
PD-1 and beyond to Activate T Cells in Cutaneous Squamous Cell Cancers: The Case for 4-1BB and VISTA Antibodies in Combination Therapy




Abstract
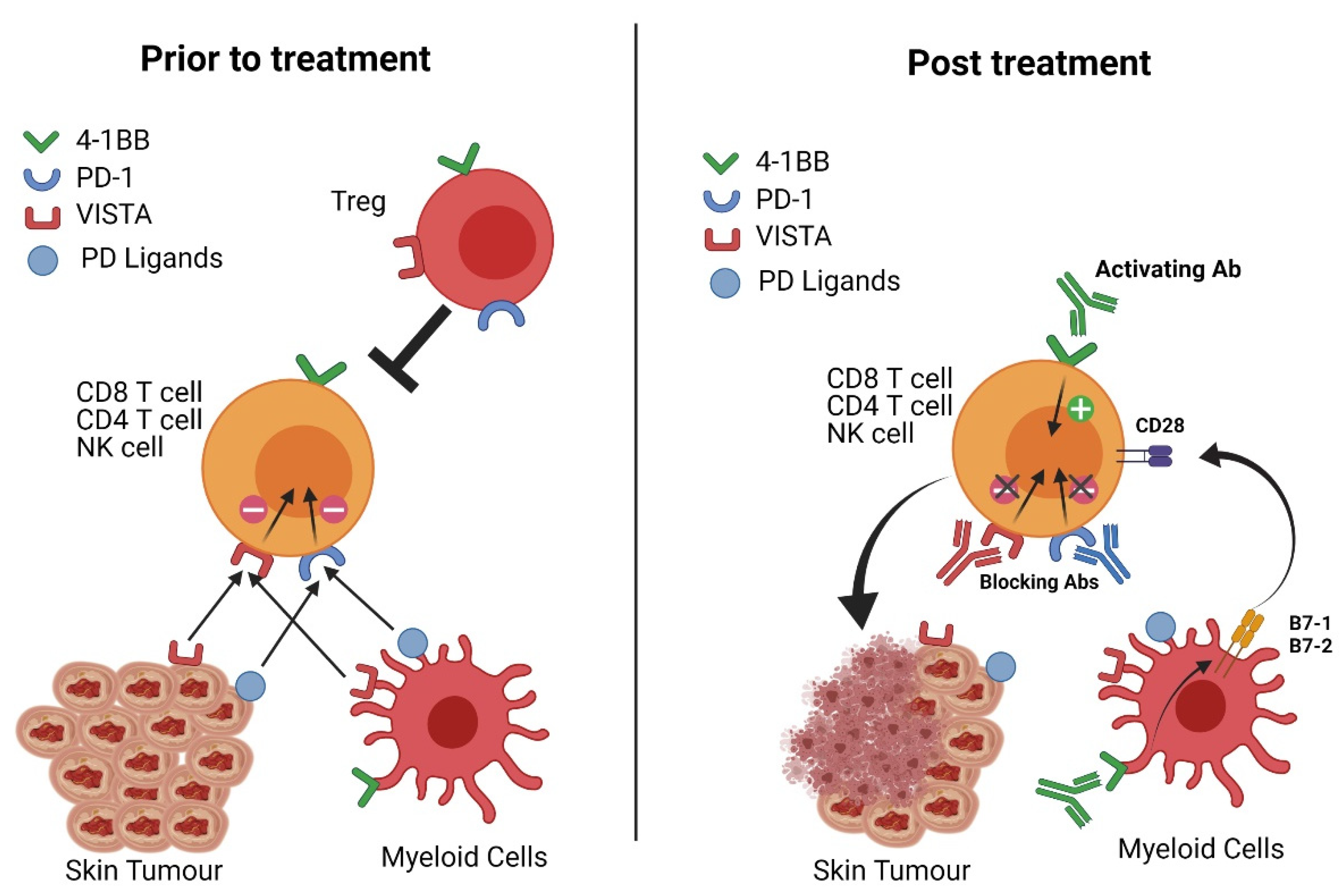
:Simple Summary
Abstract
1. Introduction
2. Natural Immunity to cSCC
3. Immunotherapy in cSCC
4. Programmed Death- 1 (PD-1)
5. V-Domain Ig Suppressor of T Cell Activation (VISTA or PD-1H)
6. 4-1BB (CD137)
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Rogers, H.W.; Weinstock, M.A.; Feldman, S.R.; Coldiron, B.M. Incidence Estimate of Nonmelanoma Skin Cancer (Keratinocyte Carcinomas) in the U.S. Population, 2012. JAMA Dermatol. 2015, 151, 1081–1086. [Google Scholar] [CrossRef]
- Misitzis, A.; Beatson, M.; Weinstock, M.A. Keratinocyte Carcinoma Mortality in the United States as Reported in Death Certificates, 2011–2017. Dermatol. Surg. 2020, 46, 1135–1140. [Google Scholar] [CrossRef]
- Pandeya, N.; Olsen, C.M.; Whiteman, D.C. The incidence and multiplicity rates of keratinocyte cancers in Australia. Med. J. Aust. 2017, 207, 339–343. [Google Scholar] [CrossRef]
- Yarchoan, M.; Hopkins, A.; Jaffee, E.M. Tumor Mutational Burden and Response Rate to PD-1 Inhibition. N. Engl. J. Med. 2017, 377, 2500–2501. [Google Scholar] [CrossRef]
- Ratushny, V.; Gober, M.D.; Hick, R.; Ridky, T.W.; Seykora, J.T. From keratinocyte to cancer: The pathogenesis and modeling of cutaneous squamous cell carcinoma. J. Clin. Investig. 2012, 122, 464–472. [Google Scholar] [CrossRef] [Green Version]
- Werner, R.N.; Sammain, A.; Erdmann, R.; Hartmann, V.; Stockfleth, E.; Nast, A. The natural history of actinic keratosis: A systematic review. Br. J. Dermatol. 2013, 169, 502–518. [Google Scholar] [CrossRef] [PubMed]
- Glogau, R.G. The risk of progression to invasive disease. J. Am. Acad. Dermatol. 2000, 42, 23–24. [Google Scholar] [CrossRef] [PubMed]
- Gutzmer, R.; Wiegand, S.; Kolbl, O.; Wermker, K.; Heppt, M.; Berking, C. Actinic Keratosis and Cutaneous Squamous Cell Carcinoma. Dtsch. Ärzteblatt Int. 2019, 116, 616–626. [Google Scholar] [CrossRef] [Green Version]
- Maubec, E. Update of the Management of Cutaneous Squamous-cell Carcinoma. Acta Derm. Venereol. 2020, 100, adv00143. [Google Scholar] [CrossRef]
- Garrett, G.L.; Blanc, P.D.; Boscardin, J.; Lloyd, A.A.; Ahmed, R.L.; Anthony, T.; Bibee, K.; Breithaupt, A.; Cannon, J.; Chen, A.; et al. Incidence of and Risk Factors for Skin Cancer in Organ Transplant Recipients in the United States. JAMA Dermatol. 2017, 153, 296–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, C.; Cheng, J.; Colegio, O.R. Cutaneous squamous cell carcinomas in solid organ transplant recipients: Emerging strategies for surveillance, staging, and treatment. Semin. Oncol. 2016, 43, 390–394. [Google Scholar] [CrossRef]
- Haeffner, A.C.; Zepter, K.; Elmets, C.A.; Wood, G.S. Analysis of tumor-infiltrating lymphocytes in cutaneous squamous cell carcinoma. Arch. Dermatol. 1997, 133, 585–590. [Google Scholar] [CrossRef] [PubMed]
- Black, A.P.; Bailey, A.; Jones, L.; Turner, R.J.; Hollowood, K.; Ogg, G.S. p53-specific CD8+ T-cell responses in individuals with cutaneous squamous cell carcinoma. Br. J. Dermatol. 2005, 153, 987–991. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, D.; Krammer, S.; Bachmann, M.R.; Mathemeier, L.; Ruzicka, T.; Bagci, I.S.; von Braunmuhl, T. Ex vivo confocal microscopy features of cutaneous squamous cell carcinoma. J. Biophotonics 2018, 11, e201700318. [Google Scholar] [CrossRef]
- Ji, A.L.; Rubin, A.J.; Thrane, K.; Jiang, S.; Reynolds, D.L.; Meyers, R.M.; Guo, M.G.; George, B.M.; Mollbrink, A.; Bergenstrahle, J.; et al. Multimodal Analysis of Composition and Spatial Architecture in Human Squamous Cell Carcinoma. Cell 2020, 182, 497–514.e22. [Google Scholar] [CrossRef]
- Avramoiu, I.; Petrescu, I.O.; Ciurea, M.E.; Bold, A.; Silosi, I.; tanTu, M.M.; Niculescu, M.; Anghel Savciu, R.E.; Mogoanta, S.S. Peritumoral inflammatory reaction in non-melanoma skin cancers—Histological and immunohistochemical study. Rom. J. Morphol. Embryol. 2016, 57, 943–950. [Google Scholar] [PubMed]
- Kai, H.; Kadono, T.; Kakinuma, T.; Tomita, M.; Ohmatsu, H.; Asano, Y.; Tada, Y.; Sugaya, M.; Sato, S. CCR10 and CCL27 are overexpressed in cutaneous squamous cell carcinoma. Pathol. Res. Pract. 2011, 207, 43–48. [Google Scholar] [CrossRef]
