
The Sex-Related Interplay between TME and Cancer: On the Critical Role of Estrogen, MicroRNAs and Autophagy

 ,
, 
Abstract
Simple Summary
Abstract
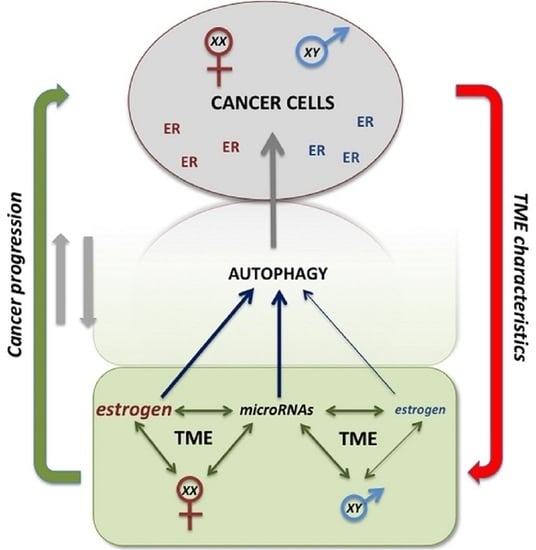
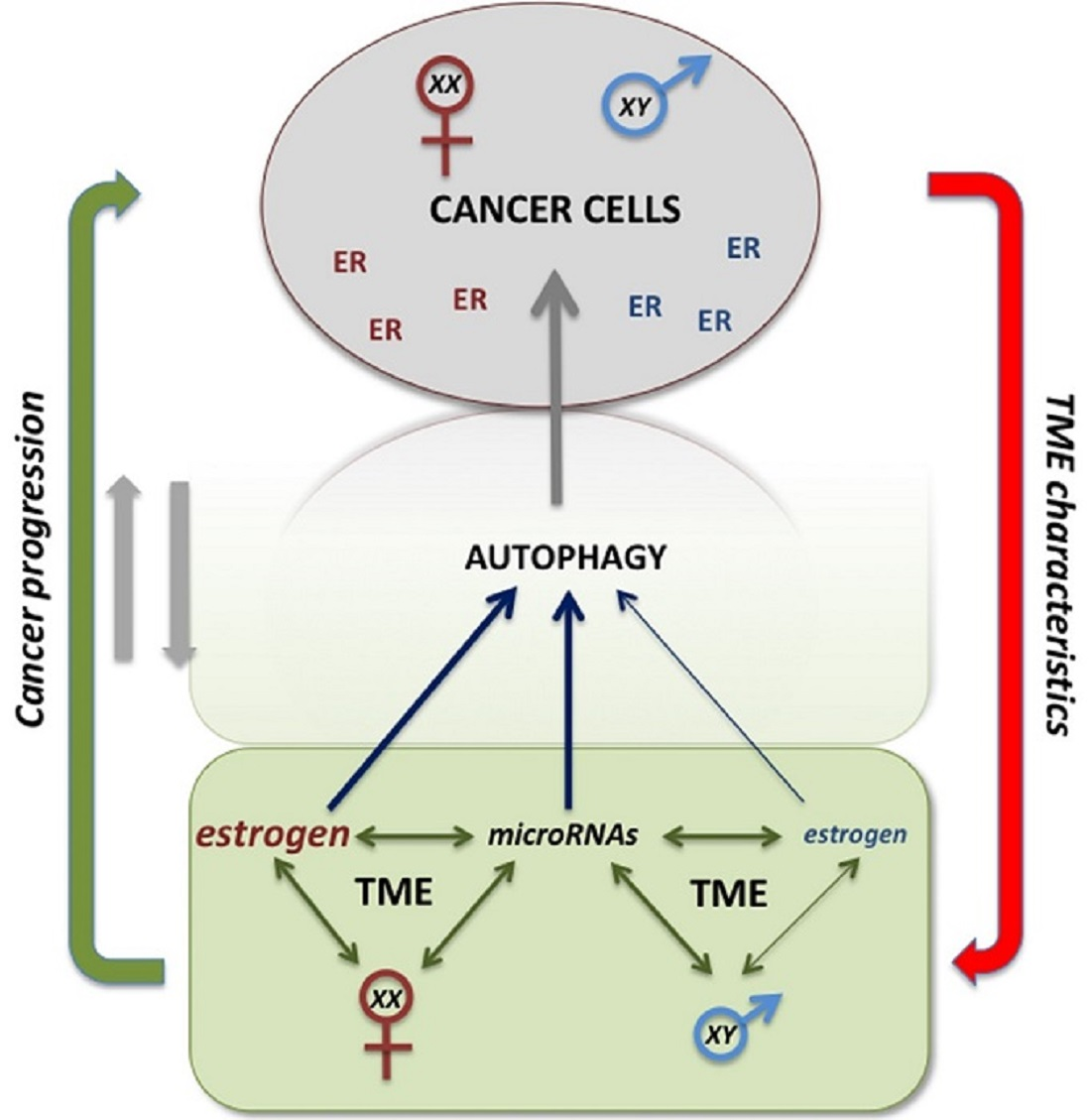
1. Introduction
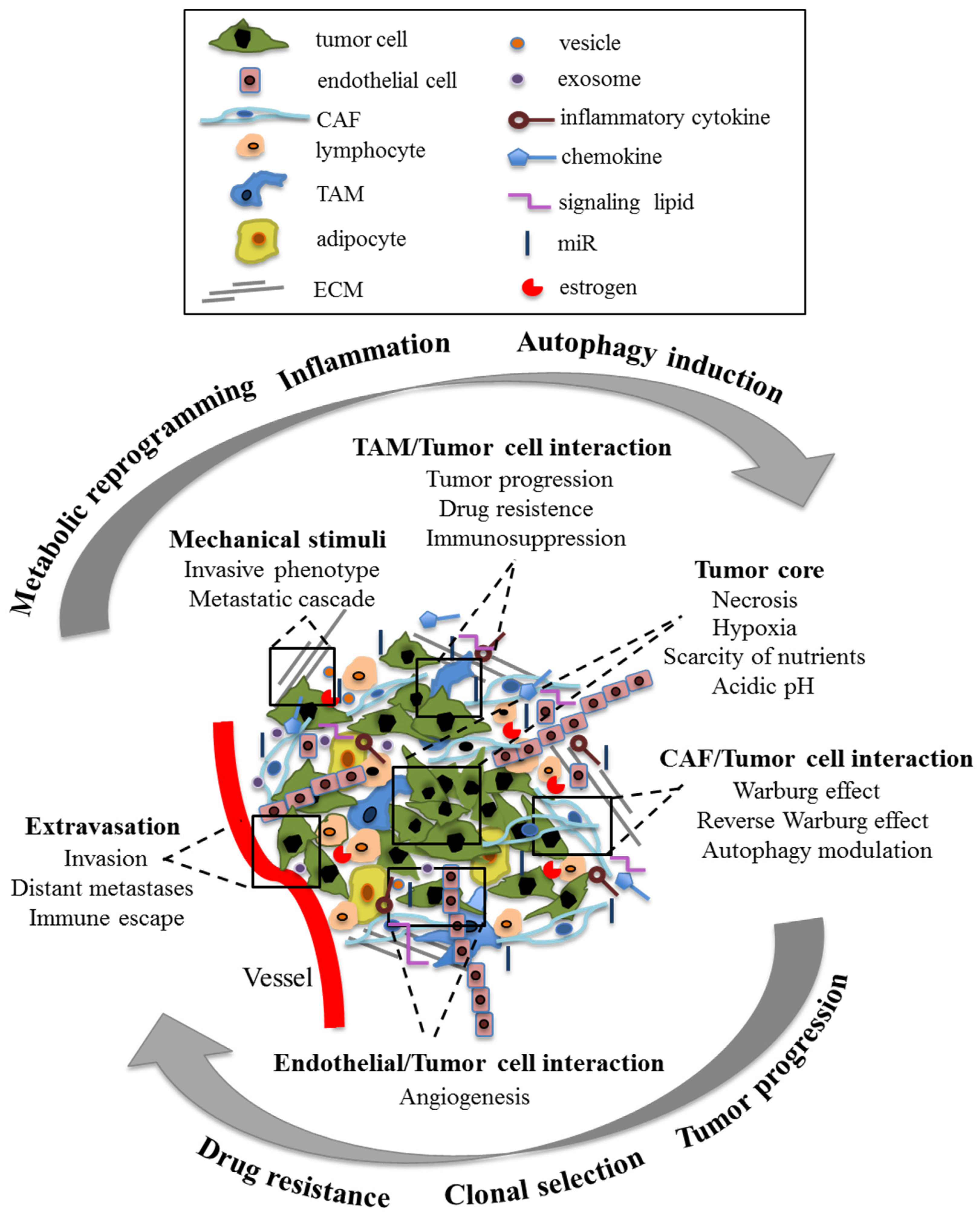
1.1. Tumor Microenvironment
1.2. Autophagy
1.3. Sex Differences in Cancer
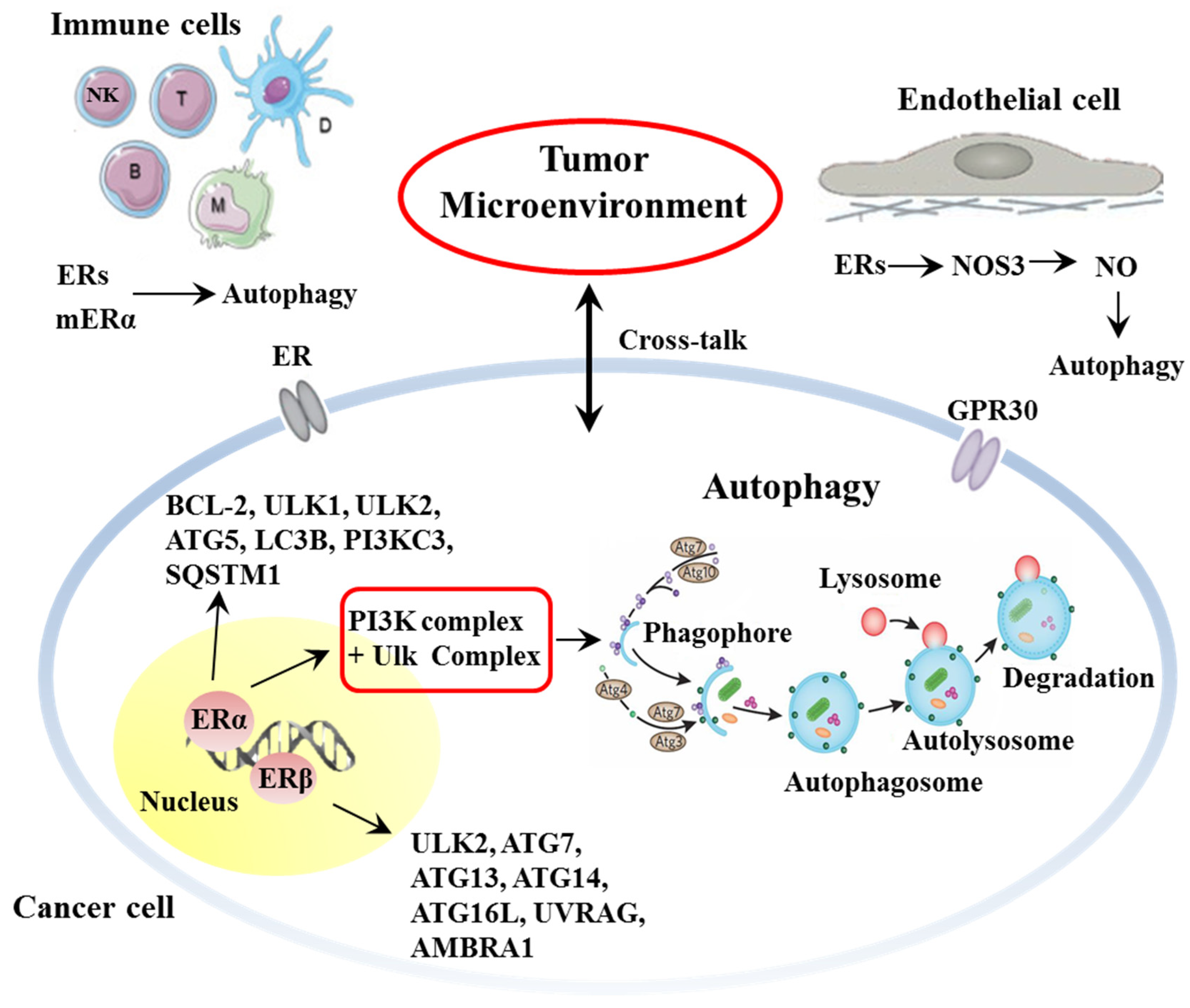
2. Estrogen and Estrogen-Mediated Autophagy as Tumor and TME Key Determinants
2.1. Estrogen and Estrogen Receptors
2.2. Estrogen and Autophagy
2.3. Estrogen and Cancer
2.3.1. Colon Cancer
2.3.2. Melanoma
2.3.3. Lymphoma
2.3.4. Lung Carcinoma
3. Sex-Specific Epigenetic Control: MicroRNAs and Autophagy
3.1. Colon Cancer
3.2. Melanoma
3.3. Lymphoma
3.4. Lung Carcinoma
4. TME–Cancer Cells Crosstalk as Possible Target for Cancer Growth Control
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| 5FU | 5-fluorouracil |
| AKT | protein Kinase B |
| AMPK | adenosine monophosphate-activated protein kinase |
| ATG | autophagy gene |
| Bcl-2 | B-cell lymphoma 2 |
| Bcl-xL | B-cell lymphoma-extra large |
| BNIP3 | Bcl-2 and adenovirus E1B 19-kDa-interacting protein 3 |
| BTG1 | B-cell Translocation Gene 1 |
| CAFs | cancer-associated fibroblasts |
| CC | colon cancer |
| CD | Crohn’s disease |
| COL10A1 | collagen α-1(X) chain |
| DAMP | damage-associated molecular pattern |
| DNMT1 | DNA Methyltransferase 1 |
| DRAM2 | damage regulated autophagy modulator 2 |
| E2 | estradiol |
| ECM | extracellular matrix |
| EMT | epithelial–mesenchymal transition |
| ER | estrogen receptor |
| ERE | estrogen response element |
| EVL | Enah/Vasp-like |
| FATS | fragile-site associated tumor suppressor |
| FOXO3 | Forkhead box O3 |
| GPR30 | G protein-coupled receptor 30 |
| HIF-1α | hypoxia-inducible factor 1-alpha |
| HL | hodgkin lymphoma |
| HMGB1 | high mobility group box 1 |
| IBD1 | inflammatory bowel disease protein 1 |
| ICI | immune checkpoint inhibitors |
| KRAS | kirsten rat sarcoma |
| LC | lung carcinoma |
| LC3 | light chain 3 |
| MAP | mitogen-activated protein |
| MAP1LC3B | microtubule-associated protein 1A/1B light chain 3B |
| mER | plama membrane estrogen receptor |
| miR | microRNA |
| mTOR | mammalian target of rapamycin |
| NF-κB | nuclear factor kappa B |
| NHL | non Hodgkin lymphoma |
| NK | natural killer |
| NO | nitric oxide |
| NOS3 | nitric oxide synthase 3 |
| NSCLC | non-small cell lung cancer |
| ODC | ornithine decarboxylase |
| PERK | PKR-like ER kinase |
| PI3K | phosphatidylinositol-3-kinase |
| PtdIns3K | phosphatidylinositol 3-kinase |
| PTEN | phosphatase and tensin homolog |
| Rheb | Ras homolog enriched in brain |
| ROS TAM | reactive oxygen species tumor associated macrophages |
| TME | tumor microenvironment |
| UC | ulcerative colitis |
| ULK | Unc-51-like autophagy activating kinase |
| XCI | X chromosome inactivation |
References
- Fouad, Y.A.; Aanei, C. Revisiting the hallmarks of cancer. Am. J. Cancer Res. 2017, 7, 1016–1036. [Google Scholar]
- Metcalf, K.J.; Alazzeh, A.; Werb, Z.; Weaver, V.M. Leveraging microenvironmental synthetic lethalities to treat cancer. J. Clin. Investig. 2021, 131, e143765. [Google Scholar] [CrossRef] [PubMed]
- Bacac, M.; Stamenkovic, I. Metastatic cancer cell. Annu. Rev. Pathol. 2008, 3, 221–247. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Malorni, W.; Matarrese, P.; Tinari, A.; Farrace, M.G.; Piacentini, M. Xeno-cannibalism: A survival “escamotage”. Autophagy 2007, 3, 75–77. [Google Scholar] [CrossRef][Green Version]
- Matarrese, P.; Ciarlo, L.; Tinari, A.; Piacentini, M.; Malorni, W. Xeno-cannibalism as an exacerbation of self-cannibalism: A possible fruitful survival strategy for cancer cells. Curr. Pharm. Des. 2008, 14, 245–252. [Google Scholar]
- Wilde, L.; Roche, M.; Domingo-Vidal, M.; Tanson, K.; Philp, N.; Curry, J.; Martinez-Outschoorn, U. Metabolic coupling and the Reverse Warburg Effect in cancer: Implications for novel biomarker and anticancer agent development. Semin. Oncol. 2017, 44, 198–203. [Google Scholar] [CrossRef]
- Kreuzaler, P.; Panina, Y.; Segal, J.; Yuneva, M. Adapt and conquer: Metabolic flexibility in cancer growth, invasion and evasion. Mol. Metab. 2020, 33, 83–101. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Abdel-Aziz, A.K.; Abdelfatah, S.; Abdellatif, M.; Abdoli, A.; Abel, S.; Abeliovich, H.; Abildgaard, M.H.; Abudu, Y.P.; Acevedo-Arozena, A.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (4th edition) (1). Autophagy 2021, 17, 1–382. [Google Scholar] [CrossRef]
- Lorin, S.; Hamai, A.; Mehrpour, M.; Codogno, P. Autophagy regulation and its role in cancer. Semin. Cancer Biol. 2013, 23, 361–379. [Google Scholar] [CrossRef]
- Galluzzi, L.; Pietrocola, F.; Bravo-San Pedro, J.M.; Amaravadi, R.K.; Baehrecke, E.H.; Cecconi, F.; Codogno, P.; Debnath, J.; Gewirtz, D.A.; Karantza, V.; et al. Autophagy in malignant transformation and cancer progression. EMBO J. 2015, 34, 856–880. [Google Scholar] [CrossRef]
- Akkoc, Y.; Peker, N.; Akcay, A.; Gozuacik, D. Autophagy and Cancer Dormancy. Front. Oncol. 2021, 11, 627023. [Google Scholar] [CrossRef]
- Yang, X.; Xu, X.; Zhu, J.; Zhang, S.; Wu, Y.; Zhao, K.; Xing, C.; Cao, J.; Zhu, H.; Li, M.; et al. miR-31 affects colorectal cancer cells by inhibiting autophagy in cancer-associated fibroblasts. Oncotarget 2016, 7, 79617–79628. [Google Scholar] [CrossRef]
- Taniguchi, K.; Karin, M. NF-kappaB, inflammation, immunity and cancer: Coming of age. Nat. Rev. Immunol 2018, 18, 309–324. [Google Scholar] [CrossRef]
- Vara-Perez, M.; Felipe-Abrio, B.; Agostinis, P. Mitophagy in Cancer: A Tale of Adaptation. Cells 2019, 8, 493. [Google Scholar] [CrossRef]
