
Stem Cells in the Exocrine Pancreas during Homeostasis, Injury, and Cancer

 and
and 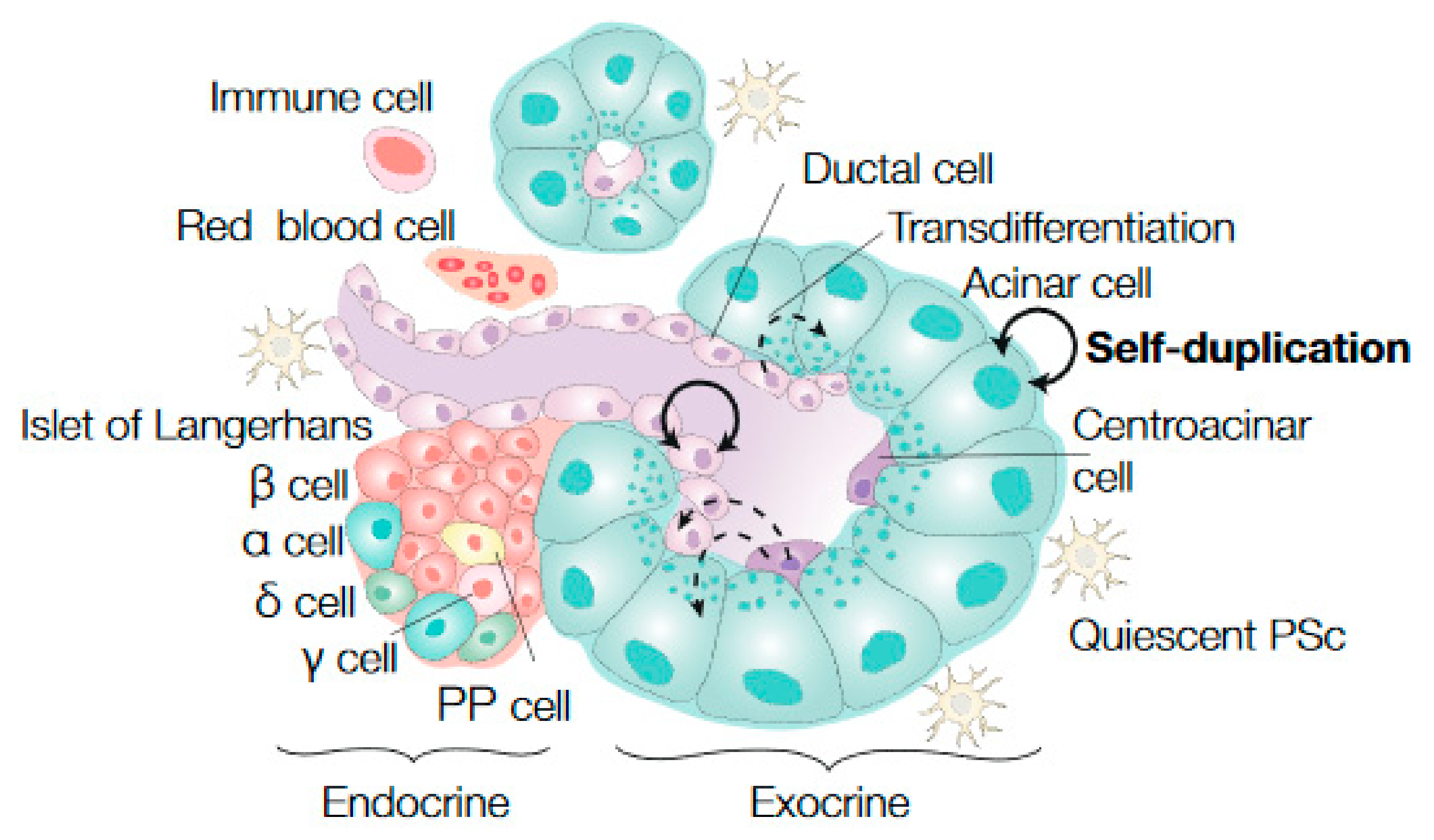{kind=link}
{kind=link}
{kind=link}
Simple Summary
Abstract
1. Introduction
Strategies to Evaluate Stem Cell Qualities
2. Stem Cell Dynamics in Healthy Pancreatic Tissue
3. Pancreatic Stem Cell Dynamics during Regeneration
3.1. Ductal Cells as Progenitor Cells during Regeneration
3.2. Acinar Cell Plasticity during Regeneration
4. Pancreatic Stem Cell Dynamics during Cancer Initiation and Cancer
4.1. The Cell of Origin in Pancreatic Cancer
4.2. Stem Cell Identification and Functionality in PanIN, PDAC, and Metastasis
4.3. Microenvironmental Impact on Stem Cell Functionality in PDAC
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Da Silva Xavier, G. The Cells of the Islets of Langerhans. J. Clin. Med. 2018, 7, 54. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; White, R.A.; Wang, T.C. Adult Pancreatic Acinar Progenitor-like Populations in Regeneration and Cancer. Trends Mol. Med. 2020. [Google Scholar] [CrossRef] [PubMed]
- Redondo, M.J.; Fain, P.R.; Eisenbarth, G.S. Genetics of type 1A diabetes. Recent Prog. Horm. Res. 2001, 56, 69–89. [Google Scholar] [CrossRef] [PubMed]
- Eisenbarth, G.S. Type I diabetes mellitus. A chronic autoimmune disease. N. Engl. J. Med. 1986, 314, 1360–1368. [Google Scholar] [CrossRef] [PubMed]
- Stumvoll, M.; Goldstein, B.J.; van Haeften, T.W. Type 2 diabetes: Principles of pathogenesis and therapy. Lancet 2005, 365, 1333–1346. [Google Scholar] [CrossRef]
- Li, D.; Xie, K.; Wolff, R.; Abbruzzese, J.L. Pancreatic cancer. Lancet 2004, 363, 1049–1057. [Google Scholar] [CrossRef]
- Hidalgo, M. Pancreatic cancer. N. Engl. J. Med. 2010, 362, 1605–1617. [Google Scholar] [CrossRef]
- Mizrahi, J.D.; Surana, R.; Valle, J.W.; Shroff, R.T. Pancreatic cancer. Lancet 2020, 395, 2008–2020. [Google Scholar] [CrossRef]
- Yao, W.; Maitra, A.; Ying, H. Recent insights into the biology of pancreatic cancer. EBioMedicine 2020, 53, 102655. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef]
- Feldmann, G.; Beaty, R.; Hruban, R.H.; Maitra, A. Molecular genetics of pancreatic intraepithelial neoplasia. J. Hepatobiliary Pancreat. Surg. 2007, 14, 224–232. [Google Scholar] [CrossRef]
- Maitra, A.; Hruban, R.H. Pancreatic cancer. Annu. Rev. Pathol. 2008, 3, 157–188. [Google Scholar] [CrossRef]
- Feig, C.; Gopinathan, A.; Neesse, A.; Chan, D.S.; Cook, N.; Tuveson, D.A. The pancreas cancer microenvironment. Clin. Cancer Res. 2012, 18, 4266–4276. [Google Scholar] [CrossRef]
- Hermann, P.C.; Huber, S.L.; Herrler, T.; Aicher, A.; Ellwart, J.W.; Guba, M.; Bruns, C.J.; Heeschen, C. Distinct populations of cancer stem cells determine tumor growth and metastatic activity in human pancreatic cancer. Cell Stem Cell 2007, 1, 313–323. [Google Scholar] [CrossRef]
- Li, C.; Heidt, D.G.; Dalerba, P.; Burant, C.F.; Zhang, L.; Adsay, V.; Wicha, M.; Clarke, M.F.; Simeone, D.M. Identification of pancreatic cancer stem cells. Cancer Res. 2007, 67, 1030–1037. [Google Scholar] [CrossRef]
- Garcia-Silva, S.; Frias-Aldeguer, J.; Heeschen, C. Stem cells & pancreatic cancer. Pancreatology 2013, 13, 110–113. [Google Scholar] [CrossRef]
- Puri, S.; Folias, A.E.; Hebrok, M. Plasticity and dedifferentiation within the pancreas: Development, homeostasis, and disease. Cell Stem Cell 2015, 16, 18–31. [Google Scholar] [CrossRef]
- Eisenberg, L.M.; Eisenberg, C.A. Stem cell plasticity, cell fusion, and transdifferentiation. Birth Defects Res. C Embryo Today 2003, 69, 209–218. [Google Scholar] [CrossRef]
- Weissman, I.L. Stem cells: Units of development, units of regeneration, and units in evolution. Cell 2000, 100, 157–168. [Google Scholar] [CrossRef]
- Morrison, S.J.; Weissman, I.L. The long-term repopulating subset of hematopoietic stem cells is deterministic and isolatable by phenotype. Immunity 1994, 1, 661–673. [Google Scholar] [CrossRef]
- Shackleton, M.; Vaillant, F.; Simpson, K.J.; Stingl, J.; Smyth, G.K.; Asselin-Labat, M.L.; Wu, L.; Lindeman, G.J.; Visvader, J.E. Generation of a functional mammary gland from a single stem cell. Nature 2006, 439, 84–88. [Google Scholar] [CrossRef]
- Stingl, J.; Eirew, P.; Ricketson, I.; Shackleton, M.; Vaillant, F.; Choi, D.; Li, H.I.; Eaves, C.J. Purification and unique properties of mammary epithelial stem cells. Nature 2006, 439, 993–997. [Google Scholar] [CrossRef]
- Cotsarelis, G.; Sun, T.T.; Lavker, R.M. Label-retaining cells reside in the bulge area of pilosebaceous unit: Implications for follicular stem cells, hair cycle, and skin carcinogenesis. Cell 1990, 61, 1329–1337. [Google Scholar] [CrossRef]
- Tumbar, T.; Guasch, G.; Greco, V.; Blanpain, C.; Lowry, W.E.; Rendl, M.; Fuchs, E. Defining the epithelial stem cell niche in skin. Science 2004, 303, 359–363. [Google Scholar] [CrossRef]
