
Repurposing Niclosamide for Targeting Pancreatic Cancer by Inhibiting Hh/Gli Non-Canonical Axis of Gsk3β
 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Cell Lines and Culture
2.3. Cell Viability Assay
2.4. Annexin-V/Propidium Iodide Labeling and Flow Cytometry Assay for Apoptosis
2.5. Cell Cycle Analysis
2.6. Western Blotting
2.7. Colony Formation Assay
2.8. Cell Migration Assays
2.9. Measurement of Intracellular and Mitochondrial Reactive Oxygen Species (ROS) Level
2.10. Confocal Microscopy
2.11. Mitochondria Imaging
2.12. Monodansylcadaverine (MDC) Staining
2.13. Fluorescence Imaging
2.14. Nuclear and Cytoplasmic Extraction
2.15. Immunoprecipitation
2.16. Statistical Analysis
3. Results
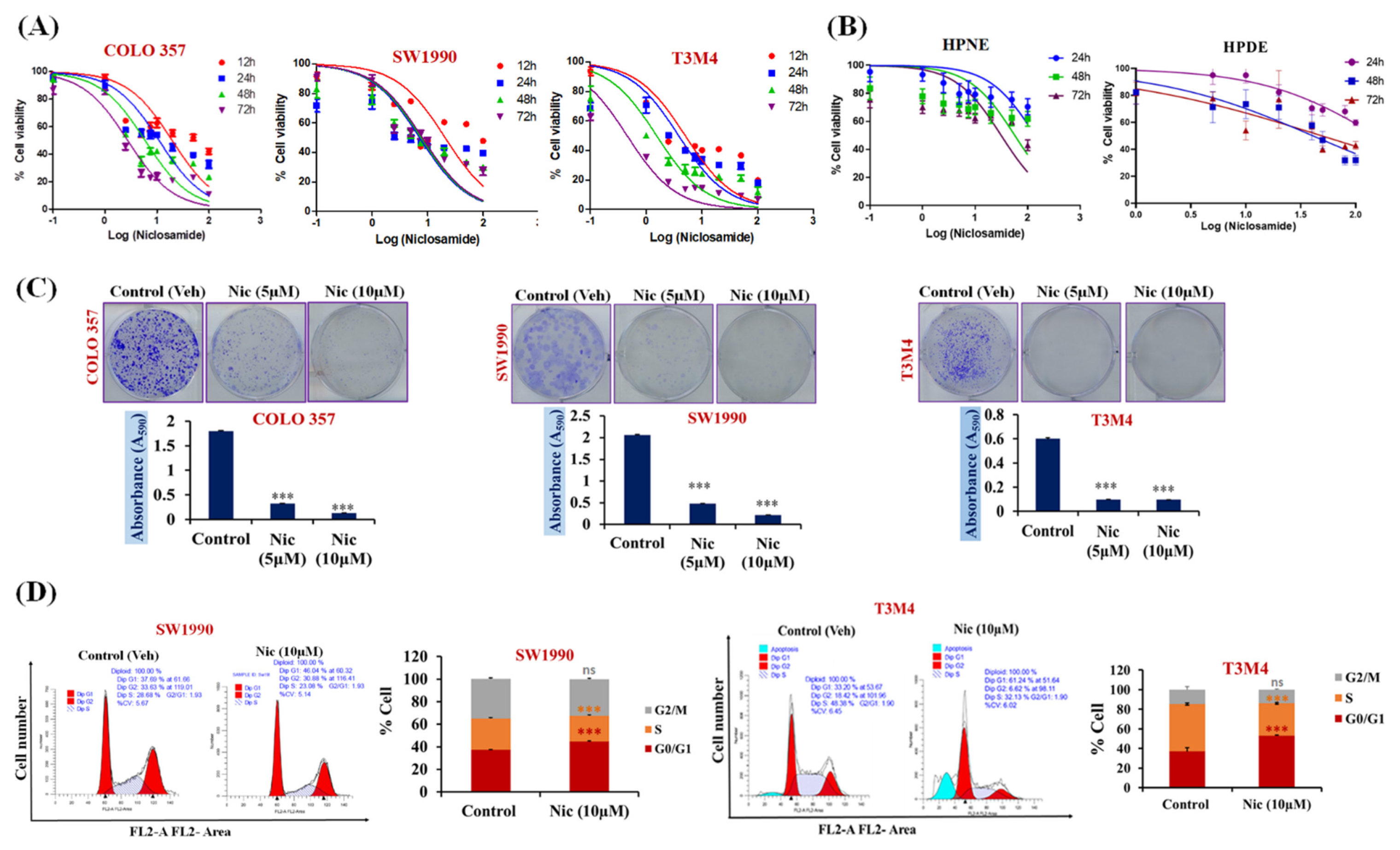
3.1. Nic Specifically Suppresses Pancreatic Cancer Cell Growth and Migration
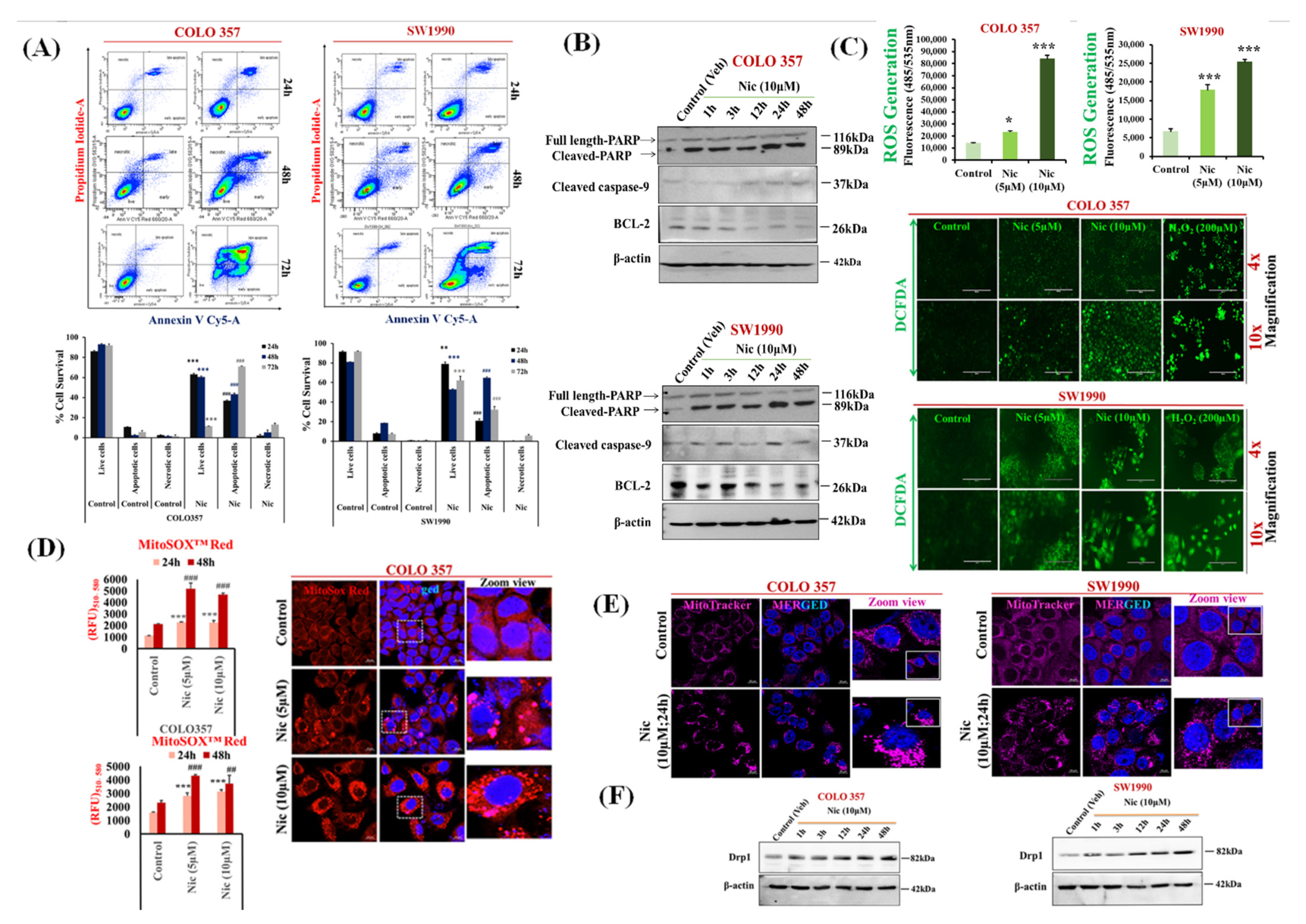
3.2. Nic Increases Intracellular and Mitochondrial Reactive Oxygen Species and Promotes Mitochondrial Fragmentation Resulting in PC Cell Apoptosis
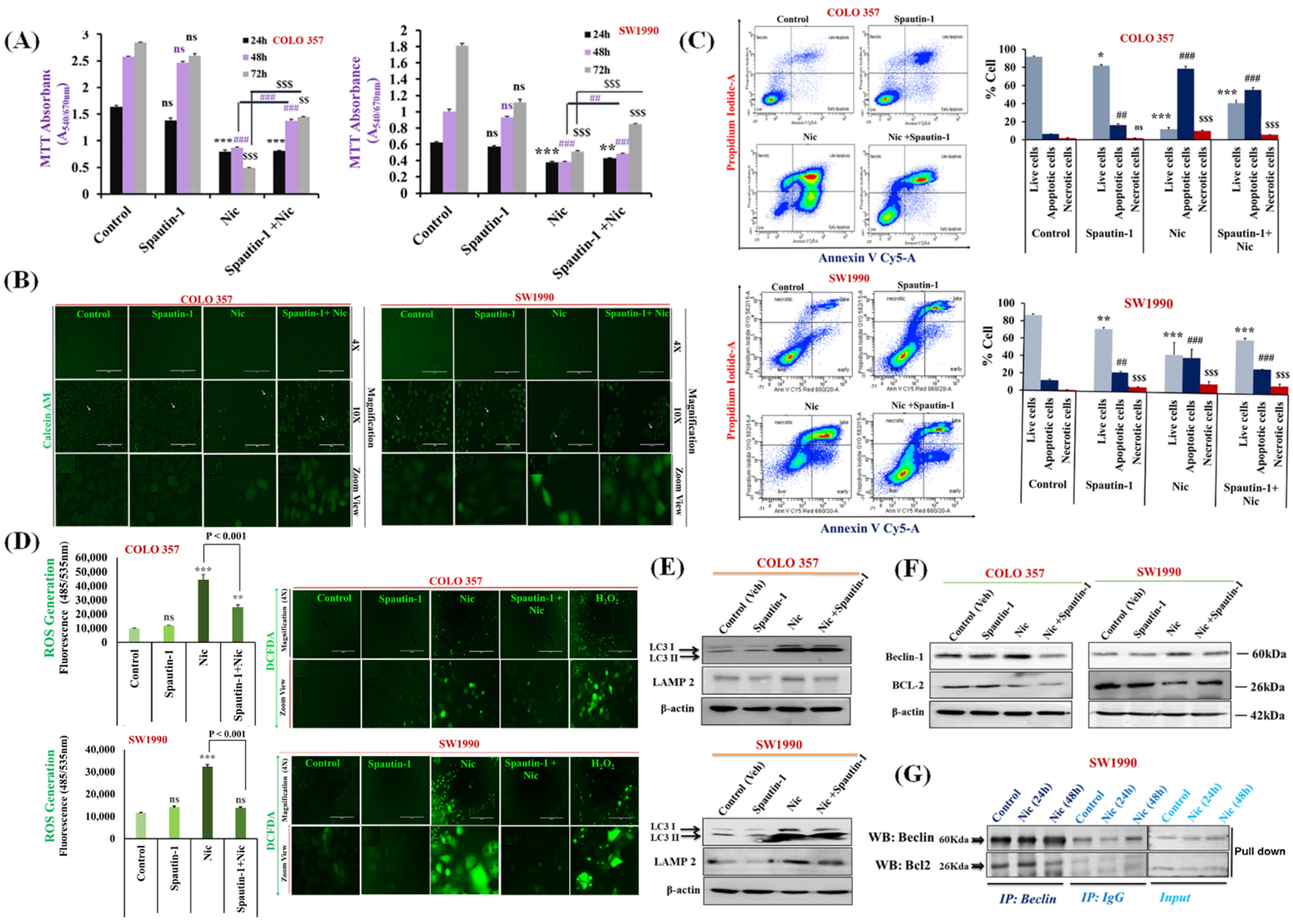
3.3. Nic Stimulates Autophagy through Inhibition of mTORC1 Signaling in Pancreatic Cancer (PC) Cells
3.4. Nic-Induced Autophagy Leads to PC Cell Death by Disrupting the Beclin1–BCl2 Interaction
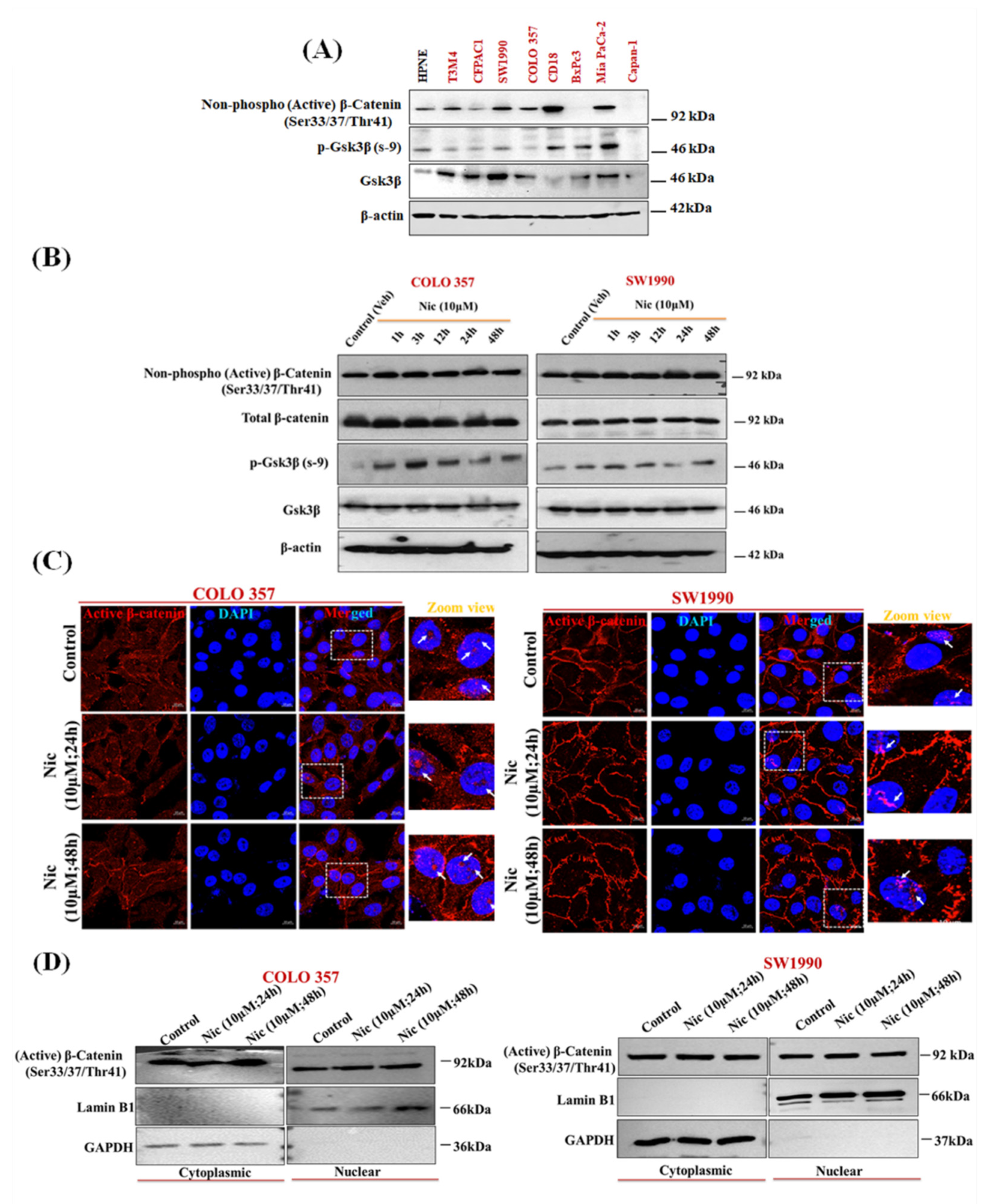
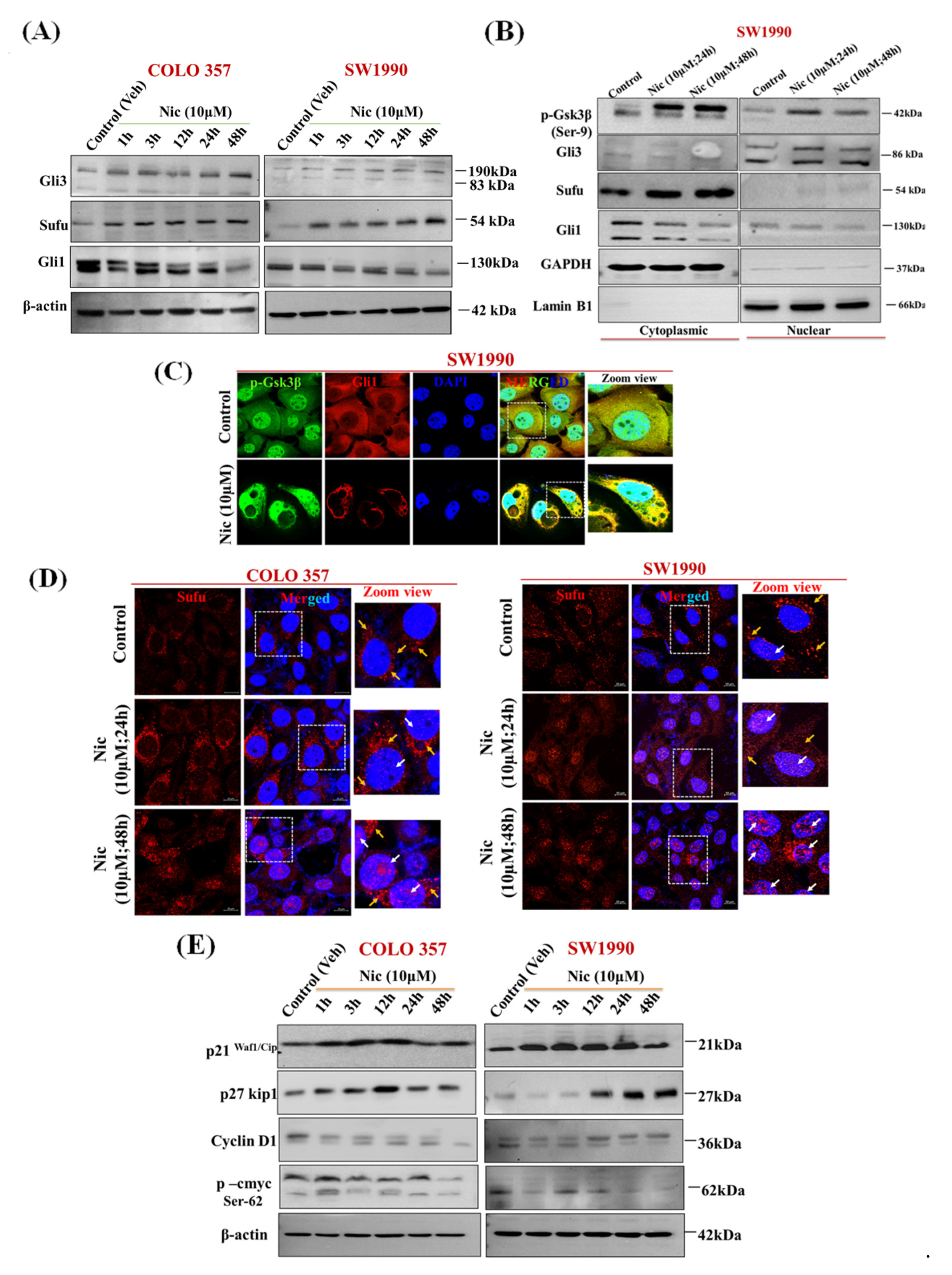
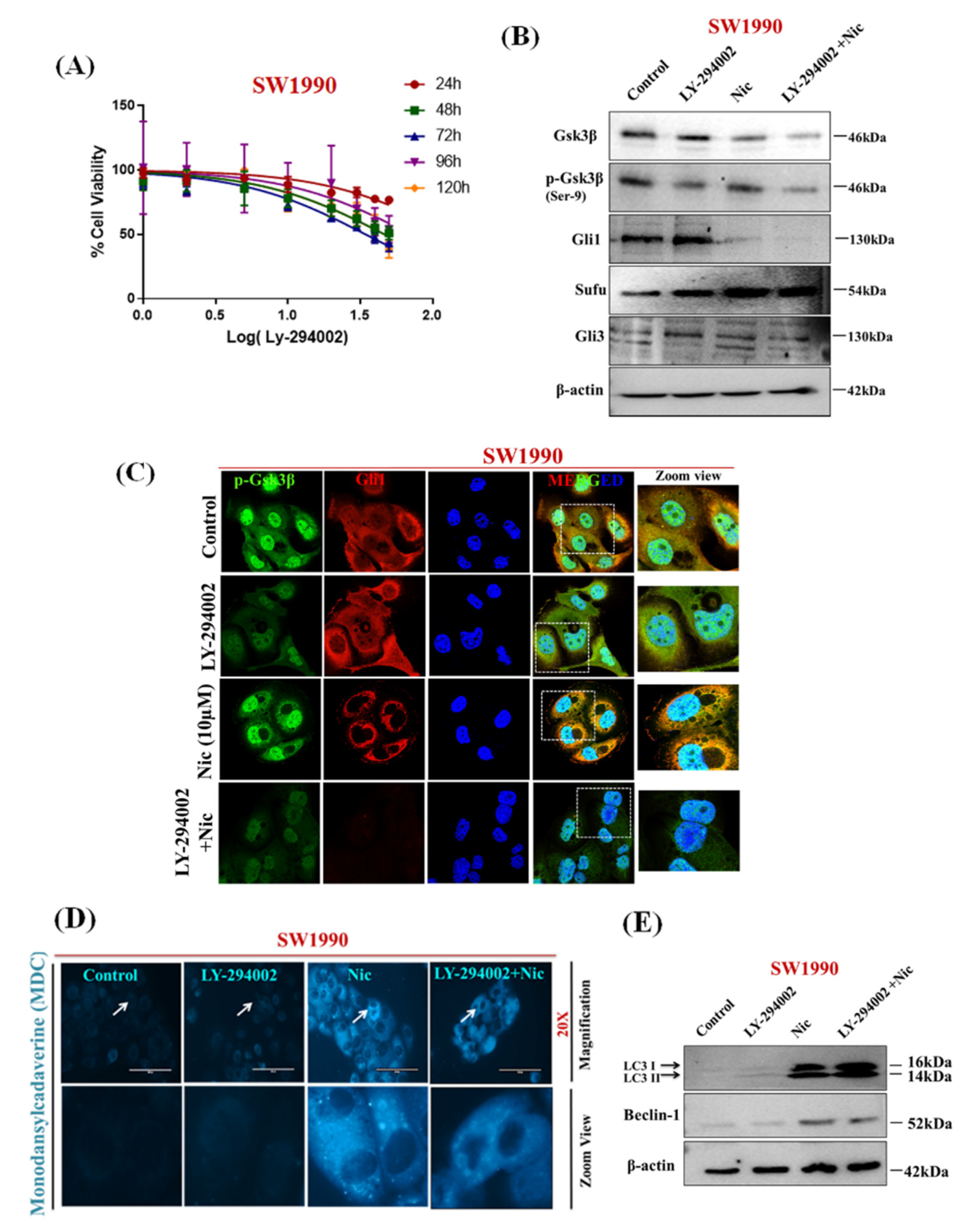
3.5. Nic Promotes Gsk3β Inactivation and Inhibits Hh/Gli Cascade via Upregulation of Sufu and Gli3, a Negative Regulator of Hh Signaling in PC Cells
3.6. Nic Induced Gsk3β Inactivation Inhibits Gli1 Activation, Negatively Impacting Hh Signaling and Promoting Autophagy-Mediated Apoptosis
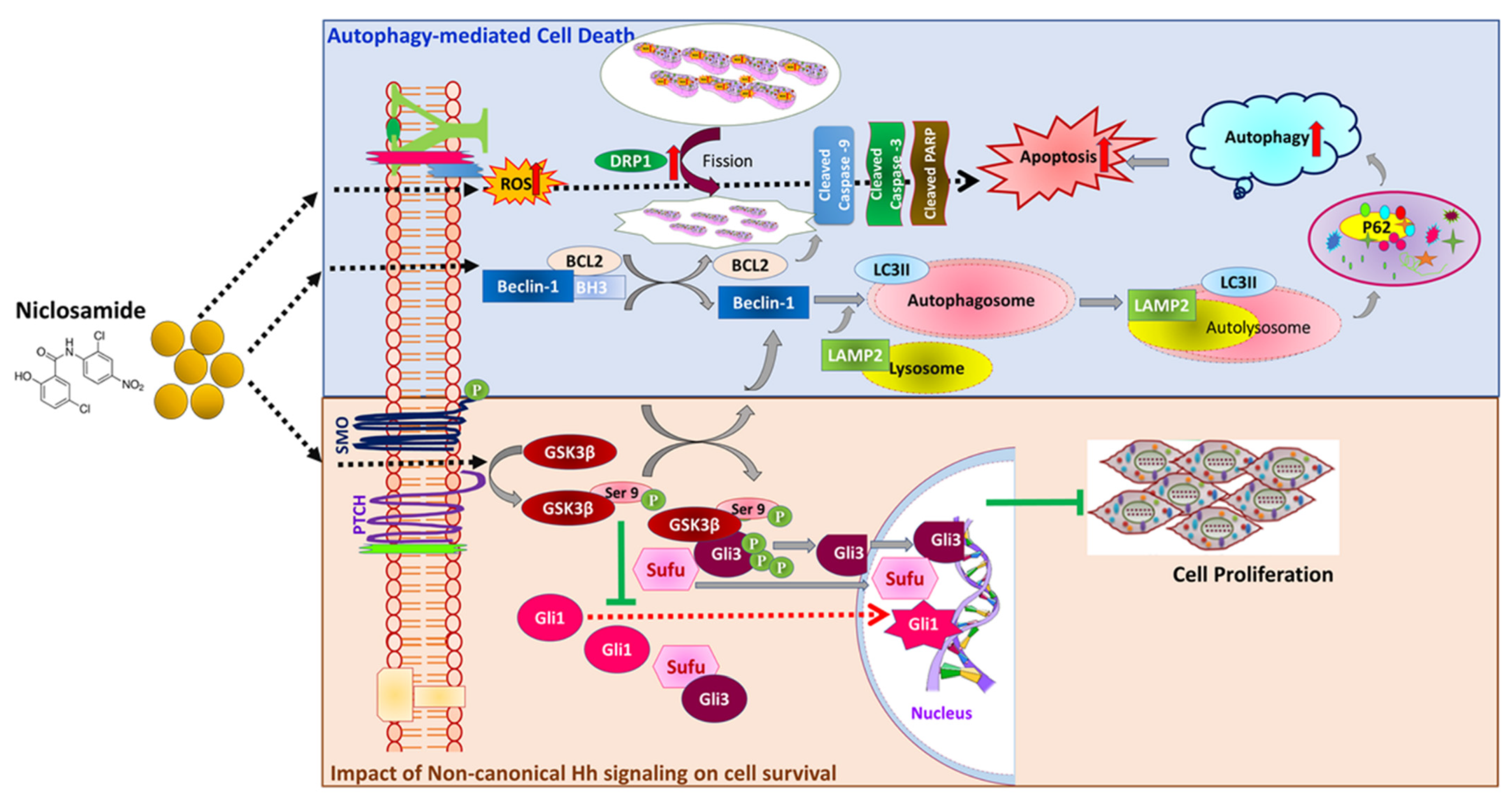
