
Depletion of DNA Polymerase Theta Inhibits Tumor Growth and Promotes Genome Instability through the cGAS-STING-ISG Pathway in Esophageal Squamous Cell Carcinoma

 , ,
, ,  and
and 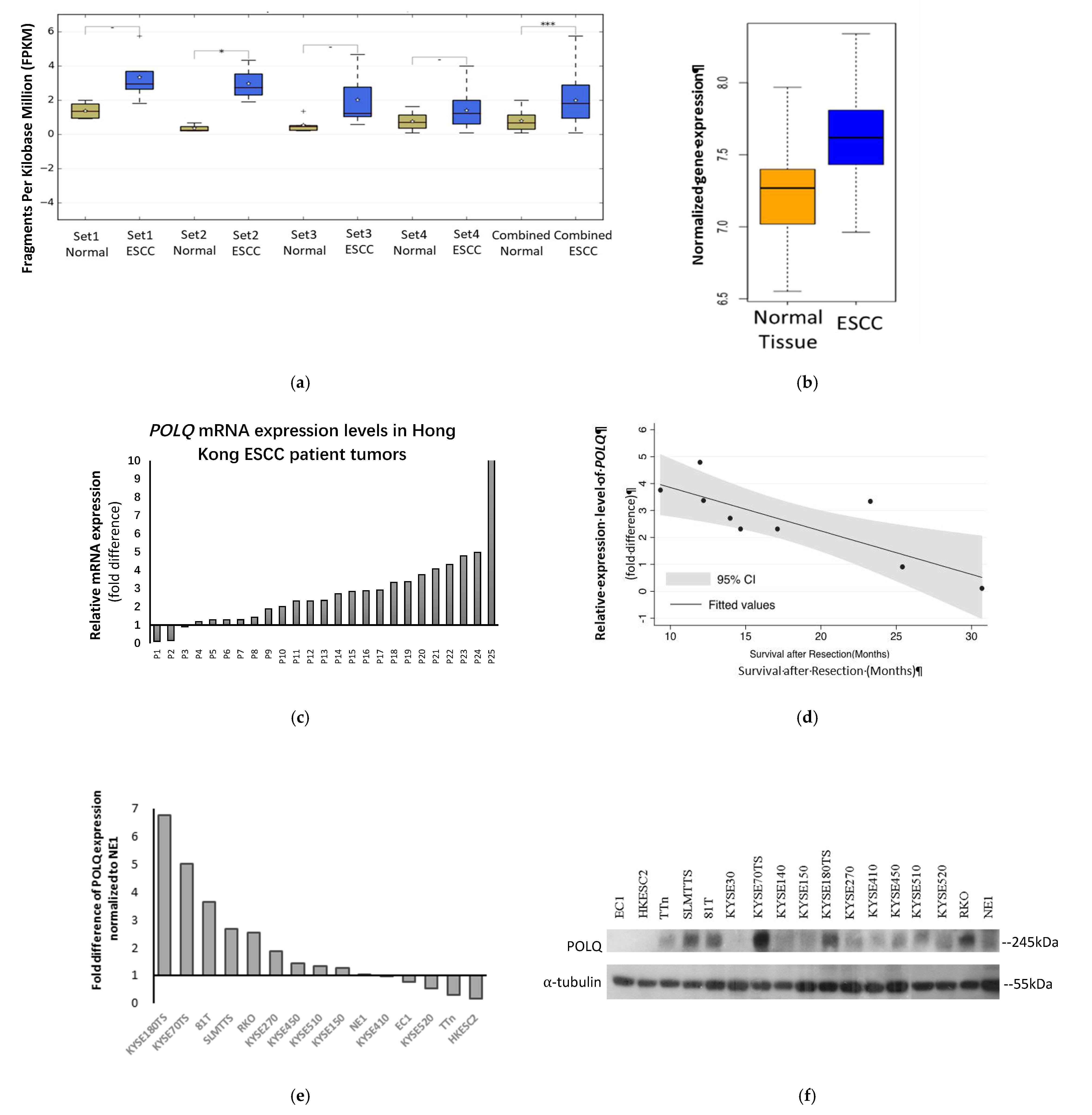{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Clinical Specimens
2.2. RNA Sequence Analysis
2.3. Cell Lines
2.4. Plasmids and Lentivirus Preparation and Infection
2.5. RNA Isolation and Real-Time Quantitative Polymerase Chain Reaction
2.6. Protein Extraction and Western Blot Analysis
2.7. MTT Assay Following Genotoxic Drug Treatments
2.8. Colony Formation Assay
2.9. Ionizing Radiation
2.10. Immunofluorescence (IF) Staining and Confocal Microscopy
2.11. Single Cell Gel Electrophoresis Assay (Comet Assay)
2.12. Enzyme-Linked Immunosorbent Assay (ELISA)
2.13. In Vivo Tumorigenicity Assay
2.14. Statistical Analysis
3. Results
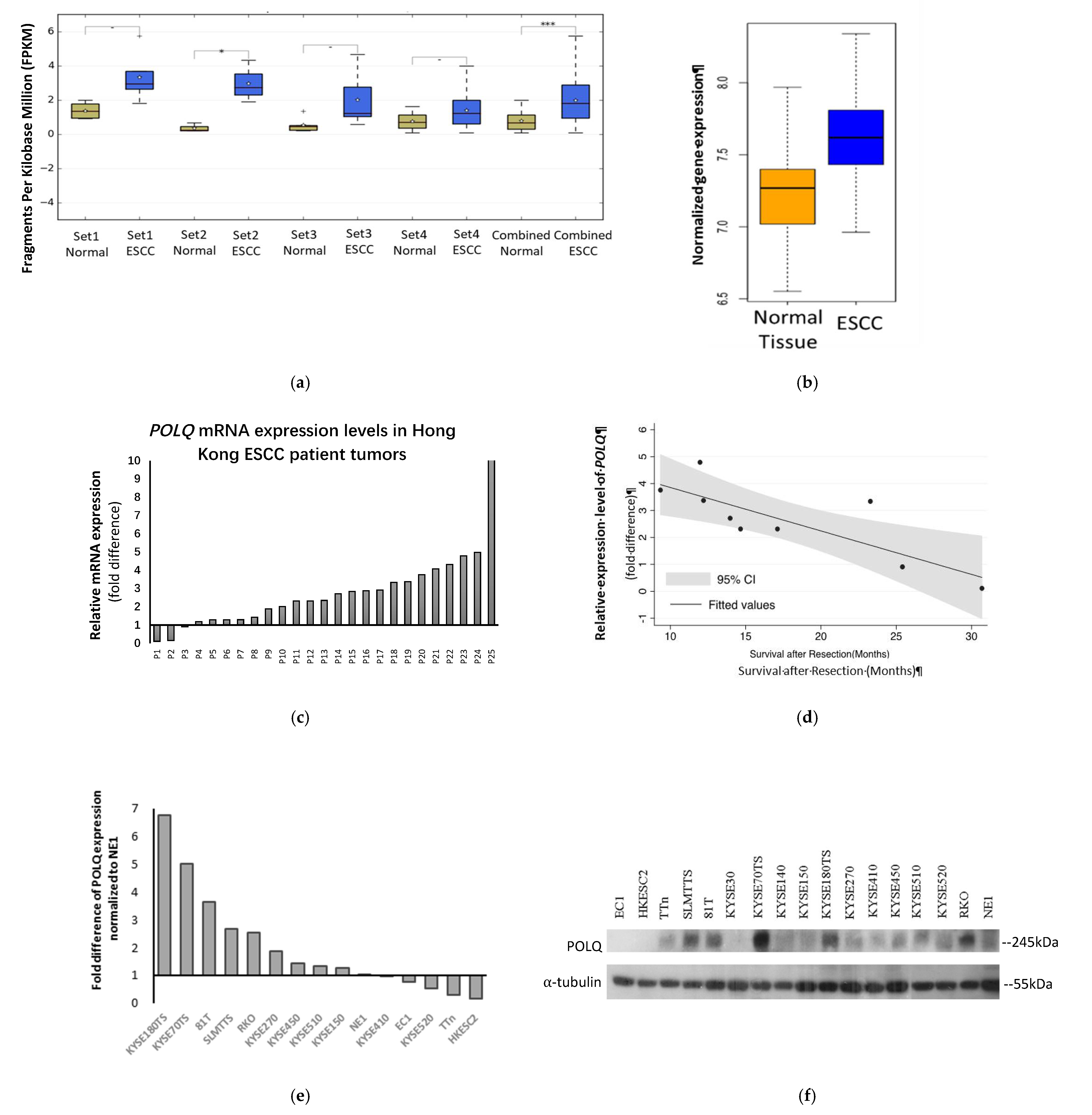
3.1. POLQ Is Upregulated in ESCC and Correlates with Unfavorable Clinical Outcome
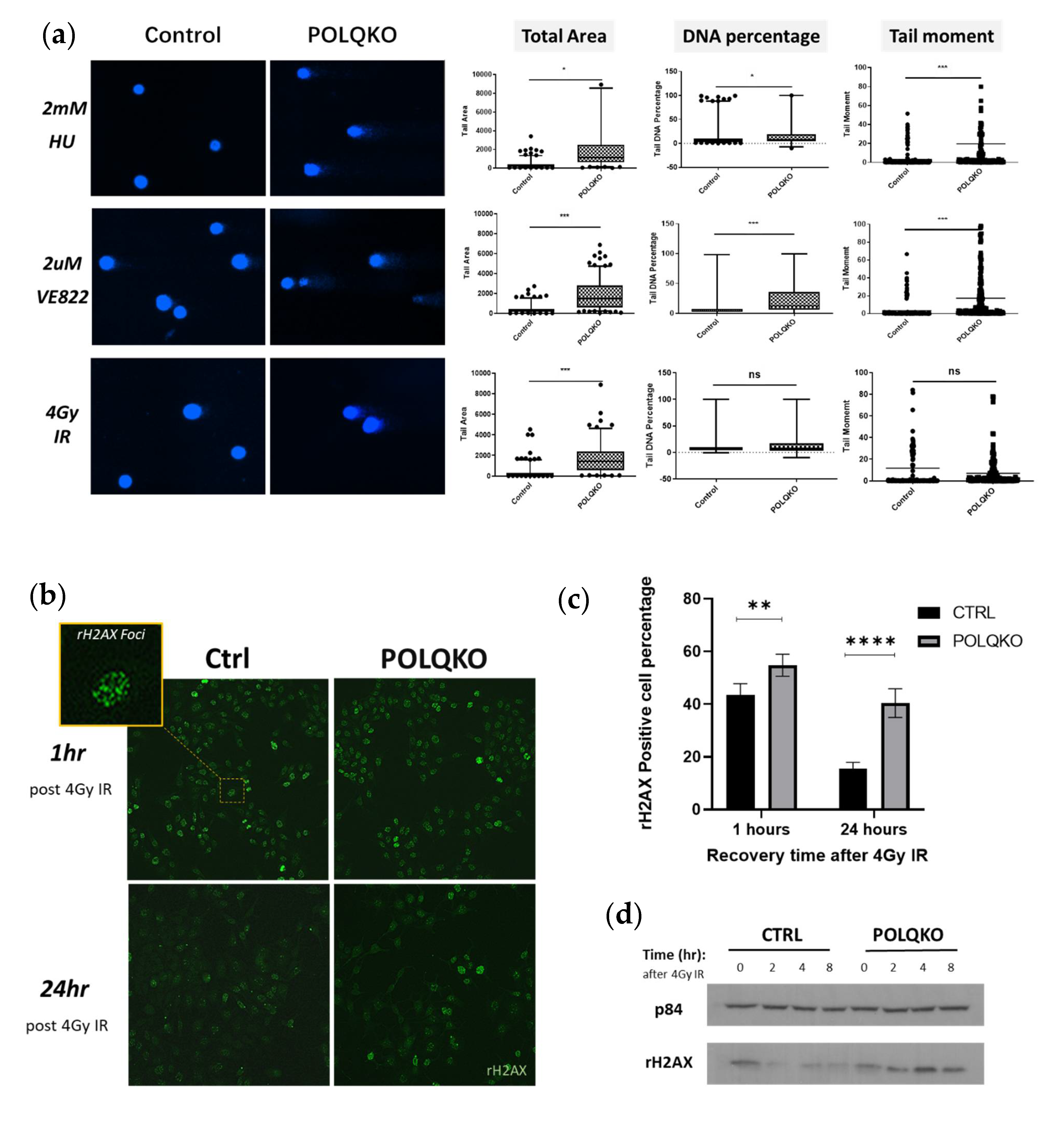
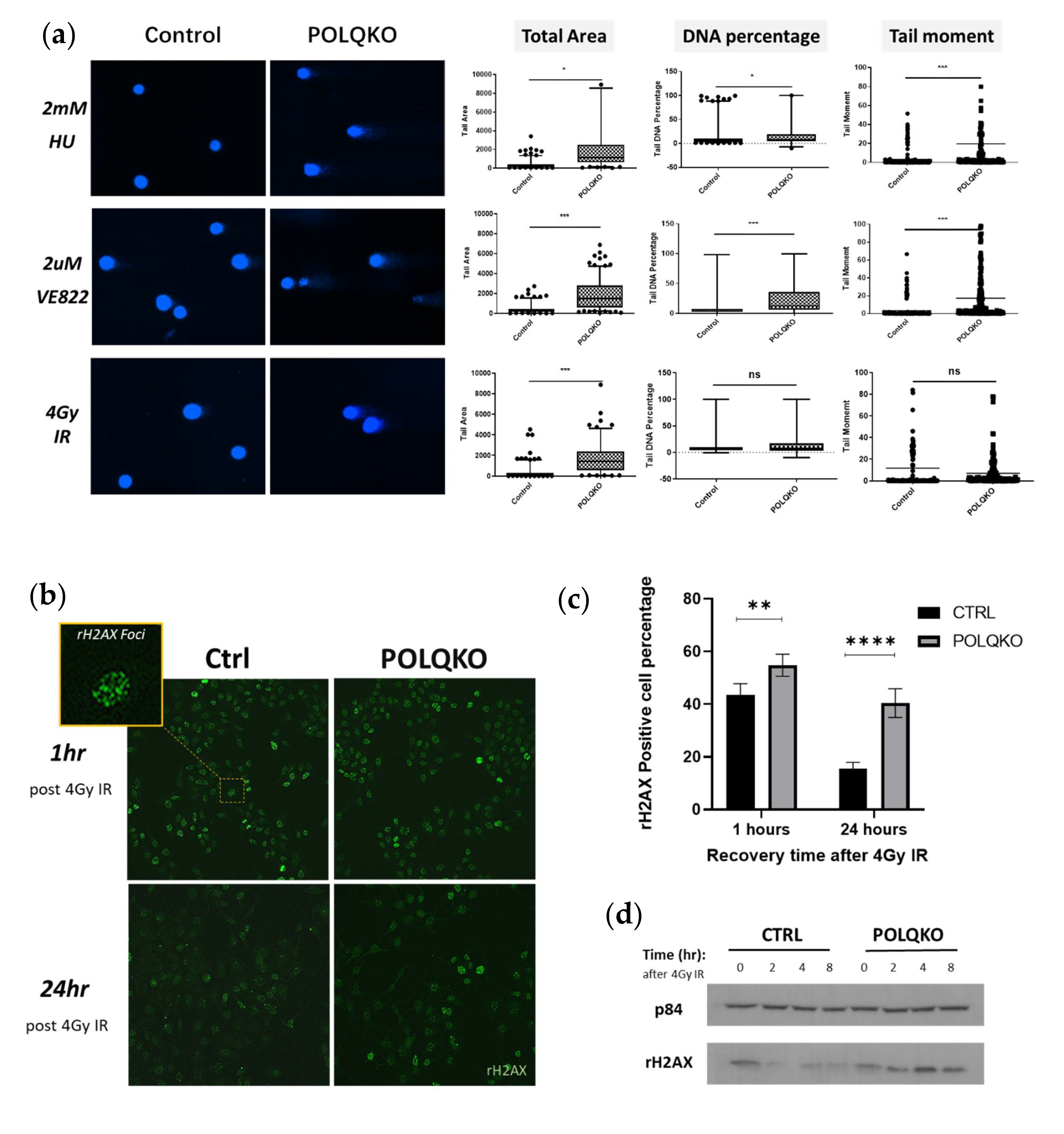
3.2. POLQ Maintains Genome Stability in ESCC Cells
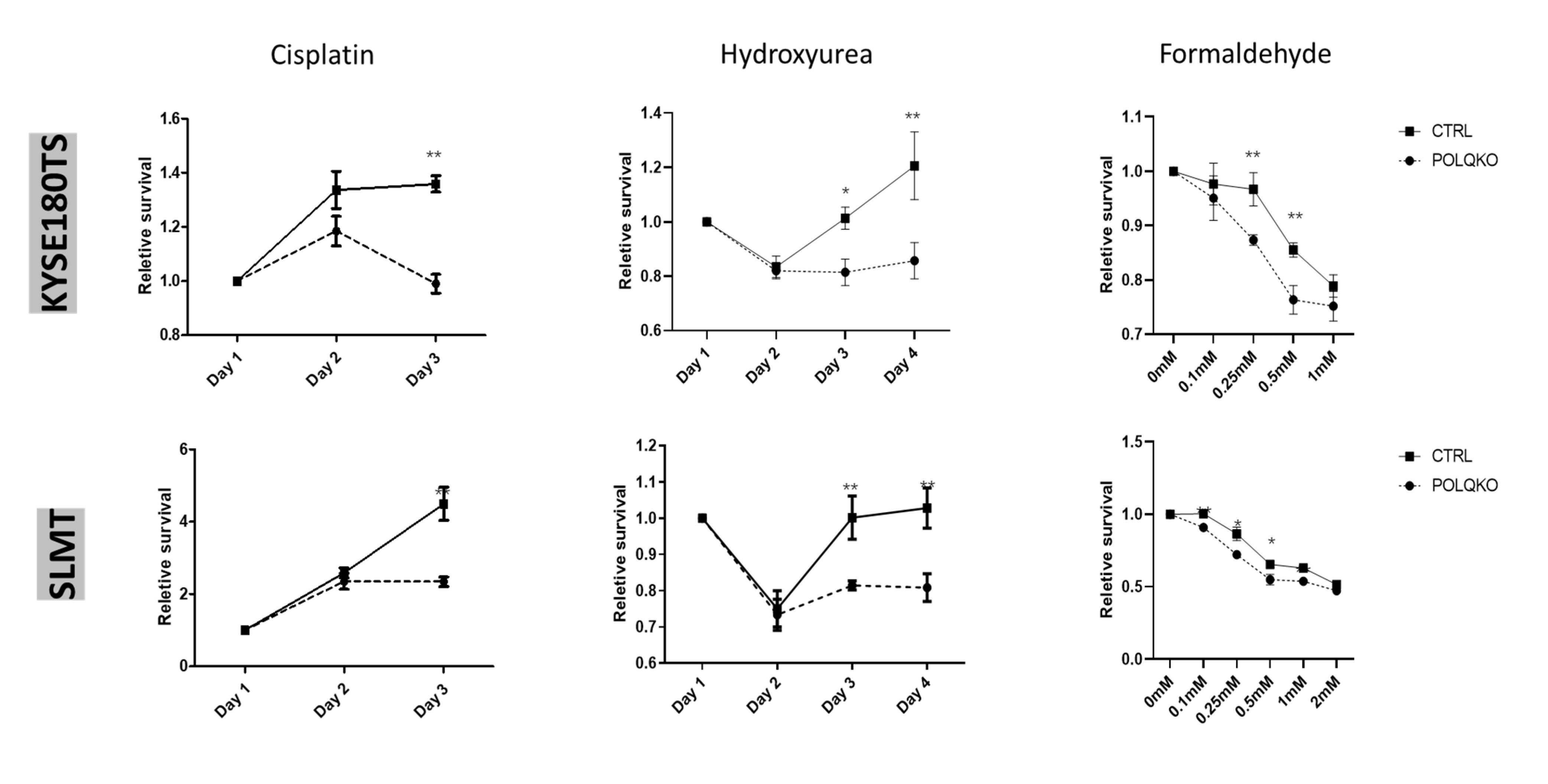
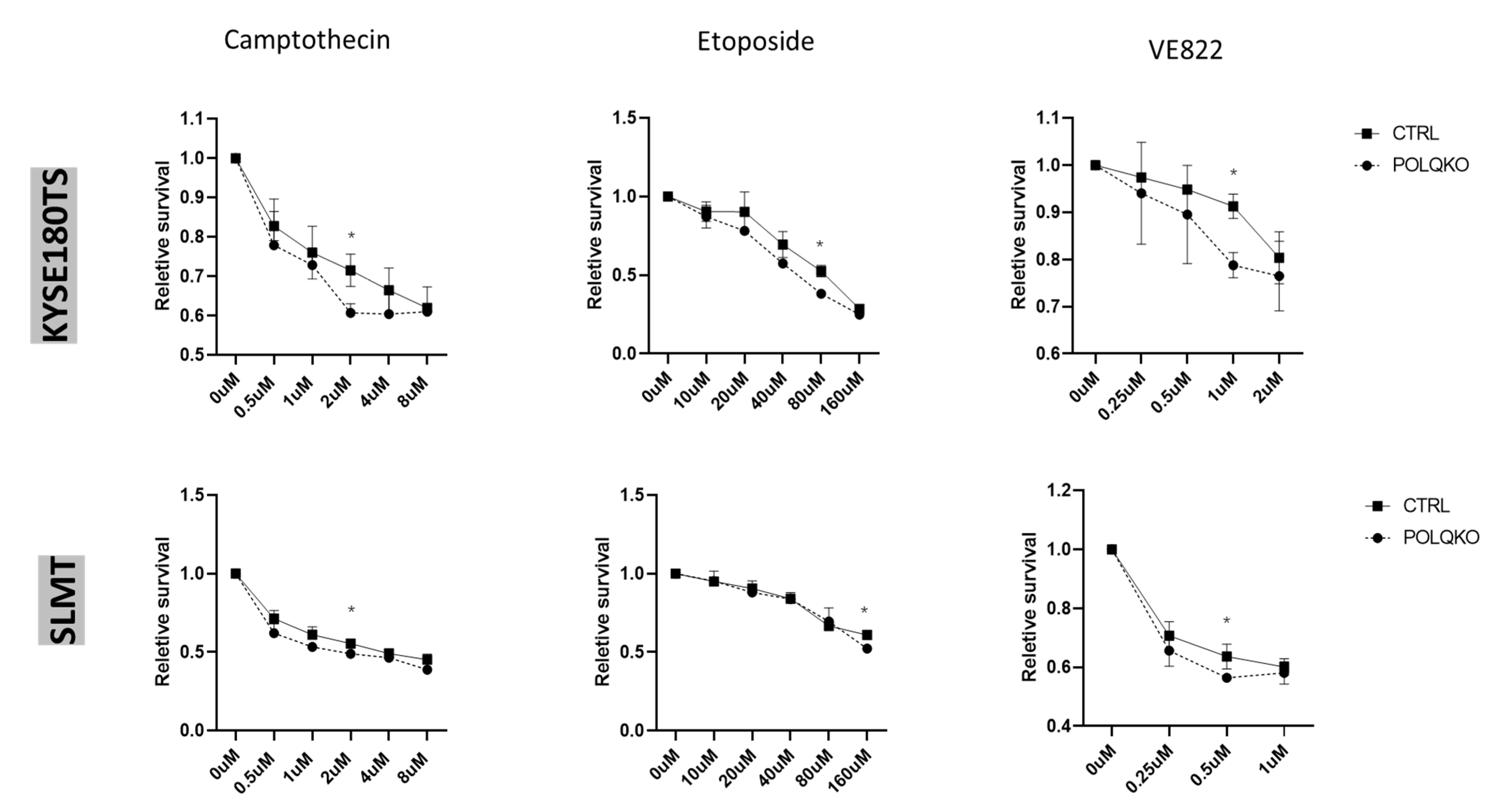
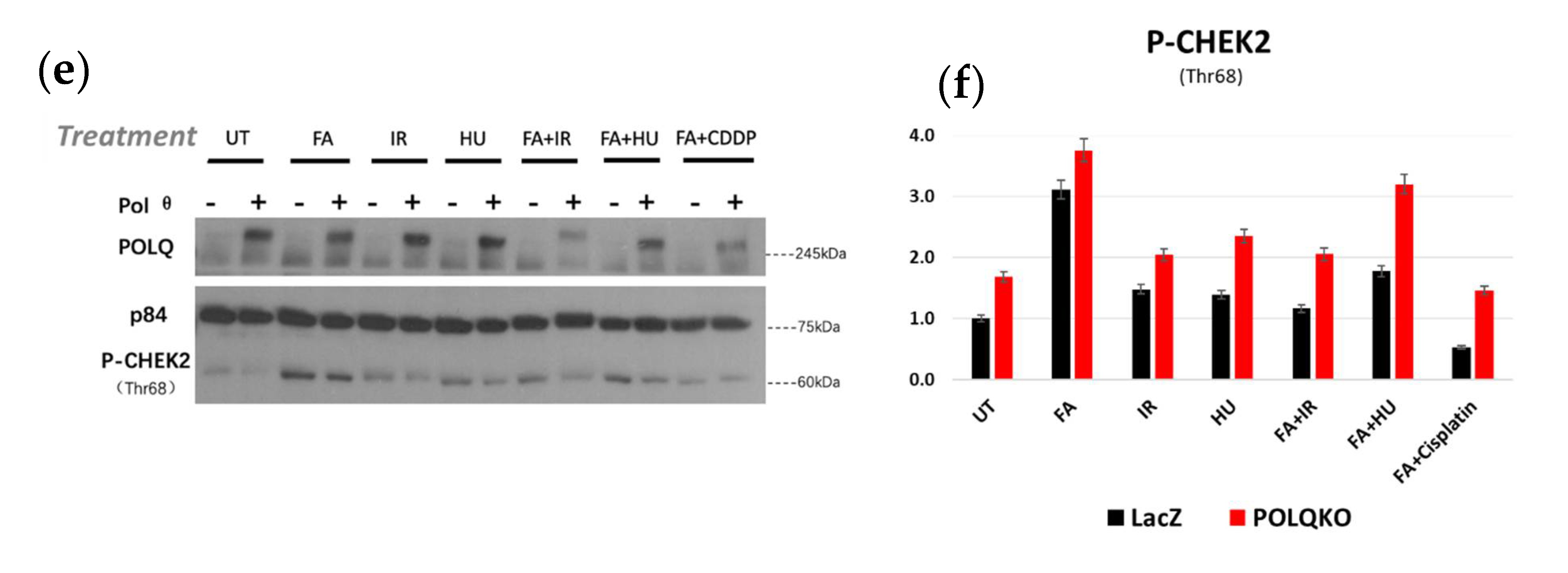
3.3. POLQ Depletion Sensitizes ESCC Cells to Mutiple Genotoxic Agents
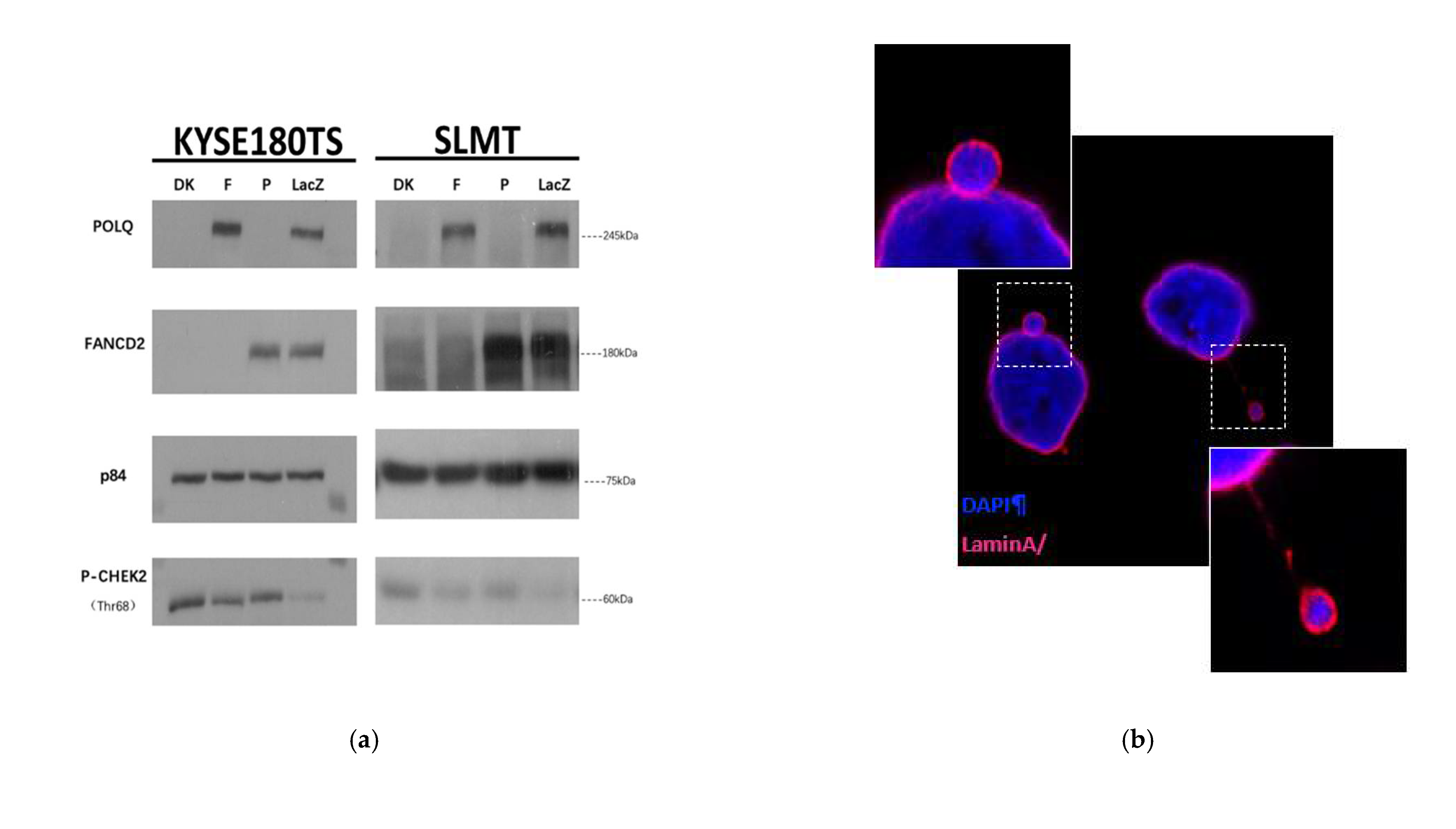
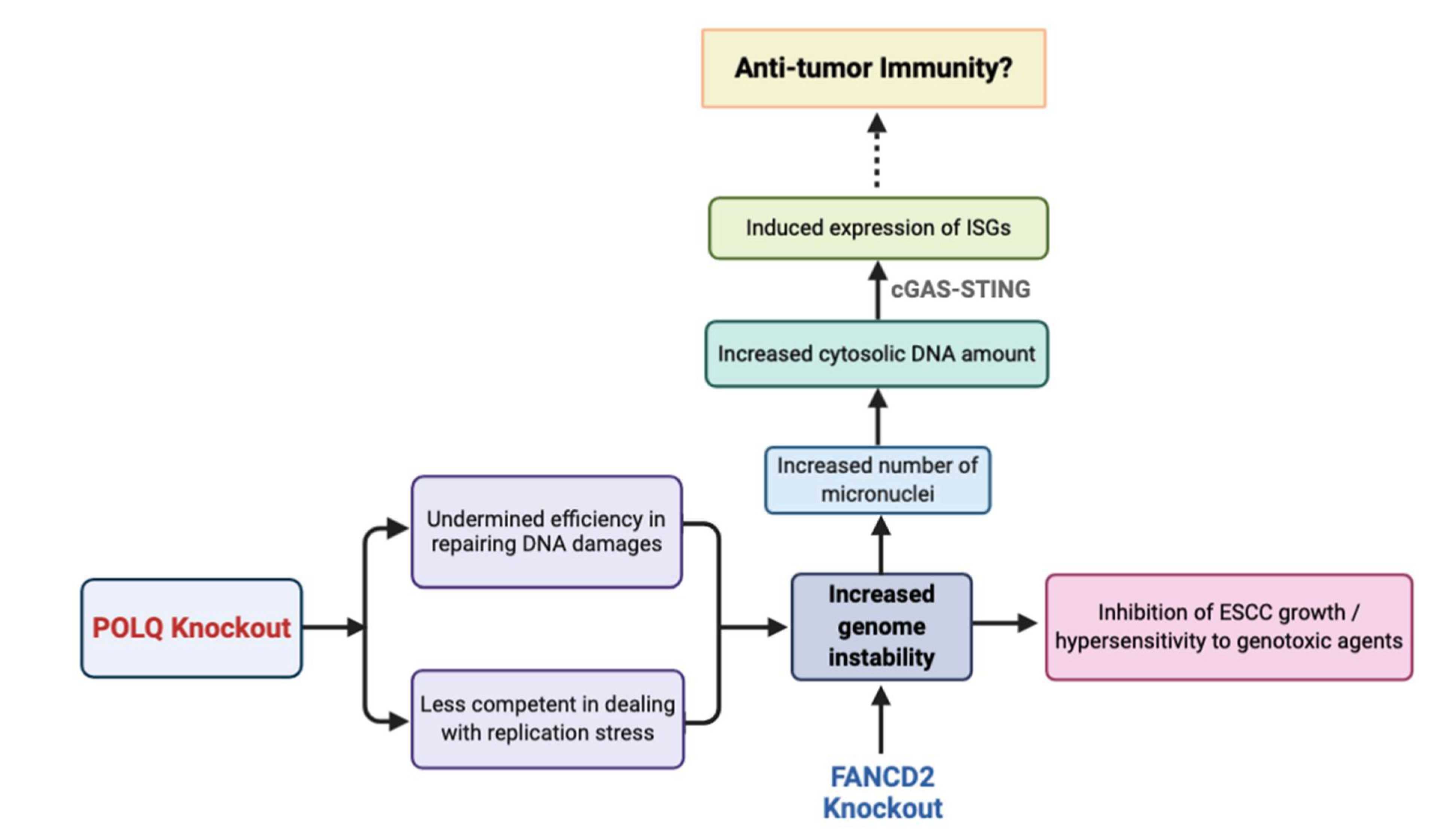
3.4. Double Knockout POLQ and FANCD2 Drastically Inhibits ESCC Growth Both In Vivo and In Vitro
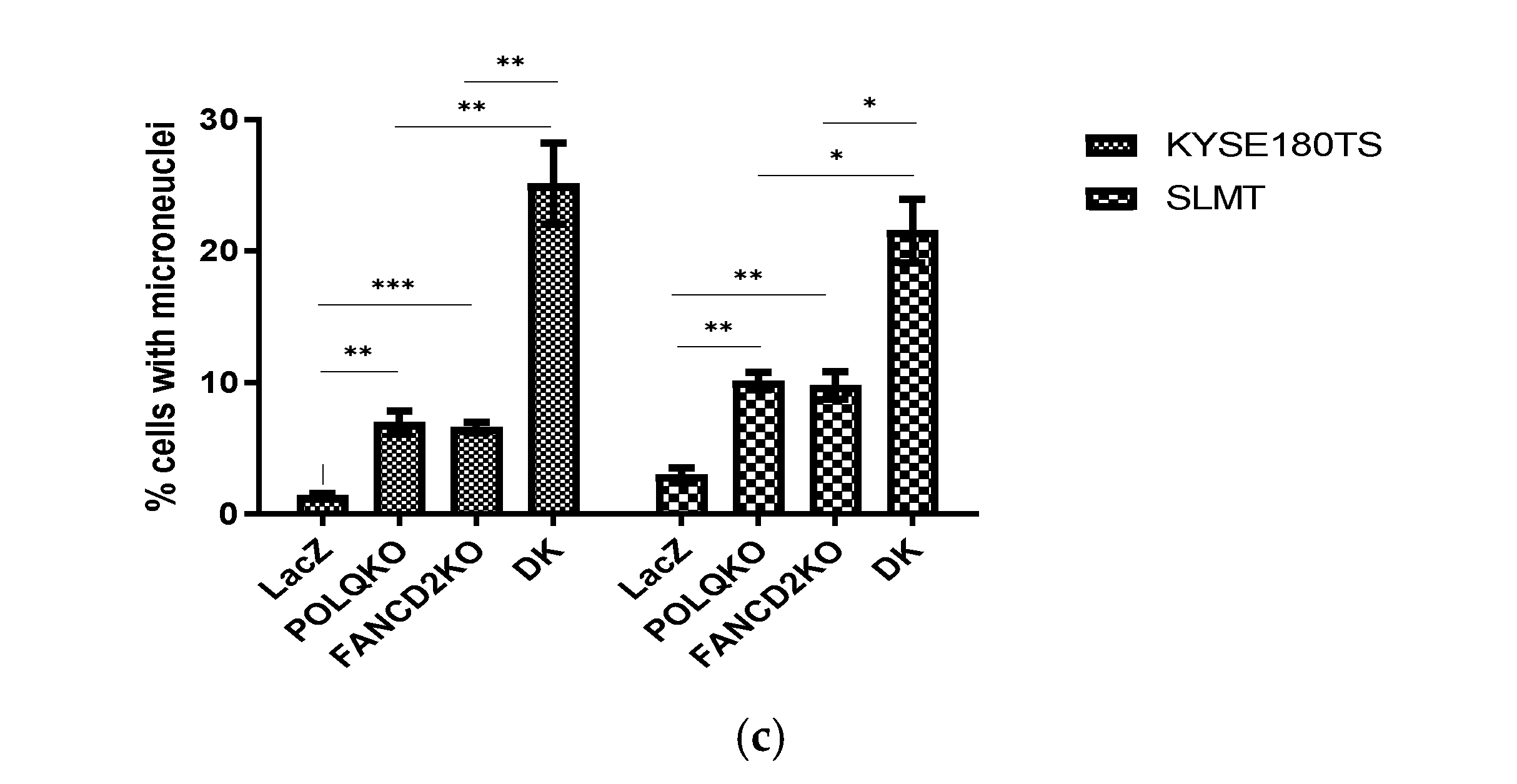
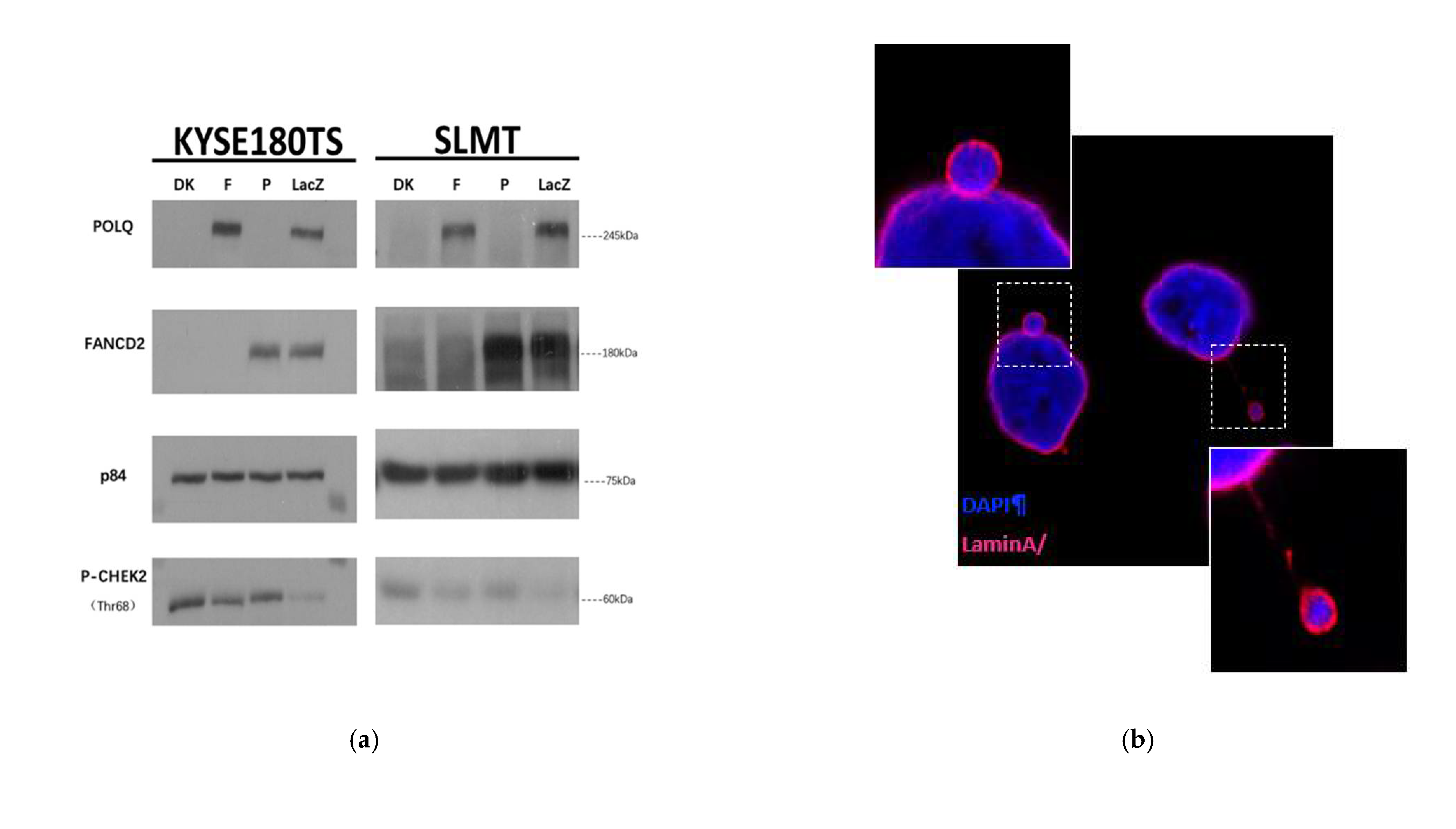
3.5. Double Knockout of POLQ and FANCD2 Significantly Induces Genome Instability and the Formation of Micronuclei
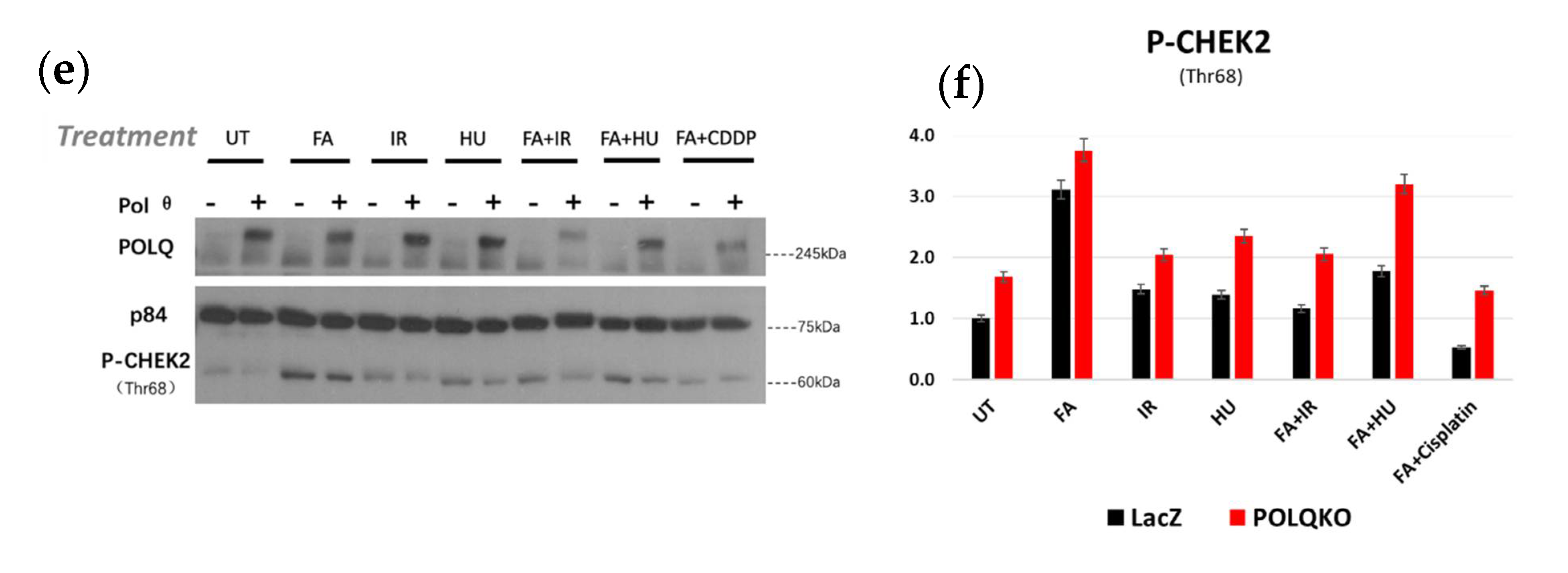
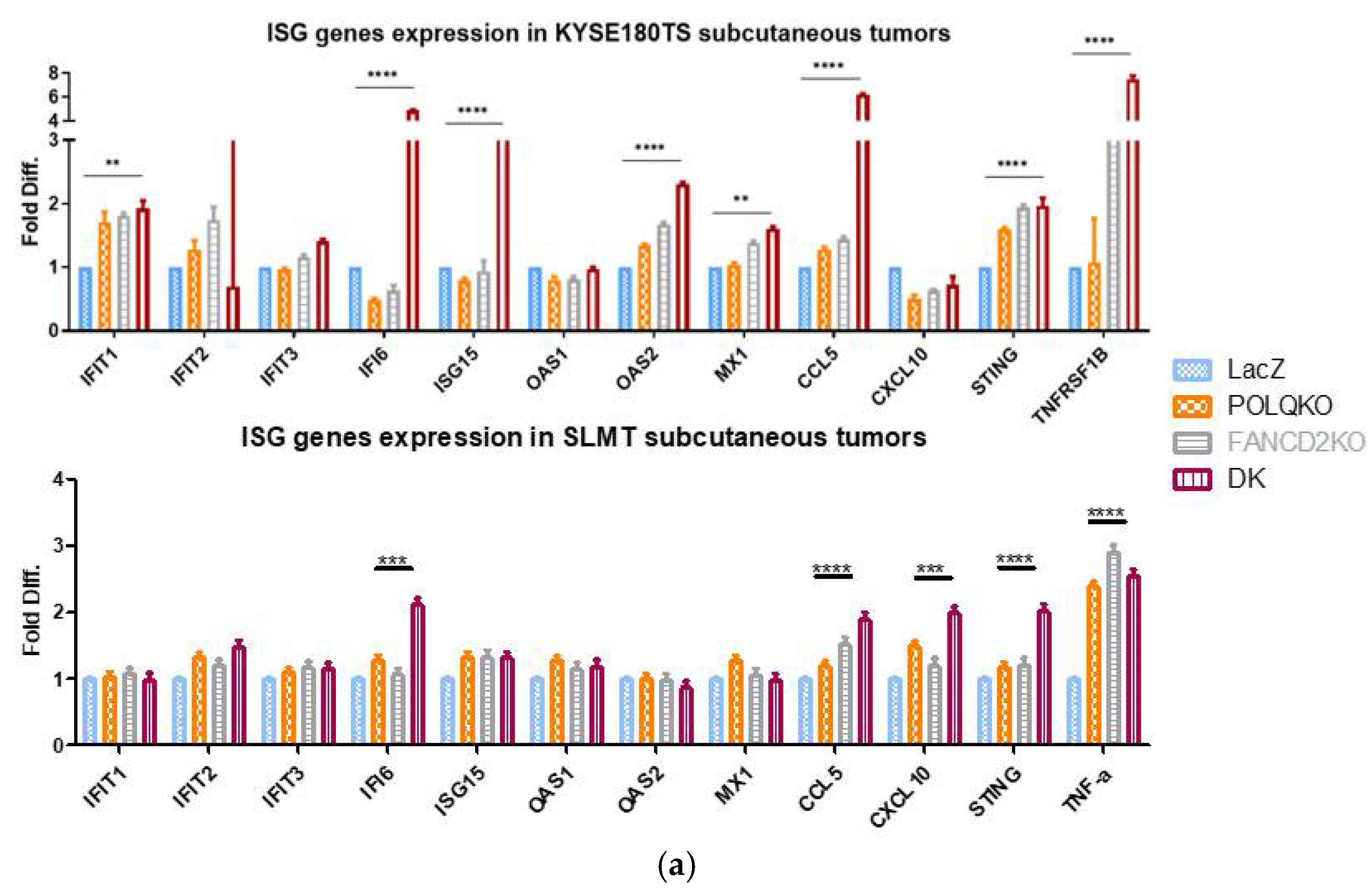
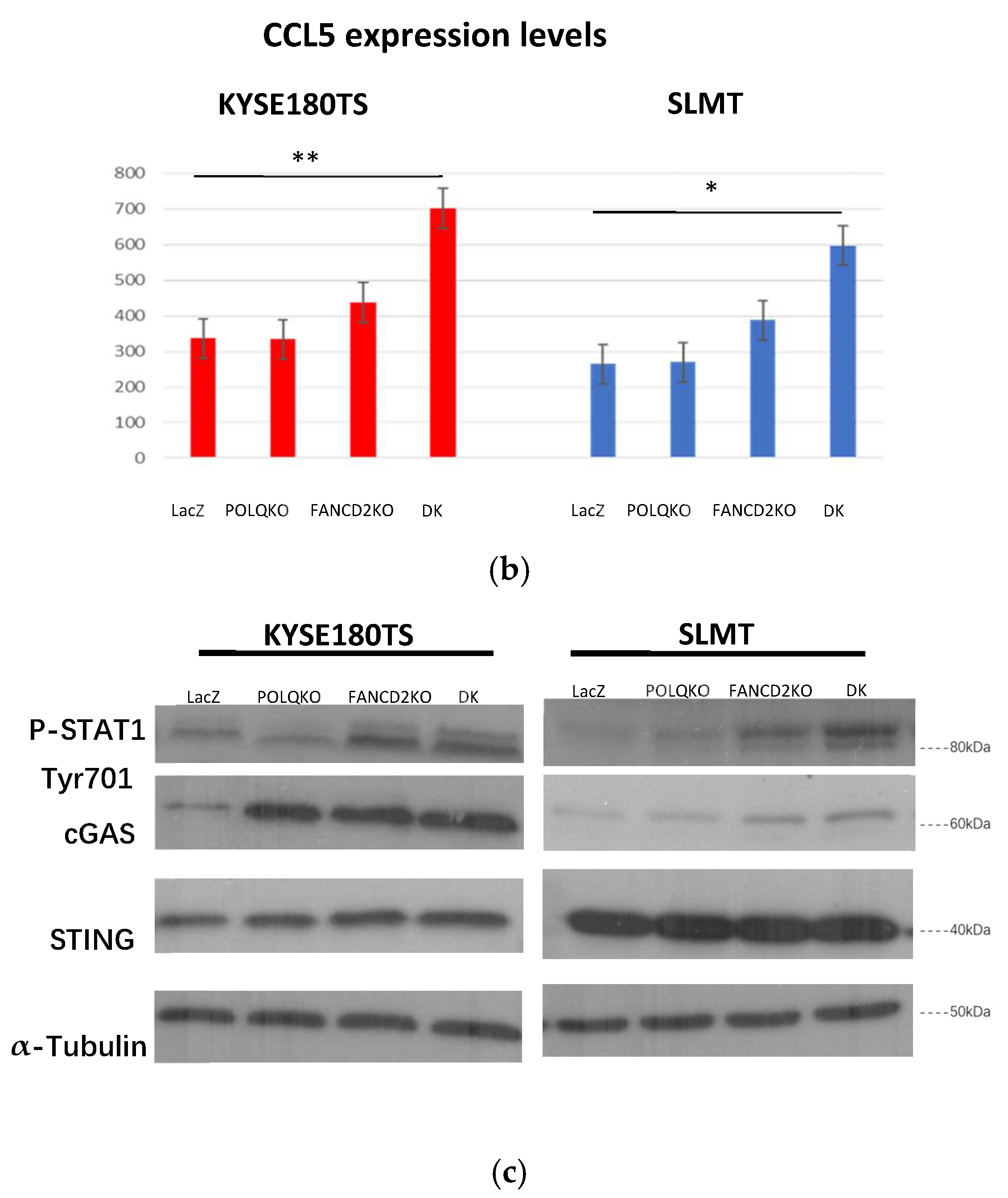
3.6. Double Knockout POLQ and FANCD2 Induces the Expression of Interferon-Stimulated Genes (ISGs) and Upregulates cGAS and STAT1 Phosphorylation
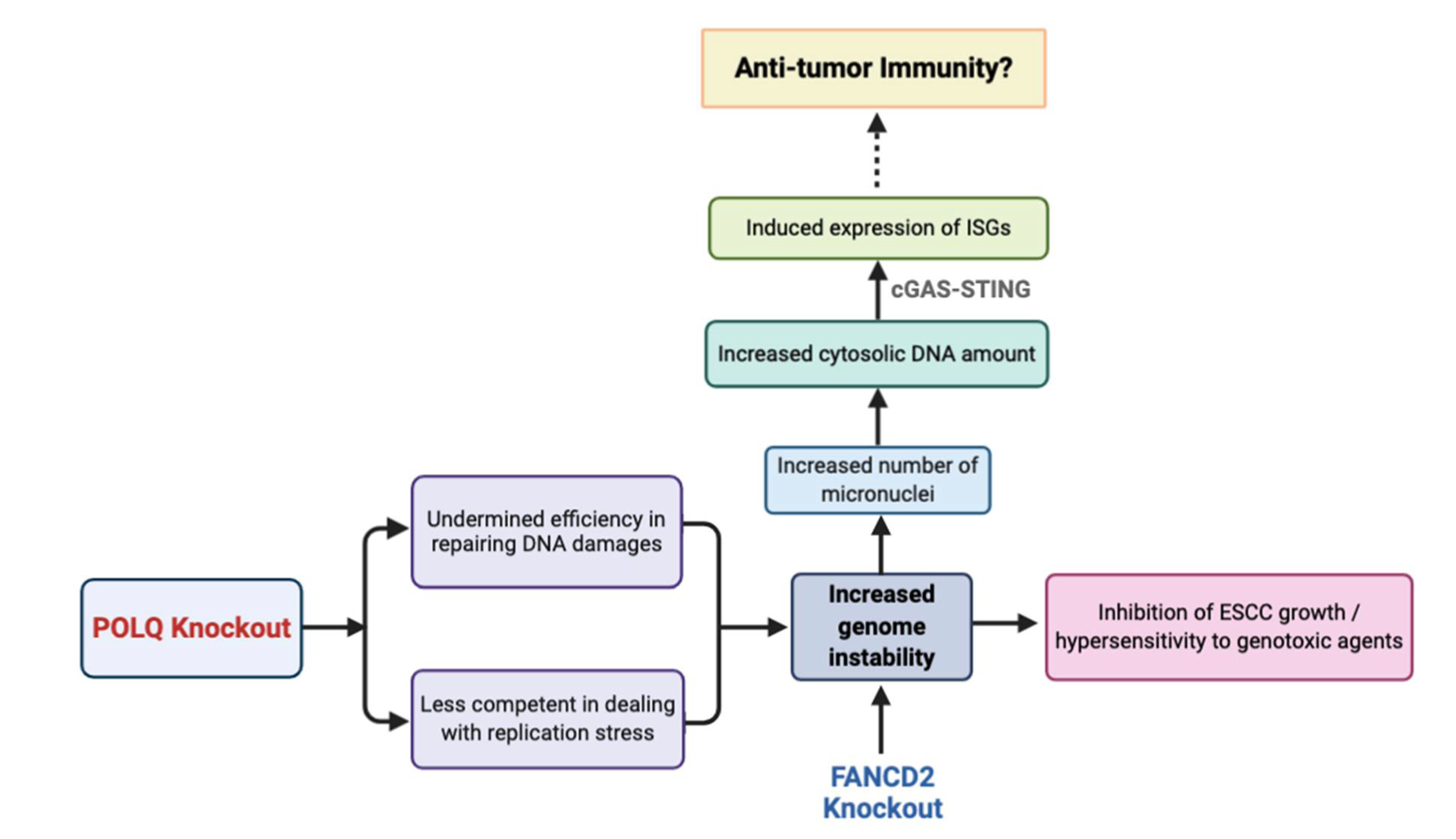
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Then, E.O.; Lopez, M.; Saleem, S.; Gayam, V.; Sunkara, T.; Culliford, A.; Gaduputi, V. Esophageal Cancer: An Updated Surveillance Epidemiology and End Results Database Analysis. World J. Oncol. 2020, 11, 55–64. [Google Scholar] [CrossRef]
- Abnet, C.C.; Arnold, M.; Wei, W.-Q. Epidemiology of Esophageal Squamous Cell Carcinoma. Gastroenterol. 2018, 154, 360–373. [Google Scholar] [CrossRef]
- Roshandel, G.; Nourouzi, A.; Pourshams, A.; Semnani, S.; Merat, S.; Khoshnia, M. Endoscopic screening for esophageal squamous cell carcinoma. Arch. Iran. Med. 2013, 16, 351–357. [Google Scholar]