- Lysa, B.; Tartler, U.; Wolf, R.; Arenberger, P.; Benninghoff, B.; Ruzicka, T.; Hengge, U.R.; Walz, M. Gene expression in actinic keratoses: Pharmacological modulation by imiquimod. Br. J. Dermatol. 2004, 151, 1150–1159. [Google Scholar] [CrossRef]
- Nasti, T.H.; Timares, L. Inflammasome activation of IL-1 family mediators in response to cutaneous photodamage. Photochem. Photobiol. 2012, 88, 1111–1125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Awad, F.; Assrawi, E.; Louvrier, C.; Jumeau, C.; Giurgea, I.; Amselem, S.; Karabina, S.A. Photoaging and skin cancer: Is the inflammasome the missing link? Mech. Ageing Dev. 2018, 172, 131–137. [Google Scholar] [CrossRef]
- Meier, K.; Drexler, S.K.; Eberle, F.C.; Lefort, K.; Yazdi, A.S. Silencing of ASC in Cutaneous Squamous Cell Carcinoma. PLoS ONE 2016, 11, e0164742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sand, J.; Fenini, G.; Grossi, S.; Hennig, P.; Di Filippo, M.; Levesque, M.; Werner, S.; French, L.E.; Beer, H.D. The NLRP1 Inflammasome Pathway Is Silenced in Cutaneous Squamous Cell Carcinoma. J. Investig. Dermatol. 2019, 139, 1788–1797.e6. [Google Scholar] [CrossRef] [PubMed]
- Hart, P.H.; Norval, M. Ultraviolet radiation-induced immunosuppression and its relevance for skin carcinogenesis. Photochem. Photobiol. Sci. 2018, 17, 1872–1884. [Google Scholar] [CrossRef] [PubMed]
- Clifford, J.L.; Walch, E.; Yang, X.; Xu, X.; Alberts, D.S.; Clayman, G.L.; El-Naggar, A.K.; Lotan, R.; Lippman, S.M. Suppression of type I interferon signaling proteins is an early event in squamous skin carcinogenesis. Clin. Cancer Res. 2002, 8, 2067–2072. [Google Scholar]
- Azzimonti, B.; Zavattaro, E.; Provasi, M.; Vidali, M.; Conca, A.; Catalano, E.; Rimondini, L.; Colombo, E.; Valente, G. Intense Foxp3+ CD25+ regulatory T-cell infiltration is associated with high-grade cutaneous squamous cell carcinoma and counterbalanced by CD8+/Foxp3+ CD25+ ratio. Br. J. Dermatol. 2015, 172, 64–73. [Google Scholar] [CrossRef]
- Lai, C.; August, S.; Behar, R.; Polak, M.; Ardern-Jones, M.; Theaker, J.; Al-Shamkhani, A.; Healy, E. Characteristics of immunosuppressive regulatory T cells in cutaneous squamous cell carcinomas and role in metastasis. Lancet 2015, 385, S59. [Google Scholar] [CrossRef]
- Lai, C.; August, S.; Albibas, A.; Behar, R.; Cho, S.Y.; Polak, M.E.; Theaker, J.; MacLeod, A.S.; French, R.R.; Glennie, M.J.; et al. OX40+ Regulatory T Cells in Cutaneous Squamous Cell Carcinoma Suppress Effector T-Cell Responses and Associate with Metastatic Potential. Clin. Cancer Res. 2016, 22, 4236–4248. [Google Scholar] [CrossRef] [Green Version]
- Freeman, A.; Bridge, J.A.; Maruthayanar, P.; Overgaard, N.H.; Jung, J.W.; Simpson, F.; Prow, T.W.; Soyer, H.P.; Frazer, I.H.; Freeman, M.; et al. Comparative immune phenotypic analysis of cutaneous Squamous Cell Carcinoma and Intraepidermal Carcinoma in immune-competent individuals: Proportional representation of CD8+ T-cells but not FoxP3+ Regulatory T-cells is associated with disease stage. PLoS ONE 2014, 9, e110928. [Google Scholar] [CrossRef]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [Green Version]
- Sharma, P.; Siddiqui, B.A.; Anandhan, S.; Yadav, S.S.; Subudhi, S.K.; Gao, J.; Goswami, S.; Allison, J.P. The Next Decade of Immune Checkpoint Therapy. Cancer Discov. 2021, 11, 838–857. [Google Scholar] [CrossRef] [PubMed]
- Vidal, D. Topical imiquimod: Mechanism of action and clinical applications. Mini Rev. Med. Chem. 2006, 6, 499–503. [Google Scholar] [CrossRef] [PubMed]
- Guedan, S.; Ruella, M.; June, C.H. Emerging Cellular Therapies for Cancer. Annu. Rev. Immunol. 2019, 37, 145–171. [Google Scholar] [CrossRef] [PubMed]
- Curran, M.A.; Glisson, B.S. New Hope for Therapeutic Cancer Vaccines in the Era of Immune Checkpoint Modulation. Annu. Rev. Med. 2019, 70, 409–424. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Allison, J.P. Dissecting the mechanisms of immune checkpoint therapy. Nat. Rev. Immunol. 2020, 20, 75–76. [Google Scholar] [CrossRef]