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mizrahi, J.D.; Surana, R.; Valle, J.W.; Shroff, R.T. Pancreatic cancer. Lancet 2020, 395, 2008–2020. [Google Scholar] [CrossRef]
- Rawla, P.; Sunkara, T.; Gaduputi, V. Epidemiology of pancreatic cancer: Global trends, etiology and risk factors. World J. Oncol. 2019, 10, 10. [Google Scholar] [CrossRef]
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting cancer incidence and deaths to 2030: The unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014, 74, 2913–2921. [Google Scholar] [CrossRef]
- Street, W. Cancer Facts & Figures 2019; American Cancer Society: Atlanta, GA, USA, 2019; Volume 76. [Google Scholar]
- Chu, L.C.; Goggins, M.G.; Fishman, E.K. Diagnosis and detection of pancreatic cancer. Cancer J. 2017, 23, 333–342. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Sanagapalli, S.; Stoita, A. Challenges in diagnosis of pancreatic cancer. World J. Gastroenterol. 2018, 24, 2047. [Google Scholar] [CrossRef] [PubMed]
- Roth, M.T.; Cardin, D.B.; Berlin, J.D. Recent advances in the treatment of pancreatic cancer. F1000Research 2020, 9, 131. [Google Scholar] [CrossRef] [PubMed]
- Ducreux, M.; Seufferlein, T.; Van Laethem, J.-L.; Laurent-Puig, P.; Smolenschi, C.; Malka, D.; Boige, V.; Hollebecque, A.; Conroy, T. Systemic treatment of pancreatic cancer revisited. Semin. Oncol. 2019, 46, 28–38. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhou, L.; Xie, N.; Nice, E.C.; Zhang, T.; Cui, Y.; Huang, C. Overcoming cancer therapeutic bottleneck by drug repurposing. Signal Transduct. Target. Ther. 2020, 5, 113. [Google Scholar] [CrossRef] [PubMed]
- Rudrapal, M.; Khairnar, S.J.; Jadhav, A.G. Drug Repurposing (DR): An Emerging Approach in Drug Discovery. In Drug Repurposing-Hypothesis, Molecular Aspects and Therapeutic Applications; IntechOpen: London, UK, 2020. [Google Scholar]