- Yousefzadeh, M.J.; Wood, R.D. DNA polymerase POLQ and cellular defense against DNA damage. DNA Repair 2013, 12, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Wood, R.D.; Doublié, S. DNA polymerase θ (POLQ), double-strand break repair, and cancer. DNA Repair 2016, 44, 22–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshimura, M.; Kohzaki, M.; Nakamura, J.; Asagoshi, K.; Sonoda, E.; Hou, E.; Prasad, R.; Wilson, S.H.; Tano, K.; Yasui, A.; et al. Vertebrate POLQ and POLbeta cooperate in base excision repair of oxidative DNA damage. Mol. Cell. 2006, 24, 115–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koole, W.; Van Schendel, R.; Karambelas, A.E.; Van Heteren, J.T.; Okihara, K.L.; Tijsterman, M. A Polymerase Theta-dependent repair pathway suppresses extensive genomic instability at endogenous G4 DNA sites. Nat. Commun. 2014, 5, 3216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ceccaldi, R.; Liu, J.C.; Amunugama, R.; Hajdu, I.; Primack, B.; Petalcorin, M.; O’Connor, K.; Konstantinopoulos, P.A.; Elledge, S.J.; Boulton, S.J.; et al. Homologous-recombination-deficient tumours are dependent on Polθ-mediated repair. Nat. Cell Biol. 2015, 518, 258–262. [Google Scholar] [CrossRef] [Green Version]
- Kawamura, K.; Bahar, R.; Seimiya, M.; Chiyo, M.; Wada, A.; Okada, S.; Hatano, M.; Tokuhisa, T.; Kimura, H.; Watanabe, S.; et al. DNA polymerase θ is preferentially expressed in lymphoid tissues and upregulated in human cancers. Inter. J. Cancer 2004, 109, 9–16. [Google Scholar] [CrossRef]
- Pillaire, M.-J.; Selves, J.; Gordien, K.; Gouraud, P.-A.; Gentil, C.; Danjoux, M.; Do, C.; Negre, V.; Bieth, A.; Guimbaud, R.; et al. A ‘DNA replication’signature of progression and negative outcome in colorectal cancer. Oncogene 2010, 29, 876–887. [Google Scholar] [CrossRef]
- Lessa, R.C.; Campos, A.H.; De Freitas, C.E.; Da Silva, F.R.; Kowalski, L.P.; Carvalho, A.; Vettore, A.L. Identification of upregulated genes in oral squamous cell carcinomas. Head Neck 2012, 35, 1475–1481. [Google Scholar] [CrossRef]
- Lemée, F.; Bergoglio, V.; Fernandez-Vidal, A.; Machado-Silva, A.; Pillaire, M.-J.; Bieth, A.; Gentil, C.; Baker, L.; Martin, A.-L.; Leduc, C.; et al. DNA polymerase θ up-regulation is associated with poor survival in breast cancer, perturbs DNA replication, and promotes genetic instability. Proc. Natl. Acad. Sci. USA 2010, 107, 13390–13395. [Google Scholar] [CrossRef] [Green Version]