- Migden, M.R.; Rischin, D.; Schmults, C.D.; Guminski, A.; Hauschild, A.; Lewis, K.D.; Chung, C.H.; Hernandez-Aya, L.; Lim, A.M.; Chang, A.L.S.; et al. PD-1 Blockade with Cemiplimab in Advanced Cutaneous Squamous-Cell Carcinoma. N. Engl. J. Med. 2018, 379, 341–351. [Google Scholar] [CrossRef] [Green Version]
- Ellmark, P.; Mangsbo, S.M.; Furebring, C.; Norlen, P.; Totterman, T.H. Tumor-directed immunotherapy can generate tumor-specific T cell responses through localized co-stimulation. Cancer Immunol. Immunother. 2017, 66, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Flies, D.B. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat. Rev. Immunol. 2013, 13, 227–242. [Google Scholar] [CrossRef]
- Ishida, Y.; Agata, Y.; Shibahara, K.; Honjo, T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J. 1992, 11, 3887–3895. [Google Scholar] [CrossRef]
- Sharpe, A.H.; Pauken, K.E. The diverse functions of the PD1 inhibitory pathway. Nat. Rev. Immunol. 2018, 18, 153–167. [Google Scholar] [CrossRef]
- Barber, D.L.; Wherry, E.J.; Masopust, D.; Zhu, B.; Allison, J.P.; Sharpe, A.H.; Freeman, G.J.; Ahmed, R. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature 2006, 439, 682–687. [Google Scholar] [CrossRef] [PubMed]
- McLane, L.M.; Abdel-Hakeem, M.S.; Wherry, E.J. CD8 T Cell Exhaustion During Chronic Viral Infection and Cancer. Annu. Rev. Immunol. 2019, 37, 457–495. [Google Scholar] [CrossRef] [Green Version]
- Hirano, F.; Kaneko, K.; Tamura, H.; Dong, H.; Wang, S.; Ichikawa, M.; Rietz, C.; Flies, D.B.; Lau, J.S.; Zhu, G.; et al. Blockade of B7-H1 and PD-1 by monoclonal antibodies potentiates cancer therapeutic immunity. Cancer Res. 2005, 65, 1089–1096. [Google Scholar] [PubMed]
- Nishimura, H.; Nose, M.; Hiai, H.; Minato, N.; Honjo, T. Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motif-carrying immunoreceptor. Immunity 1999, 11, 141–151. [Google Scholar] [CrossRef] [Green Version]
- Mallett, G.; Laurence, A.; Amarnath, S. Programmed Cell Death-1 Receptor (PD-1)-Mediated Regulation of Innate Lymphoid Cells. Int. J. Mol. Sci. 2019, 20, 2836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baumeister, S.H.; Freeman, G.J.; Dranoff, G.; Sharpe, A.H. Coinhibitory Pathways in Immunotherapy for Cancer. Annu. Rev. Immunol. 2016, 34, 539–573. [Google Scholar] [CrossRef]
- Patsoukis, N.; Wang, Q.; Strauss, L.; Boussiotis, V.A. Revisiting the PD-1 pathway. Sci. Adv. 2020, 6. [Google Scholar] [CrossRef]
- Kumagai, S.; Togashi, Y.; Kamada, T.; Sugiyama, E.; Nishinakamura, H.; Takeuchi, Y.; Vitaly, K.; Itahashi, K.; Maeda, Y.; Matsui, S.; et al. The PD-1 expression balance between effector and regulatory T cells predicts the clinical efficacy of PD-1 blockade therapies. Nat. Immunol. 2020, 21, 1346–1358. [Google Scholar] [CrossRef]
- Trefny, M.P.; Kaiser, M.; Stanczak, M.A.; Herzig, P.; Savic, S.; Wiese, M.; Lardinois, D.; Laubli, H.; Uhlenbrock, F.; Zippelius, A. PD-1(+) natural killer cells in human non-small cell lung cancer can be activated by PD-1/PD-L1 blockade. Cancer Immunol. Immunother. 2020, 69, 1505–1517. [Google Scholar] [CrossRef]
- Pesce, S.; Trabanelli, S.; Di Vito, C.; Greppi, M.; Obino, V.; Guolo, F.; Minetto, P.; Bozzo, M.; Calvi, M.; Zaghi, E.; et al. Cancer Immunotherapy by Blocking Immune Checkpoints on Innate Lymphocytes. Cancers 2020, 12, 3504. [Google Scholar] [CrossRef]
- Lee, H.; Quek, C.; Silva, I.; Tasker, A.; Batten, M.; Rizos, H.; Lim, S.Y.; Nur Gide, T.; Shang, P.; Attrill, G.H.; et al. Integrated molecular and immunophenotypic analysis of NK cells in anti-PD-1 treated metastatic melanoma patients. Oncoimmunology 2019, 8, e1537581. [Google Scholar] [CrossRef] [Green Version]
- Strauss, L.; Mahmoud, M.A.A.; Weaver, J.D.; Tijaro-Ovalle, N.M.; Christofides, A.; Wang, Q.; Pal, R.; Yuan, M.; Asara, J.; Patsoukis, N.; et al. Targeted deletion of PD-1 in myeloid cells induces antitumor immunity. Sci. Immunol. 2020, 5. [Google Scholar] [CrossRef]
- Dammeijer, F.; van Gulijk, M.; Mulder, E.E.; Lukkes, M.; Klaase, L.; van den Bosch, T.; van Nimwegen, M.; Lau, S.P.; Latupeirissa, K.; Schetters, S.; et al. The PD-1/PD-L1-Checkpoint Restrains T cell Immunity in Tumor-Draining Lymph Nodes. Cancer Cell 2020. [Google Scholar] [CrossRef] [PubMed]
