
Immune Circuits to Shape Natural Killer Cells in Cancer


{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
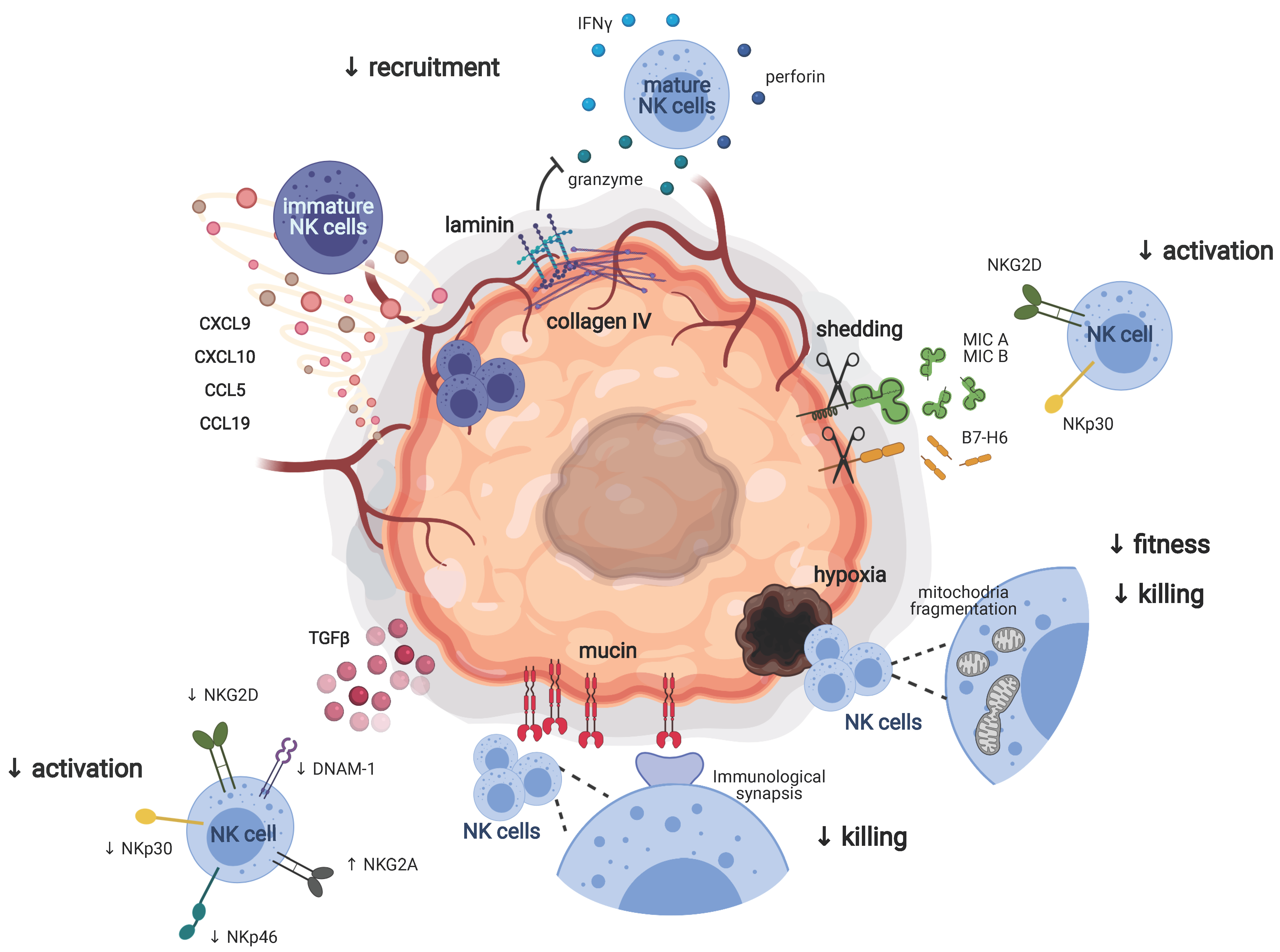
2. Adverse Environmental Factors Dampening the Antitumoral Functions of NK Cells
2.1. NK Cells in Metastases and Primary Tumors
2.2. Recruitment of NK Cells to the Tumor Site
2.3. Tumor Escape Mechanisms Driving NK Cell Tolerance
2.3.1. NK Cell Activating and Inhibitory Receptors
2.3.2. Beyond NK Cell Receptors
2.3.3. Therapeutic Approaches to Unleash NK Cells in Cancer
3. Plasticity of Innate Lymphoid Cells in Cancer
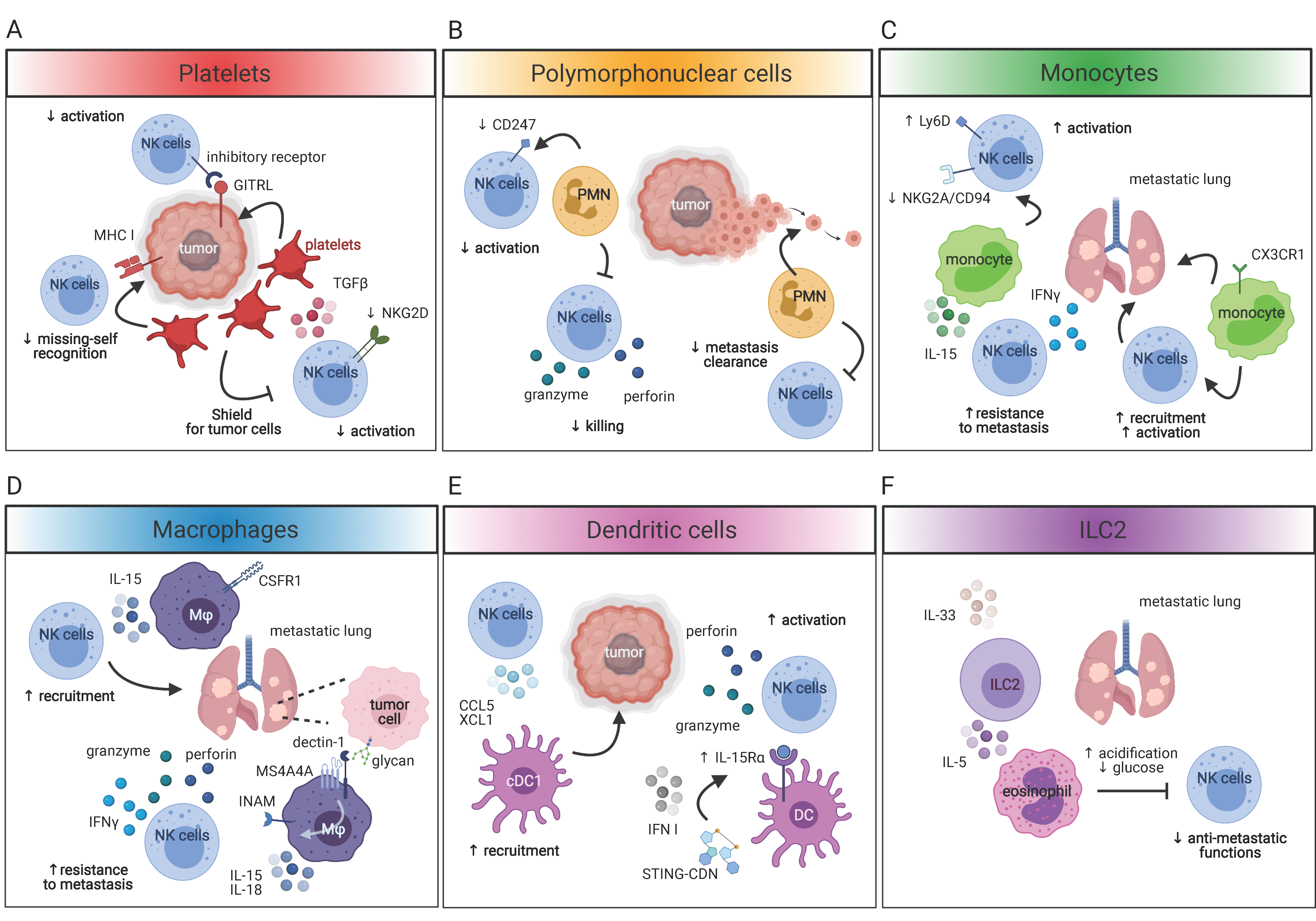
4. Immune Cell Networking in the Tumor Microenvironment
4.1. NK Cells and Platelets
4.2. NK Cells and Myeloid Cells
4.2.1. Polymorphonuclear Cells
4.2.2. Monocytes, Macrophages and DC
4.3. NK Cells and Helper-Innate Lymphoid Cells
5. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Herberman, R.B.; Nunn, M.E.; Holden, H.T.; Lavrin, D.H. Natural cytotoxic reactivity of mouse lymphoid cells against syngeneic and allogeneic tumors. II. Characterization of effector cells. Int. J. Cancer 1975, 16, 230–239. [Google Scholar] [CrossRef] [PubMed]
- Herberman, R.B.; Nunn, M.E.; Lavrin, D.H. Natural cytotoxic reactivity of mouse lymphoid cells against syngeneic acid allogeneic tumors. I. Distribution of reactivity and specificity. Int. J. Cancer 1975, 16, 216–229. [Google Scholar] [CrossRef]
- Kiessling, R.; Klein, E.; Pross, H.; Wigzell, H. “Natural” killer cells in the mouse. II. Cytotoxic cells with specificity for mouse Moloney leukemia cells. Characteristics of the killer cell. Eur. J. Immunol. 1975, 5, 117–121. [Google Scholar] [CrossRef]
- Kiessling, R.; Klein, E.; Wigzell, H. “Natural” killer cells in the mouse. I. Cytotoxic cells with specificity for mouse Moloney leukemia cells. Specificity and distribution according to genotype. Eur. J. Immunol. 1975, 5, 112–117. [Google Scholar] [CrossRef]
- Mebius, R.E.; Rennert, P.; Weissman, I.L. Developing lymph nodes collect CD4+ CD3− LTbeta+ cells that can differentiate to APC, NK cells, and follicular cells but not T or B cells. Immunity 1997, 7, 493–504. [Google Scholar] [CrossRef] [Green Version]
- Diefenbach, A.; Colonna, M.; Romagnani, C. The ILC World Revisited. Immunity 2017, 46, 327–332. [Google Scholar] [CrossRef] [Green Version]
- Spits, H.; Artis, D.; Colonna, M.; Diefenbach, A.; Di Santo, J.P.; Eberl, G.; Koyasu, S.; Locksley, R.M.; McKenzie, A.N.; Mebius, R.E.; et al. Innate lymphoid cells—A proposal for uniform nomenclature. Nat. Rev. Immunol. 2013, 13, 145–149. [Google Scholar] [CrossRef]
- Seillet, C.; Brossay, L.; Vivier, E. Natural killers or ILC1s? That is the question. Curr. Opin. Immunol. 2021, 68, 48–53. [Google Scholar] [CrossRef]
- Chiossone, L.; Dumas, P.Y.; Vienne, M.; Vivier, E. Natural killer cells and other innate lymphoid cells in cancer. Nat. Rev. Immunol. 2018, 18, 671–688. [Google Scholar] [CrossRef] [PubMed]
- Crinier, A.; Kerdiles, Y.; Vienne, M.; Cozar, B.; Vivier, E.; Berruyer, C. Multidimensional molecular controls defining NK/ILC1 identity in cancers. Semin. Immunol. 2020, 101424. [Google Scholar] [CrossRef] [PubMed]
- Molgora, M.; Cortez, V.S.; Colonna, M. Killing the Invaders: NK Cell Impact in Tumors and Anti-Tumor Therapy. Cancers 2021, 13, 595. [Google Scholar] [CrossRef]
- Diefenbach, A.; Raulet, D.H. Strategies for target cell recognition by natural killer cells. Immunol. Rev. 2001, 181, 170–184. [Google Scholar] [CrossRef] [Green Version]
- Mattiola, I.; Diefenbach, A. Innate lymphoid cells and cancer at border surfaces with the environment. Semin. Immunol. 2019, 41, 101278. [Google Scholar] [CrossRef]
- Sivori, S.; Meazza, R.; Quintarelli, C.; Carlomagno, S.; Della Chiesa, M.; Falco, M.; Moretta, L.; Locatelli, F.; Pende, D. NK Cell-Based Immunotherapy for Hematological Malignancies. J. Clin. Med. 2019, 8, 1702. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Soto, A.; Gonzalez, S.; Smyth, M.J.; Galluzzi, L. Control of Metastasis by NK Cells. Cancer Cell 2017, 32, 135–154. [Google Scholar] [CrossRef]
