.C. The Multifaceted Contributions of Mitochondria to Cellular Metabolism. Nat. Cell Biol. 2018, 20, 745–754. [Google Scholar] [CrossRef]
- Newmeyer, D.D.; Ferguson-Miller, S. Mitochondria: Releasing Power for Life and Unleashing the Machineries of Death. Cell 2003, 112, 481–490. [Google Scholar] [CrossRef] [Green Version]
- Chandra, D.; Liu, J.-W.; Tang, D.G. Early Mitochondrial Activation and Cytochrome c Up-Regulation during Apoptosis*210. J. Biol. Chem. 2002, 277, 50842–50854. [Google Scholar] [CrossRef] [Green Version]
- Desai, R.; East, D.A.; Hardy, L.; Faccenda, D.; Rigon, M.; Crosby, J.; Alvarez, M.S.; Singh, A.; Mainenti, M.; Hussey, L.K.; et al. Mitochondria Form Contact Sites with the Nucleus to Couple Prosurvival Retrograde Response. Sci. Adv. 2020, 6. [Google Scholar] [CrossRef] [PubMed]
- Bertholet, A.M.; Chouchani, E.T.; Kazak, L.; Angelin, A.; Fedorenko, A.; Long, J.Z.; Vidoni, S.; Garrity, R.; Cho, J.; Terada, N.; et al. H+ Transport Is an Integral Function of the Mitochondrial ADP/ATP Carrier. Nature 2019, 571, 515–520. [Google Scholar] [CrossRef]
- Theurey, P.; Rieusset, J. Mitochondria-Associated Membranes Response to Nutrient Availability and Role in Metabolic Diseases. Trends Endocrinol. Metab. 2017, 28, 32–45. [Google Scholar] [CrossRef] [Green Version]
- Wallace, D.C. Mitochondria and Cancer. Nat. Rev. Cancer 2012, 12, 685–698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taanman, J.-W. The Mitochondrial Genome: Structure, Transcription, Translation and Replication. Biochim. Biophys. Acta Bioenerg. 1999, 1410, 103–123. [Google Scholar] [CrossRef] [Green Version]
- Chaban, Y.; Boekema, E.J.; Dudkina, N.V. Structures of Mitochondrial Oxidative Phosphorylation Supercomplexes and Mechanisms for Their Stabilisation. Biochim. Biophys. Acta Bioenerg. 2014, 1837, 418–426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hui, S.; Ghergurovich, J.M.; Morscher, R.J.; Jang, C.; Teng, X.; Lu, W.; Esparza, L.A.; Reya, T.; Zhan, L.; Guo, J.Y.; et al. Glucose Feeds the TCA Cycle via Circulating Lactate. Nature 2017, 551, 115–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, H.; Ying, M.; Hu, X. Lactic Acidosis Switches Cancer Cells from Aerobic Glycolysis Back to Dominant Oxidative Phosphorylation. Oncotarget 2016, 7, 40621–40629. [Google Scholar] [CrossRef] [Green Version]
- Vyas, S.; Zaganjor, E.; Haigis, M.C. Mitochondria and Cancer. Cell 2016, 166, 555–566. [Google Scholar] [CrossRef] [PubMed]
- Reznik, E.; Miller, M.L.; Şenbabaoğlu, Y.; Riaz, N.; Sarungbam, J.; Tickoo, S.K.; Al-Ahmadie, H.A.; Lee, W.; Seshan, V.E.; Hakimi, A.A.; et al. Mitochondrial DNA Copy Number Variation across Human Cancers. eLife 2016, 5, e10769. [Google Scholar] [CrossRef] [PubMed]
- Petros, J.A.; Baumann, A.K.; Ruiz-Pesini, E.; Amin, M.B.; Sun, C.Q.; Hall, J.; Lim, S.; Issa, M.M.; Flanders, W.D.; Hosseini, S.H.; et al. MtDNA Mutations Increase Tumorigenicity in Prostate Cancer. Proc. Natl. Acad. Sci. USA 2005, 102, 719–724. [Google Scholar] [CrossRef] [Green Version]
- Alexeyev, M.; Shokolenko, I.; Wilson, G.; LeDoux, S. The Maintenance of Mitochondrial DNA Integrity—Critical Analysis and Update. Cold Spring Harb. Perspect. Biol. 2013, 5, a012641. [Google Scholar] [CrossRef] [Green Version]
- Chatterjee, A.; Mambo, E.; Sidransky, D. Mitochondrial DNA Mutations in Human Cancer. Oncogene 2006, 25, 4663–4674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warowicka, A.; Wołuń-Cholewa, M.; Kwaśniewska, A.; Goździcka-Józefiak, A. Alternations in Mitochondrial Genome in Carcinogenesis of HPV Positive Cervix. Exp. Mol. Pathol. 2020, 117, 104530. [Google Scholar] [CrossRef]
- Park, J.S.; Sharma, L.K.; Li, H.; Xiang, R.; Holstein, D.; Wu, J.; Lechleiter, J.; Naylor, S.L.; Deng, J.J.; Lu, J.; et al. A Heteroplasmic, Not Homoplasmic, Mitochondrial DNA Mutation Promotes Tumorigenesis via Alteration in Reactive Oxygen Species Generation and Apoptosis. Hum. Mol. Genet. 2009, 18, 1578–1589. [Google Scholar] [CrossRef]