- Muntyanu, A.; Netchiporouk, E.; Gerstein, W.; Gniadecki, R.; Litvinov, I.V. Cutaneous Immune-Related Adverse Events (irAEs) to Immune Checkpoint Inhibitors: A Dermatology Perspective on Management. J. Cutan. Med. Surg. 2020, 1203475420943260. [Google Scholar] [CrossRef]
- Rovers, J.F.J.; Bovenschen, H.J. Dermatological side effects rarely interfere with the continuation of checkpoint inhibitor immunotherapy for cancer. Int. J. Dermatol. 2020. [Google Scholar] [CrossRef]
- Simonsen, A.B.; Kaae, J.; Ellebaek, E.; Svane, I.M.; Zachariae, C. Cutaneous adverse reactions to anti-PD-1 treatment-A systematic review. J. Am. Acad. Dermatol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Mandala, M.; Tondini, C.; Merelli, B.; Massi, D. Rationale for New Checkpoint Inhibitor Combinations in Melanoma Therapy. Am. J. Clin. Dermatol. 2017, 18, 597–611. [Google Scholar] [CrossRef] [PubMed]
- Patrinely, J.R., Jr.; Dewan, A.K.; Johnson, D.B. The Role of Anti-PD-1/PD-L1 in the Treatment of Skin Cancer. BioDrugs 2020, 34, 495–503. [Google Scholar] [CrossRef]
- Schachter, J.; Ribas, A.; Long, G.V.; Arance, A.; Grob, J.J.; Mortier, L.; Daud, A.; Carlino, M.S.; McNeil, C.; Lotem, M.; et al. Pembrolizumab versus ipilimumab for advanced melanoma: Final overall survival results of a multicentre, randomised, open-label phase 3 study (KEYNOTE-006). Lancet 2017, 390, 1853–1862. [Google Scholar] [CrossRef]
- Hodi, F.S.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Rutkowski, P.; Cowey, C.L.; Lao, C.D.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Nivolumab plus ipilimumab or nivolumab alone versus ipilimumab alone in advanced melanoma (CheckMate 067): 4-year outcomes of a multicentre, randomised, phase 3 trial. Lancet Oncol. 2018, 19, 1480–1492. [Google Scholar] [CrossRef]
- Migden, M.R.; Khushalani, N.I.; Chang, A.L.S.; Lewis, K.D.; Schmults, C.D.; Hernandez-Aya, L.; Meier, F.; Schadendorf, D.; Guminski, A.; Hauschild, A.; et al. Cemiplimab in locally advanced cutaneous squamous cell carcinoma: Results from an open-label, phase 2, single-arm trial. Lancet Oncol. 2020, 21, 294–305. [Google Scholar] [CrossRef]
- Dougan, M.; Luoma, A.M.; Dougan, S.K.; Wucherpfennig, K.W. Understanding and treating the inflammatory adverse events of cancer immunotherapy. Cell 2021, 184, 1575–1588. [Google Scholar] [CrossRef] [PubMed]
- Safarzadeh, A.; Alizadeh, M.; Beyranvand, F.; Falavand Jozaaee, R.; Hajiasgharzadeh, K.; Baghbanzadeh, A.; Derakhshani, A.; Argentiero, A.; Baradaran, B.; Silvestris, N. Varied functions of immune checkpoints during cancer metastasis. Cancer Immunol. Immunother. 2020. [Google Scholar] [CrossRef]
- Anonymous. LAG3-PD-1 Inhibitor Combo Impresses in Melanoma. Cancer Discov. 2021. [Google Scholar] [CrossRef]
- Naing, A.; Meric-Bernstam, F.; Stephen, B.; Karp, D.D.; Hajjar, J.; Rodon Ahnert, J.; Piha-Paul, S.A.; Colen, R.R.; Jimenez, C.; Raghav, K.P.; et al. Phase 2 study of pembrolizumab in patients with advanced rare cancers. J. Immunother. Cancer 2020, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grob, J.J.; Gonzalez, R.; Basset-Seguin, N.; Vornicova, O.; Schachter, J.; Joshi, A.; Meyer, N.; Grange, F.; Piulats, J.M.; Bauman, J.R.; et al. Pembrolizumab Monotherapy for Recurrent or Metastatic Cutaneous Squamous Cell Carcinoma: A Single-Arm Phase II Trial (KEYNOTE-629). J. Clin. Oncol. 2020, 38, 2916–2925. [Google Scholar] [CrossRef]
- Kudchadkar, R.R.; Yushak, M.L.; Lawson, D.H.; Delman, K.A.; Lowe, M.C.; Goings, M.; McBrien, S.; Mckellar, M.; Sieja, K.; Maynard, N.; et al. Phase II trial of pembrolizumab (MK-3475) in metastatic cutaneous squamous cell carcinoma (cSCC). J. Clin. Oncol. 2018, 36. [Google Scholar] [CrossRef]
- Maubec, E.; Helfen, S.; Scheer-Senyarich, I.; Boubaya, M.; Schischmanoff, O.; Alloux, C.; Deschamps, L.; Petrow, P.; Lopez, I.; Tibi, A.; et al. CARSKIN: Pembrolizumab as first line therapy in patients with unresectable cutaneous squamous cell carcinoma (cSCC). J. Clin. Oncol. 2017, 35. [Google Scholar] [CrossRef]
- Wang, L.; Rubinstein, R.; Lines, J.L.; Wasiuk, A.; Ahonen, C.; Guo, Y.; Lu, L.F.; Gondek, D.; Wang, Y.; Fava, R.A.; et al. VISTA, a novel mouse Ig superfamily ligand that negatively regulates T cell responses. J. Exp. Med. 2011, 208, 577–592. [Google Scholar] [CrossRef]