- Lorenzo-Herrero, S.; Lopez-Soto, A.; Sordo-Bahamonde, C.; Gonzalez-Rodriguez, A.P.; Vitale, M.; Gonzalez, S. NK Cell-Based Immunotherapy in Cancer Metastasis. Cancers 2019, 11, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delahaye, N.F.; Rusakiewicz, S.; Martins, I.; Menard, C.; Roux, S.; Lyonnet, L.; Paul, P.; Sarabi, M.; Chaput, N.; Semeraro, M.; et al. Alternatively spliced NKp30 isoforms affect the prognosis of gastrointestinal stromal tumors. Nat. Med. 2011, 17, 700–707. [Google Scholar] [CrossRef] [PubMed]
- Ishigami, S.; Natsugoe, S.; Tokuda, K.; Nakajo, A.; Che, X.; Iwashige, H.; Aridome, K.; Hokita, S.; Aikou, T. Prognostic value of intratumoral natural killer cells in gastric carcinoma. Cancer 2000, 88, 577–583. [Google Scholar] [CrossRef]
- Coca, S.; Perez-Piqueras, J.; Martinez, D.; Colmenarejo, A.; Saez, M.A.; Vallejo, C.; Martos, J.A.; Moreno, M. The prognostic significance of intratumoral natural killer cells in patients with colorectal carcinoma. Cancer 1997, 79, 2320–2328. [Google Scholar] [CrossRef]
- Donskov, F.; Von der Maase, H. Impact of immune parameters on long-term survival in metastatic renal cell carcinoma. J. Clin. Oncol. 2006, 24, 1997–2005. [Google Scholar] [CrossRef] [PubMed]
- Gannon, P.O.; Poisson, A.O.; Delvoye, N.; Lapointe, R.; Mes-Masson, A.M.; Saad, F. Characterization of the intra-prostatic immune cell infiltration in androgen-deprived prostate cancer patients. J. Immunol. Methods 2009, 348, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Roder, J.C.; Haliotis, T.; Klein, M.; Korec, S.; Jett, J.R.; Ortaldo, J.; Heberman, R.B.; Katz, P.; Fauci, A.S. A new immunodeficiency disorder in humans involving NK cells. Nature 1980, 284, 553–555. [Google Scholar] [CrossRef]
- Sullivan, J.L.; Byron, K.S.; Brewster, F.E.; Purtilo, D.T. Deficient natural killer cell activity in x-linked lymphoproliferative syndrome. Science 1980, 210, 543–545. [Google Scholar] [CrossRef] [PubMed]
- Ebbo, M.; Gerard, L.; Carpentier, S.; Vely, F.; Cypowyj, S.; Farnarier, C.; Vince, N.; Malphettes, M.; Fieschi, C.; Oksenhendler, E.; et al. Low Circulating Natural Killer Cell Counts are Associated With Severe Disease in Patients With Common Variable Immunodeficiency. EBioMedicine 2016, 6, 222–230. [Google Scholar] [CrossRef] [Green Version]
- Vely, F.; Barlogis, V.; Vallentin, B.; Neven, B.; Piperoglou, C.; Ebbo, M.; Perchet, T.; Petit, M.; Yessaad, N.; Touzot, F.; et al. Evidence of innate lymphoid cell redundancy in humans. Nat. Immunol. 2016, 17, 1291–1299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tartter, P.I.; Steinberg, B.; Barron, D.M.; Martinelli, G. The prognostic significance of natural killer cytotoxicity in patients with colorectal cancer. Arch. Surg. 1987, 122, 1264–1268. [Google Scholar] [CrossRef]
- Muntasell, A.; Rojo, F.; Servitja, S.; Rubio-Perez, C.; Cabo, M.; Tamborero, D.; Costa-Garcia, M.; Martinez-Garcia, M.; Menendez, S.; Vazquez, I.; et al. NK Cell Infiltrates and HLA Class I Expression in Primary HER2(+) Breast Cancer Predict and Uncouple Pathological Response and Disease-free Survival. Clin. Cancer Res. 2019, 25, 1535–1545. [Google Scholar] [CrossRef] [Green Version]
- Halama, N.; Braun, M.; Kahlert, C.; Spille, A.; Quack, C.; Rahbari, N.; Koch, M.; Weitz, J.; Kloor, M.; Zoernig, I.; et al. Natural killer cells are scarce in colorectal carcinoma tissue despite high levels of chemokines and cytokines. Clin. Cancer Res. 2011, 17, 678–689. [Google Scholar] [CrossRef] [Green Version]
- Ali, T.H.; Pisanti, S.; Ciaglia, E.; Mortarini, R.; Anichini, A.; Garofalo, C.; Tallerico, R.; Santinami, M.; Gulletta, E.; Ietto, C.; et al. Enrichment of CD56(dim)KIR + CD57 + highly cytotoxic NK cells in tumour-infiltrated lymph nodes of melanoma patients. Nat. Commun. 2014, 5, 5639. [Google Scholar] [CrossRef]
- Erdag, G.; Schaefer, J.T.; Smolkin, M.E.; Deacon, D.H.; Shea, S.M.; Dengel, L.T.; Patterson, J.W.; Slingluff, C.L., Jr. Immunotype and immunohistologic characteristics of tumor-infiltrating immune cells are associated with clinical outcome in metastatic melanoma. Cancer Res. 2012, 72, 1070–1080. [Google Scholar] [CrossRef] [Green Version]
- Lavin, Y.; Kobayashi, S.; Leader, A.; Amir, E.D.; Elefant, N.; Bigenwald, C.; Remark, R.; Sweeney, R.; Becker, C.D.; Levine, J.H.; et al. Innate Immune Landscape in Early Lung Adenocarcinoma by Paired Single-Cell Analyses. Cell 2017, 169, 750–765. [Google Scholar] [CrossRef] [Green Version]