- Dasgupta, S.; Hoque, M.O.; Upadhyay, S.; Sidransky, D. Mitochondrial Cytochrome B Gene Mutation Promotes Tumor Growth in Bladder Cancer. Cancer Res. 2008, 68, 700–706. [Google Scholar] [CrossRef] [Green Version]
- Hertweck, K.L.; Dasgupta, S. The Landscape of MtDNA Modifications in Cancer: A Tale of Two Cities. Front. Oncol. 2017, 7. [Google Scholar] [CrossRef]
- Kopinski, P.K.; Janssen, K.A.; Schaefer, P.M.; Trefely, S.; Perry, C.E.; Potluri, P.; Tintos-Hernandez, J.A.; Singh, L.N.; Karch, K.R.; Campbell, S.L.; et al. Regulation of Nuclear Epigenome by Mitochondrial DNA Heteroplasmy. Proc. Natl. Acad. Sci. USA 2019, 116, 16028–16035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smiraglia, D.J.; Kulawiec, M.; Bistulfi, G.L.; Gupta, S.G.; Singh, K.K. A Novel Role for Mitochondria in Regulating Epigenetic Modification in the Nucleus. Cancer Biol. 2008, 7, 1182–1190. [Google Scholar] [CrossRef] [Green Version]
- Han, S.; Liu, Y.; Cai, S.J.; Qian, M.; Ding, J.; Larion, M.; Gilbert, M.R.; Yang, C. IDH Mutation in Glioma: Molecular Mechanisms and Potential Therapeutic Targets. Br. J. Cancer 2020, 122, 1580–1589. [Google Scholar] [CrossRef]
- Bardella, C.; Pollard, P.J.; Tomlinson, I. SDH Mutations in Cancer. Biochim. Biophys. Acta 2011, 1807, 1432–1443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frezza, C.; Zheng, L.; Folger, O.; Rajagopalan, K.N.; MacKenzie, E.D.; Jerby, L.; Micaroni, M.; Chaneton, B.; Adam, J.; Hedley, A.; et al. Haem Oxygenase Is Synthetically Lethal with the Tumour Suppressor Fumarate Hydratase. Nature 2011, 477, 225–228. [Google Scholar] [CrossRef]
- Tseng, P.-L.; Wu, W.-H.; Hu, T.-H.; Chen, C.-W.; Cheng, H.-C.; Li, C.-F.; Tsai, W.-H.; Tsai, H.-J.; Hsieh, M.-C.; Chuang, J.-H.; et al. Decreased Succinate Dehydrogenase B in Human Hepatocellular Carcinoma Accelerates Tumor Malignancy by Inducing the Warburg Effect. Sci. Rep. 2018, 8. [Google Scholar] [CrossRef] [PubMed]
- Selak, M.A.; Armour, S.M.; MacKenzie, E.D.; Boulahbel, H.; Watson, D.G.; Mansfield, K.D.; Pan, Y.; Simon, M.C.; Thompson, C.B.; Gottlieb, E. Succinate Links TCA Cycle Dysfunction to Oncogenesis by Inhibiting HIF-Alpha Prolyl Hydroxylase. Cancer Cell 2005, 7, 77–85. [Google Scholar] [CrossRef] [Green Version]
- Isaacs, J.S.; Jung, Y.J.; Mole, D.R.; Lee, S.; Torres-Cabala, C.; Chung, Y.-L.; Merino, M.; Trepel, J.; Zbar, B.; Toro, J.; et al. HIF Overexpression Correlates with Biallelic Loss of Fumarate Hydratase in Renal Cancer: Novel Role of Fumarate in Regulation of HIF Stability. Cancer Cell 2005, 8, 143–153. [Google Scholar] [CrossRef] [Green Version]
- McBrayer, S.K.; Mayers, J.R.; DiNatale, G.J.; Shi, D.D.; Khanal, J.; Chakraborty, A.A.; Sarosiek, K.A.; Briggs, K.J.; Robbins, A.K.; Sewastianik, T.; et al. Transaminase Inhibition by 2-Hydroxyglutarate Impairs Glutamate Biosynthesis and Redox Homeostasis in Glioma. Cell 2018, 175, 101–116.e25. [Google Scholar] [CrossRef] [Green Version]
- Ishii, T.; Yasuda, K.; Akatsuka, A.; Hino, O.; Hartman, P.S.; Ishii, N. A Mutation in the SDHC Gene of Complex II Increases Oxidative Stress, Resulting in Apoptosis and Tumorigenesis. Cancer Res. 2005, 65, 203–209. [Google Scholar] [PubMed]
- Purohit, V.; Simeone, D.M.; Lyssiotis, C.A. Metabolic Regulation of Redox Balance in Cancer. Cancers 2019, 11, 955. [Google Scholar] [CrossRef] [Green Version]
- Zheng, L.; Cardaci, S.; Jerby, L.; MacKenzie, E.D.; Sciacovelli, M.; Johnson, T.I.; Gaude, E.; King, A.; Leach, J.D.G.; Edrada-Ebel, R.; et al. Fumarate Induces Redox-Dependent Senescence by Modifying Glutathione Metabolism. Nat. Commun. 2015, 6, 6001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanamori, M.; Higa, T.; Sonoda, Y.; Murakami, S.; Dodo, M.; Kitamura, H.; Taguchi, K.; Shibata, T.; Watanabe, M.; Suzuki, H.; et al. Activation of the NRF2 Pathway and Its Impact on the Prognosis of Anaplastic Glioma Patients. Neuro-Oncology 2015, 17, 555–565. [Google Scholar] [CrossRef] [Green Version]