- Flies, D.B.; Wang, S.; Xu, H.; Chen, L. Cutting edge: A monoclonal antibody specific for the programmed death-1 homolog prevents graft-versus-host disease in mouse models. J. Immunol. 2011, 187, 1537–1541. [Google Scholar] [CrossRef]
- Wang, L.; Jia, B.; Claxton, D.F.; Ehmann, W.C.; Rybka, W.B.; Mineishi, S.; Naik, S.; Khawaja, M.R.; Sivik, J.; Han, J.; et al. VISTA is highly expressed on MDSCs and mediates an inhibition of T cell response in patients with AML. Oncoimmunology 2018, 7, e1469594. [Google Scholar] [CrossRef]
- Huang, X.; Zhang, X.; Li, E.; Zhang, G.; Wang, X.; Tang, T.; Bai, X.; Liang, T. VISTA: An immune regulatory protein checking tumor and immune cells in cancer immunotherapy. J. Hematol. Oncol. 2020, 13, 83. [Google Scholar] [CrossRef] [PubMed]
- Johnston, R.J.; Su, L.J.; Pinckney, J.; Critton, D.; Boyer, E.; Krishnakumar, A.; Corbett, M.; Rankin, A.L.; Dibella, R.; Campbell, L.; et al. VISTA is an acidic pH-selective ligand for PSGL-1. Nature 2019, 574, 565–570. [Google Scholar] [CrossRef]
- Wang, J.; Wu, G.; Manick, B.; Hernandez, V.; Renelt, M.; Erickson, C.; Guan, J.; Singh, R.; Rollins, S.; Solorz, A.; et al. VSIG-3 as a ligand of VISTA inhibits human T-cell function. Immunology 2019, 156, 74–85. [Google Scholar] [CrossRef] [Green Version]
- ElTanbouly, M.A.; Croteau, W.; Noelle, R.J.; Lines, J.L. VISTA: A novel immunotherapy target for normalizing innate and adaptive immunity. Semin. Immunol. 2019, 42, 101308. [Google Scholar] [CrossRef]
- Yasinska, I.M.; Meyer, N.H.; Schlichtner, S.; Hussain, R.; Siligardi, G.; Casely-Hayford, M.; Fiedler, W.; Wellbrock, J.; Desmet, C.; Calzolai, L.; et al. Ligand-Receptor Interactions of Galectin-9 and VISTA Suppress Human T Lymphocyte Cytotoxic Activity. Front. Immunol. 2020, 11, 580557. [Google Scholar] [CrossRef]
- Flies, D.B.; Han, X.; Higuchi, T.; Zheng, L.; Sun, J.; Ye, J.J.; Chen, L. Coinhibitory receptor PD-1H preferentially suppresses CD4(+) T cell-mediated immunity. J. Clin. Investig. 2014, 124, 1966–1975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Le Mercier, I.; Putra, J.; Chen, W.; Liu, J.; Schenk, A.D.; Nowak, E.C.; Suriawinata, A.A.; Li, J.; Noelle, R.J. Disruption of the immune-checkpoint VISTA gene imparts a proinflammatory phenotype with predisposition to the development of autoimmunity. Proc. Natl. Acad. Sci. USA 2014, 111, 14846–14851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- ElTanbouly, M.A.; Schaafsma, E.; Noelle, R.J.; Lines, J.L. VISTA: Coming of age as a multi-lineage immune checkpoint. Clin. Exp. Immunol. 2020, 200, 120–130. [Google Scholar] [CrossRef] [Green Version]
- Broughton, T.W.K.; ElTanbouly, M.A.; Schaafsma, E.; Deng, J.; Sarde, A.; Croteau, W.; Li, J.; Nowak, E.C.; Mabaera, R.; Smits, N.C.; et al. Defining the Signature of VISTA on Myeloid Cell Chemokine Responsiveness. Front. Immunol. 2019, 10, 2641. [Google Scholar] [CrossRef] [Green Version]
- Le Mercier, I.; Chen, W.; Lines, J.L.; Day, M.; Li, J.; Sergent, P.; Noelle, R.J.; Wang, L. VISTA Regulates the Development of Protective Antitumor Immunity. Cancer Res. 2014, 74, 1933–1944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, L.; Deng, W.W.; Huang, C.F.; Bu, L.L.; Yu, G.T.; Mao, L.; Zhang, W.F.; Liu, B.; Sun, Z.J. Expression of VISTA correlated with immunosuppression and synergized with CD8 to predict survival in human oral squamous cell carcinoma. Cancer Immunol. Immunother. 2017, 66, 627–636. [Google Scholar] [CrossRef]
- Kuklinski, L.F.; Yan, S.; Li, Z.; Fisher, J.L.; Cheng, C.; Noelle, R.J.; Angeles, C.V.; Turk, M.J.; Ernstoff, M.S. VISTA expression on tumor-infiltrating inflammatory cells in primary cutaneous melanoma correlates with poor disease-specific survival. Cancer Immunol. Immunother. 2018, 67, 1113–1121. [Google Scholar] [CrossRef]
- Kakavand, H.; Jackett, L.A.; Menzies, A.M.; Gide, T.N.; Carlino, M.S.; Saw, R.P.M.; Thompson, J.F.; Wilmott, J.S.; Long, G.V.; Scolyer, R.A. Negative immune checkpoint regulation by VISTA: A mechanism of acquired resistance to anti-PD-1 therapy in metastatic melanoma patients. Mod. Pathol. 2017, 30, 1666–1676. [Google Scholar] [CrossRef]