- Ko, J.M.; Ning, L.; Zhao, X.; Chai, A.W.; Lei, L.C.; Choi, S.S.A.; Tao, L.; Law, S.; Kwong, A.; Lee, N.P.; et al. BRCA2 loss-of-function germline mutations are associated with esophageal squamous cell carcinoma risk in Chinese. Int. J. Cancer 2020, 146, 1042–1051. [Google Scholar] [CrossRef]
- Parkes, E.E.; Walker, S.M.; Taggart, L.E.; McCabe, N.; Knight, L.A.; Wilkinson, R.; McCloskey, K.D.; Buckley, N.; Savage, K.I.; Salto-Tellez, M.; et al. Activation of STING-Dependent Innate Immune Signaling By S-Phase-Specific DNA Damage in Breast Cancer. J. Natl. Cancer Inst. 2017, 109. [Google Scholar] [CrossRef] [Green Version]
- Pantelidou, C.; Sonzogni, O.; Taveira, M.D.O.; Mehta, A.K.; Kothari, A.; Wang, D.; Visal, T.; Li, M.K.; Pinto, J.; Castrillon, J.A.; et al. PARP Inhibitor Efficacy Depends on CD8+ T-cell Recruitment via Intratumoral STING Pathway Activation in BRCA-Deficient Models of Triple-Negative Breast Cancer. Cancer Discov. 2019, 9, 722–737. [Google Scholar] [CrossRef] [Green Version]
- Reisländer, T.; Lombardi, E.P.; Groelly, F.; Miar, A.; Porru, M.; Di Vito, S.; Wright, B.; Lockstone, H.; Biroccio, A.; Harris, A.; et al. BRCA2 abrogation triggers innate immune responses potentiated by treatment with PARP inhibitors. Nat. Commun. 2019, 10, 1–13. [Google Scholar] [CrossRef] [Green Version]
- MacKenzie, K.J.; Carroll, P.; Martin, C.-A.; Murina, O.; Fluteau, A.; Simpson, D.J.; Olova, N.; Sutcliffe, H.; Rainger, J.K.; Leitch, A.; et al. cGAS surveillance of micronuclei links genome instability to innate immunity. Nat. Cell Biol. 2017, 548, 461–465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erdal, E.; Haider, S.; Rehwinkel, J.; Harris, A.L.; McHugh, P.J. A prosurvival DNA damage-induced cytoplasmic interferon response is mediated by end resection factors and is limited by Trex1. Genes Dev. 2017, 31, 353–369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bose, D. cGAS/STING Pathway in Cancer: Jekyll and Hyde Story of Cancer Immune Response. Int. J. Mol. Sci. 2017, 18, 2456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hellström, K.E.; Hellström, I. Cellular Immunity Against Tumor Antigens. Adv. Cancer Res. 1969, 12, 167–223. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Sun, L.; Chen, Z.J. Regulation and function of the cGAS-STING pathway of cytosolic DNA sensing. Nat. Immunol. 2016, 17, 1142–1149. [Google Scholar] [CrossRef]
- Wang, H.; Hu, S.; Chen, X.; Shi, H.; Chen, C.; Sun, L.; Chen, Z.J. cGAS is essential for the antitumor effect of immune checkpoint blockade. Proc. Natl. Acad. Sci. USA 2017, 114, 1637–1642. [Google Scholar] [CrossRef] [Green Version]
