
Global Proteomic Profiling of Pediatric AML: A Pilot Study

 , ,
, ,  ,
, 
Abstract
:Simple Summary
Abstract
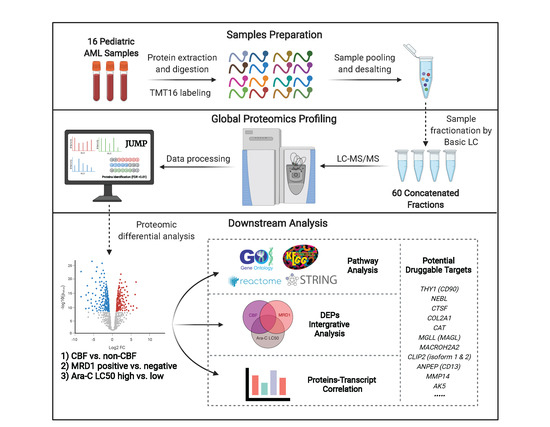
1. Introduction
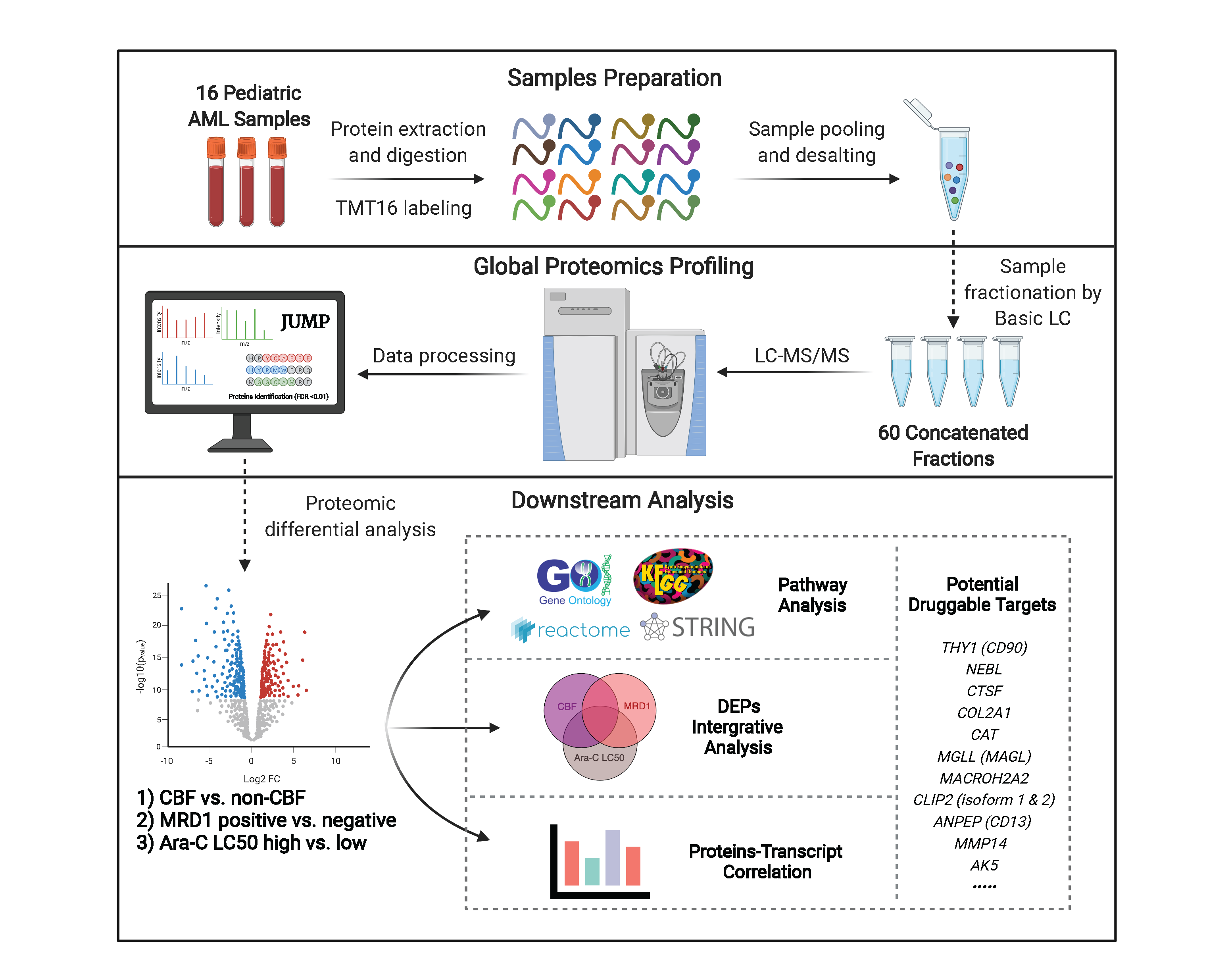
2. Materials and Methods
2.1. Study Cohort, Proteomic and Transcriptomic Expression Profiling
2.2. Differential Expression Analysis of Proteomics Data and Integrative Analysis
2.3. Protein–Protein Interaction (PPI), Pathway Analysis, and Gene Set Enrichment Pathway (GSEA) of CBF Status Comparison
2.4. Correlation Analysis between Proteomics and Transcriptomics Data
3. Results
3.1. Study Cohort and Proteomic Profiling
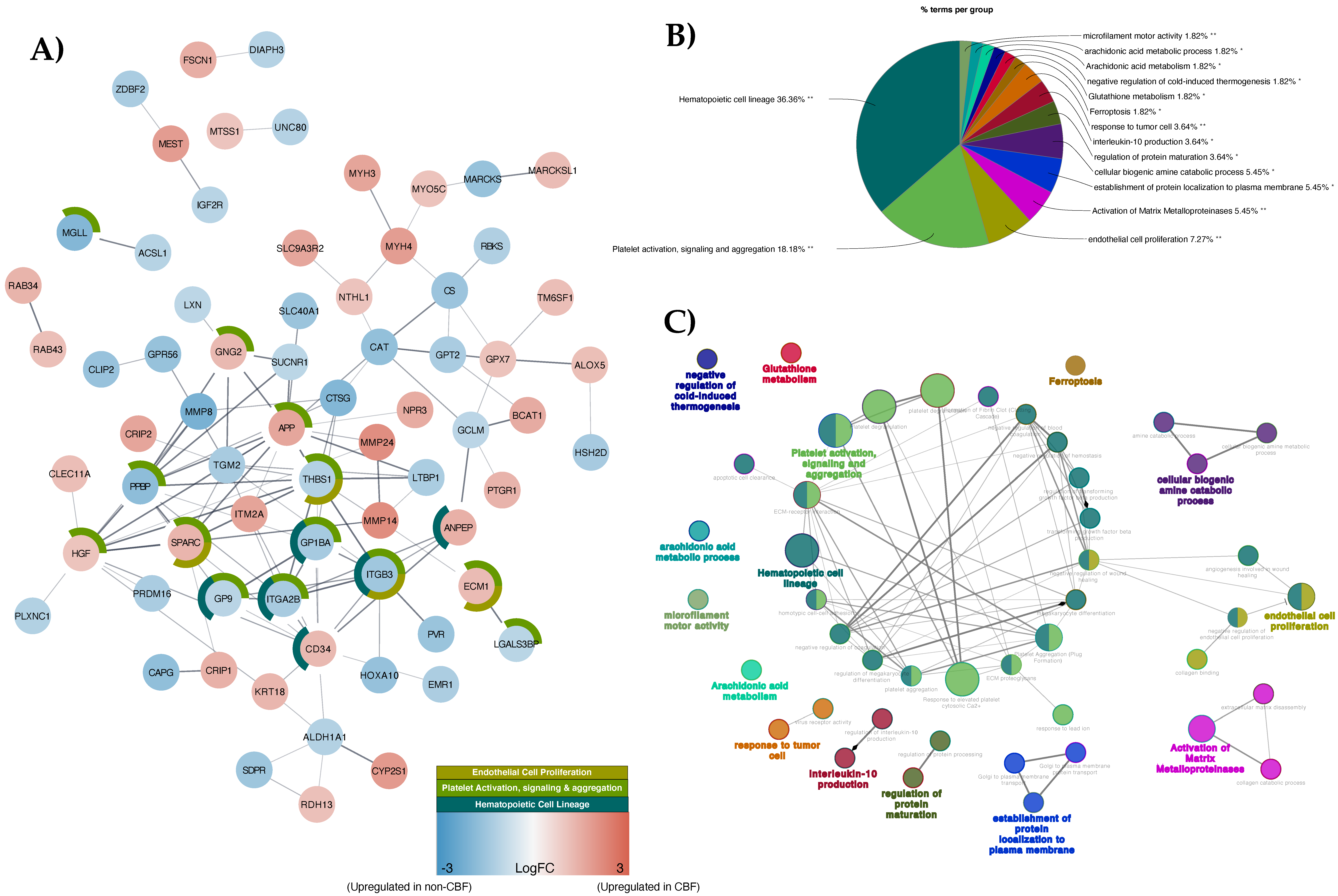
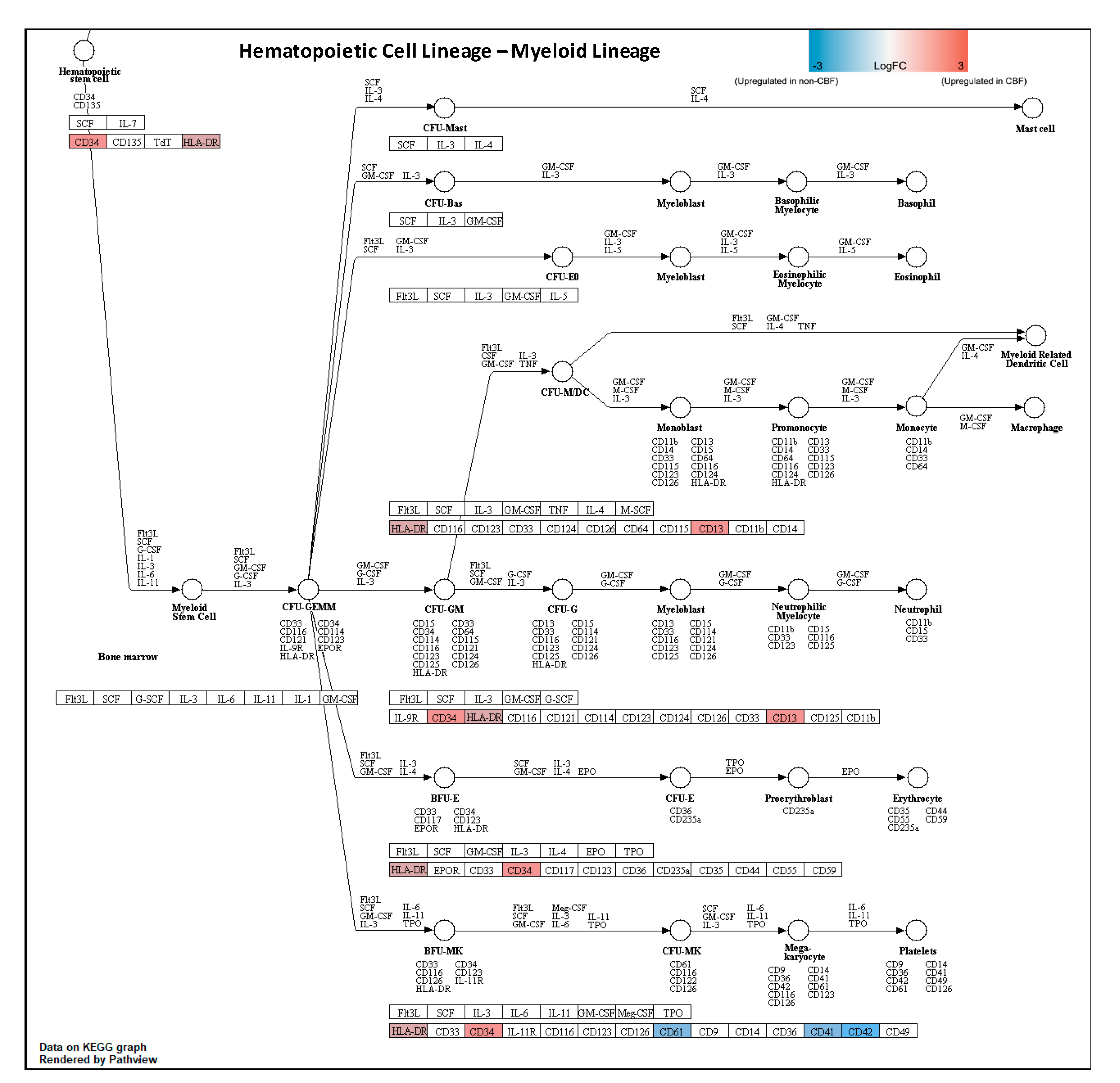
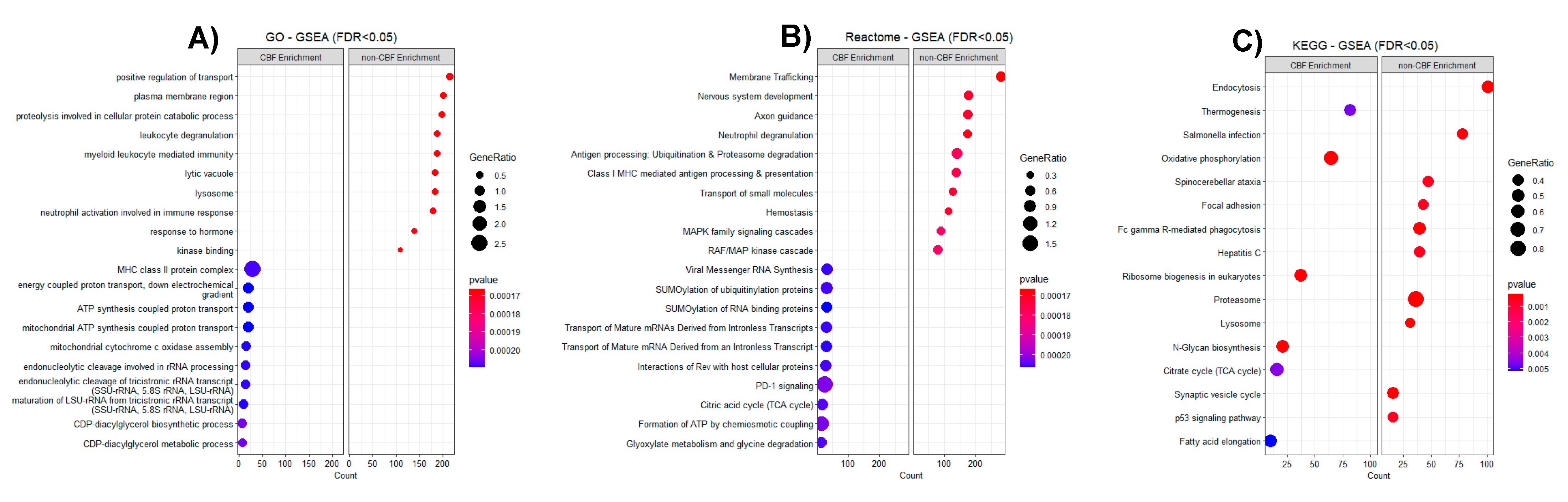
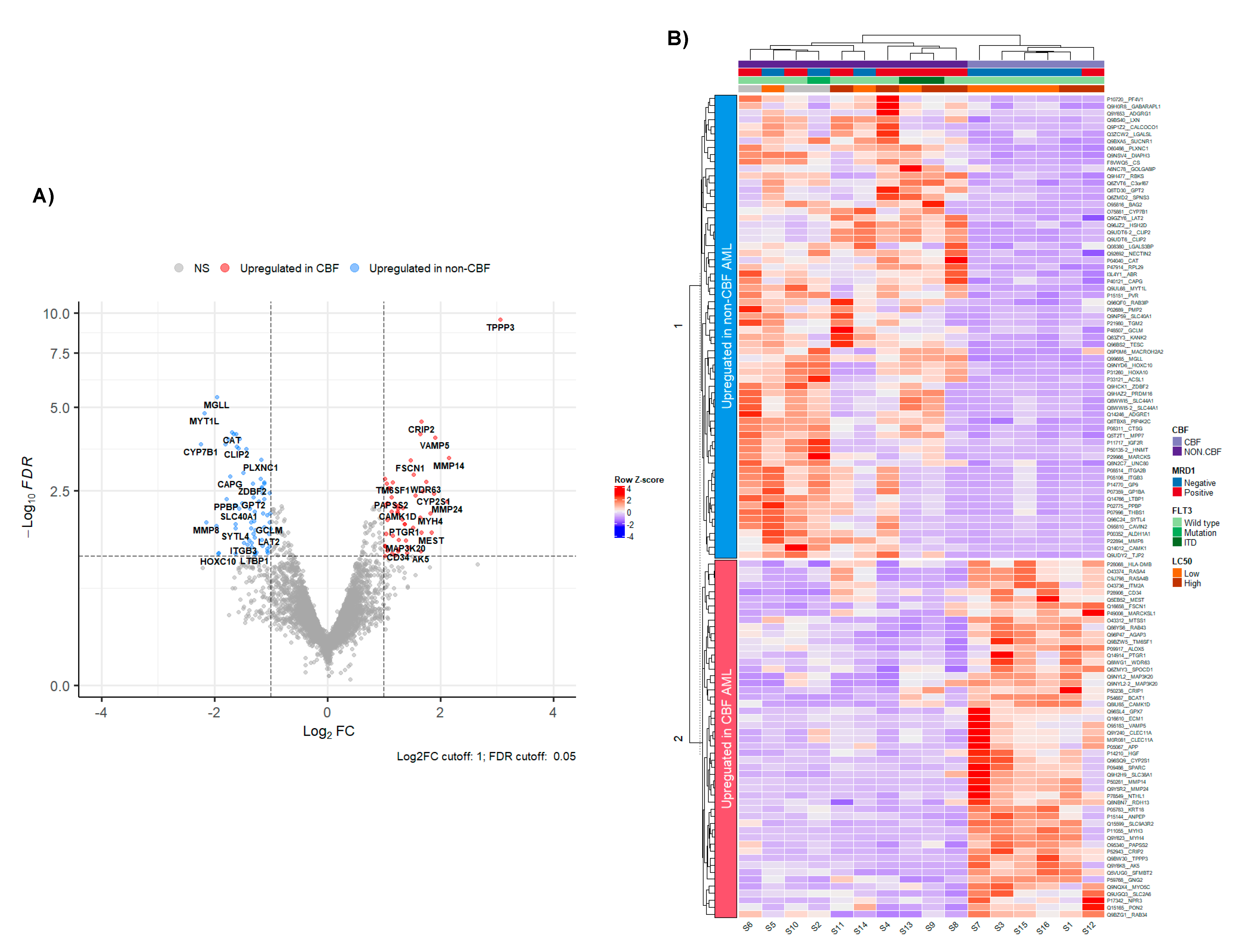
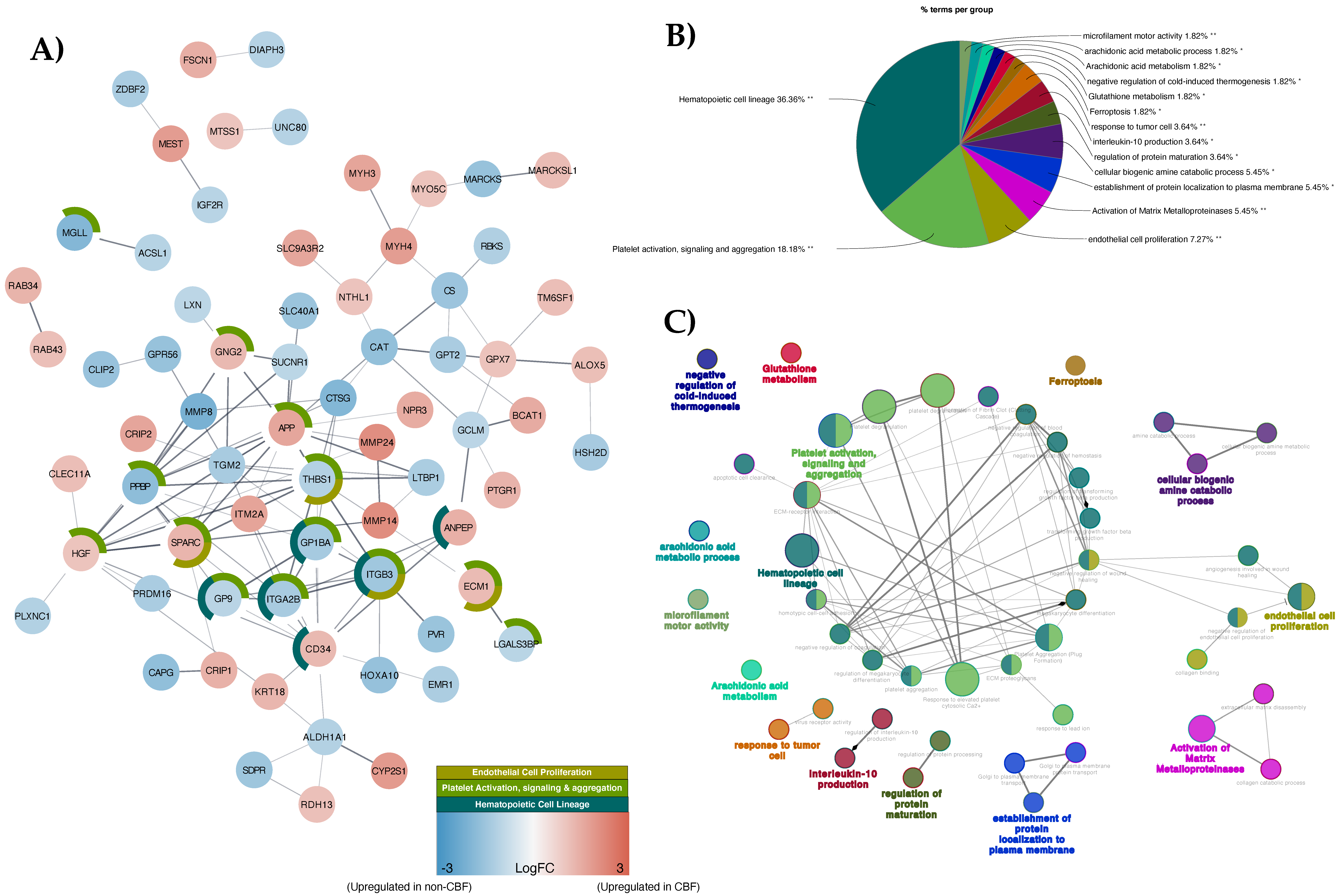
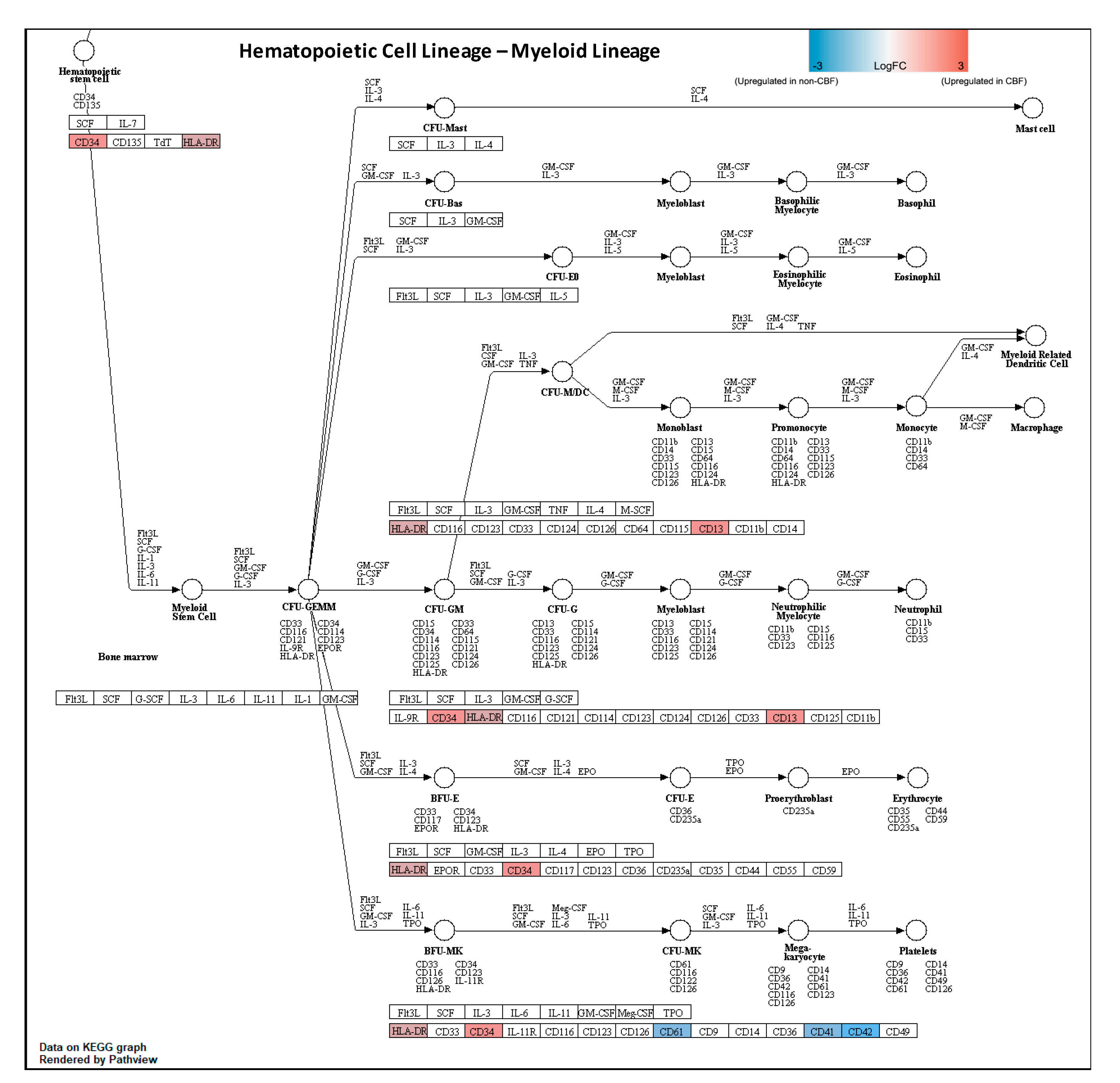
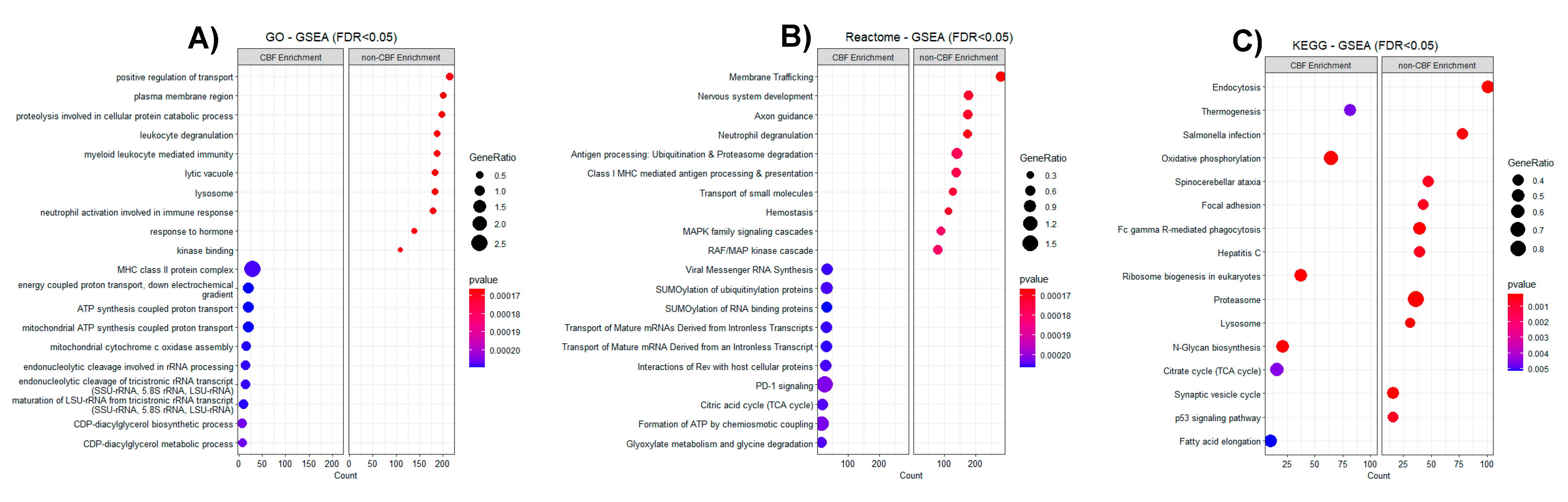
3.2. Proteomic Profiling of CBF Compared to Non-CBF AML Patients and Functional Analysis
3.3. Proteomic Profiling by MRD1 Status and by In Vitro Ara-C LC50 Level
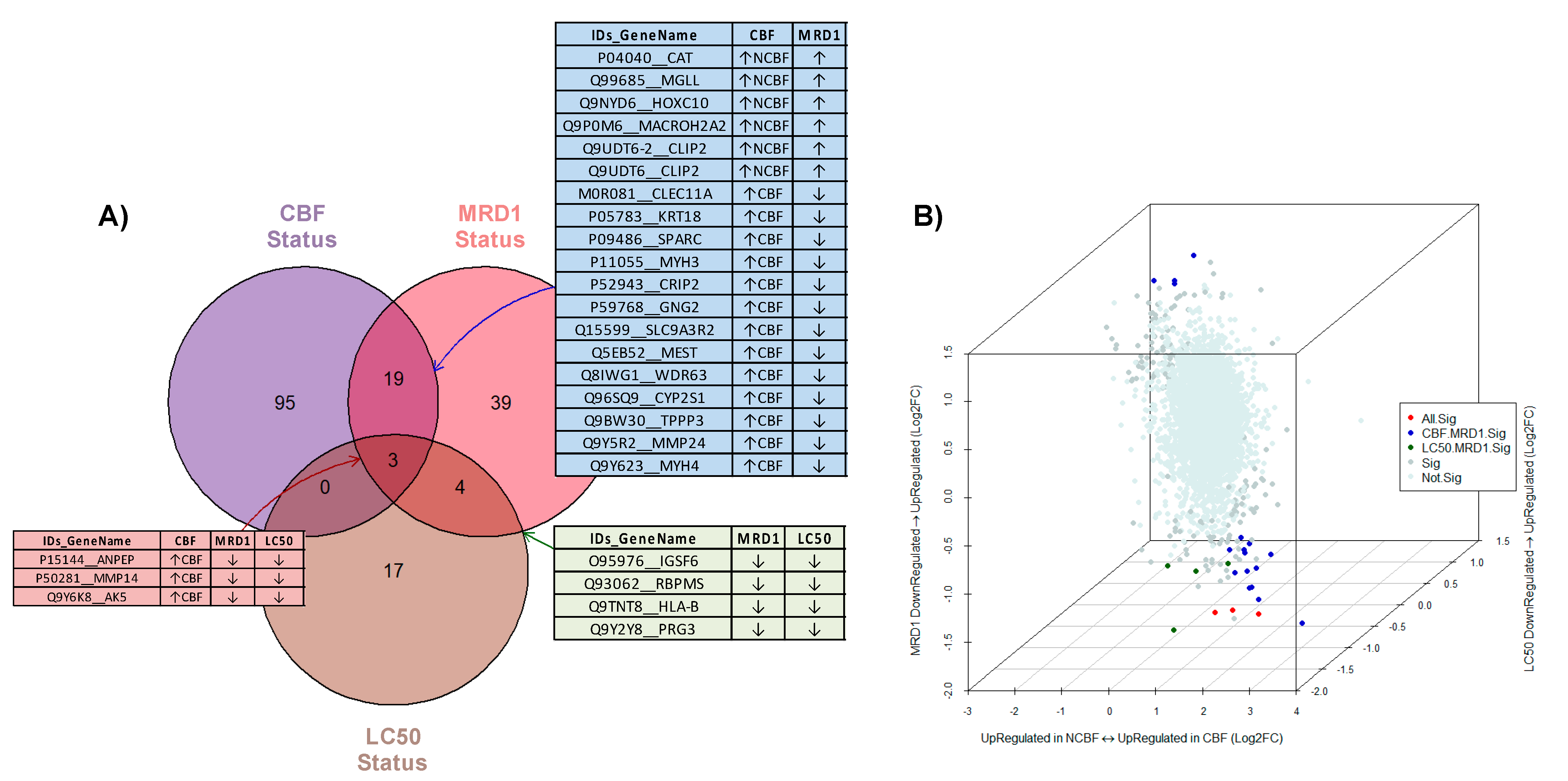
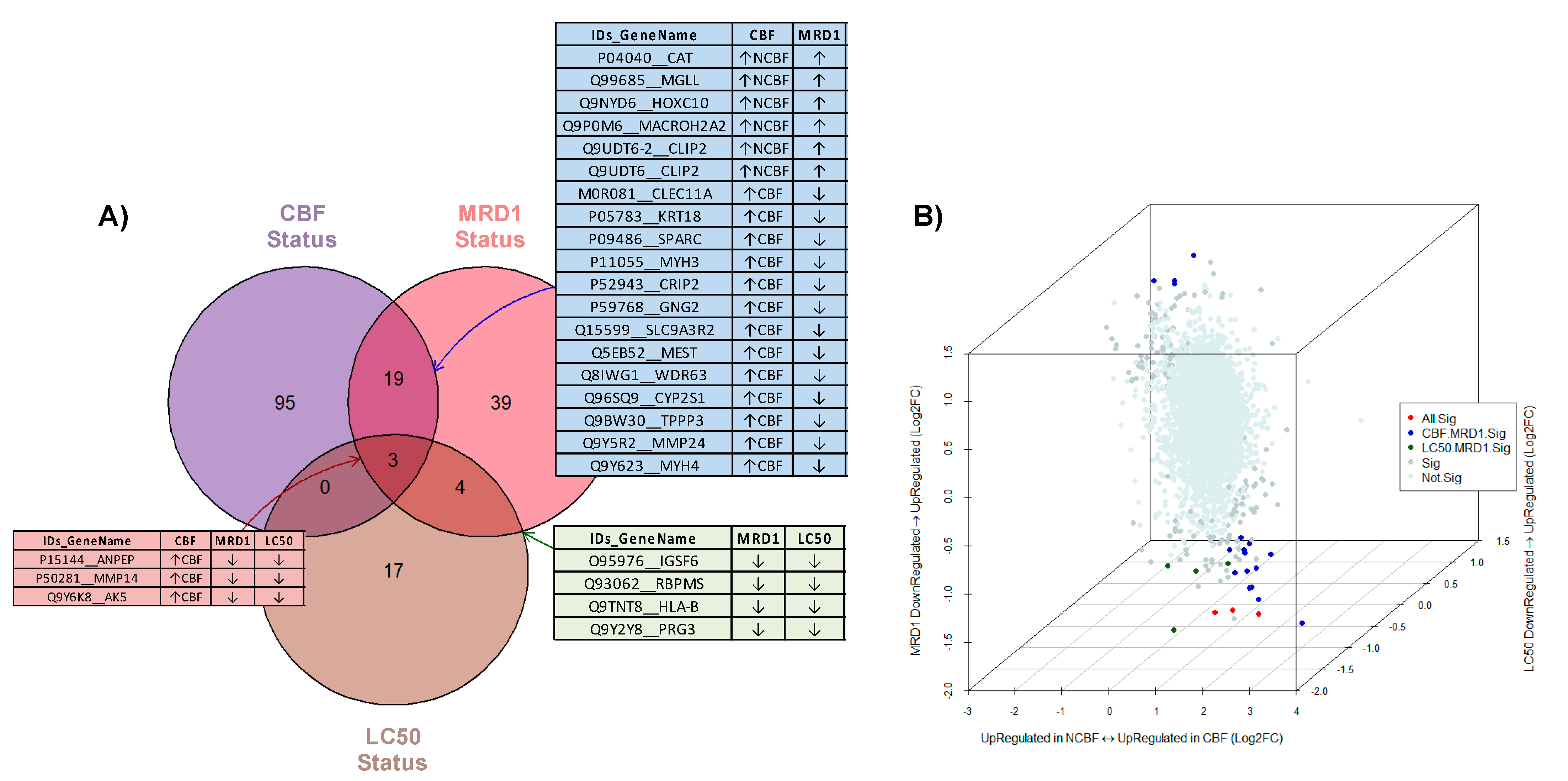
3.4. Integrative Analysis of Three Comparison Strategies (CBF, MRD1, and Ara-C LC50)
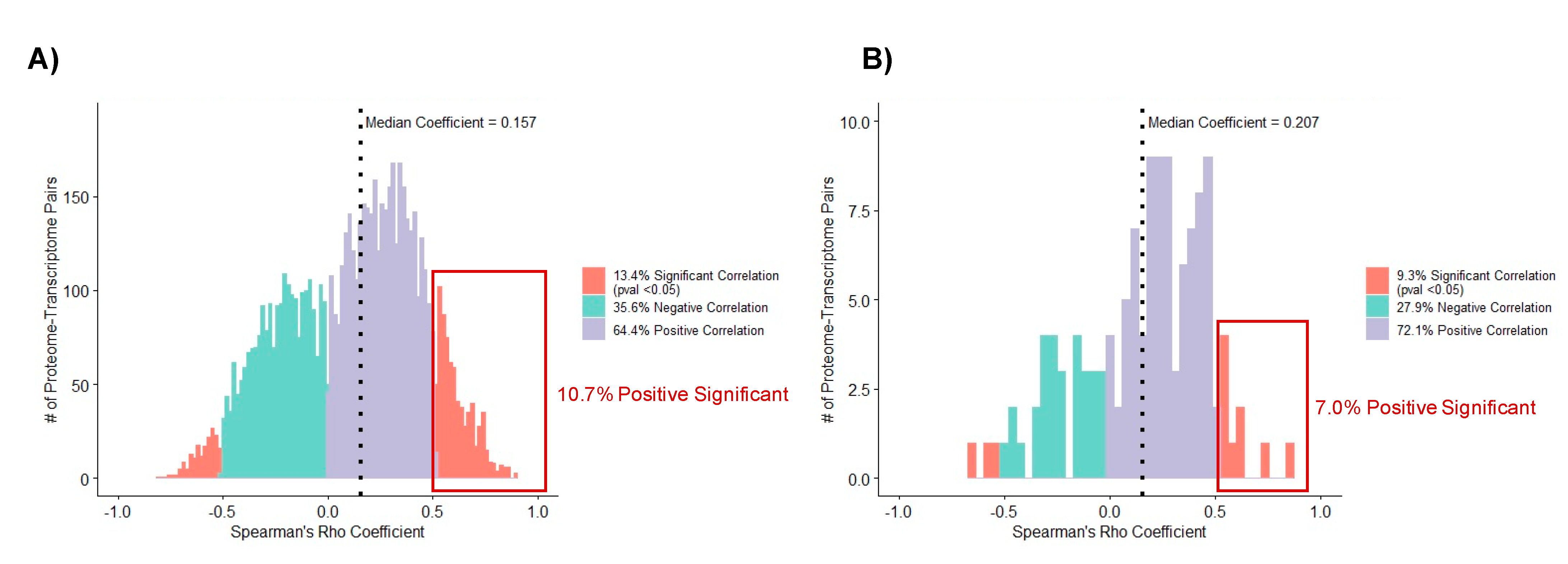
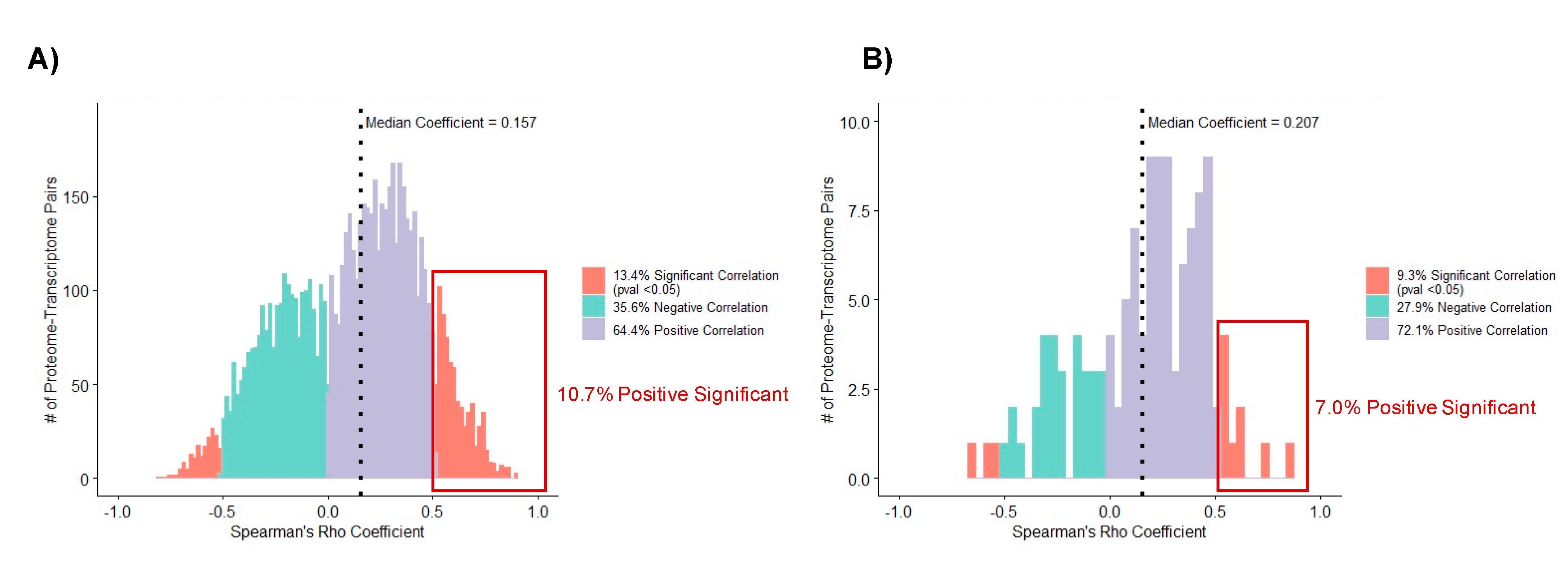
3.5. Correlation Analysis of Matched Proteome and Transcriptome
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ward, E.; DeSantis, C.; Robbins, A.; Kohler, B.; Jemal, A. Childhood and adolescent cancer statistics, 2014. CA A Cancer J. Clin. 2014, 64, 83–103. [Google Scholar] [CrossRef] [PubMed]
- Faber, Z.; Chen, X.; Gedman, A.L.; Boggs, K.; Cheng, J.; Ma, J.; Radtke, I.; Chao, J.-R.; Walsh, M.P.; Song, G.; et al. The genomic landscape of core-binding factor acute myeloid leukemias. Nat. Genet. 2016, 48, 1551–1556. [Google Scholar] [CrossRef] [PubMed]
- Solh, M.; Yohe, S.; Weisdorf, D.; Ustun, C. Core-binding factor acute myeloid leukemia: Heterogeneity, monitoring, and therapy. Am. J. Hematol. 2014, 89, 1121–1131. [Google Scholar] [CrossRef]