- Bernardini, J.P.; Lazarou, M.; Dewson, G. Parkin and mitophagy in cancer. Oncogene 2017, 36, 1315–1327. [Google Scholar] [CrossRef]
- Poillet-Perez, L.; Xie, X.; Zhan, L.; Yang, Y.; Sharp, D.W.; Hu, Z.S.; Su, X.; Maganti, A.; Jiang, C.; Lu, W.; et al. Autophagy maintains tumour growth through circulating arginine. Nature 2018, 563, 569–573. [Google Scholar] [CrossRef]
- Abdrakhmanov, A.; Kulikov, A.V.; Luchkina, E.A.; Zhivotovsky, B.; Gogvadze, V. Involvementofmitophagy in cisplatin-induced cell death regulation. Biol. Chem. 2019, 400, 161–170. [Google Scholar] [CrossRef]
- Kimmelman, A.C.; White, E. Autophagy and Tumor Metabolism. Cell Metab. 2017, 25, 1037–1043. [Google Scholar] [CrossRef]
- Rohani, N.; Hao, L.; Alexis, M.S.; Joughin, B.A.; Krismer, K.; Moufarrej, M.N.; Soltis, A.R.; Lauffenburger, D.A.; Yaffe, M.B.; Burge, C.B.; et al. Acidification of Tumor at Stromal Boundaries Drives Transcriptome Alterations Associated with Aggressive Phenotypes. Cancer Res. 2019, 79, 1952–1966. [Google Scholar] [CrossRef]
- Robey, I.F.; Baggett, B.K.; Kirkpatrick, N.D.; Roe, D.J.; Dosescu, J.; Sloane, B.F.; Hashim, A.I.; Morse, D.L.; Raghunand, N.; Gatenby, R.A.; et al. Bicarbonate increases tumor pH and inhibits spontaneous metastases. Cancer Res. 2009, 69, 2260–2268. [Google Scholar] [CrossRef]
- Potzl, J.; Roser, D.; Bankel, L.; Homberg, N.; Geishauser, A.; Brenner, C.D.; Weigand, M.; Rocken, M.; Mocikat, R. Reversal of tumor acidosis by systemic buffering reactivates NK cells to express IFN-gamma and induces NK cell-dependent lymphoma control without other immunotherapies. Int. J. Cancer 2017, 140, 2125–2133. [Google Scholar] [CrossRef]
- Ghafouri-Fard, S.; Shoorei, H.; Mohaqiq, M.; Majidpoor, J.; Moosavi, M.A.; Taheri, M. Exploring the role of non-coding RNAs in autophagy. Autophagy 2021, 1–22. [Google Scholar] [CrossRef]
- Raue, R.; Frank, A.C.; Syed, S.N.; Brune, B. Therapeutic Targeting of MicroRNAs in the Tumor Microenvironment. Int. J. Mol. Sci. 2021, 22, 2210. [Google Scholar] [CrossRef]
- Clocchiatti, A.; Cora, E.; Zhang, Y.; Dotto, G.P. Sexual dimorphism in cancer. Nat. Rev. Cancer 2016, 16, 330–339. [Google Scholar] [CrossRef]
- Haupt, S.; Caramia, F.; Klein, S.L.; Rubin, J.B.; Haupt, Y. Sex disparities matter in cancer development and therapy. Nat. Rev. Cancer 2021, 21, 393–407. [Google Scholar] [CrossRef]
- Costa, A.R.; Lanca de Oliveira, M.; Cruz, I.; Goncalves, I.; Cascalheira, J.F.; Santos, C.R.A. The Sex Bias of Cancer. Trends Endocrinol. Metab. 2020, 31, 785–799. [Google Scholar] [CrossRef]
- Gabriele, L.; Buoncervello, M.; Ascione, B.; Bellenghi, M.; Matarrese, P.; Care, A. The gender perspective in cancer research and therapy: Novel insights and on-going hypotheses. Ann. Ist. Super. Sanita 2016, 52, 213–222. [Google Scholar]
- Irelli, A.; Sirufo, M.M.; D’Ugo, C.; Ginaldi, L.; De Martinis, M. Sex and Gender Influences on Cancer Immunotherapy Response. Biomedicines 2020, 8, 232. [Google Scholar] [CrossRef]
- Wagner, A.D.; Oertelt-Prigione, S.; Adjei, A.; Buclin, T.; Cristina, V.; Csajka, C.; Coukos, G.; Dafni, U.; Dotto, G.P.; Ducreux, M.; et al. Gender medicine and oncology: Report and consensus of an ESMO workshop. Ann. Oncol. 2019, 30, 1914–1924. [Google Scholar] [CrossRef]
- Li, C.H.; Haider, S.; Shiah, Y.J.; Thai, K.; Boutros, P.C. Sex Differences in Cancer Driver Genes and Biomarkers. Cancer Res. 2018, 78, 5527–5537. [Google Scholar] [CrossRef]
- Muscogiuri, G.; Altieri, B.; Albertelli, M.; Dotto, A.; Modica, R.; Barrea, L.; Fanciulli, G.; Feola, T.; Baldelli, R.; Ruggeri, R.M.; et al. Epidemiology of pancreatic neuroendocrine neoplasms: A gender perspective. Endocrine 2020, 69, 441–450. [Google Scholar] [CrossRef]
- Park, J.; Shin, H.; Song, H.; Lim, H.J. Autophagic regulation in steroid hormone-responsive systems. Steroids 2016, 115, 177–181. [Google Scholar] [CrossRef]
- Du, L.; Hickey, R.W.; Bayir, H.; Watkins, S.C.; Tyurin, V.A.; Guo, F.; Kochanek, P.M.; Jenkins, L.W.; Ren, J.; Gibson, G.; et al. Starving neurons show sex difference in autophagy. J. Biol. Chem. 2009, 284, 2383–2396. [Google Scholar] [CrossRef]
- Lista, P.; Straface, E.; Brunelleschi, S.; Franconi, F.; Malorni, W. On the role of autophagy in human diseases: A gender perspective. J. Cell Mol. Med. 2011, 15, 1443–1457. [Google Scholar] [CrossRef]
- Maselli, A.; Matarrese, P.; Straface, E.; Canu, S.; Franconi, F.; Malorni, W. Cell sex: A new look at cell fate studies. FASEB J. 2009, 23, 978–984. [Google Scholar] [CrossRef]
- Muralimanoharan, S.; Li, C.; Nakayasu, E.S.; Casey, C.P.; Metz, T.O.; Nathanielsz, P.W.; Maloyan, A. Sexual dimorphism in the fetal cardiac response to maternal nutrient restriction. J. Mol. Cell Cardiol. 2017, 108, 181–193. [Google Scholar] [CrossRef]
- Straface, E.; Vona, R.; Gambardella, L.; Ascione, B.; Marino, M.; Bulzomi, P.; Canu, S.; Coinu, R.; Rosano, G.; Malorni, W.; et al. Cell sex determines anoikis resistance in vascular smooth muscle cells. FEBS Lett. 2009, 583, 3448–3454. [Google Scholar] [CrossRef]
- Shang, D.; Wang, L.; Klionsky, D.J.; Cheng, H.; Zhou, R. Sex differences in autophagy-mediated diseases: Toward precision medicine. Autophagy 2020, 17, 1065–1076. [Google Scholar] [CrossRef]
- Cui, C.; Yang, W.; Shi, J.; Zhou, Y.; Yang, J.; Cui, Q. Identification and Analysis of Human Sex-biased MicroRNAs. Genom. Proteom. Bioinform. 2018, 16, 200–211. [Google Scholar] [CrossRef]
- Kukurba, K.R.; Parsana, P.; Balliu, B.; Smith, K.S.; Zappala, Z.; Knowles, D.A.; Fave, M.J.; Davis, J.R.; Li, X.; Zhu, X.; et al. Impact of the X Chromosome and sex on regulatory variation. Genome Res. 2016, 26, 768–777. [Google Scholar] [CrossRef]