- Gilbert, M.R.; Liu, Y.; Neltner, J.; Pu, H.; Morris, A.; Sunkara, M.; Pittman, T.; Kyprianou, N.; Horbinski, C. Autophagy and Oxidative Stress in Gliomas with IDH1 Mutations. Acta Neuropathol. 2014, 127, 221–233. [Google Scholar] [CrossRef]
- Guzy, R.D.; Sharma, B.; Bell, E.; Chandel, N.S.; Schumacker, P.T. Loss of the SdhB, but Not the SdhA, Subunit of Complex II Triggers Reactive Oxygen Species-Dependent Hypoxia-Inducible Factor Activation and Tumorigenesis. Mol. Cell Biol. 2008, 28, 718–731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sulkowski, P.L.; Sundaram, R.K.; Oeck, S.; Corso, C.D.; Liu, Y.; Noorbakhsh, S.; Niger, M.; Boeke, M.; Ueno, D.; Kalathil, A.N.; et al. Krebs Cycle-Deficient Hereditary Cancer Syndromes Are Defined by Homologous Recombination DNA Repair Defects. Nat. Genet. 2018, 50, 1086–1092. [Google Scholar] [CrossRef]
- Sciacovelli, M.; Gonçalves, E.; Johnson, T.I.; Zecchini, V.R.; da Costa, A.S.H.; Gaude, E.; Drubbel, A.V.; Theobald, S.J.; Abbo, S.R.; Tran, M.G.B.; et al. Fumarate Is an Epigenetic Modifier That Elicits Epithelial-to-Mesenchymal Transition. Nature 2016, 537, 544–547. [Google Scholar] [CrossRef]
- Letouzé, E.; Martinelli, C.; Loriot, C.; Burnichon, N.; Abermil, N.; Ottolenghi, C.; Janin, M.; Menara, M.; Nguyen, A.T.; Benit, P.; et al. SDH Mutations Establish a Hypermethylator Phenotype in Paraganglioma. Cancer Cell 2013, 23, 739–752. [Google Scholar] [CrossRef] [Green Version]
- Xu, W.; Yang, H.; Liu, Y.; Yang, Y.; Wang, P.; Kim, S.-H.; Ito, S.; Yang, C.; Wang, P.; Xiao, M.-T.; et al. Oncometabolite 2-Hydroxyglutarate Is a Competitive Inhibitor of α-Ketoglutarate-Dependent Dioxygenases. Cancer Cell 2011, 19, 17–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warburg, O.; Wind, F.; Negelein, E. The Metabolism of Tumors in the Body. J. Gen. Physiol. 1927, 8, 519–530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warburg, O. On the Origin of Cancer Cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Kubota, K. From Tumor Biology to Clinical Pet: A Review of Positron Emission Tomography (PET) in Oncology. Ann. Nucl. Med. 2001, 15, 471–486. [Google Scholar] [CrossRef]
- Luengo, A.; Li, Z.; Gui, D.Y.; Sullivan, L.B.; Zagorulya, M.; Do, B.T.; Ferreira, R.; Naamati, A.; Ali, A.; Lewis, C.A.; et al. Increased Demand for NAD+ Relative to ATP Drives Aerobic Glycolysis. Mol. Cell 2021, 81, 691–707.e6. [Google Scholar] [CrossRef]
- Faubert, B.; Li, K.Y.; Cai, L.; Hensley, C.T.; Kim, J.; Zacharias, L.G.; Yang, C.; Do, Q.N.; Doucette, S.; Burguete, D.; et al. Lactate Metabolism in Human Lung Tumors. Cell 2017, 171, 358–371.e9. [Google Scholar] [CrossRef] [Green Version]
- Pfeiffer, T.; Schuster, S.; Bonhoeffer, S. Cooperation and Competition in the Evolution of ATP-Producing Pathways. Science 2001, 292, 504–507. [Google Scholar] [CrossRef] [Green Version]
- Cairns, R.A.; Harris, I.S.; Mak, T.W. Regulation of Cancer Cell Metabolism. Nat. Rev. Cancer 2011, 11, 85–95. [Google Scholar] [CrossRef] [Green Version]
- Zheng, J. Energy Metabolism of Cancer: Glycolysis versus Oxidative Phosphorylation (Review). Oncol. Lett. 2012, 4, 1151–1157. [Google Scholar] [CrossRef] [Green Version]
- Koppenol, W.H.; Bounds, P.L.; Dang, C.V. Otto Warburg’s Contributions to Current Concepts of Cancer Metabolism. Nat. Rev. Cancer 2011, 11, 325–337. [Google Scholar] [CrossRef]
- Fantin, V.R.; St-Pierre, J.; Leder, P. Attenuation of LDH-A Expression Uncovers a Link between Glycolysis, Mitochondrial Physiology, and Tumor Maintenance. Cancer Cell 2006, 9, 425–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Sun, X.-X.; Qian, D.Z.; Dai, M.-S. Molecular Crosstalk Between MYC and HIF in Cancer. Front. Cell Dev. Biol. 2020, 8. [Google Scholar] [CrossRef]