- Choi, J.W.; Kim, Y.J.; Yun, K.A.; Won, C.H.; Lee, M.W.; Choi, J.H.; Chang, S.E.; Lee, W.J. The prognostic significance of VISTA and CD33-positive myeloid cells in cutaneous melanoma and their relationship with PD-1 expression. Sci. Rep. 2020, 10, 14372. [Google Scholar] [CrossRef] [PubMed]
- Yoon, K.W.; Byun, S.; Kwon, E.; Hwang, S.Y.; Chu, K.; Hiraki, M.; Jo, S.H.; Weins, A.; Hakroush, S.; Cebulla, A.; et al. Control of signaling-mediated clearance of apoptotic cells by the tumor suppressor p53. Science 2015, 349, 1261669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kondo, Y.; Ohno, T.; Nishii, N.; Harada, K.; Yagita, H.; Azuma, M. Differential contribution of three immune checkpoint (VISTA, CTLA-4, PD-1) pathways to antitumor responses against squamous cell carcinoma. Oral Oncol. 2016, 57, 54–60. [Google Scholar] [CrossRef] [PubMed]
- Piao, J.; Kamimura, Y.; Iwai, H.; Cao, Y.; Kikuchi, K.; Hashiguchi, M.; Masunaga, T.; Jiang, H.; Tamura, K.; Sakaguchi, S.; et al. Enhancement of T-cell-mediated anti-tumour immunity via the ectopically expressed glucocorticoid-induced tumour necrosis factor receptor-related receptor ligand (GITRL) on tumours. Immunology 2009, 127, 489–499. [Google Scholar] [CrossRef]
- Liu, J.; Yuan, Y.; Chen, W.; Putra, J.; Suriawinata, A.A.; Schenk, A.D.; Miller, H.E.; Guleria, I.; Barth, R.J.; Huang, Y.H.; et al. Immune-checkpoint proteins VISTA and PD-1 nonredundantly regulate murine T-cell responses. Proc. Natl. Acad. Sci. USA 2015, 112, 6682–6687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartkowiak, T.; Curran, M.A. 4-1BB Agonists: Multi-Potent Potentiators of Tumor Immunity. Front. Oncol. 2015, 5, 117. [Google Scholar] [CrossRef] [Green Version]
- Chester, C.; Sanmamed, M.F.; Wang, J.; Melero, I. Immunotherapy targeting 4-1BB: Mechanistic rationale, clinical results, and future strategies. Blood 2018, 131, 49–57. [Google Scholar] [CrossRef]
- Heinisch, I.V.; Bizer, C.; Volgger, W.; Simon, H.U. Functional CD137 receptors are expressed by eosinophils from patients with IgE-mediated allergic responses but not by eosinophils from patients with non-IgE-mediated eosinophilic disorders. J. Allergy Clin. Immunol. 2001, 108, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Shuford, W.W.; Klussman, K.; Tritchler, D.D.; Loo, D.T.; Chalupny, J.; Siadak, A.W.; Brown, T.J.; Emswiler, J.; Raecho, H.; Larsen, C.P.; et al. 4-1BB costimulatory signals preferentially induce CD8+ T cell proliferation and lead to the amplification in vivo of cytotoxic T cell responses. J. Exp. Med. 1997, 186, 47–55. [Google Scholar] [CrossRef]
- Stelekati, E.; Bahri, R.; D’Orlando, O.; Orinska, Z.; Mittrucker, H.W.; Langenhaun, R.; Glatzel, M.; Bollinger, A.; Paus, R.; Bulfone-Paus, S. Mast cell-mediated antigen presentation regulates CD8+ T cell effector functions. Immunity 2009, 31, 665–676. [Google Scholar] [CrossRef]
- Vinay, D.S.; Choi, B.K.; Bae, J.S.; Kim, W.Y.; Gebhardt, B.M.; Kwon, B.S. CD137-deficient mice have reduced NK/NKT cell numbers and function, are resistant to lipopolysaccharide-induced shock syndromes, and have lower IL-4 responses. J. Immunol. 2004, 173, 4218–4229. [Google Scholar] [CrossRef] [Green Version]
- Chester, C.; Ambulkar, S.; Kohrt, H.E. 4-1BB agonism: Adding the accelerator to cancer immunotherapy. Cancer Immunol. Immunother. 2016, 65, 1243–1248. [Google Scholar] [CrossRef] [Green Version]
- Kuang, Y.; Weng, X.; Liu, X.; Zhu, H.; Chen, Z.; Chen, H. Effects of 4-1BB signaling on the biological function of murine dendritic cells. Oncol. Lett. 2012, 3, 477–481. [Google Scholar] [CrossRef] [Green Version]
- Zhang, P.; Gao, F.; Wang, Q.; Wang, X.; Zhu, F.; Ma, C.; Sun, W.; Zhang, L. Agonistic anti-4-1BB antibody promotes the expansion of natural regulatory T cells while maintaining Foxp3 expression. Scand. J. Immunol. 2007, 66, 435–440. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Lin, X.; Chen, H.M.; Wu, Q.; Subudhi, S.K.; Chen, L.; Fu, Y.X. Administration of agonistic anti-4-1BB monoclonal antibody leads to the amelioration of experimental autoimmune encephalomyelitis. J. Immunol. 2002, 168, 1457–1465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foell, J.; McCausland, M.; Burch, J.; Corriazzi, N.; Yan, X.J.; Suwyn, C.; O’Neil, S.P.; Hoffmann, M.K.; Mittler, R.S. CD137-mediated T cell co-stimulation terminates existing autoimmune disease in SLE-prone NZB/NZW F1 mice. Ann. N. Y. Acad. Sci. 2003, 987, 230–235. [Google Scholar] [CrossRef] [PubMed]