- Farrar, J.E.; Schuback, H.L.; Ries, R.E.; Wai, D.; Hampton, O.A.; Trevino, L.R.; Alonzo, T.A.; Auvil, J.M.G.; Davidsen, T.M.; Gesuwan, P.; et al. Genomic Profiling of Pediatric Acute Myeloid Leukemia Reveals a Changing Mutational Landscape from Disease Diagnosis to Relapse. Cancer Res. 2016, 76, 2197–2205. [Google Scholar] [CrossRef] [Green Version]
- Marks, L.J.; Oberg, J.A.; Glade-Bender, J.L.; Hsiao, S.J.; Pendrick, D.; Sireci, A.N.; Andrews, S.J.; Mansukhani, M.M.; Kung, A.L.; Sulis, M.L. Molecular Profiling of High-Risk Pediatric Acute Myeloid Leukemia. Blood 2016, 128, 5250. [Google Scholar] [CrossRef]
- Ross, M.E.; Mahfouz, R.; Onciu, M.; Liu, H.-C.; Zhou, X.; Song, G.; Shurtleff, S.A.; Pounds, S.; Cheng, C.; Ma, J.; et al. Gene expression profiling of pediatric acute myelogenous leukemia. Blood 2004, 104, 3679–3687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Çelik, H.; Lindblad, K.E.; Popescu, B.; Gui, G.; Goswami, M.; Valdez, J.; DeStefano, C.; Lai, C.; Thompson, J.; Ghannam, J.Y.; et al. Highly multiplexed proteomic assessment of human bone marrow in acute myeloid leukemia. Blood Adv. 2020, 4, 367–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raffel, S.; Klimmeck, D.; Falcone, M.; Demir, A.; Pouya, A.; Zeisberger, P.; Lutz, C.; Tinelli, M.; Bischel, O.; Bullinger, L.; et al. Quantitative proteomics reveals specific metabolic features of acute myeloid leukemia stem cells. Blood 2020, 136, 1507–1519. [Google Scholar] [CrossRef]
- Chebouba, L.; Boughaci, D.; Guziolowski, C. Proteomics Versus Clinical Data and Stochastic Local Search Based Feature Selection for Acute Myeloid Leukemia Patients’ Classification. J. Med. Syst. 2018, 42, 129. [Google Scholar] [CrossRef]
- Forthun, R.B.; Aasebø, E.; Rasinger, J.D.; Bedringaas, S.L.; Berven, F.; Selheim, F.; Bruserud, Ø.; Gjertsen, B.T. Phosphoprotein DIGE profiles reflect blast differentiation, cytogenetic risk stratification, FLT3/NPM1 mutations and therapy response in acute myeloid leukaemia. J. Proteom. 2018, 173, 32–41. [Google Scholar] [CrossRef]
- Kadia, T.M.; Kantarjian, H.; Kornblau, S.; Borthakur, G.; Faderl, S.; Freireich, E.J.; Luthra, R.; Garcia-Manero, G.; Pierce, S.; Cortes, J.; et al. Clinical and proteomic characterization of acute myeloid leukemia with mutated RAS. Cancer 2012, 118, 5550–5559. [Google Scholar] [CrossRef] [Green Version]