- Nagao, A.; Kobayashi, M.; Koyasu, S.; Chow, C.C.T.; Harada, H. HIF-1-Dependent Reprogramming of Glucose Metabolic Pathway of Cancer Cells and Its Therapeutic Significance. Int. J. Mol. Sci. 2019, 20, 238. [Google Scholar] [CrossRef] [Green Version]
- Prigione, A.; Rohwer, N.; Hoffmann, S.; Mlody, B.; Drews, K.; Bukowiecki, R.; Blümlein, K.; Wanker, E.E.; Ralser, M.; Cramer, T.; et al. HIF1α Modulates Cell Fate Reprogramming through Early Glycolytic Shift and Upregulation of PDK1-3 and PKM2. Stem Cells 2014, 32, 364–376. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Liu, J.; Liang, Y.; Wu, R.; Zhao, Y.; Hong, X.; Lin, M.; Yu, H.; Liu, L.; Levine, A.J.; et al. Tumour-Associated Mutant P53 Drives the Warburg Effect. Nat. Commun. 2013, 4, 2935. [Google Scholar] [CrossRef] [Green Version]
- Bensaad, K.; Tsuruta, A.; Selak, M.A.; Vidal, M.N.C.; Nakano, K.; Bartrons, R.; Gottlieb, E.; Vousden, K.H. TIGAR, a P53-Inducible Regulator of Glycolysis and Apoptosis. Cell 2006, 126, 107–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lonetto, G.; Koifman, G.; Silberman, A.; Attery, A.; Solomon, H.; Levin-Zaidman, S.; Goldfinger, N.; Porat, Z.; Erez, A.; Rotter, V. Mutant P53-Dependent Mitochondrial Metabolic Alterations in a Mesenchymal Stem Cell-Based Model of Progressive Malignancy. Cell Death Differ. 2019, 26, 1566–1581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scott, I.; Youle, R.J. Mitochondrial Fission and Fusion. Essays Biochem. 2010, 47, 85–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, J.; Guo, Y.; Xue, B.; Shi, P.; Chen, Y.; Su, Q.P.; Hao, H.; Zhao, S.; Wu, C.; Yu, L.; et al. ER-Mitochondria Contacts Promote MtDNA Nucleoids Active Transportation via Mitochondrial Dynamic Tubulation. Nat. Commun. 2020, 11, 4471. [Google Scholar] [CrossRef]
- Westermann, B. Bioenergetic Role of Mitochondrial Fusion and Fission. Biochim. Biophys. Acta Bioenerg. 2012, 1817, 1833–1838. [Google Scholar] [CrossRef] [Green Version]
- Zhan, L.; Cao, H.; Wang, G.; Lyu, Y.; Sun, X.; An, J.; Wu, Z.; Huang, Q.; Liu, B.; Xing, J. Drp1-Mediated Mitochondrial Fission Promotes Cell Proliferation through Crosstalk of P53 and NF-ΚB Pathways in Hepatocellular Carcinoma. Oncotarget 2016, 7, 65001–65011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hackenbrock, C.R. Ultrastructural Bases for Metabolically Linked Mechanical Activity in Mitochondria. I. Reversible Ultrastructural Changes with Change in Metabolic Steady State in Isolated Liver Mitochondria. J. Cell Biol. 1966, 30, 269–297. [Google Scholar] [CrossRef] [Green Version]
- Gao, T.; Zhang, X.; Zhao, J.; Zhou, F.; Wang, Y.; Zhao, Z.; Xing, J.; Chen, B.; Li, J.; Liu, S. SIK2 Promotes Reprogramming of Glucose Metabolism through PI3K/AKT/HIF-1α Pathway and Drp1-Mediated Mitochondrial Fission in Ovarian Cancer. Cancer Lett. 2020, 469, 89–101. [Google Scholar] [CrossRef]
- Yao, C.-H.; Wang, R.; Wang, Y.; Kung, C.-P.; Weber, J.D.; Patti, G.J. Mitochondrial Fusion Supports Increased Oxidative Phosphorylation during Cell Proliferation. eLife 2019, 8, e41351. [Google Scholar] [CrossRef] [PubMed]
- Liesa, M.; Shirihai, O.S. Mitochondrial Dynamics in the Regulation of Nutrient Utilization and Energy Expenditure. Cell Metab. 2013, 17, 491–506. [Google Scholar] [CrossRef] [Green Version]
- Pagliuso, A.; Cossart, P.; Stavru, F. The Ever-Growing Complexity of the Mitochondrial Fission Machinery. Cell. Mol. Life Sci. 2018, 75, 355–374. [Google Scholar] [CrossRef] [Green Version]
- Rambold, A.S.; Kostelecky, B.; Elia, N.; Lippincott-Schwartz, J. Tubular Network Formation Protects Mitochondria from Autophagosomal Degradation during Nutrient Starvation. Proc. Natl. Acad. Sci. USA 2011, 108, 10190–10195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Si, L.; Fu, J.; Liu, W.; Hayashi, T.; Nie, Y.; Mizuno, K.; Hattori, S.; Fujisaki, H.; Onodera, S.; Ikejima, T. Silibinin Inhibits Migration and Invasion of Breast Cancer MDA-MB-231 Cells through Induction of Mitochondrial Fusion. Mol. Cell Biochem. 2020, 463, 189–201. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Nguyen, N.D.; Huang, Y.; Lin, D.; Fujimoto, T.N.; Molkentine, J.M.; Deorukhkar, A.; Kang, Y.; San Lucas, F.A.; Fernandes, C.J.; et al. Mitochondrial Fusion Exploits a Therapeutic Vulnerability of Pancreatic Cancer. JCI Insight 2019, 5. [Google Scholar] [CrossRef] [PubMed]