- Morris, R.J.; Liu, Y.; Marles, L.; Yang, Z.; Trempus, C.; Li, S.; Lin, J.S.; Sawicki, J.A.; Cotsarelis, G. Capturing and profiling adult hair follicle stem cells. Nat. Biotechnol. 2004, 22, 411–417. [Google Scholar] [CrossRef]
- Barker, N.; van Es, J.H.; Kuipers, J.; Kujala, P.; van den Born, M.; Cozijnsen, M.; Haegebarth, A.; Korving, J.; Begthel, H.; Peters, P.J.; et al. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature 2007, 449, 1003–1007. [Google Scholar] [CrossRef]
- Kong, B.; Michalski, C.W.; Erkan, M.; Friess, H.; Kleeff, J. From tissue turnover to the cell of origin for pancreatic cancer. Nat. Rev. Gastroenterol. Hepatol. 2011, 8, 467–472. [Google Scholar] [CrossRef]
- Stanger, B.Z.; Hebrok, M. Control of cell identity in pancreas development and regeneration. Gastroenterology 2013, 144, 1170–1179. [Google Scholar] [CrossRef]
- Ziv, O.; Glaser, B.; Dor, Y. The plastic pancreas. Dev. Cell 2013, 26, 3–7. [Google Scholar] [CrossRef]
- Vermeulen, L.; De Sousa, E.M.F.; van der Heijden, M.; Cameron, K.; de Jong, J.H.; Borovski, T.; Tuynman, J.B.; Todaro, M.; Merz, C.; Rodermond, H.; et al. Wnt activity defines colon cancer stem cells and is regulated by the microenvironment. Nat. Cell Biol. 2010, 12, 468–476. [Google Scholar] [CrossRef]
- Huch, M.; Dorrell, C.; Boj, S.F.; van Es, J.H.; Li, V.S.; van de Wetering, M.; Sato, T.; Hamer, K.; Sasaki, N.; Finegold, M.J.; et al. In vitro expansion of single Lgr5+ liver stem cells induced by Wnt-driven regeneration. Nature 2013, 494, 247–250. [Google Scholar] [CrossRef] [PubMed]
- Vermeulen, L.; Snippert, H.J. Stem cell dynamics in homeostasis and cancer of the intestine. Nat. Rev. Cancer 2014, 14, 468–480. [Google Scholar] [CrossRef] [PubMed]
- Zaret, K.S.; Grompe, M. Generation and regeneration of cells of the liver and pancreas. Science 2008, 322, 1490–1494. [Google Scholar] [CrossRef] [PubMed]
- Puri, S.; Hebrok, M. Cellular plasticity within the pancreas--lessons learned from development. Dev. Cell 2010, 18, 342–356. [Google Scholar] [CrossRef] [PubMed]
- Gu, G.; Dubauskaite, J.; Melton, D.A. Direct evidence for the pancreatic lineage: NGN3+ cells are islet progenitors and are distinct from duct progenitors. Development 2002, 129, 2447–2457. [Google Scholar] [CrossRef]
- Zhou, Q.; Law, A.C.; Rajagopal, J.; Anderson, W.J.; Gray, P.A.; Melton, D.A. A multipotent progenitor domain guides pancreatic organogenesis. Dev. Cell 2007, 13, 103–114. [Google Scholar] [CrossRef]
- Stanger, B.Z.; Tanaka, A.J.; Melton, D.A. Organ size is limited by the number of embryonic progenitor cells in the pancreas but not the liver. Nature 2007, 445, 886–891. [Google Scholar] [CrossRef]
- Solar, M.; Cardalda, C.; Houbracken, I.; Martin, M.; Maestro, M.A.; De Medts, N.; Xu, X.; Grau, V.; Heimberg, H.; Bouwens, L.; et al. Pancreatic exocrine duct cells give rise to insulin-producing beta cells during embryogenesis but not after birth. Dev. Cell 2009, 17, 849–860. [Google Scholar] [CrossRef]
- Seymour, P.A.; Freude, K.K.; Tran, M.N.; Mayes, E.E.; Jensen, J.; Kist, R.; Scherer, G.; Sander, M. SOX9 is required for maintenance of the pancreatic progenitor cell pool. Proc. Natl. Acad. Sci. USA 2007, 104, 1865–1870. [Google Scholar] [CrossRef]
- Schaffer, A.E.; Freude, K.K.; Nelson, S.B.; Sander, M. Nkx6 transcription factors and Ptf1a function as antagonistic lineage determinants in multipotent pancreatic progenitors. Dev. Cell 2010, 18, 1022–1029. [Google Scholar] [CrossRef]
- Kopp, J.L.; Dubois, C.L.; Schaffer, A.E.; Hao, E.; Shih, H.P.; Seymour, P.A.; Ma, J.; Sander, M. Sox9+ ductal cells are multipotent progenitors throughout development but do not produce new endocrine cells in the normal or injured adult pancreas. Development 2011, 138, 653–665. [Google Scholar] [CrossRef]
- Larsen, H.L.; Martin-Coll, L.; Nielsen, A.V.; Wright, C.V.E.; Trusina, A.; Kim, Y.H.; Grapin-Botton, A. Stochastic priming and spatial cues orchestrate heterogeneous clonal contribution to mouse pancreas organogenesis. Nat. Commun. 2017, 8, 605. [Google Scholar] [CrossRef]
- Sznurkowska, M.K.; Hannezo, E.; Azzarelli, R.; Rulands, S.; Nestorowa, S.; Hindley, C.J.; Nichols, J.; Gottgens, B.; Huch, M.; Philpott, A.; et al. Defining Lineage Potential and Fate Behavior of Precursors during Pancreas Development. Dev. Cell 2018, 46, 360–375. [Google Scholar] [CrossRef]
- Liu, Z.; Sneddon, J.B. Pancreatic development: One cell at a (pseudo)time. EMBO J. 2019, 38. [Google Scholar] [CrossRef]
- Byrnes, L.E.; Wong, D.M.; Subramaniam, M.; Meyer, N.P.; Gilchrist, C.L.; Knox, S.M.; Tward, A.D.; Chun, J.Y.; Sneddon, J.B. Lineage dynamics of murine pancreatic development at single-cell resolution. Nat. Commun. 2018, 9, 1–17. [Google Scholar] [CrossRef]
- Liu, J.; Banerjee, A.; Herring, C.A.; Attalla, J.; Hu, R.; Xu, Y.; Shao, Q.; Simmons, A.J.; Dadi, P.K.; Wang, S.; et al. Neurog3-Independent Methylation Is the Earliest Detectable Mark Distinguishing Pancreatic Progenitor Identity. Dev. Cell 2019, 48, 49–63. [Google Scholar] [CrossRef]