- Kornblau, S.M.; Tibes, R.; Qiu, Y.H.; Chen, W.; Kantarjian, H.M.; Andreeff, M.; Coombes, K.R.; Mills, G.B. Functional proteomic profiling of AML predicts response and survival. Blood 2009, 113, 154–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoff, F.W.; Hu, C.W.; Qiu, Y.; Ligeralde, A.; Yoo, S.-Y.; Mahmud, H.; de Bont, E.S.J.M.; Qutub, A.A.; Horton, T.M.; Kornblau, S.M. Recognition of recurrent protein expression patterns in pediatric acute myeloid leukemia identified new therapeutic targets. Mol. Cancer Res. 2018, 16, 1275–1286. [Google Scholar] [CrossRef] [Green Version]
- Bolouri, H.; Farrar, J.E.; Triche, T., Jr.; Ries, R.E.; Lim, E.L.; Alonzo, T.A.; Ma, Y.; Moore, R.; Mungall, A.J.; Marra, M.; et al. The molecular landscape of pediatric acute myeloid leukemia reveals recurrent structural alterations and age-specific mutational interactions. Nat. Med. 2018, 24, 103–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubnitz, J.; Inaba, H.; Dahl, G.; Ribeiro, R.C.; Bowman, W.P.; Taub, J.; Pounds, S.; Razzouk, B.; Lacayo, N.J.; Cao, X.; et al. Minimal residual disease-directed therapy for childhood acute myeloid leukaemia: Results of the AML02 multicentre trial. Lancet Oncol. 2010, 11, 543–552. [Google Scholar] [CrossRef] [Green Version]
- Lamba, J.K.; Crews, K.R.; Pounds, S.; Cao, X.; Gandhi, V.; Plunkett, W.; Razzouk, B.; Lamba, V.; Baker, S.; Raimondi, S.C.; et al. Identification of predictive markers of cytarabine response in AML by integrative analysis of gene-expression profiles with multiple phenotypes. Pharmacogenomics 2011, 12, 327–339. [Google Scholar] [CrossRef] [Green Version]
- Bargal, S.A.; Rafiee, R.; Crews, K.R.; Wu, H.; Cao, X.; Rubnitz, J.E.; Ribeiro, R.C.; Downing, J.R.; Pounds, S.B.; Lamba, J.K. Genome-wide association analysis identifies SNPs predictive of in vitro leukemic cell sensitivity to cytarabine in pediatric AML. Oncotarget 2018, 9, 34859–34875. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Kavdia, K.; Dey, K.K.; Pagala, V.R.; Kodali, K.; Liu, D.; Lee, D.G.; Sun, H.; Chepyala, S.R.; Cho, J.