- Rehman, J.; Zhang, H.J.; Toth, P.T.; Zhang, Y.; Marsboom, G.; Hong, Z.; Salgia, R.; Husain, A.N.; Wietholt, C.; Archer, S.L. Inhibition of Mitochondrial Fission Prevents Cell Cycle Progression in Lung Cancer. FASEB J. 2012, 26, 2175–2186. [Google Scholar] [CrossRef] [Green Version]
- Tseng, H.-C.; Lin, C.-C.; Hsiao, L.-D.; Yang, C.-M. Lysophosphatidylcholine-Induced Mitochondrial Fission Contributes to Collagen Production in Human Cardiac Fibroblasts[S]. J. Lipid Res. 2019, 60, 1573–1589. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Zhan, L.; Cao, H.; Li, J.; Lyu, Y.; Guo, X.; Zhang, J.; Ji, L.; Ren, T.; An, J.; et al. Increased Mitochondrial Fission Promotes Autophagy and Hepatocellular Carcinoma Cell Survival through the ROS-Modulated Coordinated Regulation of the NFKB and TP53 Pathways. Autophagy 2016, 12, 999–1014. [Google Scholar] [CrossRef]
- Picard, M.; Shirihai, O.S.; Gentil, B.J.; Burelle, Y. Mitochondrial Morphology Transitions and Functions: Implications for Retrograde Signaling? Am. J. Physiol. Regul. Integr. Comp. Physiol. 2013, 304, R393–R406. [Google Scholar] [CrossRef] [Green Version]
- Willems, P.H.G.M.; Rossignol, R.; Dieteren, C.E.J.; Murphy, M.P.; Koopman, W.J.H. Redox Homeostasis and Mitochondrial Dynamics. Cell Metab. 2015, 22, 207–218. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Q.; Li, H.; Li, Y.; Tan, M.; Fan, S.; Cao, C.; Meng, F.; Zhu, L.; Zhao, L.; Guan, M.-X.; et al. Inhibiting Neddylation Modification Alters Mitochondrial Morphology and Reprograms Energy Metabolism in Cancer Cells. JCI Insight 2019, 4, e121582. [Google Scholar] [CrossRef] [Green Version]
- Tsushima, K.; Bugger, H.; Wende, A.R.; Soto, J.; Jenson, G.A.; Tor, A.R.; McGlauflin, R.; Kenny, H.C.; Zhang, Y.; Souvenir, R.; et al. Mitochondrial Reactive Oxygen Species in Lipotoxic Hearts Induce Post-Translational Modifications of AKAP121, DRP1, and OPA1 That Promote Mitochondrial Fission. Circ. Res. 2018, 122, 58–73. [Google Scholar] [CrossRef]
- Duan, H.; Lei, Z.; Xu, F.; Pan, T.; Lu, D.; Ding, P.; Zhu, C.; Pan, C.; Zhang, S. PARK2 Suppresses Proliferation and Tumorigenicity in Non-Small Cell Lung Cancer. Front. Oncol. 2019, 9, 790. [Google Scholar] [CrossRef]
- Li, C.; Zhang, Y.; Cheng, X.; Yuan, H.; Zhu, S.; Liu, J.; Wen, Q.; Xie, Y.; Liu, J.; Kroemer, G.; et al. PINK1 and PARK2 Suppress Pancreatic Tumorigenesis through Control of Mitochondrial Iron-Mediated Immunometabolism. Dev. Cell 2018, 46, 441–455.e8. [Google Scholar] [CrossRef] [Green Version]
- Bernardini, J.P.; Lazarou, M.; Dewson, G. Parkin and Mitophagy in Cancer. Oncogene 2017, 36, 1315–1327. [Google Scholar] [CrossRef]
- Liu, L.; Feng, D.; Chen, G.; Chen, M.; Zheng, Q.; Song, P.; Ma, Q.; Zhu, C.; Wang, R.; Qi, W.; et al. Mitochondrial Outer-Membrane Protein FUNDC1 Mediates Hypoxia-Induced Mitophagy in Mammalian Cells. Nat. Cell Biol. 2012, 14, 177–185. [Google Scholar] [CrossRef]
- Panigrahi, D.P.; Praharaj, P.P.; Bhol, C.S.; Mahapatra, K.K.; Patra, S.; Behera, B.P.; Mishra, S.R.; Bhutia, S.K. The Emerging, Multifaceted Role of Mitophagy in Cancer and Cancer Therapeutics. Semin. Cancer Biol. 2020, 66, 45–58. [Google Scholar] [CrossRef]
- Ma, Y.; Wang, L.; Jia, R. The Role of Mitochondrial Dynamics in Human Cancers. Am. J. Cancer Res. 2020, 10, 1278–1293. [Google Scholar]
- Zhang, J.; Ney, P.A. Role of BNIP3 and NIX in Cell Death, Autophagy, and Mitophagy. Cell Death Differ. 2009, 16, 939–946. [Google Scholar] [CrossRef] [Green Version]