- Pinheiro, I.; Dejager, L.; Libert, C. X-chromosome-located microRNAs in immunity: Might they explain male/female differences? The X chromosome-genomic context may affect X-located miRNAs and downstream signaling, thereby contributing to the enhanced immune response of females. Bioessays 2011, 33, 791–802. [Google Scholar] [CrossRef]
- Deng, X.; Berletch, J.B.; Nguyen, D.K.; Disteche, C.M. X chromosome regulation: Diverse patterns in development, tissues and disease. Nat. Rev. Genet. 2014, 15, 367–378. [Google Scholar] [CrossRef] [PubMed]
- Matarrese, P.; Tieri, P.; Anticoli, S.; Ascione, B.; Conte, M.; Franceschi, C.; Malorni, W.; Salvioli, S.; Ruggieri, A. X-chromosome-linked miR548am-5p is a key regulator of sex disparity in the susceptibility to mitochondria-mediated apoptosis. Cell Death Dis. 2019, 10, 673. [Google Scholar] [CrossRef] [PubMed]
- Tukiainen, T.; Villani, A.C.; Yen, A.; Rivas, M.A.; Marshall, J.L.; Satija, R.; Aguirre, M.; Gauthier, L.; Fleharty, M.; Kirby, A.; et al. Landscape of X chromosome inactivation across human tissues. Nature 2017, 550, 244–248. [Google Scholar] [CrossRef] [PubMed]
- Shireman, J.M.; Ahmed, A.U. Separate and not equal: Sex differences in JAM-A tumor suppression in glioblastoma. Neuro-Oncology 2020, 22, 1555–1556. [Google Scholar] [CrossRef]
- Nilsson, S.; Makela, S.; Treuter, E.; Tujague, M.; Thomsen, J.; Andersson, G.; Enmark, E.; Pettersson, K.; Warner, M.; Gustafsson, J.A. Mechanisms of estrogen action. Physiol. Rev. 2001, 81, 1535–1565. [Google Scholar] [CrossRef]
- Green, S.; Walter, P.; Kumar, V.; Krust, A.; Bornert, J.M.; Argos, P.; Chambon, P. Human oestrogen receptor cDNA: Sequence, expression and homology to v-erb-A. Nature 1986, 320, 134–139. [Google Scholar] [CrossRef]
- Greene, G.L.; Gilna, P.; Waterfield, M.; Baker, A.; Hort, Y.; Shine, J. Sequence and expression of human estrogen receptor complementary DNA. Science 1986, 231, 1150–1154. [Google Scholar] [CrossRef]
- Kuiper, G.G.; Enmark, E.; Pelto-Huikko, M.; Nilsson, S.; Gustafsson, J.A. Cloning of a novel receptor expressed in rat prostate and ovary. Proc. Natl. Acad. Sci. USA 1996, 93, 5925–5930. [Google Scholar] [CrossRef]
- Mosselman, S.; Polman, J.; Dijkema, R. ER beta: Identification and characterization of a novel human estrogen receptor. FEBS Lett. 1996, 392, 49–53. [Google Scholar] [CrossRef]
- Márquez, D.C.; Chen, H.W.; Curran, E.M.; Welshons, W.V.; Pietras, R.J. Estrogen receptors in membrane lipid rafts and signal transduction in breast cancer. Mol. Cell. Endocrinol. 2006, 246, 91–100. [Google Scholar] [CrossRef]
- Levin, E.R.; Hammes, S.R. Nuclear receptors outside the nucleus: Extranuclear signalling by steroid receptors. Nat. Rev. Mol. Cell Biol. 2016, 17, 783–797. [Google Scholar] [CrossRef]
- Marino, M.; Ascenzi, P. Membrane association of estrogen receptor alpha and beta influences 17beta-estradiol-mediated cancer cell proliferation. Steroids 2008, 73, 853–858. [Google Scholar] [CrossRef]
- Gonzalez de Valdivia, E.; Broselid, S.; Kahn, R.; Olde, B.; Leeb-Lundberg, L.M.F. G protein-coupled estrogen receptor 1 (GPER1)/GPR30 increases ERK1/2 activity through PDZ motif-dependent and -independent mechanisms. J. Biol. Chem. 2017, 292, 9932–9943. [Google Scholar] [CrossRef]
- Schiebler, T.H.; Danner, K.G. The effect of sex hormones on the proximal tubules in the rat kidney. Cell Tissue Res. 1978, 192, 527–549. [Google Scholar] [CrossRef]
- Guido, C.; Panza, S.; Santoro, M.; Avena, P.; Panno, M.L.; Perrotta, I.; Giordano, F.; Casaburi, I.; Catalano, S.; De Amicis, F.; et al. Estrogen receptor beta (ERbeta) produces autophagy and necroptosis in human seminoma cell line through the binding of the Sp1 on the phosphatase and tensin homolog deleted from chromosome 10 (PTEN) promoter gene. Cell Cycle 2012, 11, 2911–2921. [Google Scholar] [CrossRef]
- Yang, Y.H.; Chen, K.; Li, B.; Chen, J.W.; Zheng, X.F.; Wang, Y.R.; Jiang, S.D.; Jiang, L.S. Estradiol inhibits osteoblast apoptosis via promotion of autophagy through the ER-ERK-mTOR pathway. Apoptosis 2013, 18, 1363–1375. [Google Scholar] [CrossRef]
- Hsieh, D.J.; Kuo, W.W.; Lai, Y.P.; Shibu, M.A.; Shen, C.Y.; Pai, P.; Yeh, Y.L.; Lin, J.Y.; Viswanadha, V.P.; Huang, C.Y. 17beta-Estradiol and/or Estrogen Receptor beta Attenuate the Autophagic and Apoptotic Effects Induced by Prolonged Hypoxia Through HIF-1alpha-Mediated BNIP3 and IGFBP-3 Signaling Blockage. Cell Physiol. Biochem. 2015, 36, 274–284. [Google Scholar] [CrossRef]
- Lin, C.W.; Chen, B.; Huang, K.L.; Dai, Y.S.; Teng, H.L. Inhibition of Autophagy by Estradiol Promotes Locomotor Recovery after Spinal Cord Injury in Rats. Neurosci. Bull. 2016, 32, 137–144. [Google Scholar] [CrossRef]
- Wang, F.; Xiao, J.; Shen, Y.; Yao, F.; Chen, Y. Estrogen protects cardiomyocytes against lipopolysaccharide by inhibiting autophagy. Mol. Med. Rep. 2014, 10, 1509–1512. [Google Scholar] [CrossRef]
- Dong, L.; Wang, W.; Wang, F.; Stoner, M.; Reed, J.C.; Harigai, M.; Samudio, I.; Kladde, M.P.; Vyhlidal, C.; Safe, S. Mechanisms of transcriptional activation of bcl-2 gene expression by 17beta-estradiol in breast cancer cells. J. Biol. Chem. 1999, 274, 32099–32107. [Google Scholar] [CrossRef]
- Hua, S.; Kallen, C.B.; Dhar, R.; Baquero, M.T.; Mason, C.E.; Russell, B.A.; Shah, P.K.; Liu, J.; Khramtsov, A.; Tretiakova, M.S.; et al. Genomic analysis of estrogen cascade reveals histone variant H2A.Z associated with breast cancer progression. Mol. Syst. Biol. 2008, 4, 188. [Google Scholar] [CrossRef]