-H.; et al. High-throughput and Deep-proteome Profiling by 16-plex Tandem Mass Tag Labeling Coupled with Two-dimensional Chromatography and Mass Spectrometry. J. Vis. Exp. 2020, e61684. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Wang, X.; Li, Y.; Wu, Z.; Wang, H.; Tan, H.; Peng, J. JUMP: A tag-based database search tool for peptide identification with high sensitivity and accuracy. Mol. Cell Proteom. 2014, 13, 3663–3673. [Google Scholar] [CrossRef] [Green Version]
- Niu, M.; Cho, J.-H.; Kodali, K.; Pagala, V.; High, A.A.; Wang, H.; Wu, Z.; Li, Y.; Bi, W.; Zhang, H.; et al. Extensive Peptide Fractionation and y1 Ion-Based Interference Detection Method for Enabling Accurate Quantification by Isobaric Labeling and Mass Spectrometry. Anal. Chem. 2017, 89, 2956–2963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huber, W.; Von Heydebreck, A.; Sültmann, H.; Poustka, A.; Vingron, M. Variance stabilization applied to microarray data calibration and to the quantification of differential expression. Bioinformatics 2002, 18, S96–S104. [Google Scholar] [CrossRef]
- Välikangas, T.; Suomi, T.; Elo, L.L. A systematic evaluation of normalization methods in quantitative label-free proteomics. Briefings Bioinform. 2016, 19, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Graw, S.; Tang, J.; Zafar, M.K.; Byrd, A.K.; Bolden, C.; Peterson, E.C.; Byrum, S.D. proteiNorm—A User-Friendly Tool for Normalization and Analysis of TMT and Label-Free Protein Quantification. ACS Omega 2020, 5, 25625–25633. [Google Scholar] [CrossRef]
- Jahn, N.; Terzer, T.; Sträng, E.; Dolnik, A.; Cocciardi, S.; Panina, E.; Corbacioglu, A.; Herzig, J.; Weber, D.; Schrade, A.; et al. Genomic heterogeneity in core-binding factor acute myeloid leukemia and its clinical implication. Blood Adv. 2020, 4, 6342–6352. [Google Scholar] [CrossRef]