- Ray, S.K.; Mukherjee, S. Mitophagy in Carcinogenesis and Tumor Progression- A New Paradigm with Emerging Importance. Anticancer Agents Med. Chem. 2021. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, M.; Marusawa, H.; Wang, H.-Q.; Iwai, A.; Ikeuchi, K.; Imai, Y.; Kataoka, A.; Nukina, N.; Takahashi, R.; Chiba, T. Parkin as a Tumor Suppressor Gene for Hepatocellular Carcinoma. Oncogene 2008, 27, 6002–6011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chourasia, A.H.; Tracy, K.; Frankenberger, C.; Boland, M.L.; Sharifi, M.N.; Drake, L.E.; Sachleben, J.R.; Asara, J.M.; Locasale, J.W.; Karczmar, G.S.; et al. Mitophagy Defects Arising from BNip3 Loss Promote Mammary Tumor Progression to Metastasis. EMBO Rep. 2015, 16, 1145–1163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.-Y.; Wang, W.-H.; Che, L.; Lan, Y.; Zhang, L.-Y.; Zhan, D.-L.; Huang, Z.-Y.; Lin, Z.-N.; Lin, Y.-C. BNIP3L-Dependent Mitophagy Promotes HBx-Induced Cancer Stemness of Hepatocellular Carcinoma Cells via Glycolysis Metabolism Reprogramming. Cancers 2020, 12, 655. [Google Scholar] [CrossRef] [Green Version]
- Gang, H.; Dhingra, R.; Lin, J.; Hai, Y.; Aviv, Y.; Margulets, V.; Hamedani, M.; Thanasupawat, T.; Leygue, E.; Klonisch, T.; et al. PDK2-Mediated Alternative Splicing Switches Bnip3 from Cell Death to Cell Survival. J. Cell Biol. 2015, 210, 1101–1115. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Zhang, C.; Zhao, Y.; Yue, X.; Wu, H.; Huang, S.; Chen, J.; Tomsky, K.; Xie, H.; Khella, C.A.; et al. Parkin Targets HIF-1α for Ubiquitination and Degradation to Inhibit Breast Tumor Progression. Nat. Commun. 2017, 8, 1823. [Google Scholar] [CrossRef]
- Andréasson, C.; Ott, M.; Büttner, S. Mitochondria Orchestrate Proteostatic and Metabolic Stress Responses. EMBO Rep. 2019, 20, e47865. [Google Scholar] [CrossRef] [PubMed]
- Amuthan, G.; Biswas, G.; Zhang, S.-Y.; Klein-Szanto, A.; Vijayasarathy, C.; Avadhani, N.G. Mitochondria-to-Nucleus Stress Signaling Induces Phenotypic Changes, Tumor Progression and Cell Invasion. EMBO J. 2001, 20, 1910–1920. [Google Scholar] [CrossRef] [Green Version]
- Carden, T.; Singh, B.; Mooga, V.; Bajpai, P.; Singh, K.K. Epigenetic Modification of MiR-663 Controls Mitochondria-to-Nucleus Retrograde Signaling and Tumor Progression. J. Biol. Chem. 2017, 292, 20694–20706. [Google Scholar] [CrossRef] [Green Version]
- Guha, M.; Tang, W.; Sondheimer, N.; Avadhani, N.G. Role of Calcineurin, HnRNPA2 and Akt in Mitochondrial Respiratory Stress-Mediated Transcription Activation of Nuclear Gene Targets. Biochim. Biophys Acta 2010, 1797, 1055–1065. [Google Scholar] [CrossRef] [Green Version]
- Mishra, P.; Chan, D.C. Metabolic Regulation of Mitochondrial Dynamics. J. Cell Biol. 2016, 212, 379–387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonekamp, N.A.; Peter, B.; Hillen, H.S.; Felser, A.; Bergbrede, T.; Choidas, A.; Horn, M.; Unger, A.; Di Lucrezia, R.; Atanassov, I.; et al. Small-Molecule Inhibitors of Human Mitochondrial DNA Transcription. Nature 2020, 588, 712–716. [Google Scholar] [CrossRef]
- Skrtić, M.; Sriskanthadevan, S.; Jhas, B.; Gebbia, M.; Wang, X.; Wang, Z.; Hurren, R.; Jitkova, Y.; Gronda, M.; Maclean, N.; et al. Inhibition of Mitochondrial Translation as a Therapeutic Strategy for Human Acute Myeloid Leukemia. Cancer Cell 2011, 20, 674–688. [Google Scholar] [CrossRef] [Green Version]
- Hu, B.; Guo, Y. Inhibition of Mitochondrial Translation as a Therapeutic Strategy for Human Ovarian Cancer to Overcome Chemoresistance. Biochem. Biophys. Res. Commun. 2019, 509, 373–378. [Google Scholar] [CrossRef]
- Kuntz, E.M.; Baquero, P.; Michie, A.M.; Dunn, K.; Tardito, S.; Holyoake, T.L.; Helgason, G.V.; Gottlieb, E. Targeting Mitochondrial Oxidative Phosphorylation Eradicates Therapy-Resistant Chronic Myeloid Leukemia Stem Cells. Nat. Med. 2017, 23, 1234–1240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reed, G.A.; Schiller, G.J.; Kambhampati, S.; Tallman, M.S.; Douer, D.; Minden, M.D.; Yee, K.W.; Gupta, V.; Brandwein, J.; Jitkova, Y.; et al. A Phase 1 Study of Intravenous Infusions of Tigecycline in Patients with Acute Myeloid Leukemia. Cancer Med. 2016, 5, 3031–3040. [Google Scholar] [CrossRef] [PubMed]