- Shi, F.D.; Ljunggren, H.G.; La Cava, A.; Van Kaer, L. Organ-specific features of natural killer cells. Nat. Rev. Immunol. 2011, 11, 658–671. [Google Scholar] [CrossRef] [PubMed]
- Carrega, P.; Bonaccorsi, I.; Di Carlo, E.; Morandi, B.; Paul, P.; Rizzello, V.; Cipollone, G.; Navarra, G.; Mingari, M.C.; Moretta, L.; et al. CD56(bright)perforin(low) noncytotoxic human NK cells are abundant in both healthy and neoplastic solid tissues and recirculate to secondary lymphoid organs via afferent lymph. J. Immunol. 2014, 192, 3805–3815. [Google Scholar] [CrossRef] [Green Version]
- Gregoire, C.; Chasson, L.; Luci, C.; Tomasello, E.; Geissmann, F.; Vivier, E.; Walzer, T. The trafficking of natural killer cells. Immunol. Rev. 2007, 220, 169–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brauner, H.; Elemans, M.; Lemos, S.; Broberger, C.; Holmberg, D.; Flodstrom-Tullberg, M.; Karre, K.; Hoglund, P. Distinct phenotype and function of NK cells in the pancreas of nonobese diabetic mice. J. Immunol. 2010, 184, 2272–2280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bald, T.; Krummel, M.F.; Smyth, M.J.; Barry, K.C. The NK cell-cancer cycle: Advances and new challenges in NK cell-based immunotherapies. Nat. Immunol. 2020, 21, 835–847. [Google Scholar] [CrossRef] [PubMed]
- Carrega, P.; Morandi, B.; Costa, R.; Frumento, G.; Forte, G.; Altavilla, G.; Ratto, G.B.; Mingari, M.C.; Moretta, L.; Ferlazzo, G. Natural killer cells infiltrating human nonsmall-cell lung cancer are enriched in CD56 bright CD16(-) cells and display an impaired capability to kill tumor cells. Cancer 2008, 112, 863–875. [Google Scholar] [CrossRef]
- Wendel, M.; Galani, I.E.; Suri-Payer, E.; Cerwenka, A. Natural killer cell accumulation in tumors is dependent on IFN-gamma and CXCR3 ligands. Cancer Res. 2008, 68, 8437–8445. [Google Scholar] [CrossRef] [Green Version]
- Voshtani, R.; Song, M.; Wang, H.; Li, X.; Zhang, W.; Tavallaie, M.S.; Yan, W.; Sun, J.; Wei, F.; Ma, X. Progranulin promotes melanoma progression by inhibiting natural killer cell recruitment to the tumor microenvironment. Cancer Lett. 2019, 465, 24–35. [Google Scholar] [CrossRef] [PubMed]
- Qi, L.; Zhang, Q.; Miao, Y.; Kang, W.; Tian, Z.; Xu, D.; Xiao, W.; Fang, F. Interleukin-33 activates and recruits natural killer cells to inhibit pulmonary metastatic cancer development. Int. J. Cancer 2020, 146, 1421–1434. [Google Scholar] [CrossRef]
- O’Sullivan, T.; Saddawi-Konefka, R.; Gross, E.; Tran, M.; Mayfield, S.P.; Ikeda, H.; Bui, J.D. Interleukin-17D mediates tumor rejection through recruitment of natural killer cells. Cell Rep. 2014, 7, 989–998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, S.; Deng, Y.; Hao, J.W.; Li, Y.; Liu, B.; Yu, Y.; Shi, F.D.; Zhou, Q.H. NK cell phenotypic modulation in lung cancer environment. PLoS ONE 2014, 9, e109976. [Google Scholar] [CrossRef] [Green Version]
- De Andrade, L.F.; Lu, Y.; Luoma, A.; Ito, Y.; Pan, D.; Pyrdol, J.W.; Yoon, C.H.; Yuan, G.C.; Wucherpfennig, K.W. Discovery of specialized NK cell populations infiltrating human melanoma metastases. JCI Insight 2019, 4. [Google Scholar] [CrossRef] [Green Version]
- Peng, L.S.; Zhang, J.Y.; Teng, Y.S.; Zhao, Y.L.; Wang, T.T.; Mao, F.Y.; Lv, Y.P.; Cheng, P.; Li, W.H.; Chen, N.; et al. Tumor-Associated Monocytes/Macrophages Impair NK-Cell Function via TGFβ1 in Human Gastric Cancer. Cancer Immunol. Res. 2017, 5, 248–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Q.F.; Yin, W.W.; Xia, Y.; Yi, Y.Y.; He, Q.F.; Wang, X.; Ren, H.; Zhang, D.Z. Liver-infiltrating CD11b(-)CD27(-) NK subsets account for NK-cell dysfunction in patients with hepatocellular carcinoma and are associated with tumor progression. Cell Mol. Immunol. 2017, 14, 819–829. [Google Scholar] [CrossRef]
- Balsamo, M.; Vermi, W.; Parodi, M.; Pietra, G.; Manzini, C.; Queirolo, P.; Lonardi, S.; Augugliaro, R.; Moretta, A.; Facchetti, F.; et al. Melanoma cells become resistant to NK-cell-mediated killing when exposed to NK-cell numbers compatible with NK-cell infiltration in the tumor. Eur. J. Immunol. 2012, 42, 1833–1842. [Google Scholar] [CrossRef] [PubMed]
- Huntington, N.D.; Cursons, J.; Rautela, J. The cancer-natural killer cell immunity cycle. Nat. Rev. Cancer 2020, 20, 437–454. [Google Scholar] [CrossRef] [PubMed]