- Turei, D.; Foldvari-Nagy, L.; Fazekas, D.; Modos, D.; Kubisch, J.; Kadlecsik, T.; Demeter, A.; Lenti, K.; Csermely, P.; Vellai, T.; et al. Autophagy Regulatory Network—A systems-level bioinformatics resource for studying the mechanism and regulation of autophagy. Autophagy 2015, 11, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Felzen, V.; Hiebel, C.; Koziollek-Drechsler, I.; Reissig, S.; Wolfrum, U.; Kogel, D.; Brandts, C.; Behl, C.; Morawe, T. Estrogen receptor alpha regulates non-canonical autophagy that provides stress resistance to neuroblastoma and breast cancer cells and involves BAG3 function. Cell Death Dis. 2015, 6, e1812. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.M.; Yang, M.F.; Yu, W.; Tao, H.M. Molecular mechanisms of estrogen receptor beta-induced apoptosis and autophagy in tumors: Implication for treating osteosarcoma. J. Int. Med. Res. 2019, 47, 4644–4655. [Google Scholar] [CrossRef] [PubMed]
- Solakidi, S.; Psarra, A.M.; Sekeris, C.E. Differential subcellular distribution of estrogen receptor isoforms: Localization of ERalpha in the nucleoli and ERbeta in the mitochondria of human osteosarcoma SaOS-2 and hepatocarcinoma HepG2 cell lines. Biochim. Biophys. Acta 2005, 1745, 382–392. [Google Scholar] [CrossRef]
- Solakidi, S.; Psarra, A.M.; Sekeris, C.E. Differential distribution of glucocorticoid and estrogen receptor isoforms: Localization of GRbeta and ERalpha in nucleoli and GRalpha and ERbeta in the mitochondria of human osteosarcoma SaOS-2 and hepatocarcinoma HepG2 cell lines. J. Musculoskelet. Neuronal Interact. 2007, 7, 240–245. [Google Scholar]
- Yang, S.H.; Liu, R.; Perez, E.J.; Wen, Y.; Stevens, S.M., Jr.; Valencia, T.; Brun-Zinkernagel, A.M.; Prokai, L.; Will, Y.; Dykens, J.; et al. Mitochondrial localization of estrogen receptor beta. Proc. Natl. Acad. Sci. USA 2004, 101, 4130–4135. [Google Scholar] [CrossRef]
- Cheng, S.B.; Quinn, J.A.; Graeber, C.T.; Filardo, E.J. Down-modulation of the G-protein-coupled estrogen receptor, GPER, from the cell surface occurs via a trans-Golgi-proteasome pathway. J. Biol. Chem. 2011, 286, 22441–22455. [Google Scholar] [CrossRef]
- Totta, P.; Pesiri, V.; Marino, M.; Acconcia, F. Lysosomal function is involved in 17beta-estradiol-induced estrogen receptor alpha degradation and cell proliferation. PLoS ONE 2014, 9, e94880. [Google Scholar] [CrossRef]
- Somasundaram, A.; Rothenberger, N.J.; Stabile, L.P. The Impact of Estrogen in the Tumor Microenvironment. Adv. Exp. Med. Biol. 2020, 1277, 33–52. [Google Scholar]
- Orshal, J.M.; Khalil, R.A. Gender, sex hormones, and vascular tone. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2004, 286, R233–R249. [Google Scholar] [CrossRef]
- He, H.; Feng, Y.S.; Zang, L.H.; Liu, W.W.; Ding, L.Q.; Chen, L.X.; Kang, N.; Hayashi, T.; Tashiro, S.; Onodera, S.; et al. Nitric oxide induces apoptosis and autophagy; autophagy down-regulates NO synthesis in physalin A-treated A375-S2 human melanoma cells. Food Chem. Toxicol. 2014, 71, 128–135. [Google Scholar] [CrossRef]
- Jiang, G.M.; Tan, Y.; Wang, H.; Peng, L.; Chen, H.T.; Meng, X.J.; Li, L.L.; Liu, Y.; Li, W.F.; Shan, H. The relationship between autophagy and the immune system and its applications for tumor immunotherapy. Mol. Cancer 2019, 18, 17. [Google Scholar] [CrossRef]
- Straub, R.H. The Complex Role of Estrogens in Inflammation. Endocr. Rev. 2007, 28, 521–574. [Google Scholar] [CrossRef]
- Pierdominici, M.; Maselli, A.; Colasanti, T.; Giammarioli, A.M.; Delunardo, F.; Vacirca, D.; Sanchez, M.; Giovannetti, A.; Malorni, W.; Ortona, E. Estrogen receptor profiles in human peripheral blood lymphocytes. Immunol. Lett. 2010, 132, 79–85. [Google Scholar] [CrossRef]
- Pierdominici, M.; Maselli, A.; Locatelli, S.L.; Ciarlo, L.; Careddu, G.; Patrizio, M.; Ascione, B.; Tinari, A.; Carlo-Stella, C.; Malorni, W.; et al. Estrogen receptor beta ligation inhibits Hodgkin lymphoma growth by inducing autophagy. Oncotarget 2017, 8, 8522–8535. [Google Scholar] [CrossRef]
- Shepherd, R.; Cheung, A.S.; Pang, K.; Saffery, R.; Novakovic, B. Sexual Dimorphism in Innate Immunity: The Role of Sex Hormones and Epigenetics. Front. Immunol. 2021, 11, 604000. [Google Scholar] [CrossRef]
- Kovats, S.; Carreras, E. Regulation of dendritic cell differentiation and function by estrogen receptor ligands. Cell. Immunol. 2008, 252, 81–90. [Google Scholar] [CrossRef]
- Moulton, V.R. Sex Hormones in Acquired Immunity and Autoimmune Disease. Front. Immunol. 2018, 9, 2279. [Google Scholar] [CrossRef] [PubMed]
- Ortona, E.; Delunardo, F.; Maselli, A.; Pierdominici, M.; Malorni, W. Sex hormones and gender disparity in immunity and autoimmunity. Ital. J. Gend. Specif. Med. 2015, 1, 45–50. [Google Scholar]
- Dupuis, M.; Maselli, A.; Pagano, M.T.; Pierdominici, M.; Ortona, E. Immune response and autoimmune diseases: A matter of sex. Ital. J. Gend. Specif. Med. 2019, 5, 11–20. [Google Scholar]
- Ozdemir, B.C.; Dotto, G.P. Sex Hormones and Anticancer Immunity. Clin. Cancer Res. 2019, 25, 4603–4610. [Google Scholar] [CrossRef]
- Klein, S.L.; Morgan, R. The impact of sex and gender on immunotherapy outcomes. Biol. Sex Differ. 2020, 11, 24. [Google Scholar] [CrossRef]
- Wei, Y.; Huang, C.; Wu, H.; Huang, J. Estrogen Receptor Beta (ERbeta) Mediated-CyclinD1 Degradation via Autophagy Plays an Anti-Proliferation Role in Colon Cells. Int. J. Biol. Sci. 2019, 15, 942–952. [Google Scholar] [CrossRef]
- Kohli, L.; Kaza, N.; Coric, T.; Byer, S.J.; Brossier, N.M.; Klocke, B.J.; Bjornsti, M.A.; Carroll, S.L.; Roth, K.A. 4-Hydroxytamoxifen induces autophagic death through K-Ras degradation. Cancer Res. 2013, 73, 4395–4405. [Google Scholar] [CrossRef]
- Marzagalli, M.; Casati, L.; Moretti, R.M.; Montagnani Marelli, M.; Limonta, P. Estrogen Receptor beta Agonists Differentially Affect the Growth of Human Melanoma Cell Lines. PLoS ONE 2015, 10, e0134396. [Google Scholar] [CrossRef]
- Chatterjee, S.J.; Pandey, S. Chemo-resistant melanoma sensitized by tamoxifen to low dose curcumin treatment through induction of apoptosis and autophagy. Cancer Biol. Ther. 2011, 11, 216–228. [Google Scholar] [CrossRef]