- Parvathaneni, V.; Kulkarni, N.S.; Muth, A.; Gupta, V. Drug repurposing: A promising tool to accelerate the drug discovery process. Drug Discov. Today 2019, 24, 2076–2085. [Google Scholar] [CrossRef]
- Pearson, R.D.; Hewlett, E.L. Niclosamide therapy for tapeworm infections. Ann. Intern. Med. 1985, 102, 550. [Google Scholar] [CrossRef]
- Al-Hadiya, B.M. Niclosamide: Comprehensive profile. Profiles Drug Subst. Excip. Relat. Methodol. 2005, 32, 67–96. [Google Scholar]
- Frayha, G.J.; Smyth, J.; Gobert, J.G.; Savel, J. The mechanisms of action of antiprotozoal and anthelmintic drugs in man. Gen. Pharmacol. Vasc. Syst. 1997, 28, 273–299. [Google Scholar] [CrossRef]
- Chen, W.; Mook, R.A., Jr.; Premont, R.T.; Wang, J. Niclosamide: Beyond an antihelminthic drug. Cell Signal. 2018, 41, 89–96. [Google Scholar] [CrossRef] [PubMed]
- Satoh, K.; Zhang, L.; Zhang, Y.; Chelluri, R.; Boufraqech, M.; Nilubol, N.; Patel, D.; Shen, M.; Kebebew, E. Identification of niclosamide as a novel anticancer agent for adrenocortical carcinoma. Clin. Cancer Res. 2016, 22, 3458–3466. [Google Scholar] [CrossRef] [PubMed]
- Arend, R.C.; Londoño-Joshi, A.I.; Samant, R.S.; Li, Y.; Conner, M.; Hidalgo, B.; Alvarez, R.D.; Landen, C.N.; Straughn, J.M.; Buchsbaum, D.J. Inhibition of Wnt/β-catenin pathway by niclosamide: A therapeutic target for ovarian cancer. Gynecol. Oncol. 2014, 134, 112–120. [Google Scholar] [CrossRef]
- Wieland, A.; Trageser, D.; Gogolok, S.; Reinartz, R.; Höfer, H.; Keller, M.; Leinhaas, A.; Schelle, R.; Normann, S.; Klaas, L. Anticancer effects of niclosamide in human glioblastoma. Clin. Cancer Res. 2013, 19, 4124–4136. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Ren, X.-R.; Piao, H.; Zhao, S.; Osada, T.; Premont, R.T.; Mook, R.A.; Morse, M.A.; Lyerly, H.K.; Chen, W. Niclosamide-induced Wnt signaling inhibition in colorectal cancer is mediated by autophagy. Biochem. J. 2019, 476, 535–546. [Google Scholar] [CrossRef] [PubMed]