- Leung, A.C.C.; Wong, V.C.L.; Yang, L.C.; Chan, P.L.; Daigo, Y.; Nakamura, Y.; Qi, R.Z.; Miller, L.D.; Liu, E.T.-B.; Wang, L.D.; et al. Frequent decreased expression of candidate tumor suppressor gene, DEC1, and its anchorage-independent growth properties and impact on global gene expression in esophageal carcinoma. Int. J. Cancer 2008, 122, 587–594. [Google Scholar] [CrossRef]
- Kim, D.; Pertea, G.; Trapnell, C.; Pimentel, H.; Kelley, R.; Salzberg, S.L. TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013, 14, R36. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.; Cao, J.; Wu, W.; Zhu, Q.; Tang, Y.; Zhu, C.; Dai, J.; Li, Z.; Wang, J.; Xue, L.; et al. Genome-wide copy number variation analysis identified ANO1 as a novel oncogene and prognostic biomarker in esophageal squamous cell cancer. Carcinog. 2019, 40, 1198–1208. [Google Scholar] [CrossRef] [PubMed]
- Young, L.; Sung, J.; Stacey, G.; Masters, J.R. Detection of Mycoplasma in cell cultures. Nat. Protoc. 2010, 5, 929–934. [Google Scholar] [CrossRef] [PubMed]
- Yu, V.Z.; Ko, J.M.Y.; Ning, L.; Dai, W.; Law, S.; Lung, M.L. Endoplasmic reticulum-localized ECM1b suppresses tumor growth and regulates MYC and MTORC1 through modulating MTORC2 activation in esophageal squamous cell carcinoma. Cancer Lett. 2019, 461, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Yu, V.Z.; Wong, V.C.-L.; Dai, W.; Ko, J.M.-Y.; Lam, A.K.-Y.; Chan, K.W.; Samant, R.S.; Lung, H.L.; Shuen, W.H.; Law, S.; et al. Nuclear Localization of DNAJB6 Is Associated With Survival of Patients With Esophageal Cancer and Reduces AKT Signaling and Proliferation of Cancer Cells. Gastroenterol. 2015, 149, 1825–1836.e5. [Google Scholar] [CrossRef] [Green Version]
- Kearns, N.A.; Genga, R.M.J.; Enuameh, M.S.; Garber, M.; Wolfe, S.A.; Maehr, R. Cas9 effector-mediated regulation of transcription and differentiation in human pluripotent stem cells. Development 2014, 141, 219–223. [Google Scholar] [CrossRef] [Green Version]
- Lo, P.H.Y.; Ko, J.M.Y.; Yu, Z.Y.; Law, S.; Wang, L.D.; Li, J.-L.; Srivastava, G.; Tsao, S.W.; Stanbridge, E.J.; Lung, M.L. The LIM domain protein, CRIP2, promotes apoptosis in esophageal squamous cell carcinoma. Cancer Lett. 2012, 316, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Lung, H.L.; Bangarusamy, D.K.; Xie, D.; Cheung, A.K.L.; Cheng, Y.; Kumaran, M.K.; Miller, L.; Liu, E.T.-B.; Guan, X.-Y.; Sham, J.S.; et al. THY1 is a candidate tumour suppressor gene with decreased expression in metastatic nasopharyngeal carcinoma. Oncogene 2005, 24, 6525–6532. [Google Scholar] [CrossRef] [Green Version]