- Del Carratore, F.; Jankevics, A.; Eisinga, R.; Heskes, T.; Hong, F.; Breitling, R. RankProd 2.0: A refactored bioconductor package for detecting differentially expressed features in molecular profiling datasets. Bioinformatics 2017, 33, 2774–2775. [Google Scholar] [CrossRef] [Green Version]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [Green Version]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene Ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fabregat, A.; Jupe, S.; Matthews, L.; Sidiropoulos, K.; Gillespie, M.; Garapati, P.; Haw, R.; Jassal, B.; Korninger, F.; May, B.; et al. The Reactome Pathway Knowledgebase. Nucleic Acids Res. 2018, 46, D649–D655. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016, 44, D457–D462. [Google Scholar] [CrossRef] [Green Version]
- Bindea, G.; Mlecnik, B.; Hackl, H.; Charoentong, P.; Tosolini, M.; Kirilovsky, A.; Fridman, W.-H.; Pagès, F.; Trajanoski, Z.; Galon, J. ClueGO: A Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 2009, 25, 1091–1093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, W.; Brouwer, C. Pathview: An R/Bioconductor package for pathway-based data integration and visualization. Bioinformatics 2013, 29, 1830–1831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, G.; Wang, L.-G.; Han, Y.; He, Q.-Y. clusterProfiler: An R Package for Comparing Biological Themes Among Gene Clusters. OMICS J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Göllner, S.; Oellerich, T.; Agrawal-Singh, S.; Schenk, T.; Klein, H.-U.; Rohde, C.; Pabst, C.; Sauer, T.; Lerdrup, M.; Tavor, S.; et al. Loss of the histone methyltransferase EZH2 induces resistance to multiple drugs in acute myeloid leukemia. Nat. Med. 2017, 23, 69–78. [Google Scholar] [CrossRef]
- Patel, C.; Stenke, L.; Varma, S.; Lindberg, M.L.; Björkholm, M.; Sjöberg, J.; Viktorsson, K.; Lewensohn, R.; Landgren, O.; Gottesman, M.M.; et al. Multidrug resistance in relapsed acute myeloid leukemia: Evidence of biological heterogeneity. Cancer 2013, 119, 3076–3083. [Google Scholar] [CrossRef] [Green Version]
- Man, C.H.; Lam, S.S.Y.; Sun, M.K.H.; Chow, H.C.H.; Gill, H.; Kwong, Y.L.; Leung, A.Y.H. A novel tescalcin-sodium/hydrogen exchange axis underlying sorafenib resistance in FLT3-ITD+ AML. Blood 2014, 123, 2530–2539. [Google Scholar] [CrossRef] [Green Version]
- Roushangar, R.; Mias, G.I. Multi-study reanalysis of 2,213 acute myeloid leukemia patients reveals age- and sex-dependent gene expression signatures. Sci. Rep. 2019, 9, 12413. [Google Scholar] [CrossRef] [Green Version]