- Londoño-Joshi, A.I.; Arend, R.C.; Aristizabal, L.; Lu, W.; Samant, R.S.; Metge, B.J.; Hidalgo, B.; Grizzle, W.E.; Conner, M.; Forero-Torres, A. Effect of niclosamide on basal-like breast cancers. Mol. Cancer Ther. 2014, 13, 800–811. [Google Scholar] [CrossRef]
- Kleszcz, R.; Paluszczak, J.; Baer-Dubowska, W. The effect of niclosamide on the head and neck carcinoma cells survival and the expression of Wnt/β-catenin signaling and glycolysis pathway components. Acta Pol. Pharm. Drug Res 2019, 76, 661–669. [Google Scholar] [CrossRef]
- Zhao, J.; He, Q.; Gong, Z.; Chen, S.; Cui, L. Niclosamide suppresses renal cell carcinoma by inhibiting Wnt/β-catenin and inducing mitochondrial dysfunctions. SpringerPlus 2016, 5, 1436. [Google Scholar] [CrossRef]
- Wang, C.; Zhou, X.; Xu, H.; Shi, X.; Zhao, J.; Yang, M.; Zhang, L.; Jin, X.; Hu, Y.; Li, X. Niclosamide inhibits cell growth and enhances drug sensitivity of hepatocellular carcinoma cells via STAT3 signaling pathway. J. Cancer 2018, 9, 4150. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Zheng, H.; Hu, W.; Zhou, B.; Dai, X.; Zhang, Y.; Liu, Z.; Wu, X.; Zhao, C.; Liang, G. Niclosamide inhibition of STAT3 synergizes with erlotinib in human colon cancer. OncoTargets Ther. 2017, 10, 1767. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Wang, L.; Shen, H.; Lin, H.; Li, D. Anthelminthic drug niclosamide sensitizes the responsiveness of cervical cancer cells to paclitaxel via oxidative stress-mediated mTOR inhibition. Biochem. Biophys. Res. Commun. 2017, 484, 416–421. [Google Scholar] [CrossRef]
