Circulating Tumor DNA as a Marker for Treatment Response in Metastatic Melanoma Patients Using Next-Generation Sequencing—A Prospective Feasibility Study

 , , , ,
, , , ,
Abstract
:Simple Summary
Abstract

1. Introduction
2. Materials and Methods
2.1. Patients
2.2. Tissue Samples
2.3. Plasma Sample Preparation and Circulating Cell Free DNA(cfDNA) Extractions
2.4. Routine Laboratory Parameter Determination
2.5. Disease Characteristics and Response Assessment
2.6. Statistical Analysis
3. Results
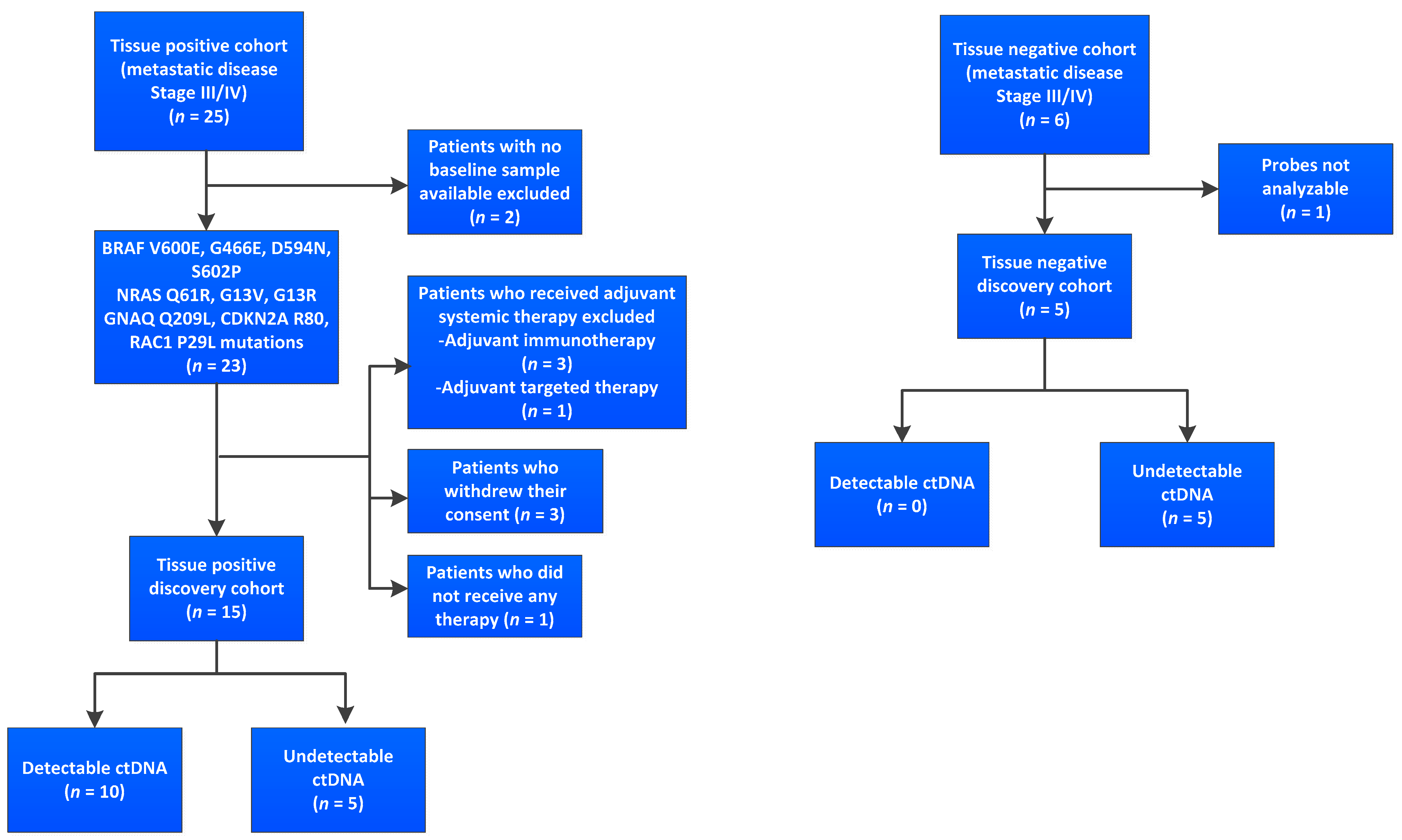
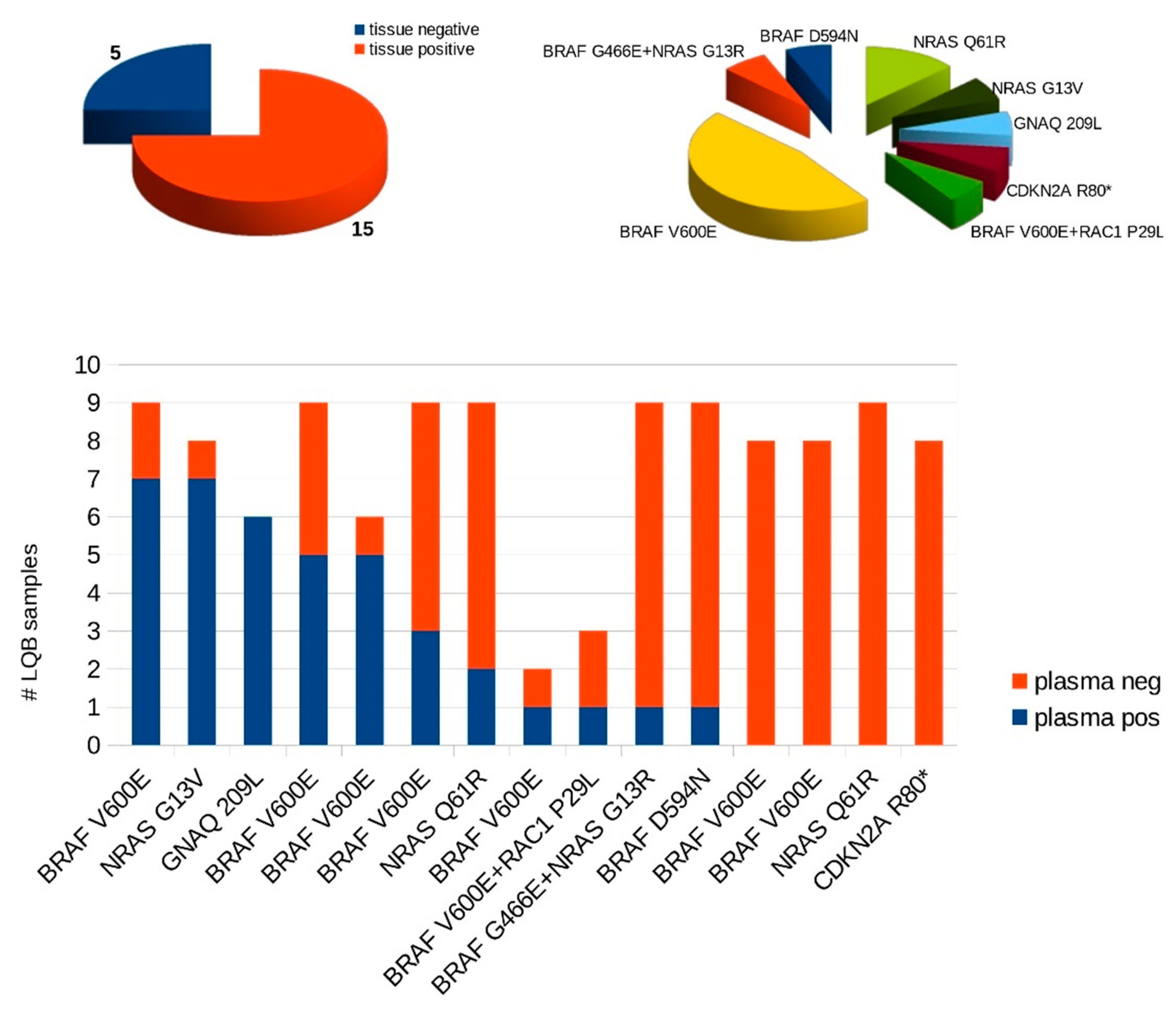
3.1. Cohort and Sample Characteristics
3.2. Evaluation of the