- Guerra, N.; Tan, Y.X.; Joncker, N.T.; Choy, A.; Gallardo, F.; Xiong, N.; Knoblaugh, S.; Cado, D.; Greenberg, N.M.; Raulet, D.H. NKG2D-deficient mice are defective in tumor surveillance in models of spontaneous malignancy. Immunity 2008, 28, 571–580. [Google Scholar] [CrossRef] [Green Version]
- Kaiser, B.K.; Yim, D.; Chow, I.T.; Gonzalez, S.; Dai, Z.; Mann, H.H.; Strong, R.K.; Groh, V.; Spies, T. Disulphide-isomerase-enabled shedding of tumour-associated NKG2D ligands. Nature 2007, 447, 482–486. [Google Scholar] [CrossRef]
- Zingoni, A.; Molfetta, R.; Fionda, C.; Soriani, A.; Paolini, R.; Cippitelli, M.; Cerboni, C.; Santoni, A. NKG2D and Its Ligands: “One for All, All for One”. Front. Immunol. 2018, 9, 476. [Google Scholar] [CrossRef]
- Groh, V.; Wu, J.; Yee, C.; Spies, T. Tumour-derived soluble MIC ligands impair expression of NKG2D and T-cell activation. Nature 2002, 419, 734–738. [Google Scholar] [CrossRef]
- Fiegler, N.; Textor, S.; Arnold, A.; Rolle, A.; Oehme, I.; Breuhahn, K.; Moldenhauer, G.; Witzens-Harig, M.; Cerwenka, A. Downregulation of the activating NKp30 ligand B7-H6 by HDAC inhibitors impairs tumor cell recognition by NK cells. Blood 2013, 122, 684–693. [Google Scholar] [CrossRef] [PubMed]
- Schlecker, E.; Fiegler, N.; Arnold, A.; Altevogt, P.; Rose-John, S.; Moldenhauer, G.; Sucker, A.; Paschen, A.; Von Strandmann, E.P.; Textor, S.; et al. Metalloprotease-mediated tumor cell shedding of B7-H6, the ligand of the natural killer cell-activating receptor NKp30. Cancer Res. 2014, 74, 3429–3440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glasner, A.; Ghadially, H.; Gur, C.; Stanietsky, N.; Tsukerman, P.; Enk, J.; Mandelboim, O. Recognition and prevention of tumor metastasis by the NK receptor NKp46/NCR1. J. Immunol. 2012, 188, 2509–2515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cagnano, E.; Hershkovitz, O.; Zilka, A.; Bar-Ilan, A.; Golder, A.; Sion-Vardy, N.; Bogdanov-Berezovsky, A.; Mandelboim, O.; Benharroch, D.; Porgador, A. Expression of ligands to NKp46 in benign and malignant melanocytes. J. Investig. Dermatol. 2008, 128, 972–979. [Google Scholar] [CrossRef] [Green Version]
- Niehrs, A.; Garcia-Beltran, W.F.; Norman, P.J.; Watson, G.M.; Holzemer, A.; Chapel, A.; Richert, L.; Pommerening-Roser, A.; Korner, C.; Ozawa, M.; et al. A subset of HLA-DP molecules serve as ligands for the natural cytotoxicity receptor NKp44. Nat. Immunol. 2019, 20, 1129–1137. [Google Scholar] [CrossRef]
- Bottino, C.; Castriconi, R.; Pende, D.; Rivera, P.; Nanni, M.; Carnemolla, B.; Cantoni, C.; Grassi, J.; Marcenaro, S.; Reymond, N.; et al. Identification of PVR (CD155) and Nectin-2 (CD112) as cell surface ligands for the human DNAM-1 (CD226) activating molecule. J. Exp. Med. 2003, 198, 557–567. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Bi, J.; Zheng, X.; Chen, Y.; Wang, H.; Wu, W.; Wang, Z.; Wu, Q.; Peng, H.; Wei, H.; et al. Blockade of the checkpoint receptor TIGIT prevents NK cell exhaustion and elicits potent anti-tumor immunity. Nat. Immunol. 2018, 19, 723–732. [Google Scholar] [CrossRef]
- Mamessier, E.; Sylvain, A.; Thibult, M.L.; Houvenaeghel, G.; Jacquemier, J.; Castellano, R.; Goncalves, A.; Andre, P.; Romagne, F.; Thibault, G.; et al. Human breast cancer cells enhance self tolerance by promoting evasion from NK cell antitumor immunity. J. Clin. Investig. 2011, 121, 3609–3622. [Google Scholar] [CrossRef] [Green Version]
- Castriconi, R.; Dondero, A.; Bellora, F.; Moretta, L.; Castellano, A.; Locatelli, F.; Corrias, M.V.; Moretta, A.; Bottino, C. Neuroblastoma-derived TGF-β1 modulates the chemokine receptor repertoire of human resting NK cells. J. Immunol. 2013, 190, 5321–5328. [Google Scholar] [CrossRef] [Green Version]
- Zheng, X.; Qian, Y.; Fu, B.; Jiao, D.; Jiang, Y.; Chen, P.; Shen, Y.; Zhang, H.; Sun, R.; Tian, Z.; et al. Mitochondrial fragmentation limits NK cell-based tumor immunosurveillance. Nat. Immunol. 2019, 20, 1656–1667. [Google Scholar] [CrossRef]
- Terren, I.; Orrantia, A.; Vitalle, J.; Zenarruzabeitia, O.; Borrego, F. NK Cell Metabolism and Tumor Microenvironment. Front. Immunol. 2019, 10, 2278. [Google Scholar] [CrossRef] [PubMed]