- Suliman, M.A.; Zhang, Z.; Na, H.; Ribeiro, A.L.; Zhang, Y.; Niang, B.; Hamid, A.S.; Zhang, H.; Xu, L.; Zuo, Y. Niclosamide inhibits colon cancer progression through downregulation of the Notch pathway and upregulation of the tumor suppressor miR-200 family. Int. J. Mol. Med. 2016, 38, 776–784. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Lu, Z.; Ding, K.; Li, J.; Du, X.; Chen, C.; Sun, X.; Wu, Y.; Zhou, J.; Pan, J. Antineoplastic mechanisms of niclosamide in acute myelogenous leukemia stem cells: Inactivation of the NF-κB pathway and generation of reactive oxygen species. Cancer Res. 2010, 70, 2516–2527. [Google Scholar] [CrossRef] [PubMed]
- Fujii, A.; Masuda, T.; Iwata, M.; Tobo, T.; Wakiyama, H.; Koike, K.; Kosai, K.; Nakano, T.; Kuramitsu, S.; Kitagawa, A. The novel driver gene ASAP2 is a potential druggable target in pancreatic cancer. Cancer Sci. 2021, 112, 1655. [Google Scholar] [CrossRef] [PubMed]
- McCubrey, J.A.; Rakus, D.; Gizak, A.; Steelman, L.S.; Abrams, S.L.; Lertpiriyapong, K.; Fitzgerald, T.L.; Yang, L.V.; Montalto, G.; Cervello, M. Effects of mutations in Wnt/β-catenin, hedgehog, Notch and PI3K pathways on GSK-3 activity—Diverse effects on cell growth, metabolism and cancer. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2016, 1863, 2942–2976. [Google Scholar] [CrossRef]
- Awaji, M.; Saxena, S.; Wu, L.; Prajapati, D.R.; Purohit, A.; Varney, M.L.; Kumar, S.; Rachagani, S.; Ly, Q.P.; Jain, M. CXCR2 signaling promotes secretory cancer-associated fibroblasts in pancreatic ductal adenocarcinoma. FASEB J. 2020, 34, 9405–9418. [Google Scholar] [CrossRef]
- Kyriazis, A.P.; McCombs, W.B.; Sandberg, A.A.; Kyriazis, A.A.; Sloane, N.H.; Lepera, R. Establishment and characterization of human pancreatic adenocarcinoma cell line SW-1990 in tissue culture and the nude mouse. Cancer Res. 1983, 43, 4393–4401. [Google Scholar]