- Stanescu, D.E.; Yu, R.; Won, K.-J.; Stoffers, D.A. Single cell transcriptomic profiling of mouse pancreatic progenitors. Physiol. Genom. 2017, 49, 105–114. [Google Scholar] [CrossRef]
- Yu, X.X.; Qiu, W.L.; Yang, L.; Zhang, Y.; He, M.Y.; Li, L.C.; Xu, C.R. Defining multistep cell fate decision pathways during pancreatic development at single-cell resolution. EMBO J. 2019, 38. [Google Scholar] [CrossRef]
- Houbracken, I.; Bouwens, L. Acinar cells in the neonatal pancreas grow by self-duplication and not by neogenesis from duct cells. Sci Rep. 2017, 7, 12643. [Google Scholar] [CrossRef]
- Sangiorgi, E.; Capecchi, M.R. Bmi1 lineage tracing identifies a self-renewing pancreatic acinar cell subpopulation capable of maintaining pancreatic organ homeostasis. Proc. Natl. Acad. Sci. USA 2009, 106, 7101–7106. [Google Scholar] [CrossRef]
- Wollny, D.; Zhao, S.; Everlien, I.; Lun, X.; Brunken, J.; Brune, D.; Ziebell, F.; Tabansky, I.; Weichert, W.; Marciniak-Czochra, A.; et al. Single-Cell Analysis Uncovers Clonal Acinar Cell Heterogeneity in the Adult Pancreas. Dev. Cell 2016, 39, 289–301. [Google Scholar] [CrossRef]
- Desai, B.M.; Oliver-Krasinski, J.; De Leon, D.D.; Farzad, C.; Hong, N.; Leach, S.D.; Stoffers, D.A. Preexisting pancreatic acinar cells contribute to acinar cell, but not islet beta cell, regeneration. J. Clin. Investig. 2007, 117, 971–977. [Google Scholar] [CrossRef]
- Strobel, O.; Dor, Y.; Alsina, J.; Stirman, A.; Lauwers, G.; Trainor, A.; Castillo, C.F.; Warshaw, A.L.; Thayer, S.P. In vivo lineage tracing defines the role of acinar-to-ductal transdifferentiation in inflammatory ductal metaplasia. Gastroenterology 2007, 133, 1999–2009. [Google Scholar] [CrossRef]
- Kopinke, D.; Brailsford, M.; Shea, J.E.; Leavitt, R.; Scaife, C.L.; Murtaugh, L.C. Lineage tracing reveals the dynamic contribution of Hes1+ cells to the developing and adult pancreas. Development 2011, 138, 431–441. [Google Scholar] [CrossRef]
- Furuyama, K.; Kawaguchi, Y.; Akiyama, H.; Horiguchi, M.; Kodama, S.; Kuhara, T.; Hosokawa, S.; Elbahrawy, A.; Soeda, T.; Koizumi, M.; et al. Continuous cell supply from a Sox9-expressing progenitor zone in adult liver, exocrine pancreas and intestine. Nat. Genet. 2011, 43, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Mameishvili, E.; Serafimidis, I.; Iwaszkiewicz, S.; Lesche, M.; Reinhardt, S.; Bolicke, N.; Buttner, M.; Stellas, D.; Papadimitropoulou, A.; Szabolcs, M.; et al. Aldh1b1 expression defines progenitor cells in the adult pancreas and is required for Kras-induced pancreatic cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 20679–20688. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Huang, Y.F.; Kek, C.; Bulavin, D.V. Apoptosis differently affects lineage tracing of Lgr5 and Bmi1 intestinal stem cell populations. Cell Stem Cell 2013, 12, 298–303. [Google Scholar] [CrossRef]
- Huh, W.J.; Khurana, S.S.; Geahlen, J.H.; Kohli, K.; Waller, R.A.; Mills, J.C. Tamoxifen induces rapid, reversible atrophy, and metaplasia in mouse stomach. Gastroenterology 2012, 142, 21–24.e27. [Google Scholar] [CrossRef] [PubMed]
- Ahn, S.H.; Granger, A.; Rankin, M.M.; Lam, C.J.; Cox, A.R.; Kushner, J.A. Tamoxifen suppresses pancreatic beta-cell proliferation in mice. PLoS ONE 2019, 14, e0214829. [Google Scholar] [CrossRef] [PubMed]
- Seaberg, R.M.; Smukler, S.R.; Kieffer, T.J.; Enikolopov, G.; Asghar, Z.; Wheeler, M.B.; Korbutt, G.; van der Kooy, D. Clonal identification of multipotent precursors from adult mouse pancreas that generate neural and pancreatic lineages. Nat. Biotechnol. 2004, 22, 1115–1124. [Google Scholar] [CrossRef]
- Smukler, S.R.; Arntfield, M.E.; Razavi, R.; Bikopoulos, G.; Karpowicz, P.; Seaberg, R.; Dai, F.; Lee, S.; Ahrens, R.; Fraser, P.E.; et al. The adult mouse and human pancreas contain rare multipotent stem cells that express insulin. Cell Stem Cell 2011, 8, 281–293. [Google Scholar] [CrossRef]
- Rovira, M.; Scott, S.G.; Liss, A.S.; Jensen, J.; Thayer, S.P.; Leach, S.D. Isolation and characterization of centroacinar/terminal ductal progenitor cells in adult mouse pancreas. Proc. Natl. Acad. Sci. USA 2010, 107, 75–80. [Google Scholar] [CrossRef]
- Zulewski, H.; Abraham, E.J.; Gerlach, M.J.; Daniel, P.B.; Moritz, W.; Muller, B.; Vallejo, M.; Thomas, M.K.; Habener, J.F. Multipotential nestin-positive stem cells isolated from adult pancreatic islets differentiate ex vivo into pancreatic endocrine, exocrine, and hepatic phenotypes. Diabetes 2001, 50, 521–533. [Google Scholar] [CrossRef]
- Huch, M.; Bonfanti, P.; Boj, S.F.; Sato, T.; Loomans, C.J.; van de Wetering, M.; Sojoodi, M.; Li, V.S.; Schuijers, J.; Gracanin, A.; et al. Unlimited in vitro expansion of adult bi-potent pancreas progenitors through the Lgr5/R-spondin axis. EMBO J. 2013, 32, 2708–2721. [Google Scholar] [CrossRef]