- Fernandez-Vidal, A.; Guitton-Sert, L.; Cadoret, J.-C.; Drac, M.; Schwob, E.; Baldacci, G.; Cazaux, C.; Hoffmann, J.-S. A role for DNA polymerase θ in the timing of DNA replication. Nat. Commun. 2014, 5, 4285. [Google Scholar] [CrossRef] [Green Version]
- Olive, P.L.; Banáth, J.P. The comet assay: A method to measure DNA damage in individual cells. Nat. Protoc. 2006, 1, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Gyori, B.M.; Venkatachalam, G.; Thiagarajan, P.; Hsu, D.; Clement, M.-V. OpenComet: An automated tool for comet assay image analysis. Redox Biol. 2014, 2, 457–465. [Google Scholar] [CrossRef] [Green Version]
- Langie, S.A.S.; Azqueta, A.; Collins, A.R. The comet assay: Past, present, and future. Front. Genet. 2015, 6, 266. [Google Scholar] [CrossRef] [Green Version]
- Zhou, C.; Li, Z.; Diao, H.; Yu, Y.; Zhu, W.; Dai, Y.; Chen, F.F.; Yang, J. DNA damage evaluated by gammaH2AX foci formation by a selective group of chemical/physical stressors. Mutation Res. 2006, 604, 8–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lei, L.C.; Yu, V.Z.; Ko, J.M.Y.; Ning, L.; Lung, M.L. FANCD2 Confers a Malignant Phenotype in Esophageal Squamous Cell Carcinoma by Regulating Cell Cycle Progression. Cancers 2020, 12, 2545. [Google Scholar] [CrossRef] [PubMed]
- Michl, J.; Zimmer, J.; Buffa, F.; McDermott, U.; Tarsounas, M. FANCD2 limits replication stress and genome instability in cells lacking BRCA2. Nat. Struct. Mol. Biol. 2016, 23, 755–757. [Google Scholar] [CrossRef]
- Schneider, W.M.; Chevillotte, M.D.; Rice, C.M. Interferon-Stimulated Genes: A Complex Web of Host Defenses. Annu. Rev. Immunol. 2014, 32, 513–545. [Google Scholar] [CrossRef] [Green Version]
- Kais, Z.; Rondinelli, B.; Holmes, A.; O’Leary, C.; Kozono, D.; D’Andrea, A.D.; Ceccaldi, R. FANCD2 Maintains Fork Stability in BRCA1/2-Deficient Tumors and Promotes Alternative End-Joining DNA Repair. Cell Rep. 2016, 15, 2488–2499. [Google Scholar] [CrossRef] [Green Version]
- Kee, Y.; D’Andrea, A.D. Expanded roles of the Fanconi anemia pathway in preserving genomic stability. Genes Dev. 2010, 24, 1680–1694. [Google Scholar] [CrossRef] [Green Version]
- Harding, S.; Benci, J.; Irianto, J.; Discher, D.E.; Minn, A.J.; Greenberg, R.A. Mitotic progression following DNA damage enables pattern recognition within micronuclei. Nat. Cell Biol. 2017, 548, 466–470. [Google Scholar] [CrossRef] [Green Version]