- Zhang, S.-N.; Huang, F.-T.; Huang, Y.-J.; Zhong, W.; Yu, Z. Characterization of a cancer stem cell-like side population derived from human pancreatic adenocarcinoma cells. Tumori J. 2010, 96, 985–992. [Google Scholar] [CrossRef]
- Morgan, R.; Woods, L.; Moore, G.; Quinn, L.; McGavran, L.; Gordon, S. Human cell line (COLO357) of metastatic pancreatic adenocarcinoma. Int. J. Cancer 1980, 25, 591–598. [Google Scholar] [CrossRef]
- Sipos, B.; Möser, S.; Kalthoff, H.; Török, V.; Löhr, M.; Klöppel, G. A comprehensive characterization of pancreatic ductal carcinoma cell lines: Towards the establishment of an in vitro research platform. Virchows Arch. 2003, 442, 444–452. [Google Scholar] [CrossRef]
- Zinn, R.; Otterbein, H.; Lehnert, H.; Ungefroren, H. RAC1B: A guardian of the epithelial phenotype and protector against epithelial-mesenchymal transition. Cells 2019, 8, 1569. [Google Scholar] [CrossRef] [PubMed]
- Kaushal, J.B.; Popli, P.; Sankhwar, P.; Shukla, V.; Dwivedi, A. Sonic hedgehog protects endometrial hyperplasial cells against oxidative stress via suppressing mitochondrial fission protein dynamin-like GTPase (Drp1). Free Radic. Biol. Med. 2018, 129, 582–599. [Google Scholar] [CrossRef]
- Munafó, D.B.; Colombo, M.I. A novel assay to study autophagy: Regulation of autophagosome vacuole size by amino acid deprivation. J. Cell Sci. 2001, 114, 3619–3629. [Google Scholar] [CrossRef]
- Chien, S. Targeting the KRAS Mutation for More Effective Cancer Treatment. Available online: https://www.mdanderson.org/cancerwise/targeting-the-kras-mutation-for-more-effective-cancer-treatment.h00-159458478.html (accessed on 13 April 2021).