- Gubbels, J.A.; Felder, M.; Horibata, S.; Belisle, J.A.; Kapur, A.; Holden, H.; Petrie, S.; Migneault, M.; Rancourt, C.; Connor, J.P.; et al. MUC16 provides immune protection by inhibiting synapse formation between NK and ovarian tumor cells. Mol. Cancer 2010, 9, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrari de Andrade, L.; Tay, R.E.; Pan, D.; Luoma, A.M.; Ito, Y.; Badrinath, S.; Tsoucas, D.; Franz, B.; May, K.F., Jr.; Harvey, C.J.; et al. Antibody-mediated inhibition of MICA and MICB shedding promotes NK cell-driven tumor immunity. Science 2018, 359, 1537–1542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gauthier, L.; Morel, A.; Anceriz, N.; Rossi, B.; Blanchard-Alvarez, A.; Grondin, G.; Trichard, S.; Cesari, C.; Sapet, M.; Bosco, F.; et al. Multifunctional Natural Killer Cell Engagers Targeting NKp46 Trigger Protective Tumor Immunity. Cell 2019, 177, 1701–1713. [Google Scholar] [CrossRef] [PubMed]
- Andre, P.; Denis, C.; Soulas, C.; Bourbon-Caillet, C.; Lopez, J.; Arnoux, T.; Blery, M.; Bonnafous, C.; Gauthier, L.; Morel, A.; et al. Anti-NKG2A mAb Is a Checkpoint Inhibitor that Promotes Anti-tumor Immunity by Unleashing Both T and NK Cells. Cell 2018, 175, 1731–1743. [Google Scholar] [CrossRef] [Green Version]
- Nath, P.R.; Gangaplara, A.; Pal-Nath, D.; Mandal, A.; Maric, D.; Sipes, J.M.; Cam, M.; Shevach, E.M.; Roberts, D.D. CD47 Expression in Natural Killer Cells Regulates Homeostasis and Modulates Immune Response to Lymphocytic Choriomeningitis Virus. Front. Immunol. 2018, 9, 2985. [Google Scholar] [CrossRef] [Green Version]
- Nath, P.R.; Pal-Nath, D.; Mandal, A.; Cam, M.C.; Schwartz, A.L.; Roberts, D.D. Natural Killer Cell Recruitment and Activation Are Regulated by CD47 Expression in the Tumor Microenvironment. Cancer Immunol. Res. 2019, 7, 1547–1561. [Google Scholar] [CrossRef] [Green Version]
- Hatfield, S.M.; Kjaergaard, J.; Lukashev, D.; Schreiber, T.H.; Belikoff, B.; Abbott, R.; Sethumadhavan, S.; Philbrook, P.; Ko, K.; Cannici, R.; et al. Immunological mechanisms of the antitumor effects of supplemental oxygenation. Sci. Transl. Med. 2015, 7, 277ra230. [Google Scholar] [CrossRef] [Green Version]
- Vijayan, D.; Young, A.; Teng, M.W.L.; Smyth, M.J. Targeting immunosuppressive adenosine in cancer. Nat. Rev. Cancer 2017, 17, 709–724. [Google Scholar] [CrossRef]
- Young, A.; Ngiow, S.F.; Gao, Y.; Patch, A.M.; Barkauskas, D.S.; Messaoudene, M.; Lin, G.; Coudert, J.D.; Stannard, K.A.; Zitvogel, L.; et al. A2AR Adenosine Signaling Suppresses Natural Killer Cell Maturation in the Tumor Microenvironment. Cancer Res. 2018, 78, 1003–1016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allard, B.; Allard, D.; Buisseret, L.; Stagg, J. The adenosine pathway in immuno-oncology. Nat. Rev. Clin. Oncol 2020, 17, 611–629. [Google Scholar] [CrossRef]
- Delconte, R.B.; Kolesnik, T.B.; Dagley, L.F.; Rautela, J.; Shi, W.; Putz, E.M.; Stannard, K.; Zhang, J.G.; Teh, C.; Firth, M.; et al. CIS is a potent checkpoint in NK cell-mediated tumor immunity. Nat. Immunol. 2016, 17, 816–824. [Google Scholar] [CrossRef]
- Molgora, M.; Bonavita, E.; Ponzetta, A.; Riva, F.; Barbagallo, M.; Jaillon, S.; Popovic, B.; Bernardini, G.; Magrini, E.; Gianni, F.; et al. IL-1R8 is a checkpoint in NK cells regulating anti-tumour and anti-viral activity. Nature 2017, 551, 110–114. [Google Scholar] [CrossRef]
- Bernink, J.H.; Peters, C.P.; Munneke, M.; Te Velde, A.A.; Meijer, S.L.; Weijer, K.; Hreggvidsdottir, H.S.; Heinsbroek, S.E.; Legrand, N.; Buskens, C.J.; et al. Human type 1 innate lymphoid cells accumulate in inflamed mucosal tissues. Nat. Immunol. 2013, 14, 221–229. [Google Scholar] [CrossRef]
- Bal, S.M.; Bernink, J.H.; Nagasawa, M.; Groot, J.; Shikhagaie, M.M.; Golebski, K.; Van Drunen, C.M.; Lutter, R.; Jonkers, R.E.; Hombrink, P.; et al. IL-1beta, IL-4 and IL-12 control the fate of group 2 innate lymphoid cells in human airway inflammation in the lungs. Nat. Immunol. 2016, 17, 636–645. [Google Scholar] [CrossRef]
- Gao, Y.; Souza-Fonseca-Guimaraes, F.; Bald, T.; Ng, S.S.; Young, A.; Ngiow, S.F.; Rautela, J.; Straube, J.; Waddell, N.; Blake, S.J.; et al. Tumor immunoevasion by the conversion of effector NK cells into type 1 innate lymphoid cells. Nat. Immunol. 2017, 18, 1004–1015. [Google Scholar] [CrossRef]