- Leoncini, E.; Ricciardi, W.; Cadoni, G.; Arzani, D.; Petrelli, L.; Paludetti, G.; Brennan, P.; Luce, D.; Stucker, I.; Matsuo, K.; et al. Adult height and head and neck cancer: A pooled analysis within the INHANCE Consortium. Eur. J. Epidemiol. 2013, 29, 35–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allera-Moreau, C.; Rouquette, I.; Lepage, B.; Oumouhou, N.; Walschaerts, M.; Leconte, E.; Schilling, V.; Gordien, K.; Brouchet, L.; Delisle, M.B.; et al. DNA replication stress response involving PLK1, CDC6, POLQ, RAD51 and CLASPIN upregulation prognoses the outcome of early/mid-stage non-small cell lung cancer patients. Oncogenesis 2012, 1, e30. [Google Scholar] [CrossRef] [PubMed]
- Sallmyr, A.; Tomkinson, A.E. Repair of DNA double-strand breaks by mammalian alternative end-joining pathways. J. Biol. Chem. 2018, 293, 10536–10546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schimmel, J.; van Schendel, R.; Dunnen, J.T.D.; Tijsterman, M. Templated Insertions: A Smoking Gun for Polymerase Theta-Mediated End Joining. Trends Genet. 2019, 35, 632–644. [Google Scholar] [CrossRef] [PubMed]
- Seki, M.; Masutani, C.; Yang, L.W.; Schuffert, A.; Iwai, S.; Bahar, I.; Wood, R.D. High-efficiency bypass of DNA damage by human DNA polymerase Q. EMBO J. 2004, 23, 4484–4494. [Google Scholar] [CrossRef] [Green Version]
- Goff, J.P.; Shields, D.S.; Seki, M.; Choi, S.; Epperly, M.W.; Dixon, T.; Wang, H.; Bakkenist, C.J.; Dertinger, S.D.; Torous, D.K.; et al. Lack of DNA polymerase θ (POLQ) radiosensitizes bone marrow stromal cells in vitro and increases retic-ulocyte micronuclei after total-body irradiation. Radiat. Res. 2009, 172, 165–174. [Google Scholar] [CrossRef] [Green Version]
- Shima, N.; Munroe, R.J.; Schimenti, J.C. The Mouse Genomic Instability Mutation chaos1 Is an Allele of Polq That Exhibits Genetic Interaction with Atm. Mol. Cell. Biol. 2004, 24, 10381–10389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelso, A.A.; Lopezcolorado, F.W.; Bhargava, R.; Stark, J.M. Distinct roles of RAD52 and POLQ in chromosomal break repair and replication stress response. PLoS Genet. 2019, 15, e1008319. [Google Scholar] [CrossRef] [Green Version]
- Yousefzadeh, M.J.; Wyatt, D.; Takata, K.-I.; Mu, Y.; Hensley, S.C.; Tomida, J.; Bylund, G.O.; Doublie, S.; Johansson, E.; Ramsden, D.A.; et al. Mechanism of Suppression of Chromosomal Instability by DNA Polymerase POLQ. PLoS Genet. 2014, 10, e1004654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Higgins, G.S.; Prevo, R.; Lee, Y.-F.; Helleday, T.; Muschel, R.J.; Taylor, T.; Yoshimura, M.; Hickson, I.D.; Bernhard, E.J.; McKenna, W.J. A small interfering RNA screen of genes involved in DNA repair identifies tumor-specific radiosensi-tization by POLQ knockdown. Cancer Res. 2010, 70, 2984–2993. [Google Scholar] [CrossRef] [Green Version]