- Ahn, S.Y.; Yang, J.H.; Kim, N.H.; Lee, K.; Cha, Y.H.; Yun, J.S.; Kang, H.E.; Lee, Y.; Choi, J.; Kim, H.S. Anti-helminthic niclosamide inhibits Ras-driven oncogenic transformation via activation of GSK-3. Oncotarget 2017, 8, 31856. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Hu, B.; Guo, Q.; Chen, L. Treatment of pancreatic cancer in a nude mouse model using high-intensity focused ultrasound. Exp. Ther. Med. 2012, 5, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Meitner, P.A.; Kajiji, S.M.; LaPosta-Frazier, N.; Bogaars, H.A.; Jolly, G.A.; Dexter, D.L.; Calabresi, P.; Tumer, M.D. “COLO357”, a human pancreatic adenosquamous carcinoma: Growth in artificial capillary culture and in nude mice. Cancer Res. 1983, 43, 5978–5985. [Google Scholar] [PubMed]
- Tzanakakis, G.N.; Margioris, A.N.; Tsatsakis, A.M.; Vezeridis, M.P. The metastatic potential of human pancreatic cell lines in the liver of nude mice correlates well with cathepsin B activity. Int. J. Gastrointest. Cancer 2003, 34, 27–38. [Google Scholar] [CrossRef]
- Wu, C.-C.; Bratton, S.B. Regulation of the intrinsic apoptosis pathway by reactive oxygen species. Antioxid. Redox Signal. 2013, 19, 546–558. [Google Scholar] [CrossRef]
- Suen, D.-F.; Norris, K.L.; Youle, R.J. Mitochondrial dynamics and apoptosis. Genes Dev. 2008, 22, 1577–1590. [Google Scholar] [CrossRef]
- Roca-Agujetas, V.; de Dios, C.; Lestón, L.; Marí, M.; Morales, A.; Colell, A. Recent insights into the mitochondrial role in autophagy and its regulation by oxidative stress. Oxidative Med. Cell. Longev. 2019, 2019, 3809308. [Google Scholar] [CrossRef]
- Azad, M.B.; Chen, Y.; Gibson, S.B. Regulation of autophagy by reactive oxygen species (ROS): Implications for cancer progression and treatment. Antioxid. Redox Signal. 2009, 11, 777–790. [Google Scholar] [CrossRef]
- Newton, P.T. New insights into niclosamide action: Autophagy activation in colorectal cancer. Biochem. J. 2019, 476, 779–781. [Google Scholar] [CrossRef] [PubMed]
- Chai, W.-H.; Li, Y.-R.; Lin, S.-H.; Chao, Y.-H.; Chen, C.-H.; Chan, P.-C.; Lin, C.-H. Antihelminthic niclosamide induces autophagy and delayed apoptosis in human non-small lung cancer cells in vitro and in vivo. Anticancer Res. 2020, 40, 1405–1417. [Google Scholar] [CrossRef] [PubMed]
- Funderburk, S.F.; Wang, Q.J.; Yue, Z. The Beclin 1–VPS34 complex–at the crossroads of autophagy and beyond. Trends Cell Biol. 2010, 20, 355–362. [Google Scholar] [CrossRef] [PubMed]
- Eskelinen, E.-L.; Illert, A.L.; Tanaka, Y.; Schwarzmann, G.n.; Blanz, J.; Von Figura, K.; Saftig, P. Role of LAMP-2 in lysosome biogenesis and autophagy. Mol. Biol. Cell 2002, 13, 3355–3368. [Google Scholar] [CrossRef]
- Demishtein, A.; Fraiberg, M.; Berko, D.; Tirosh, B.; Elazar, Z.; Navon, A. SQSTM1/p62-mediated autophagy compensates for loss of proteasome polyubiquitin recruiting capacity. Autophagy 2017, 13, 1697–1708. [Google Scholar] [CrossRef] [PubMed]
- Dossou, A.S.; Basu, A. The emerging roles of mTORC1 in macromanaging autophagy. Cancers 2019, 11, 1422. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, B.D.; Diering, G.H.; Bidinosti, M.A.; Dalal, K.; Alain, T.; Balgi, A.D.; Forestieri, R.; Nodwell, M.; Rajadurai, C.V.; Gunaratnam, C. Structure-activity analysis of niclosamide reveals potential role for cytoplasmic pH in control of mammalian target of rapamycin complex 1 (mTORC1) signaling. J. Biol. Chem. 2012, 287, 17530–17545. [Google Scholar] [CrossRef]
- Saxton, R.A.; Sabatini, D.M. mTOR signaling in growth, metabolism, and disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef]
- Pesce, E.; Sondo, E.; Ferrera, L.; Tomati, V.; Caci, E.; Scudieri, P.; Musante, I.; Renda, M.; Baatallah, N.; Servel, N. The autophagy inhibitor spautin-1 antagonizes rescue of mutant CFTR through an autophagy-independent and USP13-mediated mechanism. Front. Pharmacol. 2018, 9, 1464. [Google Scholar] [CrossRef]
- Roden, M.M.; Lee, K.-H.; Panelli, M.C.; Marincola, F.M. A novel cytolysis assay using fluorescent labeling and quantitative fluorescent scanning technology. J. Immunol. Methods 1999, 226, 29–41. [Google Scholar] [CrossRef]
- Marchand, B.; Arsenault, D.; Raymond-Fleury, A.; Boisvert, F.-M.; Boucher, M.-J. Glycogen synthase kinase-3 (GSK3) inhibition induces prosurvival autophagic signals in human pancreatic cancer cells. J. Biol. Chem. 2015, 290, 5592–5605. [Google Scholar] [CrossRef]
- Kaushal, J.B.; Sankhwar, P.; Kumari, S.; Popli, P.; Shukla, V.; Hussain, M.K.; Hajela, K.; Dwivedi, A. The regulation of Hh/Gli1 signaling cascade involves Gsk3β-mediated mechanism in estrogen-derived endometrial hyperplasia. Sci. Rep. 2017, 7, 6557. [Google Scholar] [CrossRef] [PubMed]
- Takenaka, K.; Kise, Y.; Miki, H. GSK3β positively regulates Hedgehog signaling through Sufu in mammalian cells. Biochem. Biophys. Res. Commun. 2007, 353, 501–508. [Google Scholar] [CrossRef]
- Stone, D.M.; Murone, M.; Luoh, S.; Ye, W.; Armanini, M.P.; Gurney, A.; Phillips, H.; Brush, J.; Goddard, A.; de Sauvage, F.J. Characterization of the human suppressor of fused, a negative regulator of the zinc-finger transcription factor Gli. J. Cell Sci. 1999, 112, 4437–4448. [Google Scholar] [CrossRef] [PubMed]
- Honselmann, K.C.; Pross, M.; Jung, C.M.F.; Wellner, U.F.; Deichmann, S.; Keck, T.; Bausch, D. Regulation mechanisms of the hedgehog pathway in pancreatic cancer: A review. JOP J. Pancreas 2015, 16, 25–32. [Google Scholar]