- Yakimchuk, K.; Jondal, M.; Okret, S. Estrogen receptor alpha and beta in the normal immune system and in lymphoid malignancies. Mol. Cell. Endocrinol. 2013, 375, 121–129. [Google Scholar] [CrossRef]
- Bustos, V.; Nolan, A.M.; Nijhuis, A.; Harvey, H.; Parker, A.; Poulsom, R.; McBryan, J.; Thomas, W.; Silver, A.; Harvey, B.J. GPER mediates differential effects of estrogen on colon cancer cell proliferation and migration under normoxic and hypoxic conditions. Oncotarget 2017, 8, 84258–84275. [Google Scholar] [CrossRef]
- Konstantinopoulos, P.A.; Kominea, A.; Vandoros, G.; Sykiotis, G.P.; Andricopoulos, P.; Varakis, I.; Sotiropoulou-Bonikou, G.; Papavassiliou, A.G. Oestrogen receptor beta (ERbeta) is abundantly expressed in normal colonic mucosa, but declines in colon adenocarcinoma paralleling the tumour’s dedifferentiation. Eur. J. Cancer 2003, 39, 1251–1258. [Google Scholar] [CrossRef]
- Williams, C.; DiLeo, A.; Niv, Y.; Gustafsson, J.A. Estrogen receptor beta as target for colorectal cancer prevention. Cancer Lett. 2016, 372, 48–56. [Google Scholar] [CrossRef]
- Campbell-Thompson, M.; Lynch, I.J.; Bhardwaj, B. Expression of estrogen receptor (ER) subtypes and ERbeta isoforms in colon cancer. Cancer Res. 2001, 61, 632–640. [Google Scholar]
- Janik, M.E.; Belkot, K.; Przybylo, M. Is oestrogen an important player in melanoma progression? Contemp. Oncol. 2014, 18, 302–306. [Google Scholar] [CrossRef]
- Mitkov, M.; Joseph, R.; Copland, J., 3rd. Steroid hormone influence on melanomagenesis. Mol. Cell. Endocrinol. 2015, 417, 94–102. [Google Scholar] [CrossRef]
- Santen, R.J.; Santner, S.J.; Harvey, H.A.; Lipton, A.; Simmonds, M.; Feil, P.D.; Manders, E.; Davis, T.S. Marked heterogeneity of aromatase activity in human malignant melanoma tissue. Eur. J. Cancer Clin. Oncol. 1988, 24, 1811–1816. [Google Scholar] [CrossRef]
- Mitchell, D.L.; Fernandez, A.A.; Garcia, R.; Paniker, L.; Lin, K.; Hanninen, A.; Zigelsky, K.; May, M.; Nuttall, M.; Lo, H.H.; et al. Acute exposure to ultraviolet-B radiation modulates sex steroid hormones and receptor expression in the skin and may contribute to the sex bias of melanoma in a fish model. Pigment Cell Melanoma Res. 2014, 27, 408–417. [Google Scholar] [CrossRef]
- de Giorgi, V.; Gori, A.; Grazzini, M.; Rossari, S.; Scarfi, F.; Corciova, S.; Verdelli, A.; Lotti, T.; Massi, D. Estrogens, estrogen receptors and melanoma. Expert Rev. Anticancer Ther. 2011, 11, 739–747. [Google Scholar] [CrossRef]
- de Giorgi, V.; Mavilia, C.; Massi, D.; Gozzini, A.; Aragona, P.; Tanini, A.; Sestini, S.; Paglierani, M.; Boddi, V.; Brandi, M.L.; et al. Estrogen receptor expression in cutaneous melanoma: A real-time reverse transcriptase-polymerase chain reaction and immunohistochemical study. Arch. Dermatol. 2009, 145, 30–36. [Google Scholar] [CrossRef]
- Ohata, C.; Tadokoro, T.; Itami, S. Expression of estrogen receptor beta in normal skin, melanocytic nevi and malignant melanomas. J. Dermatol. 2008, 35, 215–221. [Google Scholar] [CrossRef]
- Schmidt, A.N.; Nanney, L.B.; Boyd, A.S.; King, L.E., Jr.; Ellis, D.L. Oestrogen receptor-beta expression in melanocytic lesions. Exp. Dermatol. 2006, 15, 971–980. [Google Scholar] [CrossRef]
- de Giorgi, V.; Gori, A.; Gandini, S.; Papi, F.; Grazzini, M.; Rossari, S.; Simoni, A.; Maio, V.; Massi, D. Oestrogen receptor beta and melanoma: A comparative study. Br. J. Dermatol. 2013, 168, 513–519. [Google Scholar] [CrossRef]
- Joosse, A.; de Vries, E.; Eckel, R.; Nijsten, T.; Eggermont, A.M.M.; Hölzel, D.; Coebergh, J.W.W.; Engel, J.; Munich Melanoma Group. Gender Differences in Melanoma Progression and Survival: Female patients have a decreased risk of metastasis. J. Investig. Dermatol. 2011, 131, 719–726. [Google Scholar] [CrossRef]
- Marzagalli, M.; Montagnani Marelli, M.; Casati, L.; Fontana, F.; Moretti, R.M.; Limonta, P. Estrogen Receptor beta in Melanoma: From Molecular Insights to Potential Clinical Utility. Front. Endocrinol. 2016, 7, 140. [Google Scholar] [CrossRef]
- Yakimchuk, K.; Hasni, M.S.; Guan, J.; Chao, M.P.; Sander, B.; Okret, S. Inhibition of lymphoma vascularization and dissemination by estrogen receptor beta agonists. Blood 2014, 123, 2054–2061. [Google Scholar] [CrossRef] [PubMed]
- Yakimchuk, K.; Iravani, M.; Hasni, M.S.; Rhonnstad, P.; Nilsson, S.; Jondal, M.; Okret, S. Effect of ligand-activated estrogen receptor beta on lymphoma growth in vitro and in vivo. Leukemia 2011, 25, 1103–1110. [Google Scholar] [CrossRef]
- Albain, K.S.; Unger, J.M.; Gotay, C.C.; Davies, J.S. Toxicity and survival by sex in patients with advanced non-small cell lung carcinoma (NSCLC) on modern Southwest Oncology Group (SWOG) trials. J. Clin. Oncol. 2007, 25, 7549. [Google Scholar] [CrossRef]
- Hsu, L.H.; Liu, K.J.; Tsai, M.F.; Wu, C.R.; Feng, A.C.; Chu, N.M.; Kao, S.H. Estrogen adversely affects the prognosis of patients with lung adenocarcinoma. Cancer Sci. 2015, 106, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Lara, V.; Pena-Mirabal, E.; Baez-Saldana, R.; Esparza-Silva, A.L.; Garcia-Zepeda, E.; Cerbon Cervantes, M.A.; Diaz, D.; Fortoul, T.I. Estrogen receptor beta and CXCR4/CXCL12 expression: Differences by sex and hormonal status in lung adenocarcinoma. Arch. Med. Res. 2014, 45, 158–169. [Google Scholar] [CrossRef] [PubMed]
- Stabile, L.P.; Dacic, S.; Land, S.R.; Lenzner, D.E.; Dhir, R.; Acquafondata, M.; Landreneau, R.J.; Grandis, J.R.; Siegfried, J.M. Combined analysis of estrogen receptor beta-1 and progesterone receptor expression identifies lung cancer patients with poor outcome. Clin. Cancer Res. 2011, 17, 154–164. [Google Scholar] [CrossRef]