- Mateos-Gomez, P.A.; Gong, F.; Nair, N.; Miller, K.M.; Denchi, E.L.; Sfeir, A. Mammalian polymerase θ promotes alternative NHEJ and suppresses recombination. Nat. Cell Biol. 2015, 518, 254–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoon, J.-H.; McArthur, M.J.; Park, J.; Basu, D.; Wakamiya, M.; Prakash, L.; Prakash, S. Error-Prone Replication through UV Lesions by DNA Polymerase θ Protects against Skin Cancers. Cell 2019, 176, 1295–1309.e15. [Google Scholar] [CrossRef] [Green Version]
- Franchet, C.; Hoffmann, J.-S. When RAD52 Allows Mitosis to Accept Unscheduled DNA Synthesis. Cancers 2019, 12, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maiorano, D.; El Etri, J.; Franchet, C.; Hoffmann, J.-S. Translesion Synthesis or Repair by Specialized DNA Polymerases Limits Excessive Genomic Instability upon Replication Stress. Int. J. Mol. Sci. 2021, 22, 3924. [Google Scholar] [CrossRef]
- Moldovan, G.-L.; D’Andrea, A.D. How the Fanconi Anemia Pathway Guards the Genome. Annu. Rev. Genet. 2009, 43, 223–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, C.-H.; Chen, P.; Li, J.; Lan, T.; Chen, Y.-C.; Qian, H.; Chen, K.; Li, M.-Y. Co-inhibition of pol θ and HR genes efficiently synergize with cisplatin to suppress cisplatin-resistant lung cancer cells survival. Oncotarget 2016, 7, 65157–65170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parmar, K.; Kim, J.; Sykes, S.M.; Shimamura, A.; Stuckert, P.; Zhu, K.; Hamilton, A.; Deloach, M.K.; Kutok, J.L.; Akashi, K.; et al. Hematopoietic Stem Cell Defects in Mice with Deficiency of Fancd2 or Usp1. STEM CELLS 2010, 28, 1186–1195. [Google Scholar] [CrossRef] [Green Version]
- Wilhelm, T.; Olziersky, A.-M.; Harry, D.; De Sousa, F.; Vassal, H.; Eskat, A.; Meraldi, P. Mild replication stress causes chromosome mis-segregation via premature centriole disengagement. Nat. Commun. 2019, 10, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, B.; Gnawali, N.; Hinman, A.W.; Mattingly, A.J.; Osimani, A.; Cimini, D. Chromosomes missegregated into micronuclei contribute to chromosomal instability by missegregating at the next division. Oncotarget 2019, 10, 2660–2674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schoggins, J.W.; Wilson, S.J.; Panis, M.; Murphy, M.Y.; Jones, C.T.; Bieniasz, P.; Rice, C.M. A diverse range of gene products are effectors of the type I interferon antiviral response. Nat. Cell Biol. 2011, 472, 481–485. [Google Scholar] [CrossRef]