- Jin, L.; Feng, T.; Shih, H.P.; Zerda, R.; Luo, A.; Hsu, J.; Mahdavi, A.; Sander, M.; Tirrell, D.A.; Riggs, A.D.; et al. Colony-forming cells in the adult mouse pancreas are expandable in Matrigel and form endocrine/acinar colonies in laminin hydrogel. Proc. Natl. Acad. Sci. USA 2013, 110, 3907–3912. [Google Scholar] [CrossRef]
- Loomans, C.J.M.; Williams Giuliani, N.; Balak, J.; Ringnalda, F.; van Gurp, L.; Huch, M.; Boj, S.F.; Sato, T.; Kester, L.; de Sousa Lopes, S.M.C.; et al. Expansion of Adult Human Pancreatic Tissue Yields Organoids Harboring Progenitor Cells with Endocrine Differentiation Potential. Stem Cell Rep. 2018, 10, 712–724. [Google Scholar] [CrossRef]
- Fukuda, A.; Wang, S.C.; Morris, J.P., 4th; Folias, A.E.; Liou, A.; Kim, G.E.; Akira, S.; Boucher, K.M.; Firpo, M.A.; Mulvihill, S.J.; et al. Stat3 and MMP7 contribute to pancreatic ductal adenocarcinoma initiation and progression. Cancer Cell 2011, 19, 441–455. [Google Scholar] [CrossRef]
- Nagashio, Y.; Ueno, H.; Imamura, M.; Asaumi, H.; Watanabe, S.; Yamaguchi, T.; Taguchi, M.; Tashiro, M.; Otsuki, M. Inhibition of transforming growth factor beta decreases pancreatic fibrosis and protects the pancreas against chronic injury in mice. Lab. Investig. 2004, 84, 1610–1618. [Google Scholar] [CrossRef]
- Yanger, K.; Stanger, B.Z. Facultative stem cells in liver and pancreas: Fact and fancy. Dev. Dyn. 2011, 240, 521–529. [Google Scholar] [CrossRef]
- Gu, G.; Brown, J.R.; Melton, D.A. Direct lineage tracing reveals the ontogeny of pancreatic cell fates during mouse embryogenesis. Mech. Dev. 2003, 120, 35–43. [Google Scholar] [CrossRef]
- Sharma, A.; Zangen, D.H.; Reitz, P.; Taneja, M.; Lissauer, M.E.; Miller, C.P.; Weir, G.C.; Habener, J.F.; Bonner-Weir, S. The homeodomain protein IDX-1 increases after an early burst of proliferation during pancreatic regeneration. Diabetes 1999, 48, 507–513. [Google Scholar] [CrossRef]
- Xu, X.; D’Hoker, J.; Stange, G.; Bonne, S.; De Leu, N.; Xiao, X.; Van de Casteele, M.; Mellitzer, G.; Ling, Z.; Pipeleers, D.; et al. Beta cells can be generated from endogenous progenitors in injured adult mouse pancreas. Cell 2008, 132, 197–207. [Google Scholar] [CrossRef]
- Jensen, J.N.; Cameron, E.; Garay, M.V.; Starkey, T.W.; Gianani, R.; Jensen, J. Recapitulation of elements of embryonic development in adult mouse pancreatic regeneration. Gastroenterology 2005, 128, 728–741. [Google Scholar] [CrossRef] [PubMed]
- Criscimanna, A.; Speicher, J.A.; Houshmand, G.; Shiota, C.; Prasadan, K.; Ji, B.; Logsdon, C.D.; Gittes, G.K.; Esni, F. Duct cells contribute to regeneration of endocrine and acinar cells following pancreatic damage in adult mice. Gastroenterology 2011, 141, 1451–1462. [Google Scholar] [CrossRef] [PubMed]
- Pasca di Magliano, M.; Biankin, A.V.; Heiser, P.W.; Cano, D.A.; Gutierrez, P.J.; Deramaudt, T.; Segara, D.; Dawson, A.C.; Kench, J.G.; Henshall, S.M.; et al. Common activation of canonical Wnt signaling in pancreatic adenocarcinoma. PLoS ONE 2007, 2, e1155. [Google Scholar] [CrossRef] [PubMed]
- Murtaugh, L.C.; Law, A.C.; Dor, Y.; Melton, D.A. Beta-catenin is essential for pancreatic acinar but not islet development. Development 2005, 132, 4663–4674. [Google Scholar] [CrossRef]
- Heiser, P.W.; Lau, J.; Taketo, M.M.; Herrera, P.L.; Hebrok, M. Stabilization of beta-catenin impacts pancreas growth. Development 2006, 133, 2023–2032. [Google Scholar] [CrossRef]
- Kopp, J.L.; Dubois, C.L.; Hao, E.; Thorel, F.; Herrera, P.L.; Sander, M. Progenitor cell domains in the developing and adult pancreas. Cell Cycle 2011, 10, 1921–1927. [Google Scholar] [CrossRef]
- Wang, R.N.; Kloppel, G.; Bouwens, L. Duct- to islet-cell differentiation and islet growth in the pancreas of duct-ligated adult rats. Diabetologia 1995, 38, 1405–1411. [Google Scholar] [CrossRef]
- Inada, A.; Nienaber, C.; Katsuta, H.; Fujitani, Y.; Levine, J.; Morita, R.; Sharma, A.; Bonner-Weir, S. Carbonic anhydrase II-positive pancreatic cells are progenitors for both endocrine and exocrine pancreas after birth. Proc. Natl. Acad. Sci. USA 2008, 105, 19915–19919. [Google Scholar] [CrossRef]
- Van de Casteele, M.; Leuckx, G.; Baeyens, L.; Cai, Y.; Yuchi, Y.; Coppens, V.; De Groef, S.; Eriksson, M.; Svensson, C.; Ahlgren, U.; et al. Neurogenin 3+ cells contribute to beta-cell neogenesis and proliferation in injured adult mouse pancreas. Cell Death Dis. 2013, 4, e523. [Google Scholar] [CrossRef]
- Van de Casteele, M.; Leuckx, G.; Cai, Y.; Yuchi, Y.; Coppens, V.; De Groef, S.; Van Gassen, N.; Baeyens, L.; Heremans, Y.; Wright, C.V.; et al. Partial duct ligation: Beta-cell proliferation and beyond. Diabetes 2014, 63, 2567–2577. [Google Scholar] [CrossRef]