- Ascierto, P.A.; Simeone, E.; Sznol, M.; Fu, Y.X.; Melero, I. Clinical experiences with anti-CD137 and anti-PD1 therapeutic antibodies. Semin. Oncol. 2010, 37, 508–516. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Lee, L.F.; Fisher, T.S.; Jessen, B.; Elliott, M.; Evering, W.; Logronio, K.; Tu, G.H.; Tsaparikos, K.; Li, X.; et al. Combination of 4-1BB agonist and PD-1 antagonist promotes antitumor effector/memory CD8 T cells in a poorly immunogenic tumor model. Cancer Immunol. Res. 2015, 3, 149–160. [Google Scholar] [CrossRef] [Green Version]
- Kocak, E.; Lute, K.; Chang, X.; May, K.F., Jr.; Exten, K.R.; Zhang, H.; Abdessalam, S.F.; Lehman, A.M.; Jarjoura, D.; Zheng, P.; et al. Combination therapy with anti-CTL antigen-4 and anti-4-1BB antibodies enhances cancer immunity and reduces autoimmunity. Cancer Res. 2006, 66, 7276–7284. [Google Scholar] [CrossRef] [Green Version]
- Qi, X.; Li, F.; Wu, Y.; Cheng, C.; Han, P.; Wang, J.; Yang, X. Optimization of 4-1BB antibody for cancer immunotherapy by balancing agonistic strength with FcgammaR affinity. Nat. Commun. 2019, 10, 2141. [Google Scholar] [CrossRef]
- Claus, C.; Ferrara, C.; Xu, W.; Sam, J.; Lang, S.; Uhlenbrock, F.; Albrecht, R.; Herter, S.; Schlenker, R.; Husser, T.; et al. Tumor-targeted 4-1BB agonists for combination with T cell bispecific antibodies as off-the-shelf therapy. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Xu, J.; Wu, J. The emerging role of co-stimulatory molecules and their agonistic mAb-based combination therapies in melanoma. Int. Immunopharmacol. 2020, 89, 107097. [Google Scholar] [CrossRef] [PubMed]
- Ye, Q.; Song, D.G.; Poussin, M.; Yamamoto, T.; Best, A.; Li, C.; Coukos, G.; Powell, D.J., Jr. CD137 accurately identifies and enriches for naturally occurring tumor-reactive T cells in tumor. Clin. Cancer Res. 2014, 20, 44–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shitaoka, K.; Hamana, H.; Kishi, H.; Hayakawa, Y.; Kobayashi, E.; Sukegawa, K.; Piao, X.; Lyu, F.; Nagata, T.; Sugiyama, D.; et al. Identification of Tumoricidal TCRs from Tumor-Infiltrating Lymphocytes by Single-Cell Analysis. Cancer Immunol. Res. 2018, 6, 378–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Segal, N.H.; Logan, T.F.; Hodi, F.S.; McDermott, D.; Melero, I.; Hamid, O.; Schmidt, H.; Robert, C.; Chiarion-Sileni, V.; Ascierto, P.A.; et al. Results from an Integrated Safety Analysis of Urelumab, an Agonist Anti-CD137 Monoclonal Antibody. Clin. Cancer Res. 2017, 23, 1929–1936. [Google Scholar] [CrossRef] [Green Version]
- Segal, N.H.; He, A.R.; Doi, T.; Levy, R.; Bhatia, S.; Pishvaian, M.J.; Cesari, R.; Chen, Y.; Davis, C.B.; Huang, B.; et al. Phase I Study of Single-Agent Utomilumab (PF-05082566), a 4-1BB/CD137 Agonist, in Patients with Advanced Cancer. Clin. Cancer Res. 2018, 24, 1816–1823. [Google Scholar] [CrossRef] [Green Version]
- Zelin, E.; Zalaudek, I.; Agozzino, M.; Dianzani, C.; Dri, A.; Di Meo, N.; Giuffrida, R.; Marangi, G.F.; Neagu, N.; Persichetti, P.; et al. Neoadjuvant Therapy for Non-melanoma Skin Cancer: Updated Therapeutic Approaches for Basal, Squamous, and Merkel Cell Carcinoma. Curr. Treat. Options Oncol. 2021, 22, 35. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Therapy Target | Therapy | Indication | Clinical Trial # | Clinical Trial Status | Reference |
|---|---|---|---|---|---|
| PD-1 | Pembrolizumab | Multiple, including SCC patients with poor prognosis and progression on standardized therapies | NCT02721732 | Phase II | [64] |
| PD-1 | Pembrolizumab | Recurrent and/or metastatic cSCC | NCT03284424 | Phase II | [65] |
| PD-1 | Pembrolizumab | Recurrent cSCC in patients not curable by surgery or radiation | NCT02964559 | Phase II | [66] |
| PD-1 | Pembrolizumab Radiation | Postoperative radiotherapy for cSCC of head and neck | NCT03057613 | Phase II/completed | |
| PD-1 | Pembrolizumab | Unresectable/metastatic squamous cell carcinoma | NCT02883556 | Phase II | [67] |