- MacMicking, J.D. Interferon-inducible effector mechanisms in cell-autonomous immunity. Nat. Rev. Immunol. 2012, 12, 367–382. [Google Scholar] [CrossRef] [PubMed]
- Zitvogel, L.; Galluzzi, L.; Kepp, O.; Smyth, M.; Kroemer, G. Type I interferons in anticancer immunity. Nat. Rev. Immunol. 2015, 15, 405–414. [Google Scholar] [CrossRef]
- Zschaler, J.; Schlorke, D.; Arnhold, J. Differences in innate immune response between man and mouse. Crit. Rev. Immunol. 2014, 34, 433–454. [Google Scholar] [CrossRef] [Green Version]
- Conlon, J.; Burdette, D.L.; Sharma, S.; Bhat, N.; Thompson, M.; Jiang, Z.; Rathinam, V.A.K.; Monks, B.; Jin, T.; Xiao, T.S.; et al. Mouse, but not Human STING, Binds and Signals in Response to the Vascular Disrupting Agent 5,6-Dimethylxanthenone-4-Acetic Acid. J. Immunol. 2013, 190, 5216–5225. [Google Scholar] [CrossRef] [Green Version]
- Bose, D.; Neumann, A.; Timmermann, B.; Meinke, S.; Heyd, F. Differential Interleukin-2 Transcription Kinetics Render Mouse but Not Human T Cells Vulnerable to Splicing Inhibition Early after Activation. Mol. Cell. Biol. 2019, 39, 00035-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, J.; Ko, J.M.-Y.; Dai, W.; Yu, V.Z.; Ng, H.Y.; Hoffmann, J.-S.; Lung, M.L. Depletion of DNA Polymerase Theta Inhibits Tumor Growth and Promotes Genome Instability through the cGAS-STING-ISG Pathway in Esophageal Squamous Cell Carcinoma. Cancers 2021, 13, 3204. https://doi.org/10.3390/cancers13133204
Li J, Ko JM-Y, Dai W, Yu VZ, Ng HY, Hoffmann J-S, Lung ML. Depletion of DNA Polymerase Theta Inhibits Tumor Growth and Promotes Genome Instability through the cGAS-STING-ISG Pathway in Esophageal Squamous Cell Carcinoma. Cancers. 2021; 13(13):3204. https://doi.org/10.3390/cancers13133204
Chicago/Turabian StyleLi, Jian, Josephine Mun-Yee Ko, Wei Dai, Valen Zhuoyou Yu, Hoi Yan Ng, Jean-Sébastien Hoffmann, and Maria Li Lung. 2021. "Depletion of DNA Polymerase Theta Inhibits Tumor Growth and Promotes Genome Instability through the cGAS-STING-ISG Pathway in Esophageal Squamous Cell Carcinoma" Cancers 13, no. 13: 3204. https://doi.org/10.3390/cancers13133204
APA StyleLi, J., Ko, J. M.-Y., Dai, W., Yu, V. Z., Ng, H. Y., Hoffmann, J.-S., & Lung, M. L. (2021). Depletion of DNA Polymerase Theta Inhibits Tumor Growth and Promotes Genome Instability through the cGAS-STING-ISG Pathway in Esophageal Squamous Cell Carcinoma. Cancers, 13(13), 3204. https://doi.org/10.3390/cancers13133204