- Sancho, R.; Gruber, R.; Gu, G.; Behrens, A. Loss of Fbw7 reprograms adult pancreatic ductal cells into alpha, delta, and beta cells. Cell Stem Cell 2014, 15, 139–153. [Google Scholar] [CrossRef]
- Strobel, O.; Rosow, D.E.; Rakhlin, E.Y.; Lauwers, G.Y.; Trainor, A.G.; Alsina, J.; Fernandez-Del Castillo, C.; Warshaw, A.L.; Thayer, S.P. Pancreatic duct glands are distinct ductal compartments that react to chronic injury and mediate Shh-induced metaplasia. Gastroenterology 2010, 138, 1166–1177. [Google Scholar] [CrossRef]
- Yamaguchi, J.; Liss, A.S.; Sontheimer, A.; Mino-Kenudson, M.; Castillo, C.F.; Warshaw, A.L.; Thayer, S.P. Pancreatic duct glands (PDGs) are a progenitor compartment responsible for pancreatic ductal epithelial repair. Stem Cell Res. 2015, 15, 190–202. [Google Scholar] [CrossRef]
- Morris, J.P., 4th; Cano, D.A.; Sekine, S.; Wang, S.C.; Hebrok, M. Beta-catenin blocks Kras-dependent reprogramming of acini into pancreatic cancer precursor lesions in mice. J. Clin. Investig. 2010, 120, 508–520. [Google Scholar] [CrossRef]
- Siveke, J.T.; Lubeseder-Martellato, C.; Lee, M.; Mazur, P.K.; Nakhai, H.; Radtke, F.; Schmid, R.M. Notch signaling is required for exocrine regeneration after acute pancreatitis. Gastroenterology 2008, 134, 544–555. [Google Scholar] [CrossRef]
- Pan, F.C.; Bankaitis, E.D.; Boyer, D.; Xu, X.; Van de Casteele, M.; Magnuson, M.A.; Heimberg, H.; Wright, C.V. Spatiotemporal patterns of multipotentiality in Ptf1a-expressing cells during pancreas organogenesis and injury-induced facultative restoration. Development 2013, 140, 751–764. [Google Scholar] [CrossRef]
- Folias, A.E.; Penaranda, C.; Su, A.L.; Bluestone, J.A.; Hebrok, M. Aberrant innate immune activation following tissue injury impairs pancreatic regeneration. PLoS ONE 2014, 9, e102125. [Google Scholar] [CrossRef]
- Westphalen, C.B.; Takemoto, Y.; Tanaka, T.; Macchini, M.; Jiang, Z.; Renz, B.W.; Chen, X.; Ormanns, S.; Nagar, K.; Tailor, Y.; et al. Dclk1 Defines Quiescent Pancreatic Progenitors that Promote Injury-Induced Regeneration and Tumorigenesis. Cell Stem Cell 2016, 18, 441–455. [Google Scholar] [CrossRef]
- Zhou, Q.; Brown, J.; Kanarek, A.; Rajagopal, J.; Melton, D.A. In vivo reprogramming of adult pancreatic exocrine cells to beta-cells. Nature 2008, 455, 627–632. [Google Scholar] [CrossRef] [PubMed]
- Blaine, S.A.; Ray, K.C.; Anunobi, R.; Gannon, M.A.; Washington, M.K.; Means, A.L. Adult pancreatic acinar cells give rise to ducts but not endocrine cells in response to growth factor signaling. Development 2010, 137, 2289–2296. [Google Scholar] [CrossRef] [PubMed]
- Roy, N.; Hebrok, M. Regulation of Cellular Identity in Cancer. Dev. Cell 2015, 35, 674–684. [Google Scholar] [CrossRef] [PubMed]
- Matthaei, H.; Schulick, R.D.; Hruban, R.H.; Maitra, A. Cystic precursors to invasive pancreatic cancer. Nat. Rev. Gastroenterol. Hepatol. 2011, 8, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Hingorani, S.R.; Wang, L.; Multani, A.S.; Combs, C.; Deramaudt, T.B.; Hruban, R.H.; Rustgi, A.K.; Chang, S.; Tuveson, D.A. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell 2005, 7, 469–483. [Google Scholar] [CrossRef]
- Guerra, C.; Schuhmacher, A.J.; Canamero, M.; Grippo, P.J.; Verdaguer, L.; Perez-Gallego, L.; Dubus, P.; Sandgren, E.P.; Barbacid, M. Chronic pancreatitis is essential for induction of pancreatic ductal adenocarcinoma by K-Ras oncogenes in adult mice. Cancer Cell 2007, 11, 291–302. [Google Scholar] [CrossRef]
- Bardeesy, N.; Aguirre, A.J.; Chu, G.C.; Cheng, K.H.; Lopez, L.V.; Hezel, A.F.; Feng, B.; Brennan, C.; Weissleder, R.; Mahmood, U.; et al. Both p16(Ink4a) and the p19(Arf)-p53 pathway constrain progression of pancreatic adenocarcinoma in the mouse. Proc. Natl. Acad. Sci. USA 2006, 103, 5947–5952. [Google Scholar] [CrossRef]
- Carriere, C.; Seeley, E.S.; Goetze, T.; Longnecker, D.S.; Korc, M. The Nestin progenitor lineage is the compartment of origin for pancreatic intraepithelial neoplasia. Proc. Natl. Acad. Sci. USA 2007, 104, 4437–4442. [Google Scholar] [CrossRef]
- De La, O.J.; Emerson, L.L.; Goodman, J.L.; Froebe, S.C.; Illum, B.E.; Curtis, A.B.; Murtaugh, L.C. Notch and Kras reprogram pancreatic acinar cells to ductal intraepithelial neoplasia. Proc. Natl. Acad. Sci. USA 2008, 105, 18907–18912. [Google Scholar] [CrossRef]
- Habbe, N.; Shi, G.; Meguid, R.A.; Fendrich, V.; Esni, F.; Chen, H.; Feldmann, G.; Stoffers, D.A.; Konieczny, S.F.; Leach, S.D.; et al. Spontaneous induction of murine pancreatic intraepithelial neoplasia (mPanIN) by acinar cell targeting of oncogenic Kras in adult mice. Proc. Natl. Acad. Sci. USA 2008, 105, 18913–18918. [Google Scholar] [CrossRef]
- Shi, G.; Zhu, L.; Sun, Y.; Bettencourt, R.; Damsz, B.; Hruban, R.H.; Konieczny, S.F. Loss of the acinar-restricted transcription factor Mist1 accelerates Kras-induced pancreatic intraepithelial neoplasia. Gastroenterology 2009, 136, 1368–1378. [Google Scholar] [CrossRef]