- Wunderlich, M.; Krejci, O.; Wei, J.; Mulloy, J.C. Human CD34+ cells expressing the inv(16) fusion protein exhibit a myelomonocytic phenotype with greatly enhanced proliferative ability. Blood 2006, 108, 1690–1697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, C.-H.; Xu, H.; Bark, H.; Lee, T.-B.; Yun, J.; Kang, S.-I.; Oh, Y.-K. Balance of NF-kappaB and p38 MAPK is a determinant of radiosensitivity of the AML-2 and its doxorubicin-resistant cell lines. Leuk. Res. 2007, 31, 1267–1276. [Google Scholar] [CrossRef] [PubMed]
- Prüser, J.L.; Ramer, R.; Wittig, F.; Ivanov, I.; Merkord, J.; Hinz, B. The monoacylglycerol lipase inhibitor JZL184 inhibits lung cancer cell invasion and metastasis via the CB1 cannabinoid receptor. Mol. Cancer Ther. 2021, 20, 787–802. [Google Scholar] [CrossRef]
- Jaiswal, S.; Raja Ayyannan, S. Anticancer potential of small molecule inhibitors of fatty acid amide hydrolase and monoacylglycerol lipase—A mini-review. ChemMedChem 2021. [Google Scholar] [CrossRef] [PubMed]
- Deng, H.; Li, W. Monoacylglycerol lipase inhibitors: Modulators for lipid metabolism in cancer malignancy, neurological and metabolic disorders. Acta Pharm. Sin. B 2020, 10, 582–602. [Google Scholar] [CrossRef]
- Afzal, O.; Altamimi, A.S.A.; Shahroz, M.M.; Sharma, H.K.; Riadi, Y.; Hassan, M.Q. Analgesic and Anticancer Activity of Benzoxazole Clubbed 2-Pyrrolidinones as Novel Inhibitors of Monoacylglycerol Lipase. Molecules 2021, 26, 2389. [Google Scholar] [CrossRef]
- Han, Q.; Lu, J.; Wang, J.; Ye, J.; Jiang, X.; Chen, H.; Liu, C.; Chen, L.; Lin, T.; Chen, S.; et al. H2AFY is a novel fusion partner of MECOM in acute myeloid leukemia. Cancer Genet. 2018, 222, 9–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buccisano, F.; Rossi, F.M.; Venditti, A.; DEL Poeta, G.; Cox, M.C.; Abruzzese, E.; Rupolo, M.; Berretta, M.; Degan, M.; Russo, S.; et al. CD90/Thy-1 is preferentially expressed on blast cells of high risk acute myeloid leukaemias*. Br. J. Haematol. 2004, 125, 203–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emerenciano, M.; Kowarz, E.; Karl, K.; de Almeida Lopes, B.; Scholz, B.; Bracharz, S.; Meyer, C.; Pombo-de-Oliveira, M.S.; Marschalek, R. Functional analysis of the two reciprocal fusion genes MLL-NEBL and NEBL-MLL reveal their oncogenic potential. Cancer Lett. 2013, 332, 30–34. [Google Scholar] [CrossRef] [Green Version]