- Cortez, V.S.; Ulland, T.K.; Cervantes-Barragan, L.; Bando, J.K.; Robinette, M.L.; Wang, Q.; White, A.J.; Gilfillan, S.; Cella, M.; Colonna, M. SMAD4 impedes the conversion of NK cells into ILC1-like cells by curtailing non-canonical TGF-beta signaling. Nat. Immunol. 2017, 18, 995–1003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stokic-Trtica, V.; Diefenbach, A.; Klose, C.S.N. NK Cell Development in Times of Innate Lymphoid Cell Diversity. Front. Immunol. 2020, 11, 813. [Google Scholar] [CrossRef]
- Spits, H.; Bernink, J.H.; Lanier, L. NK cells and type 1 innate lymphoid cells: Partners in host defense. Nat. Immunol. 2016, 17, 758–764. [Google Scholar] [CrossRef]
- Rautela, J.; Dagley, L.F.; De Oliveira, C.C.; Schuster, I.S.; Hediyeh-Zadeh, S.; Delconte, R.B.; Cursons, J.; Hennessy, R.; Hutchinson, D.S.; Harrison, C.; et al. Therapeutic blockade of activin-A improves NK cell function and antitumor immunity. Sci. Signal. 2019, 12. [Google Scholar] [CrossRef] [Green Version]
- Klose, C.S.N.; Flach, M.; Mohle, L.; Rogell, L.; Hoyler, T.; Ebert, K.; Fabiunke, C.; Pfeifer, D.; Sexl, V.; Fonseca-Pereira, D.; et al. Differentiation of type 1 ILCs from a common progenitor to all helper-like innate lymphoid cell lineages. Cell 2014, 157, 340–356. [Google Scholar] [CrossRef] [Green Version]
- Klose, C.S.; Kiss, E.A.; Schwierzeck, V.; Ebert, K.; Hoyler, T.; D’Hargues, Y.; Goppert, N.; Croxford, A.L.; Waisman, A.; Tanriver, Y.; et al. A T-bet gradient controls the fate and function of CCR6-RORgammat+ innate lymphoid cells. Nature 2013, 494, 261–265. [Google Scholar] [CrossRef]
- McFarland, A.P.; Yalin, A.; Wang, S.Y.; Cortez, V.S.; Landsberger, T.; Sudan, R.; Peng, V.; Miller, H.L.; Ricci, B.; David, E.; et al. Multi-tissue single-cell analysis deconstructs the complex programs of mouse natural killer and type 1 innate lymphoid cells in tissues and circulation. Immunity 2021. [Google Scholar] [CrossRef] [PubMed]
- Haemmerle, M.; Stone, R.L.; Menter, D.G.; Afshar-Kharghan, V.; Sood, A.K. The Platelet Lifeline to Cancer: Challenges and Opportunities. Cancer Cell 2018, 33, 965–983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Philippe, C.; Philippe, B.; Fouqueray, B.; Perez, J.; Lebret, M.; Baud, L. Protection from tumor necrosis factor-mediated cytolysis by platelets. Am. J. Pathol. 1993, 143, 1713–1723. [Google Scholar]
- Nieswandt, B.; Hafner, M.; Echtenacher, B.; Mannel, D.N. Lysis of tumor cells by natural killer cells in mice is impeded by platelets. Cancer Res. 1999, 59, 1295–1300. [Google Scholar] [PubMed]
- Kim, Y.J.; Borsig, L.; Varki, N.M.; Varki, A. P-selectin deficiency attenuates tumor growth and metastasis. Proc. Natl. Acad. Sci. USA 1998, 95, 9325–9330. [Google Scholar] [CrossRef] [Green Version]
- Kopp, H.G.; Placke, T.; Salih, H.R. Platelet-derived transforming growth factor-beta down-regulates NKG2D thereby inhibiting natural killer cell antitumor reactivity. Cancer Res. 2009, 69, 7775–7783. [Google Scholar] [CrossRef] [Green Version]
- Maurer, S.; Kropp, K.N.; Klein, G.; Steinle, A.; Haen, S.P.; Walz, J.S.; Hinterleitner, C.; Marklin, M.; Kopp, H.G.; Salih, H.R. Platelet-mediated shedding of NKG2D ligands impairs NK cell immune-surveillance of tumor cells. Oncoimmunology 2018, 7, e1364827. [Google Scholar] [CrossRef] [Green Version]
- Placke, T.; Orgel, M.; Schaller, M.; Jung, G.; Rammensee, H.G.; Kopp, H.G.; Salih, H.R. Platelet-derived MHC class I confers a pseudonormal phenotype to cancer cells that subverts the antitumor reactivity of natural killer immune cells. Cancer Res. 2012, 72, 440–448. [Google Scholar] [CrossRef] [Green Version]
- Placke, T.; Salih, H.R.; Kopp, H.G. GITR ligand provided by thrombopoietic cells inhibits NK cell antitumor activity. J. Immunol. 2012, 189, 154–160. [Google Scholar] [CrossRef] [Green Version]
- Palumbo, J.S.; Talmage, K.E.; Massari, J.V.; La Jeunesse, C.M.; Flick, M.J.; Kombrinck, K.W.; Hu, Z.; Barney, K.A.; Degen, J.L. Tumor cell-associated tissue factor and circulating hemostatic factors cooperate to increase metastatic potential through natural killer cell-dependent and-independent mechanisms. Blood 2007, 110, 133–141. [Google Scholar] [CrossRef] [Green Version]