- von Figura, G.; Fukuda, A.; Roy, N.; Liku, M.E.; Morris, J.P., 4th; Kim, G.E.; Russ, H.A.; Firpo, M.A.; Mulvihill, S.J.; Dawson, D.W.; et al. The chromatin regulator Brg1 suppresses formation of intraductal papillary mucinous neoplasm and pancreatic ductal adenocarcinoma. Nat. Cell Biol. 2014, 16, 255–267. [Google Scholar] [CrossRef]
- Brembeck, F.H.; Schreiber, F.S.; Deramaudt, T.B.; Craig, L.; Rhoades, B.; Swain, G.; Grippo, P.; Stoffers, D.A.; Silberg, D.G.; Rustgi, A.K. The mutant K-ras oncogene causes pancreatic periductal lymphocytic infiltration and gastric mucous neck cell hyperplasia in transgenic mice. Cancer Res. 2003, 63, 2005–2009. [Google Scholar]
- Roy, N.; Malik, S.; Villanueva, K.E.; Urano, A.; Lu, X.; Von Figura, G.; Seeley, E.S.; Dawson, D.W.; Collisson, E.A.; Hebrok, M. Brg1 promotes both tumor-suppressive and oncogenic activities at distinct stages of pancreatic cancer formation. Genes Dev. 2015, 29, 658–671. [Google Scholar] [CrossRef]
- Kopp, J.L.; von Figura, G.; Mayes, E.; Liu, F.F.; Dubois, C.L.; Morris, J.P., 4th; Pan, F.C.; Akiyama, H.; Wright, C.V.; Jensen, K.; et al. Identification of Sox9-dependent acinar-to-ductal reprogramming as the principal mechanism for initiation of pancreatic ductal adenocarcinoma. Cancer Cell 2012, 22, 737–750. [Google Scholar] [CrossRef]
- Johnson, B.L.; d’Alincourt Salazar, M.; Mackenzie-Dyck, S.; D’Apuzzo, M.; Shih, H.P.; Manuel, E.R.; Diamond, D.J. Desmoplasia and oncogene driven acinar-to-ductal metaplasia are concurrent events during acinar cell-derived pancreatic cancer initiation in young adult mice. PLoS ONE 2019, 14, e0221810. [Google Scholar] [CrossRef]
- Zhang, Y.; Morris, J.P., 4th; Yan, W.; Schofield, H.K.; Gurney, A.; Simeone, D.M.; Millar, S.E.; Hoey, T.; Hebrok, M.; Pasca di Magliano, M. Canonical wnt signaling is required for pancreatic carcinogenesis. Cancer Res. 2013, 73, 4909–4922. [Google Scholar] [CrossRef]
- Thayer, S.P.; di Magliano, M.P.; Heiser, P.W.; Nielsen, C.M.; Roberts, D.J.; Lauwers, G.Y.; Qi, Y.P.; Gysin, S.; Fernandez-del Castillo, C.; Yajnik, V.; et al. Hedgehog is an early and late mediator of pancreatic cancer tumorigenesis. Nature 2003, 425, 851–856. [Google Scholar] [CrossRef]
- Kim, S.K.; Hebrok, M. Intercellular signals regulating pancreas development and function. Genes Dev. 2001, 15, 111–127. [Google Scholar] [CrossRef]
- Olive, K.P.; Jacobetz, M.A.; Davidson, C.J.; Gopinathan, A.; McIntyre, D.; Honess, D.; Madhu, B.; Goldgraben, M.A.; Caldwell, M.E.; Allard, D.; et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science 2009, 324, 1457–1461. [Google Scholar] [CrossRef]
- Feldmann, G.; Habbe, N.; Dhara, S.; Bisht, S.; Alvarez, H.; Fendrich, V.; Beaty, R.; Mullendore, M.; Karikari, C.; Bardeesy, N.; et al. Hedgehog inhibition prolongs survival in a genetically engineered mouse model of pancreatic cancer. Gut 2008, 57, 1420–1430. [Google Scholar] [CrossRef]
- Jones, S.; Zhang, X.; Parsons, D.W.; Lin, J.C.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Kamiyama, H.; Jimeno, A.; et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science 2008, 321, 1801–1806. [Google Scholar] [CrossRef]
- Bailey, J.M.; Alsina, J.; Rasheed, Z.A.; McAllister, F.M.; Fu, Y.Y.; Plentz, R.; Zhang, H.; Pasricha, P.J.; Bardeesy, N.; Matsui, W.; et al. DCLK1 marks a morphologically distinct subpopulation of cells with stem cell properties in preinvasive pancreatic cancer. Gastroenterology 2014, 146, 245–256. [Google Scholar] [CrossRef]
- Olempska, M.; Eisenach, P.A.; Ammerpohl, O.; Ungefroren, H.; Fandrich, F.; Kalthoff, H. Detection of tumor stem cell markers in pancreatic carcinoma cell lines. Hepatobiliary Pancreat. Dis. Int. 2007, 6, 92–97. [Google Scholar]
- Kim, M.P.; Fleming, J.B.; Wang, H.; Abbruzzese, J.L.; Choi, W.; Kopetz, S.; McConkey, D.J.; Evans, D.B.; Gallick, G.E. ALDH activity selectively defines an enhanced tumor-initiating cell population relative to CD133 expression in human pancreatic adenocarcinoma. PLoS ONE 2011, 6, e20636. [Google Scholar] [CrossRef]
- Nomura, A.; Banerjee, S.; Chugh, R.; Dudeja, V.; Yamamoto, M.; Vickers, S.M.; Saluja, A.K. CD133 initiates tumors, induces epithelial-mesenchymal transition and increases metastasis in pancreatic cancer. Oncotarget 2015, 6, 8313–8322. [Google Scholar] [CrossRef]
- Huang, P.; Wang, C.Y.; Gou, S.M.; Wu, H.S.; Liu, T.; Xiong, J.X. Isolation and biological analysis of tumor stem cells from pancreatic adenocarcinoma. World J. Gastroenterol. 2008, 14, 3903–3907. [Google Scholar] [CrossRef]
- Ikenaga, N.; Ohuchida, K.; Mizumoto, K.; Yu, J.; Kayashima, T.; Hayashi, A.; Nakata, K.; Tanaka, M. Characterization of CD24 expression in intraductal papillary mucinous neoplasms and ductal carcinoma of the pancreas. Hum. Pathol. 2010, 41, 1466–1474. [Google Scholar] [CrossRef]