- Niikawa, H.; Suzuki, T.; Miki, Y.; Suzuki, S.; Nagasaki, S.; Akahira, J.; Honma, S.; Evans, D.B.; Hayashi, S.; Kondo, T.; et al. Intratumoral estrogens and estrogen receptors in human non-small cell lung carcinoma. Clin. Cancer Res. 2008, 14, 4417–4426. [Google Scholar] [CrossRef]
- Siegfried, J.M.; Hershberger, P.A.; Stabile, L.P. Estrogen receptor signaling in lung cancer. Semin. Oncol. 2009, 36, 524–531. [Google Scholar] [CrossRef]
- Ma, L.; Zhan, P.; Liu, Y.; Zhou, Z.; Zhu, Q.; Miu, Y.; Wang, X.; Jin, J.; Li, Q.; Lv, T.; et al. Prognostic value of the expression of estrogen receptor beta in patients with non-small cell lung cancer: A meta-analysis. Transl. Lung Cancer Res. 2016, 5, 202–207. [Google Scholar] [CrossRef]
- Mah, V.; Marquez, D.; Alavi, M.; Maresh, E.L.; Zhang, L.; Yoon, N.; Horvath, S.; Bagryanova, L.; Fishbein, M.C.; Chia, D.; et al. Expression levels of estrogen receptor beta in conjunction with aromatase predict survival in non-small cell lung cancer. Lung Cancer 2011, 74, 318–325. [Google Scholar] [CrossRef]
- Skjefstad, K.; Grindstad, T.; Khanehkenari, M.R.; Richardsen, E.; Donnem, T.; Kilvaer, T.; Andersen, S.; Bremnes, R.M.; Busund, L.T.; Al-Saad, S. Prognostic relevance of estrogen receptor alpha, beta and aromatase expression in non-small cell lung cancer. Steroids 2016, 113, 5–13. [Google Scholar] [CrossRef]
- Weinberg, O.K.; Marquez-Garban, D.C.; Fishbein, M.C.; Goodglick, L.; Garban, H.J.; Dubinett, S.M.; Pietras, R.J. Aromatase inhibitors in human lung cancer therapy. Cancer Res. 2005, 65, 11287–11291. [Google Scholar] [CrossRef]
- Hershberger, P.A.; Vasquez, A.C.; Kanterewicz, B.; Land, S.; Siegfried, J.M.; Nichols, M. Regulation of endogenous gene expression in human non-small cell lung cancer cells by estrogen receptor ligands. Cancer Res. 2005, 65, 1598–1605. [Google Scholar] [CrossRef]
- Siegfried, J.M.; Stabile, L.P. Estrongenic steroid hormones in lung cancer. Semin. Oncol. 2014, 41, 5–16. [Google Scholar] [CrossRef]
- Zhang, G.; Yanamala, N.; Lathrop, K.L.; Zhang, L.; Klein-Seetharaman, J.; Srinivas, H. Ligand-independent antiapoptotic function of estrogen receptor-beta in lung cancer cells. Mol. Endocrinol. 2010, 24, 1737–1747. [Google Scholar] [CrossRef]
- Qiu, L.; Hu, L.; Wang, H.; Li, J.; Ruan, X.; Sun, B.; Zhi, J.; Zheng, X.; Gu, L.; Gao, M.; et al. FATS regulates polyamine biosynthesis by promoting ODC degradation in an ERbeta-dependent manner in non-small-cell lung cancer. Cell Death Dis. 2020, 11, 839. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Jin, F.; Lian, X.; Li, M.; Wang, G.; Lan, B.; He, H.; Liu, G.D.; Wu, Y.; Sun, G.; et al. Genistein promotes ionizing radiation-induced cell death by reducing cytoplasmic Bcl-xL levels in non-small cell lung cancer. Sci. Rep. 2018, 8, 328. [Google Scholar] [CrossRef] [PubMed]
- Jansson, M.D.; Lund, A.H. MicroRNA and cancer. Mol. Oncol. 2012, 6, 590–610. [Google Scholar] [CrossRef] [PubMed]
- Kim, V.N.; Han, J.; Siomi, M.C. Biogenesis of small RNAs in animals. Nat. Rev. Mol. Cell Biol. 2009, 10, 126–139. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Y. Principles of micro-RNA production and maturation. Oncogene 2006, 25, 6156–6162. [Google Scholar] [CrossRef] [PubMed]
- Pontecorvi, G.; Bellenghi, M.; Puglisi, R.; Care, A.; Mattia, G. Tumor-derived extracellular vesicles and microRNAs: Functional roles, diagnostic, prognostic and therapeutic options. Cytokine Growth Factor Rev. 2020, 51, 75–83. [Google Scholar] [CrossRef]
- Sun, Y.; Xing, X.; Liu, Q.; Wang, Z.; Xin, Y.; Zhang, P.; Hu, C.; Liu, Y. Hypoxia-induced autophagy reduces radiosensitivity by the HIF-1alpha/miR-210/Bcl-2 pathway in colon cancer cells. Int. J. Oncol. 2015, 46, 750–756. [Google Scholar] [CrossRef]
- Zhang, H.; Tang, J.; Li, C.; Kong, J.; Wang, J.; Wu, Y.; Xu, E.; Lai, M. MiR-22 regulates 5-FU sensitivity by inhibiting autophagy and promoting apoptosis in colorectal cancer cells. Cancer Lett. 2015, 356, 781–790. [Google Scholar] [CrossRef]
- Colangelo, T.; Polcaro, G.; Ziccardi, P.; Muccillo, L.; Galgani, M.; Pucci, B.; Milone, M.R.; Budillon, A.; Santopaolo, M.; Mazzoccoli, G.; et al. miR-27a-calreticulin axis affects drug-induced immunogenic cell death in human colorectal cancer cells. Cell Death Dis. 2016, 7, e2108. [Google Scholar] [CrossRef]
- Guo, W.; Wang, H.; Yang, Y.; Guo, S.; Zhang, W.; Liu, Y.; Yi, X.; Ma, J.; Zhao, T.; Liu, L.; et al. Down-regulated miR-23a Contributes to the Metastasis of Cutaneous Melanoma by Promoting Autophagy. Theranostics 2017, 7, 2231–2249. [Google Scholar] [CrossRef]
- Yu, Y.; Xiang, N.; Lin, M.; Huang, J.W.; Zhang, J.; Cheng, B.; Ji, C. miR-26a Sensitizes Melanoma Cells to Dabrafenib Via Targeting HMGB1-Dependent Autophagy Pathways. Drug Des. Dev. Ther. 2019, 13, 3717–3726. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Gao, M.; Chen, L.; Zhou, L.; Bian, S.; Lv, Y. Licochalcone A restrains microphthalmia-associated transcription factor expression and growth by activating autophagy in melanoma cells via miR-142-3p/Rheb/mTOR pathway. Phytother. Res. 2020, 34, 349–358. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.Y.; Calin, G.A.; Yuen, K.S.; Jin, D.Y.; Chim, C.S. Epigenetic silencing of miR-342-3p in B cell lymphoma and its impact on autophagy. Clin. Epigenet. 2020, 12, 150. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Qiu, L.; Li, T.; Wang, X.; Deng, R.; Yi, H.; Su, Y.; Fan, F.Y. MiR-449a attenuates autophagy of T-cell lymphoma cells by downregulating ATG4B expression. BMB Rep. 2020, 53, 254–259. [Google Scholar] [CrossRef]
- Zhang, W.; Dong, Y.Z.; Du, X.; Peng, X.N.; Shen, Q.M. MiRNA-153-3p promotes gefitinib-sensitivity in non-small cell lung cancer by inhibiting ATG5 expression and autophagy. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 2444–2452. [Google Scholar]