- Pietrobono, S.; Gagliardi, S.; Stecca, B. Non-canonical hedgehog signaling pathway in cancer: Activation of GLI transcription factors beyond smoothened. Front. Genet. 2019, 10, 556. [Google Scholar] [CrossRef]
- Choudhry, Z.; Rikani, A.A.; Choudhry, A.M.; Tariq, S.; Zakaria, F.; Asghar, M.W.; Sarfraz, M.K.; Haider, K.; Shafiq, A.A.; Mobassarah, N.J. Sonic hedgehog signalling pathway: A complex network. Ann. Neurosci. 2014, 21, 28. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Yu, S.J.; Park, Y.G.; Kim, J.; Sohn, J. Glycogen synthase kinase 3β phosphorylates p21WAF1/CIP1 for proteasomal degradation after UV irradiation. Mol. Cell. Biol. 2007, 27, 3187–3198. [Google Scholar] [CrossRef]
- Wang, Q.; Zhou, Y.; Wang, X.; Evers, B.M. p27 Kip1 nuclear localization and cyclin-dependent kinase inhibitory activity are regulated by glycogen synthase kinase-3 in human colon cancer cells. Cell Death Differ. 2008, 15, 908–919. [Google Scholar] [CrossRef]
- Mukherji, A.; Janbandhu, V.C.; Kumar, V. GSK-3β-dependent destabilization of cyclin D1 mediates replicational stress-induced arrest of cell cycle. FEBS Lett. 2008, 582, 1111–1116. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.W.; Gallant, M.; Lamm, M.L.; Iannaccone, S.; Vieux, K.-F.; Proytcheva, M.; Hyjek, E.; Iannaccone, P.; Walterhouse, D. Noncanonical regulation of the Hedgehog mediator GLI1 by c-MYC in Burkitt lymphoma. Mol. Cancer Res. 2013, 11, 604–615. [Google Scholar] [CrossRef]
- Beurel, E.; Kornprobst, M.; Eggelpoël, B.-V.; Cadoret, A.; Capeau, J.; Desbois-Mouthon, C. GSK-3β reactivation with LY294002 sensitizes hepatoma cells to chemotherapy-induced apoptosis. Int. J. Oncol. 2005, 27, 215–222. [Google Scholar] [CrossRef]
- Katayama, E.S.; Hue, J.J.; Bajor, D.L.; Ocuin, L.M.; Ammori, J.B.; Hardacre, J.M.; Winter, J.M. A comprehensive analysis of clinical trials in pancreatic cancer: What is coming down the pike? Oncotarget 2020, 11, 3489. [Google Scholar] [CrossRef]
- Burock, S.; Daum, S.; Keilholz, U.; Neumann, K.; Walther, W.; Stein, U. Phase II trial to investigate the safety and efficacy of orally applied niclosamide in patients with metachronous or sychronous metastases of a colorectal cancer progressing after therapy: The NIKOLO trial. BMC Cancer 2018, 18, 297. [Google Scholar] [CrossRef]
- Park, S.-J.; Shin, J.-H.; Kang, H.; Hwang, J.-J.; Cho, D.-H. Niclosamide induces mitochondria fragmentation and promotes both apoptotic and autophagic cell death. BMB Rep. 2011, 44, 517–522. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.-J.; Chen, S.; Huang, K.-X.; Le, W.-D. Why should autophagic flux be assessed? Acta Pharmacol. Sin. 2013, 34, 595–599. [Google Scholar] [CrossRef] [PubMed]
- Yang, A.; Herter-Sprie, G.; Zhang, H.; Lin, E.Y.; Biancur, D.; Wang, X.; Deng, J.; Hai, J.; Yang, S.; Wong, K.-K. Autophagy sustains pancreatic cancer growth through both cell-autonomous and nonautonomous mechanisms. Cancer Discov. 2018, 8, 276–287. [Google Scholar] [CrossRef]
- Codogno, P.; Meijer, A.J. Autophagy and signaling: Their role in cell survival and cell death. Cell Death Differ. 2005, 12, 1509–1518. [Google Scholar] [CrossRef] [PubMed]
- Montagnani, V.; Stecca, B. Role of protein kinases in hedgehog pathway control and implications for cancer therapy. Cancers 2019, 11, 449. [Google Scholar] [CrossRef]
- Niewiadomski, P.; Niedziółka, S.M.; Markiewicz, Ł.; Uśpieński, T.; Baran, B.; Chojnowska, K. Gli proteins: Regulation in development and cancer. Cells 2019, 8, 147. [Google Scholar] [CrossRef] [PubMed]
- Trnski, D.; Sabol, M.; Gojević, A.; Martinić, M.; Ozretić, P.; Musani, V.; Ramić, S.; Levanat, S. GSK3β and Gli3 play a role in activation of Hedgehog-Gli pathway in human colon cancer—Targeting GSK3β downregulates the signaling pathway and reduces cell proliferation. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2015, 1852, 2574–2584. [Google Scholar] [CrossRef]
- Shimasaki, T.; Kitano, A.; Motoo, Y.; Minamoto, T. Aberrant glycogen synthase kinase 3β in the development of pancreatic cancer. J. Carcinog. 2012, 11, 15. [Google Scholar] [CrossRef]
- Ding, L.; Madamsetty, V.S.; Kiers, S.; Alekhina, O.; Ugolkov, A.; Dube, J.; Zhang, Y.; Zhang, J.-S.; Wang, E.; Dutta, S.K. Glycogen synthase kinase-3 inhibition sensitizes pancreatic cancer cells to chemotherapy by abrogating the TopBP1/ATR-mediated DNA damage response. Clin. Cancer Res. 2019, 25, 6452–6462. [Google Scholar] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kaushal, J.B.; Bhatia, R.; Kanchan, R.K.; Raut, P.; Mallapragada, S.; Ly, Q.P.; Batra, S.K.; Rachagani, S. Repurposing Niclosamide for Targeting Pancreatic Cancer by Inhibiting Hh/Gli Non-Canonical Axis of Gsk3β. Cancers 2021, 13, 3105. https://doi.org/10.3390/cancers13133105
Kaushal JB, Bhatia R, Kanchan RK, Raut P, Mallapragada S, Ly QP, Batra SK, Rachagani S. Repurposing Niclosamide for Targeting Pancreatic Cancer by Inhibiting Hh/Gli Non-Canonical Axis of Gsk3β. Cancers. 2021; 13(13):3105. https://doi.org/10.3390/cancers13133105
Chicago/Turabian StyleKaushal, Jyoti B., Rakesh Bhatia, Ranjana K. Kanchan, Pratima Raut, Surya Mallapragada, Quan P. Ly, Surinder K. Batra, and Satyanarayana Rachagani. 2021. "Repurposing Niclosamide for Targeting Pancreatic Cancer by Inhibiting Hh/Gli Non-Canonical Axis of Gsk3β" Cancers 13, no. 13: 3105. https://doi.org/10.3390/cancers13133105