- Hong, S.P.; Wen, J.; Bang, S.; Park, S.; Song, S.Y. CD44-positive cells are responsible for gemcitabine resistance in pancreatic cancer cells. Int. J. Cancer 2009, 125, 2323–2331. [Google Scholar] [CrossRef]
- Li, C.; Wu, J.J.; Hynes, M.; Dosch, J.; Sarkar, B.; Welling, T.H.; Pasca di Magliano, M.; Simeone, D.M. c-Met is a marker of pancreatic cancer stem cells and therapeutic target. Gastroenterology 2011, 141, 2218–2227. [Google Scholar] [CrossRef]
- Kawamoto, M.; Ishiwata, T.; Cho, K.; Uchida, E.; Korc, M.; Naito, Z.; Tajiri, T. Nestin expression correlates with nerve and retroperitoneal tissue invasion in pancreatic cancer. Hum. Pathol. 2009, 40, 189–198. [Google Scholar] [CrossRef] [PubMed]
- Maruno, T.; Fukuda, A.; Goto, N.; Tsuda, M.; Ikuta, K.; Hiramatsu, Y.; Ogawa, S.; Nakanishi, Y.; Yamaga, Y.; Yoshioka, T.; et al. Visualization of stem cell activity in pancreatic cancer expansion by direct lineage tracing with live imaging. eLife 2021, 10. [Google Scholar] [CrossRef] [PubMed]
- Ball, C.R.; Oppel, F.; Ehrenberg, K.R.; Dubash, T.D.; Dieter, S.M.; Hoffmann, C.M.; Abel, U.; Herbst, F.; Koch, M.; Werner, J.; et al. Succession of transiently active tumor-initiating cell clones in human pancreatic cancer xenografts. EMBO Mol. Med. 2017, 9, 918–932. [Google Scholar] [CrossRef] [PubMed]
- Maddipati, R.; Stanger, B.Z. Pancreatic Cancer Metastases Harbor Evidence of Polyclonality. Cancer Discov. 2015, 5, 1086–1097. [Google Scholar] [CrossRef] [PubMed]
- Yachida, S.; Jones, S.; Bozic, I.; Antal, T.; Leary, R.; Fu, B.; Kamiyama, M.; Hruban, R.H.; Eshleman, J.R.; Nowak, M.A.; et al. Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature 2010, 467, 1114–1117. [Google Scholar] [CrossRef]
- Lenos, K.J.; Miedema, D.M.; Lodestijn, S.C.; Nijman, L.E.; van den Bosch, T.; Romero Ros, X.; Lourenco, F.C.; Lecca, M.C.; van der Heijden, M.; van Neerven, S.M.; et al. Stem cell functionality is microenvironmentally defined during tumour expansion and therapy response in colon cancer. Nat. Cell Biol. 2018. [Google Scholar] [CrossRef]
- Takahashi, K.; Ehata, S.; Koinuma, D.; Morishita, Y.; Soda, M.; Mano, H.; Miyazono, K. Pancreatic tumor microenvironment confers highly malignant properties on pancreatic cancer cells. Oncogene 2018, 37, 2757–2772. [Google Scholar] [CrossRef]
- Whiteside, T.L. The tumor microenvironment and its role in promoting tumor growth. Oncogene 2008, 27, 5904–5912. [Google Scholar] [CrossRef]
- Berchtold, S.; Grunwald, B.; Kruger, A.; Reithmeier, A.; Hahl, T.; Cheng, T.; Feuchtinger, A.; Born, D.; Erkan, M.; Kleeff, J.; et al. Collagen type V promotes the malignant phenotype of pancreatic ductal adenocarcinoma. Cancer Lett. 2015, 356, 721–732. [Google Scholar] [CrossRef]
- Dauer, P.; Nomura, A.; Saluja, A.; Banerjee, S. Microenvironment in determining chemo-resistance in pancreatic cancer: Neighborhood matters. Pancreatology 2017, 17, 7–12. [Google Scholar] [CrossRef]
- van Mackelenbergh, M.G.; Stroes, C.I.; Spijker, R.; van Eijck, C.H.J.; Wilmink, J.W.; Bijlsma, M.F.; van Laarhoven, H.W.M. Clinical Trials Targeting the Stroma in Pancreatic Cancer: A Systematic Review and Meta-Analysis. Cancers 2019, 11, 588. [Google Scholar] [CrossRef]
- Bachem, M.G.; Schunemann, M.; Ramadani, M.; Siech, M.; Beger, H.; Buck, A.; Zhou, S.; Schmid-Kotsas, A.; Adler, G. Pancreatic carcinoma cells induce fibrosis by stimulating proliferation and matrix synthesis of stellate cells. Gastroenterology 2005, 128, 907–921. [Google Scholar] [CrossRef]
- Hwang, R.F.; Moore, T.; Arumugam, T.; Ramachandran, V.; Amos, K.D.; Rivera, A.; Ji, B.; Evans, D.B.; Logsdon, C.D. Cancer-associated stromal fibroblasts promote pancreatic tumor progression. Cancer Res. 2008, 68, 918–926. [Google Scholar] [CrossRef]
- Vonlaufen, A.; Phillips, P.A.; Xu, Z.; Goldstein, D.; Pirola, R.C.; Wilson, J.S.; Apte, M.V. Pancreatic stellate cells and pancreatic cancer cells: An unholy alliance. Cancer Res. 2008, 68, 7707–7710. [Google Scholar] [CrossRef]
- Xu, Z.; Vonlaufen, A.; Phillips, P.A.; Fiala-Beer, E.; Zhang, X.; Yang, L.; Biankin, A.V.; Goldstein, D.; Pirola, R.C.; Wilson, J.S.; et al. Role of pancreatic stellate cells in pancreatic cancer metastasis. Am. J. Pathol. 2010, 177, 2585–2596. [Google Scholar] [CrossRef]
- Kim, E.J.; Sahai, V.; Abel, E.V.; Griffith, K.A.; Greenson, J.K.; Takebe, N.; Khan, G.N.; Blau, J.L.; Craig, R.; Balis, U.G.; et al. Pilot clinical trial of hedgehog pathway inhibitor GDC-0449 (vismodegib) in combination with gemcitabine in patients with metastatic pancreatic adenocarcinoma. Clin. Cancer Res. 2014, 20, 5937–5945. [Google Scholar] [CrossRef]
- Lee, J.J.; Perera, R.M.; Wang, H.; Wu, D.C.; Liu, X.S.; Han, S.; Fitamant, J.; Jones, P.D.; Ghanta, K.S.; Kawano, S.; et al. Stromal response to Hedgehog signaling restrains pancreatic cancer progression. Proc. Natl. Acad. Sci. USA 2014, 111, E3091–E3100. [Google Scholar] [CrossRef]