- Dong, L.; Neuzil, J. Targeting Mitochondria as an Anticancer Strategy. Cancer Commun. 2019, 39, 63. [Google Scholar] [CrossRef] [Green Version]
- Wheaton, W.W.; Weinberg, S.E.; Hamanaka, R.B.; Soberanes, S.; Sullivan, L.B.; Anso, E.; Glasauer, A.; Dufour, E.; Mutlu, G.M.; Budigner, G.S.; et al. Metformin Inhibits Mitochondrial Complex I of Cancer Cells to Reduce Tumorigenesis. Elife 2014, 3, e02242. [Google Scholar] [CrossRef]
- Cai, H.; Everett, R.S.; Thakker, D.R. Efficacious Dose of Metformin for Breast Cancer Therapy Is Determined by Cation Transporter Expression in Tumours. Br. J. Pharm. 2019, 176, 2724–2735. [Google Scholar] [CrossRef] [PubMed]
- Madera, D.; Vitale-Cross, L.; Martin, D.; Schneider, A.; Molinolo, A.A.; Gangane, N.; Carey, T.E.; McHugh, J.B.; Komarck, C.M.; Walline, H.M.; et al. Prevention of Tumor Growth Driven by PIK3CA and HPV Oncogenes by Targeting MTOR Signaling with Metformin in Oral Squamous Carcinomas Expressing OCT3. Cancer Prev. Res. 2015, 8, 197–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vasan, K.; Werner, M.; Chandel, N.S. Mitochondrial Metabolism as a Target for Cancer Therapy. Cell Metab. 2020, 32, 341–352. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.C.; Carey, K.T.; McKenzie, M. Anti-Cancer Analogues ME-143 and ME-344 Exert Toxicity by Directly Inhibiting Mitochondrial NADH: Ubiquinone Oxidoreductase (Complex I). Am. J. Cancer Res. 2015, 5, 689–701. [Google Scholar] [PubMed]
- Ghosh, P.; Vidal, C.; Dey, S.; Zhang, L. Mitochondria Targeting as an Effective Strategy for Cancer Therapy. Int. J. Mol. Sci. 2020, 21, 3363. [Google Scholar] [CrossRef] [PubMed]
- Jeyaraju, D.V.; Hurren, R.; Wang, X.; MacLean, N.; Gronda, M.; Shamas-Din, A.; Minden, M.D.; Giaever, G.; Schimmer, A.D. A Novel Isoflavone, ME-344, Targets the Cytoskeleton in Acute Myeloid Leukemia. Oncotarget 2016, 7, 49777–49785. [Google Scholar] [CrossRef] [Green Version]
- Quintela-Fandino, M.; Morales, S.; Cortés-Salgado, A.; Manso, L.; Apala, J.V.; Muñoz, M.; Gasol Cudos, A.; Salla Fortuny, J.; Gion, M.; Lopez-Alonso, A.; et al. Randomized Phase 0/I Trial of the Mitochondrial Inhibitor ME-344 or Placebo Added to Bevacizumab in Early HER2-Negative Breast Cancer. Clin. Cancer Res. 2020, 26, 35–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carter, J.L.; Hege, K.; Kalpage, H.A.; Edwards, H.; Hüttemann, M.; Taub, J.W.; Ge, Y. Targeting Mitochondrial Respiration for the Treatment of Acute Myeloid Leukemia. Biochem. Pharm. 2020, 182, 114253. [Google Scholar] [CrossRef]
- Molina, J.R.; Sun, Y.; Protopopova, M.; Gera, S.; Bandi, M.; Bristow, C.; McAfoos, T.; Morlacchi, P.; Ackroyd, J.; Agip, A.-N.A.; et al. An Inhibitor of Oxidative Phosphorylation Exploits Cancer Vulnerability. Nat. Med. 2018, 24, 1036–1046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panina, S.B.; Pei, J.; Baran, N.; Konopleva, M.; Kirienko, N.V. Utilizing Synergistic Potential of Mitochondria-Targeting Drugs for Leukemia Therapy. Front. Oncol. 2020, 10, 435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stuart, S.D.; Schauble, A.; Gupta, S.; Kennedy, A.D.; Keppler, B.R.; Bingham, P.M.; Zachar, Z. A Strategically Designed Small Molecule Attacks Alpha-Ketoglutarate Dehydrogenase in Tumor Cells through a Redox Process. Cancer Metab. 2014, 2, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pardee, T.S.; Lee, K.; Luddy, J.; Maturo, C.; Rodriguez, R.; Isom, S.; Miller, L.D.; Stadelman, K.M.; Levitan, D.; Hurd, D.; et al. A Phase I Study of the First-in-Class Antimitochondrial Metabolism Agent, CPI-613, in Patients with Advanced Hematologic Malignancies. Clin. Cancer Res. 2014, 20, 5255–5264. [Google Scholar] [CrossRef] [Green Version]