- Marasca, R.; Maffei, R.; Zucchini, P.; Castelli, I.; Saviola, A.; Martinelli, S.; Ferrari, A.; Fontana, M.; Ravanetti, S.; Torelli, G. Gene expression profiling of acute promyelocytic leukaemia identifies two subtypes mainly associated with flt3 mutational status. Leukemia 2006, 20, 103–114. [Google Scholar] [CrossRef] [Green Version]
- Farge, T.; Saland, E.; De Toni, F.; Aroua, N.; Hosseini, M.; Perry, R.; Bosc, C.; Sugita, M.; Stuani, L.; Fraisse, M.; et al. Chemotherapy-Resistant Human Acute Myeloid Leukemia Cells Are Not Enriched for Leukemic Stem Cells but Require Oxidative Metabolism. Cancer Discov. 2017, 7, 716–735. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Sharma, A.; Ming, W.; Sun, X.; Liu, H. A deconvolution method and its application in analyzing the cellular fractions in acute myeloid leukemia samples. BMC Genom. 2020, 21, 652. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Feng, Z.; Ma, J.; Ling, S.; Cao, Y.; Gurung, B.; Wu, Y.; Katona, B.W.; O’Dwyer, K.P.; Siegel, D.L.; et al. Bispecific and split CAR T cells targeting CD13 and TIM3 eradicate acute myeloid leukemia. Blood 2020, 135, 713–723. [Google Scholar] [CrossRef] [PubMed]
- Pirillo, C.; Haltalli, M.; Anton, S.G.; Tini, V.; Kong, I.; Hawkins, E.; Falini, B.; Marra, A.; Duarte, D.; Celso, C.L.; et al. Identification of Therapeutic Targets for Quiescent, Chemotherapy-Resistant Human Leukemia Stem Cells. BioRxiv 2020. [Google Scholar] [CrossRef]
- Saito, Y.; Kitamura, H.; Hijikata, A.; Tomizawa-Murasawa, M.; Tanaka, S.; Takagi, S.; Uchida, N.; Suzuki, N.; Sone, A.; Najima, Y.; et al. Identification of Therapeutic Targets for Quiescent, Chemotherapy-Resistant Human Leukemia Stem Cells. Sci. Transl. Med. 2010, 2, 17ra9. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample ID | Age (Year) | Race | WBC (109/L) | CBF Status | FLT3 Status | MRD1 | Treatment Arm | Ara-C LC50 (ng/μL) |