- Poleto Spinola, L.; Vieira, G.F.; Fernandes Ferreira, R.; Calastri, M.C.J.; Tenani, G.D.; Aguiar, F.L.; Santana Ferreira Boin, I.F.; Costa, L.B.; Chaim Correia, M.F.; Zanovelo, E.M.; et al. Underexpression of miR-126-3p in Patients with Cholangiocarcinoma. Asian Pac. J. Cancer Prev. 2021, 22, 573–579. [Google Scholar] [CrossRef]
- Yang, Y.; Li, C.; Liu, T.; Dai, X.; Bazhin, A.V. Myeloid-Derived Suppressor Cells in Tumors: From Mechanisms to Antigen Specificity and Microenvironmental Regulation. Front. Immunol. 2020, 11, 1371. [Google Scholar] [CrossRef]
- Veglia, F.; Sanseviero, E.; Gabrilovich, D.I. Myeloid-derived suppressor cells in the era of increasing myeloid cell diversity. Nat. Rev. Immunol. 2021. [Google Scholar] [CrossRef]
- Bronte, V.; Brandau, S.; Chen, S.H.; Colombo, M.P.; Frey, A.B.; Greten, T.F.; Mandruzzato, S.; Murray, P.J.; Ochoa, A.; Ostrand-Rosenberg, S.; et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat. Commun. 2016, 7, 12150. [Google Scholar] [CrossRef] [Green Version]
- Vaknin, I.; Blinder, L.; Wang, L.; Gazit, R.; Shapira, E.; Genina, O.; Pines, M.; Pikarsky, E.; Baniyash, M. A common pathway mediated through Toll-like receptors leads to T- and natural killer-cell immunosuppression. Blood 2008, 111, 1437–1447. [Google Scholar] [CrossRef]
- Bruno, A.; Mortara, L.; Baci, D.; Noonan, D.M.; Albini, A. Myeloid Derived Suppressor Cells Interactions With Natural Killer Cells and Pro-angiogenic Activities: Roles in Tumor Progression. Front. Immunol. 2019, 10, 771. [Google Scholar] [CrossRef]
- Hoechst, B.; Voigtlaender, T.; Ormandy, L.; Gamrekelashvili, J.; Zhao, F.; Wedemeyer, H.; Lehner, F.; Manns, M.P.; Greten, T.F.; Korangy, F. Myeloid derived suppressor cells inhibit natural killer cells in patients with hepatocellular carcinoma via the NKp30 receptor. Hepatology 2009, 50, 799–807. [Google Scholar] [CrossRef]
- Spiegel, A.; Brooks, M.W.; Houshyar, S.; Reinhardt, F.; Ardolino, M.; Fessler, E.; Chen, M.B.; Krall, J.A.; DeCock, J.; Zervantonakis, I.K.; et al. Neutrophils Suppress Intraluminal NK Cell-Mediated Tumor Cell Clearance and Enhance Extravasation of Disseminated Carcinoma Cells. Cancer Discov. 2016, 6, 630–649. [Google Scholar] [CrossRef] [Green Version]
- Sceneay, J.; Chow, M.T.; Chen, A.; Halse, H.M.; Wong, C.S.; Andrews, D.M.; Sloan, E.K.; Parker, B.S.; Bowtell, D.D.; Smyth, M.J.; et al. Primary tumor hypoxia recruits CD11b+/Ly6Cmed/Ly6G+ immune suppressor cells and compromises NK cell cytotoxicity in the premetastatic niche. Cancer Res. 2012, 72, 3906–3911. [Google Scholar] [CrossRef] [Green Version]
- Nausch, N.; Galani, I.E.; Schlecker, E.; Cerwenka, A. Mononuclear myeloid-derived "suppressor" cells express RAE-1 and activate natural killer cells. Blood 2008, 112, 4080–4089. [Google Scholar] [CrossRef]
- Gaggero, S.; Witt, K.; Carlsten, M.; Mitra, S. Cytokines Orchestrating the Natural Killer-Myeloid Cell Crosstalk in the Tumor Microenvironment: Implications for Natural Killer Cell-Based Cancer Immunotherapy. Front. Immunol. 2020, 11, 621225. [Google Scholar] [CrossRef]
- Galluzzi, L.; Buque, A.; Kepp, O.; Zitvogel, L.; Kroemer, G. Immunogenic cell death in cancer and infectious disease. Nat. Rev. Immunol. 2017, 17, 97–111. [Google Scholar] [CrossRef]
- Chiba, S.; Ikushima, H.; Ueki, H.; Yanai, H.; Kimura, Y.; Hangai, S.; Nishio, J.; Negishi, H.; Tamura, T.; Saijo, S.; et al. Recognition of tumor cells by Dectin-1 orchestrates innate immune cells for anti-tumor responses. Elife 2014, 3, e04177. [Google Scholar] [CrossRef]
- Mattiola, I.; Tomay, F.; De Pizzol, M.; Silva-Gomes, R.; Savino, B.; Gulic, T.; Doni, A.; Lonardi, S.; Astrid Boutet, M.; Nerviani, A.; et al. The macrophage tetraspan MS4A4A enhances dectin-1-dependent NK cell-mediated resistance to metastasis. Nat. Immunol. 2019, 20, 1012–1022. [Google Scholar] [CrossRef] [PubMed]
- Soderquest, K.; Powell, N.; Luci, C.; Van Rooijen, N.; Hidalgo, A.; Geissmann, F.; Walzer, T.; Lord, G.M.; Martin-Fontecha, A. Monocytes control natural killer cell differentiation to effector phenotypes. Blood 2011, 117, 4511–4518. [Google Scholar] [CrossRef] [Green Version]
- Hanna, R.N.; Cekic, C.; Sag, D.; Tacke, R.; Thomas, G.D.; Nowyhed, H.; Herrley, E.; Rasquinha, N.; McArdle, S.; Wu, R.; et al. Patrolling monocytes control tumor metastasis to the lung. Science 2015, 350, 985–990. [Google Scholar] [CrossRef] [Green Version]