- Ozdemir, B.C.; Pentcheva-Hoang, T.; Carstens, J.L.; Zheng, X.; Wu, C.C.; Simpson, T.R.; Laklai, H.; Sugimoto, H.; Kahlert, C.; Novitskiy, S.V.; et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell 2014, 25, 719–734. [Google Scholar] [CrossRef]
- Tian, H.; Callahan, C.A.; DuPree, K.J.; Darbonne, W.C.; Ahn, C.P.; Scales, S.J.; de Sauvage, F.J. Hedgehog signaling is restricted to the stromal compartment during pancreatic carcinogenesis. Proc. Natl. Acad. Sci. USA 2009, 106, 4254–4259. [Google Scholar] [CrossRef]
- Rhim, A.D.; Oberstein, P.E.; Thomas, D.H.; Mirek, E.T.; Palermo, C.F.; Sastra, S.A.; Dekleva, E.N.; Saunders, T.; Becerra, C.P.; Tattersall, I.W.; et al. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell 2014, 25, 735–747. [Google Scholar] [CrossRef]
- Lonardo, E.; Frias-Aldeguer, J.; Hermann, P.C.; Heeschen, C. Pancreatic stellate cells form a niche for cancer stem cells and promote their self-renewal and invasiveness. Cell Cycle 2012, 11, 1282–1290. [Google Scholar] [CrossRef]
- Hamada, S.; Masamune, A.; Takikawa, T.; Suzuki, N.; Kikuta, K.; Hirota, M.; Hamada, H.; Kobune, M.; Satoh, K.; Shimosegawa, T. Pancreatic stellate cells enhance stem cell-like phenotypes in pancreatic cancer cells. Biochem. Biophys. Res. Commun. 2012, 421, 349–354. [Google Scholar] [CrossRef]
- He, W.; Wu, J.; Shi, J.; Huo, Y.M.; Dai, W.; Geng, J.; Lu, P.; Yang, M.W.; Fang, Y.; Wang, W.; et al. IL-22RA1/ STAT3 signaling promotes stemness and tumorigenicity in pancreatic cancer. Cancer Res. 2018. [Google Scholar] [CrossRef]
- Mitchem, J.B.; Brennan, D.J.; Knolhoff, B.L.; Belt, B.A.; Zhu, Y.; Sanford, D.E.; Belaygorod, L.; Carpenter, D.; Collins, L.; Piwnica-Worms, D.; et al. Targeting tumor-infiltrating macrophages decreases tumor-initiating cells, relieves immunosuppression, and improves chemotherapeutic responses. Cancer Res. 2013, 73, 1128–1141. [Google Scholar] [CrossRef]
- Russano, M.; Napolitano, A.; Ribelli, G.; Iuliani, M.; Simonetti, S.; Citarella, F.; Pantano, F.; Dell’Aquila, E.; Anesi, C.; Silvestris, N.; et al. Liquid biopsy and tumor heterogeneity in metastatic solid tumors: The potentiality of blood samples. J. Exp. Clin. Cancer Res. 2020, 39, 95. [Google Scholar] [CrossRef]
- Hopkins, A.C.; Yarchoan, M.; Durham, J.N.; Yusko, E.C.; Rytlewski, J.A.; Robins, H.S.; Laheru, D.A.; Le, D.T.; Lutz, E.R.; Jaffee, E.M. T cell receptor repertoire features associated with survival in immunotherapy-treated pancreatic ductal adenocarcinoma. JCI Insight 2018, 3. [Google Scholar] [CrossRef]
- Ohlund, D.; Handly-Santana, A.; Biffi, G.; Elyada, E.; Almeida, A.S.; Ponz-Sarvise, M.; Corbo, V.; Oni, T.E.; Hearn, S.A.; Lee, E.J.; et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J. Exp. Med. 2017, 214, 579–596. [Google Scholar] [CrossRef] [PubMed]
- Steele, N.G.; Biffi, G.; Kemp, S.B.; Zhang, Y.; Drouillard, D.; Syu, L.; Hao, Y.; Oni, T.E.; Brosnan, E.; Elyada, E.; et al. Inhibition of Hedgehog Signaling Alters Fibroblast Composition in Pancreatic Cancer. Clin. Cancer Res. 2021. [Google Scholar] [CrossRef] [PubMed]
- Biffi, G.; Oni, T.E.; Spielman, B.; Hao, Y.; Elyada, E.; Park, Y.; Preall, J.; Tuveson, D.A. IL1-Induced JAK/STAT Signaling Is Antagonized by TGFbeta to Shape CAF Heterogeneity in Pancreatic Ductal Adenocarcinoma. Cancer Discov. 2019, 9, 282–301. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Ebeling, M.C.; Chauhan, N.; Thompson, P.A.; Gara, R.K.; Ganju, A.; Yallapu, M.M.; Behrman, S.W.; Zhao, H.; Zafar, N.; et al. Ormeloxifene suppresses desmoplasia and enhances sensitivity of gemcitabine in pancreatic cancer. Cancer Res. 2015, 75, 2292–2304. [Google Scholar] [CrossRef]
- Banerjee, S.; Modi, S.; McGinn, O.; Zhao, X.; Dudeja, V.; Ramakrishnan, S.; Saluja, A.K. Impaired Synthesis of Stromal Components in Response to Minnelide Improves Vascular Function, Drug Delivery, and Survival in Pancreatic Cancer. Clin. Cancer Res. 2016, 22, 415–425. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lodestijn, S.C.; van Neerven, S.M.; Vermeulen, L.; Bijlsma, M.F. Stem Cells in the Exocrine Pancreas during Homeostasis, Injury, and Cancer. Cancers 2021, 13, 3295. https://doi.org/10.3390/cancers13133295
Lodestijn SC, van Neerven SM, Vermeulen L, Bijlsma MF. Stem Cells in the Exocrine Pancreas during Homeostasis, Injury, and Cancer. Cancers. 2021; 13(13):3295. https://doi.org/10.3390/cancers13133295
Chicago/Turabian StyleLodestijn, Sophie C., Sanne M. van Neerven, Louis Vermeulen, and Maarten F. Bijlsma. 2021. "Stem Cells in the Exocrine Pancreas during Homeostasis, Injury, and Cancer" Cancers 13, no. 13: 3295. https://doi.org/10.3390/cancers13133295
APA StyleLodestijn, S. C., van Neerven, S. M., Vermeulen, L., & Bijlsma, M. F. (2021). Stem Cells in the Exocrine Pancreas during Homeostasis, Injury, and Cancer. Cancers, 13(13), 3295. https://doi.org/10.3390/cancers13133295