|---|---|---|---|---|---|---|---|---|
| S1 | 3.71 | White | 38.9 | CBF | WT | Negative | HDAC | 1.18 |
| S2 | 21.20 | White | 70.2 | NON-CBF | Mutation | Negative | LDAC | NA |
| S3 | 10.29 | Black | 35.2 | CBF | WT | Negative | HDAC | 0.23 |
| S4 | 6.16 | White | 28.7 | NON-CBF | WT | Positive | HDAC | 0.39 |
| S5 | 12.58 | White | 24.3 | NON-CBF | WT | Negative | HDAC | 0.13 |
| S6 | 15.29 | White | 15.0 | NON-CBF | WT | Positive | LDAC | NA |
| S7 | 11.23 | White | 351.0 | CBF | WT | Negative | HDAC | 0.14 |
| S8 | 4.07 | White | 39.9 | NON-CBF | WT | Positive | LDAC | 1.79 |
| S9 | 11.70 | Black | 76.6 | NON-CBF | ITD | Positive | HDAC | 0.37 |
| S10 | 13.05 | White | 34.3 | NON-CBF | WT | Positive | LDAC | NA |
| S11 | 3.04 | White | 5.9 | NON-CBF | WT | Positive | HDAC | 0.70 |
| S12 | 5.46 | White | 24.6 | CBF | WT | Positive | HDAC | 0.34 |
| S13 | 12.69 | White | 247.9 | NON-CBF | ITD | Positive | LDAC | 0.21 |
| S14 | 5.39 | White | 6.7 | NON-CBF | WT | Negative | HDAC | 0.12 |
| S15 | 16.53 | White | 19.0 | CBF | WT | Negative | LDAC | 0.29 |
| S16 | 11.52 | White | 32.0 | CBF | WT | Negative | LDAC | 0.01 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nguyen, N.H.K.; Wu, H.; Tan, H.; Peng, J.; Rubnitz, J.E.; Cao, X.; Pounds, S.; Lamba, J.K. Global Proteomic Profiling of Pediatric AML: A Pilot Study. Cancers 2021, 13, 3161. https://doi.org/10.3390/cancers13133161
Nguyen NHK, Wu H, Tan H, Peng J, Rubnitz JE, Cao X, Pounds S, Lamba JK. Global Proteomic Profiling of Pediatric AML: A Pilot Study. Cancers. 2021; 13(13):3161. https://doi.org/10.3390/cancers13133161
Chicago/Turabian StyleNguyen, Nam H. K., Huiyun Wu, Haiyan Tan, Junmin Peng, Jeffrey E. Rubnitz, Xueyuan Cao, Stanley Pounds, and Jatinder K. Lamba. 2021. "Global Proteomic Profiling of Pediatric AML: A Pilot Study" Cancers 13, no. 13: 3161. https://doi.org/10.3390/cancers13133161
APA StyleNguyen, N. H. K., Wu, H., Tan, H., Peng, J., Rubnitz, J. E., Cao, X., Pounds, S., & Lamba, J. K. (2021). Global Proteomic Profiling of Pediatric AML: A Pilot Study. Cancers, 13(13), 3161. https://doi.org/10.3390/cancers13133161