- Kubo, H.; Mensurado, S.; Goncalves-Sousa, N.; Serre, K.; Silva-Santos, B. Primary Tumors Limit Metastasis Formation through Induction of IL15-Mediated Cross-Talk between Patrolling Monocytes and NK Cells. Cancer Immunol. Res. 2017, 5, 812–820. [Google Scholar] [CrossRef] [Green Version]
- Narasimhan, P.B.; Eggert, T.; Zhu, Y.P.; Marcovecchio, P.; Meyer, M.A.; Wu, R.; Hedrick, C.C. Patrolling Monocytes Control NK Cell Expression of Activating and Stimulatory Receptors to Curtail Lung Metastases. J. Immunol. 2020, 204, 192–198. [Google Scholar] [CrossRef]
- Beffinger, M.; Tallon de Lara, P.; Tugues, S.; Vermeer, M.; Montagnolo, Y.; Ohs, I.; Cecconi, V.; Lucchiari, G.; Gagliardi, A.; Misljencevic, N.; et al. CSF1R-dependent myeloid cells are required for NKmediated control of metastasis. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bodder, J.; Zahan, T.; Van Slooten, R.; Schreibelt, G.; De Vries, I.J.M.; Florez-Grau, G. Harnessing the cDC1-NK Cross-Talk in the Tumor Microenvironment to Battle Cancer. Front. Immunol. 2020, 11, 631713. [Google Scholar] [CrossRef]
- Nicolai, C.J.; Wolf, N.; Chang, I.C.; Kirn, G.; Marcus, A.; Ndubaku, C.O.; McWhirter, S.M.; Raulet, D.H. NK cells mediate clearance of CD8(+) T cell-resistant tumors in response to STING agonists. Sci. Immunol. 2020, 5. [Google Scholar] [CrossRef] [PubMed]
- Lucas, M.; Schachterle, W.; Oberle, K.; Aichele, P.; Diefenbach, A. Dendritic cells prime natural killer cells by trans-presenting interleukin 15. Immunity 2007, 26, 503–517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bottcher, J.P.; Bonavita, E.; Chakravarty, P.; Blees, H.; Cabeza-Cabrerizo, M.; Sammicheli, S.; Rogers, N.C.; Sahai, E.; Zelenay, S.; Reis, E.S.C. NK Cells Stimulate Recruitment of cDC1 into the Tumor Microenvironment Promoting Cancer Immune Control. Cell 2018, 172, 1022–1037. [Google Scholar] [CrossRef] [Green Version]
- Bonavita, E.; Bromley, C.P.; Jonsson, G.; Pelly, V.S.; Sahoo, S.; Walwyn-Brown, K.; Mensurado, S.; Moeini, A.; Flanagan, E.; Bell, C.R.; et al. Antagonistic Inflammatory Phenotypes Dictate Tumor Fate and Response to Immune Checkpoint Blockade. Immunity 2020, 53, 1215–1229. [Google Scholar] [CrossRef]
- Kim, T.J.; Upadhyay, V.; Kumar, V.; Lee, K.M.; Fu, Y.X. Innate lymphoid cells facilitate NK cell development through a lymphotoxin-mediated stromal microenvironment. J. Exp. Med. 2014, 211, 1421–1431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shields, J.D.; Kourtis, I.C.; Tomei, A.A.; Roberts, J.M.; Swartz, M.A. Induction of lymphoidlike stroma and immune escape by tumors that express the chemokine CCL21. Science 2010, 328, 749–752. [Google Scholar] [CrossRef] [Green Version]
- Eisenring, M.; Vom Berg, J.; Kristiansen, G.; Saller, E.; Becher, B. IL-12 initiates tumor rejection via lymphoid tissue-inducer cells bearing the natural cytotoxicity receptor NKp46. Nat. Immunol. 2010, 11, 1030–1038. [Google Scholar] [CrossRef]
- Schuijs, M.J.; Png, S.; Richard, A.C.; Tsyben, A.; Hamm, G.; Stockis, J.; Garcia, C.; Pinaud, S.; Nicholls, A.; Ros, X.R.; et al. ILC2-driven innate immune checkpoint mechanism antagonizes NK cell antimetastatic function in the lung. Nat. Immunol. 2020, 21, 998–1009. [Google Scholar] [CrossRef] [PubMed]
- Farber, D.L. Tissues, not blood, are where immune cells function. Nature 2021, 593, 506–509. [Google Scholar] [CrossRef] [PubMed]
- Dogra, P.; Rancan, C.; Ma, W.; Toth, M.; Senda, T.; Carpenter, D.J.; Kubota, M.; Matsumoto, R.; Thapa, P.; Szabo, P.A.; et al. Tissue Determinants of Human NK Cell Development, Function, and Residence. Cell 2020, 180, 749–763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mattiola, I. Immune Circuits to Shape Natural Killer Cells in Cancer. Cancers 2021, 13, 3225. https://doi.org/10.3390/cancers13133225
Mattiola I. Immune Circuits to Shape Natural Killer Cells in Cancer. Cancers. 2021; 13(13):3225. https://doi.org/10.3390/cancers13133225
Chicago/Turabian StyleMattiola, Irene. 2021. "Immune Circuits to Shape Natural Killer Cells in Cancer" Cancers 13, no. 13: 3225. https://doi.org/10.3390/cancers13133225
APA StyleMattiola, I. (2021). Immune Circuits to Shape Natural Killer Cells in Cancer. Cancers, 13(13), 3225. https://doi.org/10.3390/cancers13133225