- Lycan, T.W.; Pardee, T.S.; Petty, W.J.; Bonomi, M.; Alistar, A.; Lamar, Z.S.; Isom, S.; Chan, M.D.; Miller, A.A.; Ruiz, J. A Phase II Clinical Trial of CPI-613 in Patients with Relapsed or Refractory Small Cell Lung Carcinoma. PLoS ONE 2016, 11, e0164244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Philip, P.A.; Buyse, M.E.; Alistar, A.T.; Rocha Lima, C.M.; Luther, S.; Pardee, T.S.; Van Cutsem, E. A Phase III Open-Label Trial to Evaluate Efficacy and Safety of CPI-613 plus Modified FOLFIRINOX (MFFX) versus FOLFIRINOX (FFX) in Patients with Metastatic Adenocarcinoma of the Pancreas. Future Oncol. 2019, 15, 3189–3196. [Google Scholar] [CrossRef]
- Golub, D.; Iyengar, N.; Dogra, S.; Wong, T.; Bready, D.; Tang, K.; Modrek, A.S.; Placantonakis, D.G. Mutant Isocitrate Dehydrogenase Inhibitors as Targeted Cancer Therapeutics. Front. Oncol. 2019, 9, 417. [Google Scholar] [CrossRef] [Green Version]
- Lee, P.; Malik, D.; Perkons, N.; Huangyang, P.; Khare, S.; Rhoades, S.; Gong, Y.-Y.; Burrows, M.; Finan, J.M.; Nissim, I.; et al. Targeting Glutamine Metabolism Slows Soft Tissue Sarcoma Growth. Nat. Commun. 2020, 11, 498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altman, B.J.; Stine, Z.E.; Dang, C.V. From Krebs to Clinic: Glutamine Metabolism to Cancer Therapy. Nat. Rev. Cancer 2016, 16, 773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gross, M.I.; Demo, S.D.; Dennison, J.B.; Chen, L.; Chernov-Rogan, T.; Goyal, B.; Janes, J.R.; Laidig, G.J.; Lewis, E.R.; Li, J.; et al. Antitumor Activity of the Glutaminase Inhibitor CB-839 in Triple-Negative Breast Cancer. Mol. Cancer 2014, 13, 890–901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soth, M.J.; Le, K.; Di Francesco, M.E.; Hamilton, M.M.; Liu, G.; Burke, J.P.; Carroll, C.L.; Kovacs, J.J.; Bardenhagen, J.P.; Bristow, C.A.; et al. Discovery of IPN60090, a Clinical Stage Selective Glutaminase-1 (GLS-1) Inhibitor with Excellent Pharmacokinetic and Physicochemical Properties. J. Med. Chem. 2020, 63, 12957–12977. [Google Scholar] [CrossRef]
- Glasauer, A.; Chandel, N.S. Targeting Antioxidants for Cancer Therapy. Biochem. Pharm. 2014, 92, 90–101. [Google Scholar] [CrossRef] [PubMed]
- Sborov, D.W.; Haverkos, B.M.; Harris, P.J. Investigational Cancer Drugs Targeting Cell Metabolism in Clinical Development. Expert Opin. Investig. Drugs 2015, 24, 79–94. [Google Scholar] [CrossRef] [Green Version]
- Dong, L.; Gopalan, V.; Holland, O.; Neuzil, J. Mitocans Revisited: Mitochondrial Targeting as Efficient Anti-Cancer Therapy. Int. J. Mol. Sci. 2020, 21, 7941. [Google Scholar] [CrossRef]
- Gentric, G.; Mechta-Grigoriou, F. Tumor Cells and Cancer-Associated Fibroblasts: An Updated Metabolic Perspective. Cancers 2021, 13, 399. [Google Scholar] [CrossRef]
- Ippolito, L.; Morandi, A.; Taddei, M.L.; Parri, M.; Comito, G.; Iscaro, A.; Raspollini, M.R.; Magherini, F.; Rapizzi, E.; Masquelier, J.; et al. Cancer-Associated Fibroblasts Promote Prostate Cancer Malignancy via Metabolic Rewiring and Mitochondrial Transfer. Oncogene 2019, 38, 5339–5355. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moindjie, H.; Rodrigues-Ferreira, S.; Nahmias, C. Mitochondrial Metabolism in Carcinogenesis and Cancer Therapy. Cancers 2021, 13, 3311. https://doi.org/10.3390/cancers13133311
Moindjie H, Rodrigues-Ferreira S, Nahmias C. Mitochondrial Metabolism in Carcinogenesis and Cancer Therapy. Cancers. 2021; 13(13):3311. https://doi.org/10.3390/cancers13133311
Chicago/Turabian StyleMoindjie, Hadia, Sylvie Rodrigues-Ferreira, and Clara Nahmias. 2021. "Mitochondrial Metabolism in Carcinogenesis and Cancer Therapy" Cancers 13, no. 13: 3311. https://doi.org/10.3390/cancers13133311
APA StyleMoindjie, H., Rodrigues-Ferreira, S., & Nahmias, C. (2021). Mitochondrial Metabolism in Carcinogenesis and Cancer Therapy. Cancers, 13(13), 3311. https://doi.org/10.3390/cancers13133311