- Rothschild, S.I.; Gautschi, O.; Batliner, J.; Gugger, M.; Fey, M.F.; Tschan, M.P. MicroRNA-106a targets autophagy and enhances sensitivity of lung cancer cells to Src inhibitors. Lung Cancer 2017, 107, 73–83. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, J.; Hui, B.; Sun, W.; Li, B.; Shi, F.; Che, S.; Chai, L.; Song, L. Pristimerin enhances the effect of cisplatin by inhibiting the miR23a/Akt/GSK3beta signaling pathway and suppressing autophagy in lung cancer cells. Int. J. Mol. Med. 2019, 43, 1382–1394. [Google Scholar]
- Guo, Q.; Zheng, M.; Xu, Y.; Wang, N.; Zhao, W. MiR-384 induces apoptosis and autophagy of non-small cell lung cancer cells through the negative regulation of Collagen alpha-1(X) chain gene. Biosci. Rep. 2019, 39, BSR20181523. [Google Scholar] [CrossRef]
- Kim, E.R.; Chang, D.K. Colorectal cancer in inflammatory bowel disease: The risk, pathogenesis, prevention and diagnosis. World J. Gastroenterol. 2014, 20, 9872–9881. [Google Scholar] [CrossRef]
- Wang, S.; Huang, Y.; Zhou, C.; Wu, H.; Zhao, J.; Wu, L.; Zhao, M.; Zhang, F.; Liu, H. The Role of Autophagy and Related MicroRNAs in Inflammatory Bowel Disease. Gastroenterol. Res. Pract. 2018, 2018, 7565076. [Google Scholar] [CrossRef]
- Edvardsson, K.; Nguyen-Vu, T.; Kalasekar, S.M.; Ponten, F.; Gustafsson, J.A.; Williams, C. Estrogen receptor beta expression induces changes in the microRNA pool in human colon cancer cells. Carcinogenesis 2013, 34, 1431–1441. [Google Scholar] [CrossRef]
- Pandey, D.P.; Picard, D. miR-22 inhibits estrogen signaling by directly targeting the estrogen receptor alpha mRNA. Mol. Cell Biol. 2009, 29, 3783–3790. [Google Scholar] [CrossRef]
- Krysko, D.V.; Garg, A.D.; Kaczmarek, A.; Krysko, O.; Agostinis, P.; Vandenabeele, P. Immunogenic cell death and DAMPs in cancer therapy. Nat. Rev. Cancer 2012, 12, 860–875. [Google Scholar] [CrossRef]
- Li, X.; Mertens-Talcott, S.U.; Zhang, S.; Kim, K.; Ball, J.; Safe, S. MicroRNA-27a Indirectly Regulates Estrogen Receptor {alpha} Expression and Hormone Responsiveness in MCF-7 Breast Cancer Cells. Endocrinology 2010, 151, 2462–2473. [Google Scholar] [CrossRef]
- Zhang, J.A.; Zhou, B.R.; Xu, Y.; Chen, X.; Liu, J.; Gozali, M.; Wu, D.; Yin, Z.Q.; Luo, D. MiR-23a-depressed autophagy is a participant in PUVA- and UVB-induced premature senescence. Oncotarget 2016, 7, 37420–37435. [Google Scholar] [CrossRef]
- Amaravadi, R.K.; Lippincott-Schwartz, J.; Yin, X.M.; Weiss, W.A.; Takebe, N.; Timmer, W.; DiPaola, R.S.; Lotze, M.T.; White, E. Principles and current strategies for targeting autophagy for cancer treatment. Clin. Cancer Res. 2011, 17, 654–666. [Google Scholar] [CrossRef]
- Beatty, G.L.; Gladney, W.L. Immune escape mechanisms as a guide for cancer immunotherapy. Clin. Cancer Res. 2015, 21, 687–692. [Google Scholar] [CrossRef]
- Russell, R.C.; Yuan, H.X.; Guan, K.L. Autophagy regulation by nutrient signaling. Cell Res. 2014, 24, 42–57. [Google Scholar] [CrossRef]
- Kroemer, G.; Marino, G.; Levine, B. Autophagy and the integrated stress response. Mol. Cell 2010, 40, 280–293. [Google Scholar] [CrossRef]
- Cha, M.J.; Choi, E.; Lee, S.; Song, B.W.; Yoon, C.; Hwang, K.C. The microRNA-dependent cell fate of multipotent stromal cells differentiating to endothelial cells. Exp. Cell Res. 2016, 341, 139–146. [Google Scholar] [CrossRef]
- Dika, E.; Patrizi, A.; Lambertini, M.; Manuelpillai, N.; Fiorentino, M.; Altimari, A.; Ferracin, M.; Lauriola, M.; Fabbri, E.; Campione, E.; et al. Estrogen Receptors and Melanoma: A Review. Cells 2019, 8, 1463. [Google Scholar] [CrossRef]
- Kwiatkowski, A.V.; Gertler, F.B.; Loureiro, J.J. Function and regulation of Ena/VASP proteins. Trends Cell Biol. 2003, 13, 386–392. [Google Scholar] [CrossRef]
- Wang, H.; Wu, J.; Meng, X.; Ying, X.; Zuo, Y.; Liu, R.; Pan, Z.; Kang, T.; Huang, W. MicroRNA-342 inhibits colorectal cancer cell proliferation and invasion by directly targeting DNA methyltransferase 1. Carcinogenesis 2011, 32, 1033–1042. [Google Scholar] [CrossRef]
- Sandbothe, M.; Buurman, R.; Reich, N.; Greiwe, L.; Vajen, B.; Gurlevik, E.; Schaffer, V.; Eilers, M.; Kuhnel, F.; Vaquero, A.; et al. The microRNA-449 family inhibits TGF-beta-mediated liver cancer cell migration by targeting SOX4. J. Hepatol. 2017, 66, 1012–1021. [Google Scholar] [CrossRef] [PubMed]
- Stabile, L.P.; Lyker, J.S.; Gubish, C.T.; Zhang, W.; Grandis, J.R.; Siegfried, J.M. Combined targeting of the estrogen receptor and the epidermal growth factor receptor in non-small cell lung cancer shows enhanced antiproliferative effects. Cancer Res. 2005, 65, 1459–1470. [Google Scholar] [CrossRef] [PubMed]
- Castellano, L.; Giamas, G.; Jacob, J.; Coombes, R.C.; Lucchesi, W.; Thiruchelvam, P.; Barton, G.; Jiao, L.R.; Wait, R.; Waxman, J.; et al. The estrogen receptor-alpha-induced microRNA signature regulates itself and its transcriptional response. Proc. Natl. Acad. Sci. USA 2009, 106, 15732–15737. [Google Scholar] [CrossRef] [PubMed]
- Gomes, J.P.M.; Cardoso, C.R.P.; Varanda, E.A.; Molina, J.-M.; Fernandez, M.F.; Olea, N.S.; Carlos, I.Z.; Vilegas, W. Antitumoral, mutagenic and (anti)estrogenic activities of tingenone and pristimerin. Rev. Bras. Farmacogn. 2019, 21, 963–971. [Google Scholar] [CrossRef]
- Levy, J.M.M.; Towers, C.G.; Thorburn, A. Targeting autophagy in cancer. Nat. Rev. Cancer 2017, 17, 528–542. [Google Scholar] [CrossRef]
- Chen, H.T.; Liu, H.; Mao, M.J.; Tan, Y.; Mo, X.Q.; Meng, X.J.; Cao, M.T.; Zhong, C.Y.; Liu, Y.; Shan, H.; et al. Crosstalk between autophagy and epithelial-mesenchymal transition and its application in cancer therapy. Mol. Cancer 2019, 18, 101. [Google Scholar] [CrossRef]