| PD-1 EGFR | Pembrolizumab in combination with Cetuximab | cSCC of head and neck | NCT03082534 | Phase II/recruiting | |
| PD-1 C5a | Pembrolizumab in combination with IFX-1(anti-C5a Ab) | Locally advanced or metastatic cSCC | NCT04812535 | Phase II/not yet recruiting | |
| PD-1 | Pembrolizumab | High-risk, locally advanced cSCC following surgery and radiation | NCT03833167 | Phase III/recruiting | |
| PD-1 | Pembrolizumab | PD-1 naïve cSCC | NCT04808999 | Phase II/recruiting | |
| PD-1 | Cemiplimab | Locally advanced or metastatic cSCC | NCT02383212 | Phase I/ metastatic or locally advanced SCC phase II metastatic cSCC | [35] |
| PD-1 | Cemiplimab | Locally advanced or metastatic cSCC | NCT02760498 | Phase II/recruiting | [60] |
| PD-1 | Cemiplimab | Recurrent cSCC | NCT03889912 | Phase I/active/not recruiting | |
| PD-1 | Cemiplimab | cSCC stage II to IV | NCT04154943 | Phase II/recruiting | |
| PD-1 | Cemiplimab | Immunocompromised patients with unresectable locally recurrent and/or metastatic cSCC | NCT04242173 | Phase II recruiting | |
| PD-1 | Cemiplimab in conjunction with RP-1 (modified HSV-1) | Locally advanced/metastatic cSCC. Combination with RP-1 intratumourally | NCT04050436 | Phase II/recruiting | |
| PD-1 | Cemiplimab | Recurrent stage III-IV cSCC of head and Neck | NCT03565783 | Phase II/recruiting | |
| PD-1 SAR444245 | Cemiplimab in conjunction with SAR444245 (rhIL-2) | Locally advanced or metastatic cSSC | NCT04913220 | Phase I/II/not yet recruiting | |
| PD-1 | Cemiplimab | High-risk, stage III cSCC | NCT04632433 | Phase II/recruiting | |
| PD-1 | Cemiplimab | High-risk cSCC before and after surgery | NCT04428671 | Phase I/recruiting | |
| PD-1 | Cemiplimab | High-risk cSCC after radiation and surgery | NCT03969004 | Phase III/recruiting | |
| PD-1 TLR-9 agonist | Cemiplimab or Pembrolizumab with Cavrotolimod (TLR-9 agonist) | Advanced or metastatic cSCC. Combination with cavrotolimod intratumourally | NCT03684785 | Phase I/II/recruiting | |
| PD-1 | Cemiplimab In conjunction with Everolimus/Sirolimus/Prednisone | Advanced cSCC in participants who have previously received an allogeneic hematopoietic stem cell transplant or kidney transplant | NCT04339062 | Recruiting | |
| PD-1 | Cemiplimab | High-risk localized/locally recurrent/resectable cutaneous cSCC | NCT04315701 | Recruiting | |
| PD-1 EGFR | Cemiplimab Pembrolizumab ASP-1929 (Anti-EGFR Photoimmunotherap) | Recurrent or metastatic head and neck SCC or advanced or metastatic cSCC in EGFR-expressing tumours | NCT04305795 | Recruiting | |
| PD-1 | Nivolumab | Advanced cSCC | NCT03834233 | Active/not recruiting | |
| PD-1 CTLA-4 | Nivolumab+/− Ipilimumab | Resectable stage III-IVa cSCC | NCT04620200 | Phase II/recruiting | |
| PD-1 | Nivolumab | Locally advanced/metatstatic cSCC | NCT04204837 | Phase II/not recruiting | |
| PD-L1 EGFR | Avelumab+/− Cetuximab | Advanced cSCC | NCT03944941 | Phase II/recruiting | |
| PD-L1 | Avelumab in combination with radiation | Unresectable cSCC | NCT03737721 | Phase II/recruiting | |
| PD-L1 | Atezolimumab Cobimetnib (MEK Inhibitor) | cSCC | NCT03108131 | Active, not Recruiting | |
| PD-L1 | Atezolimumab NT-I7(rhIL-7-Fc) | cSCC | NCT03108131 | Recruiting | |
| PD-L1 | Atezolimumab | cSCC | NCT04710498 | Not yet recruiting | |
| VISTA | CI-8993 | Advanced solid tumour malignancies | NCT04475523 | Recruiting |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wright, Q.; Gonzalez Cruz, J.L.; Wells, J.W.; Leggatt, G.R. PD-1 and beyond to Activate T Cells in Cutaneous Squamous Cell Cancers: The Case for 4-1BB and VISTA Antibodies in Combination Therapy. Cancers 2021, 13, 3310. https://doi.org/10.3390/cancers13133310
Wright Q, Gonzalez Cruz JL, Wells JW, Leggatt GR. PD-1 and beyond to Activate T Cells in Cutaneous Squamous Cell Cancers: The Case for 4-1BB and VISTA Antibodies in Combination Therapy. Cancers. 2021; 13(13):3310. https://doi.org/10.3390/cancers13133310
Chicago/Turabian StyleWright, Quentin, Jazmina L. Gonzalez Cruz, James W. Wells, and Graham R. Leggatt. 2021. "PD-1 and beyond to Activate T Cells in Cutaneous Squamous Cell Cancers: The Case for 4-1BB and VISTA Antibodies in Combination Therapy" Cancers 13, no. 13: 3310. https://doi.org/10.3390/cancers13133310