- Singh, S.S.; Vats, S.; Chia, A.Y.; Tan, T.Z.; Deng, S.; Ong, M.S.; Arfuso, F.; Yap, C.T.; Goh, B.C.; Sethi, G.; et al. Dual role of autophagy in hallmarks of cancer. Oncogene 2018, 37, 1142–1158. [Google Scholar] [CrossRef]
- Qu, X.; Yu, J.; Bhagat, G.; Furuya, N.; Hibshoosh, H.; Troxel, A.; Rosen, J.; Eskelinen, E.L.; Mizushima, N.; Ohsumi, Y.; et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J. Clin. Investig. 2003, 112, 1809–1820. [Google Scholar] [CrossRef]
- Katayama, M.; Kawaguchi, T.; Berger, M.S.; Pieper, R.O. DNA damaging agent-induced autophagy produces a cytoprotective adenosine triphosphate surge in malignant glioma cells. Cell Death Differ. 2007, 14, 548–558. [Google Scholar] [CrossRef]
- Marinkovic, M.; Sprung, M.; Buljubasic, M.; Novak, I. Autophagy Modulation in Cancer: Current Knowledge on Action and Therapy. Oxidative Med. Cell. Longev. 2018, 2018, 8023821. [Google Scholar] [CrossRef]
- Buccarelli, M.; Marconi, M.; Pacioni, S.; De Pascalis, I.; D’Alessandris, Q.G.; Martini, M.; Ascione, B.; Malorni, W.; Larocca, L.M.; Pallini, R.; et al. Inhibition of autophagy increases susceptibility of glioblastoma stem cells to temozolomide by igniting ferroptosis. Cell Death Dis. 2018, 9, 841. [Google Scholar] [CrossRef]
- Sui, X.; Chen, R.; Wang, Z.; Huang, Z.; Kong, N.; Zhang, M.; Han, W.; Lou, F.; Yang, J.; Zhang, Q.; et al. Autophagy and chemotherapy resistance: A promising therapeutic target for cancer treatment. Cell Death Dis. 2013, 4, e838. [Google Scholar] [CrossRef]
- Roskoski, R., Jr. Properties of FDA-approved small molecule protein kinase inhibitors: A 2020 update. Pharmacol. Res. 2020, 152, 104609. [Google Scholar] [CrossRef]
- Haas, N.B.; Appleman, L.J.; Stein, M.; Redlinger, M.; Wilks, M.; Xu, X.; Onorati, A.; Kalavacharla, A.; Kim, T.; Zhen, C.J.; et al. Autophagy Inhibition to Augment mTOR Inhibition: A Phase I/II Trial of Everolimus and Hydroxychloroquine in Patients with Previously Treated Renal Cell Carcinoma. Clin. Cancer Res. 2019, 25, 2080–2087. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Yamada, D.; Kawai, T.; Sato, Y.; Teshima, T.; Yamada, Y.; Nakamura, M.; Suzuki, M.; Matsumoto, A.; Nakagawa, T.; et al. Different immunological effects of the molecular targeted agents sunitinib, everolimus and temsirolimus in patients with renal cell carcinoma. Int. J. Oncol. 2020, 56, 999–1013. [Google Scholar] [CrossRef]
- Bustos, S.O.; Antunes, F.; Rangel, M.C.; Chammas, R. Emerging Autophagy Functions Shape the Tumor Microenvironment and Play a Role in Cancer Progression—Implications for Cancer Therapy. Front. Oncol. 2020, 10, 606436. [Google Scholar] [CrossRef]
- Ge, Z.; Yu, H.; Yang, W.; Yang, J.; Liu, B.; Wang, X.; Liu, Z.; Liu, L. Development of Multi-Dimensional Cell Co-Culture via a Novel Microfluidic Chip Fabricated by DMD-Based Optical Projection Lithography. IEEE Trans. Nanobiosci. 2019, 18, 679–686. [Google Scholar] [CrossRef]
- Parlato, S.; Grisanti, G.; Sinibaldi, G.; Peruzzi, G.; Casciola, C.M.; Gabriele, L. Tumor-on-a-chip platforms to study cancer-immune system crosstalk in the era of immunotherapy. Lab Chip 2021, 21, 234–253. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Cancer | Effects of ERs on Autophagy | References |
|---|---|---|
| Colon cancer | ERβ induced autophagy through:
| [86,87] |
| Melanoma | ERβ induced autophagy through:
| [88] |
| Hodgkin Lymphoma | ERβ induced autophagy through:
| [78] |
| Lung carcinoma | ERβ induced autophagy in NSCLC cells through:
| [89,90] |
| Cancer | microRNAs | Targeted Proteins | Effect on Tumor Cells | References |
|---|---|---|---|---|
| Colon Cancer | miR-210 | Bcl-2 | Induces autophagy and radioresistance | [127] |
| miR-22 | BTG1 | Inhibits autophagy and promotes apoptosis | [128] | |
| miR-27a | Calreticulin | Inhibits autophagy and apoptosis | [129] | |
| Melanoma | miR-23a | ATG12 | Inhibits autophagy and reduces invasiveness | [130] |
| miR-26a | HMGB1 | Inhibits autophagy and induces apoptosis | [131] | |
| miR-142-3p | Rheb | Induces autophagy and apoptosis | [132] | |
| Hodgkin Lymphoma | miR-342-3p | MAP1LC3B, DNMT1 | Inhibits autophagy and induces cell death | [133] |
| miR-449a | ATG4B | Inhibits autophagy and induces apoptosis | [134] | |
| Lung Carcinoma | miR-153-3p | ATG5 | Inhibits autophagy and induces apoptosis | [135] |
| miR-106a | ULK1 | Inhibits autophagy and induces apoptosis | [136] | |
| miR-23a | PTEN | Induces autophagy and inhibits apoptosis | [137] | |
| miR-384 | COL10A1 | Induces autophagy and apoptosis | [138] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Matarrese, P.; Mattia, G.; Pagano, M.T.; Pontecorvi, G.; Ortona, E.; Malorni, W.; Carè, A. The Sex-Related Interplay between TME and Cancer: On the Critical Role of Estrogen, MicroRNAs and Autophagy. Cancers 2021, 13, 3287. https://doi.org/10.3390/cancers13133287
Matarrese P, Mattia G, Pagano MT, Pontecorvi G, Ortona E, Malorni W, Carè A. The Sex-Related Interplay between TME and Cancer: On the Critical Role of Estrogen, MicroRNAs and Autophagy. Cancers. 2021; 13(13):3287. https://doi.org/10.3390/cancers13133287
Chicago/Turabian StyleMatarrese, Paola, Gianfranco Mattia, Maria Teresa Pagano, Giada Pontecorvi, Elena Ortona, Walter Malorni, and Alessandra Carè. 2021. "The Sex-Related Interplay between TME and Cancer: On the Critical Role of Estrogen, MicroRNAs and Autophagy" Cancers 13, no. 13: 3287. https://doi.org/10.3390/cancers13133287
APA StyleMatarrese, P., Mattia, G., Pagano, M. T., Pontecorvi, G., Ortona, E., Malorni, W., & Carè, A. (2021). The Sex-Related Interplay between TME and Cancer: On the Critical Role of Estrogen, MicroRNAs and Autophagy. Cancers, 13(13), 3287. https://doi.org/10.3390/cancers13133287