Performance of the Custom Melanoma Panels
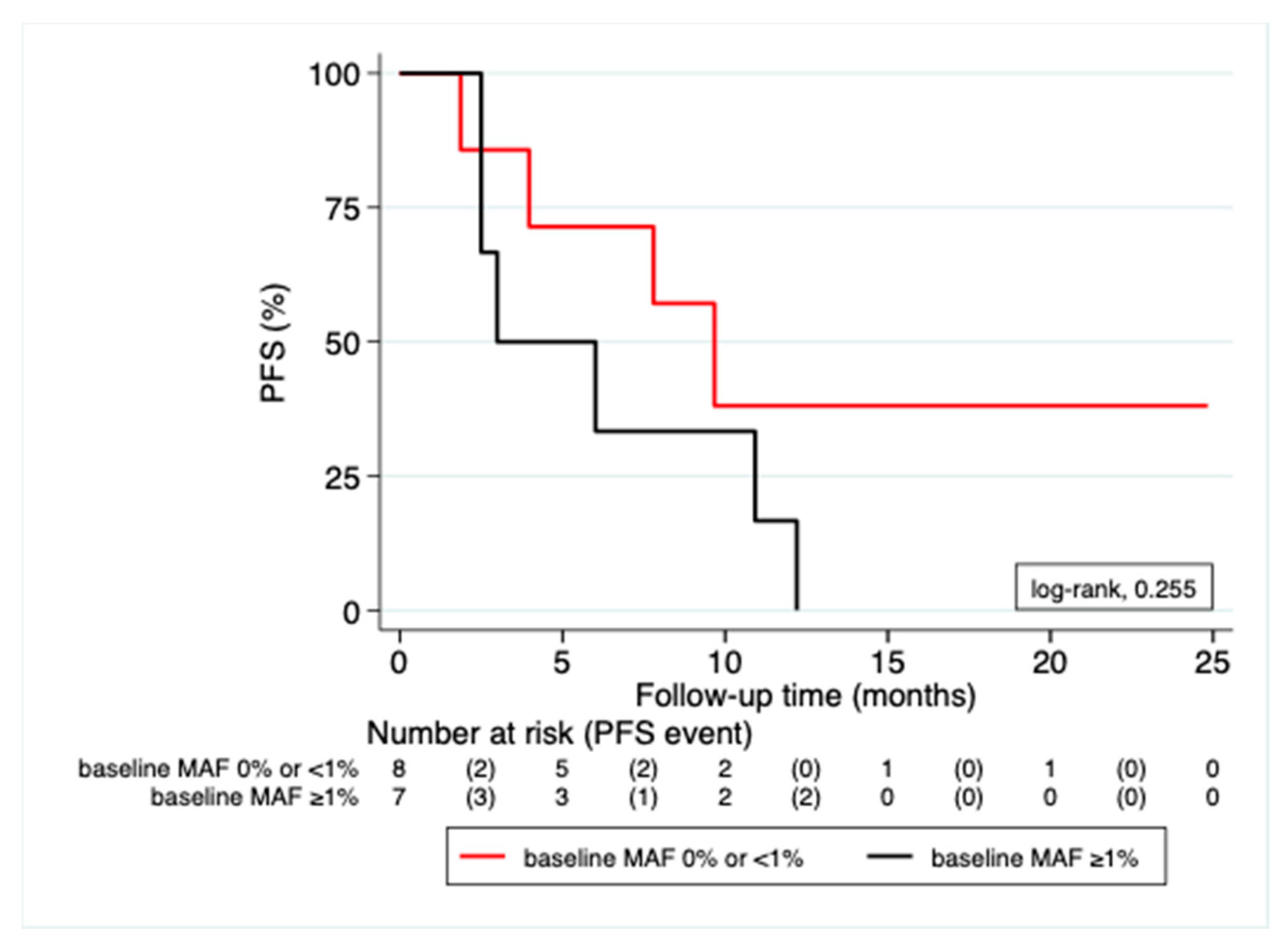
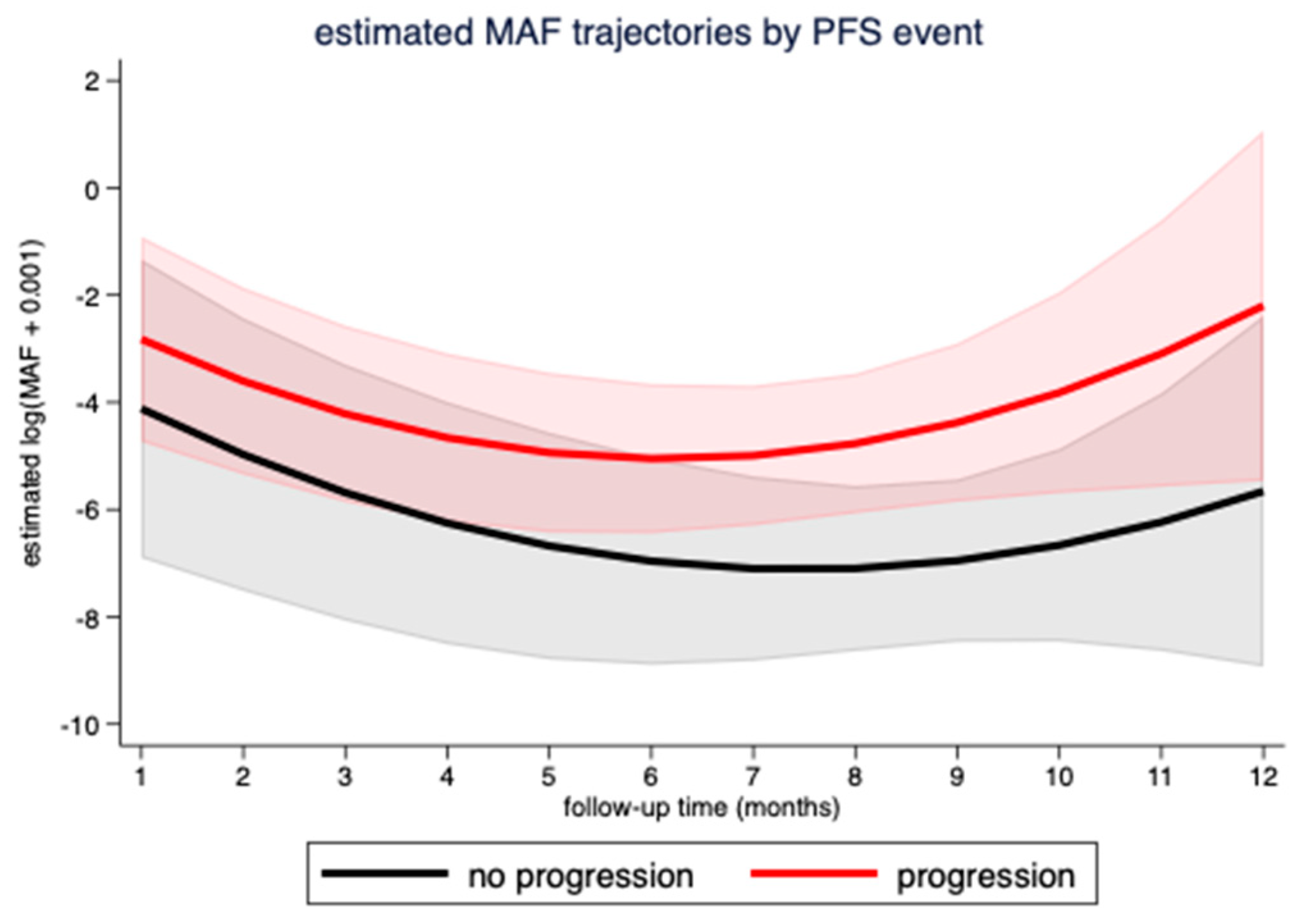
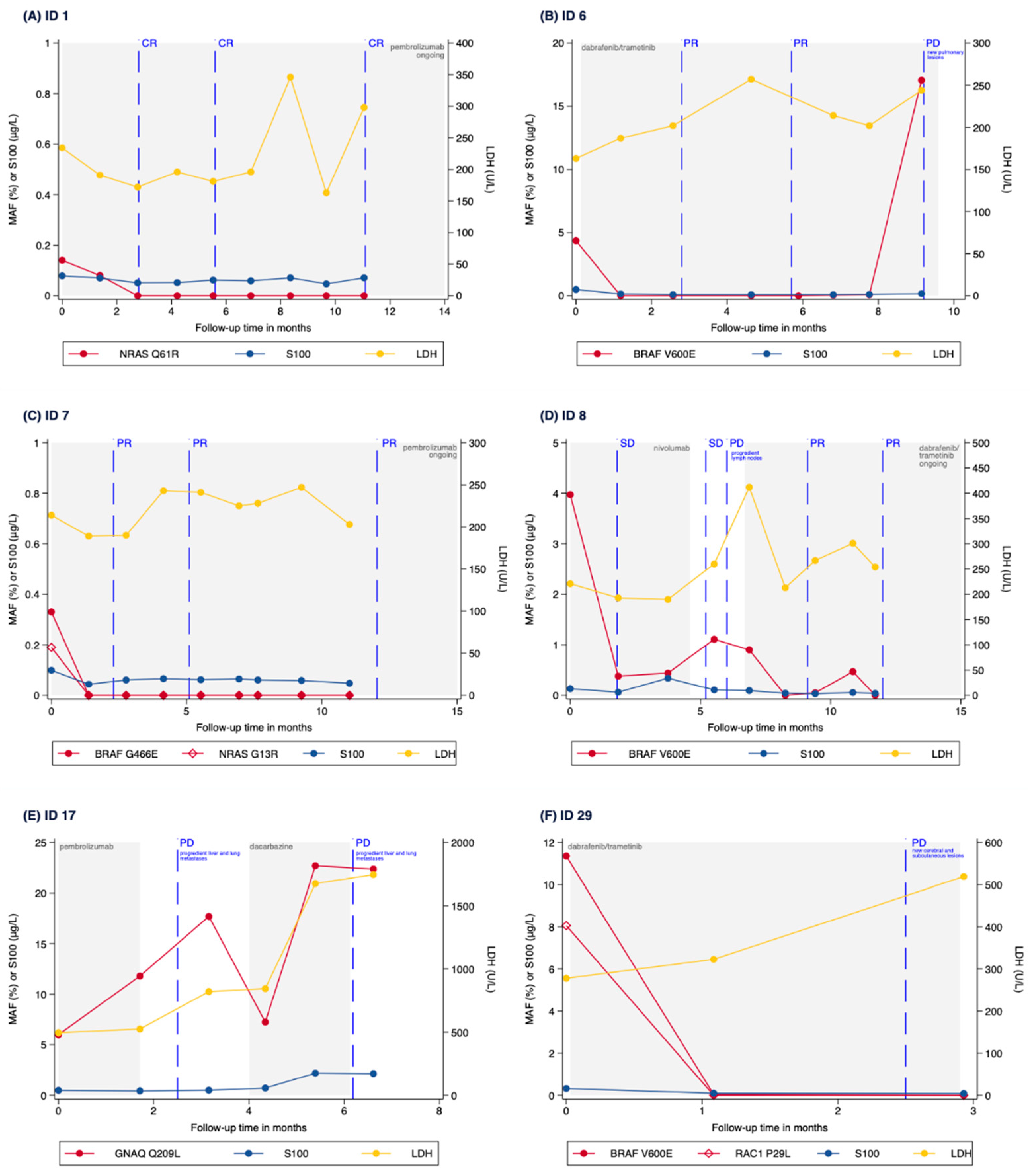
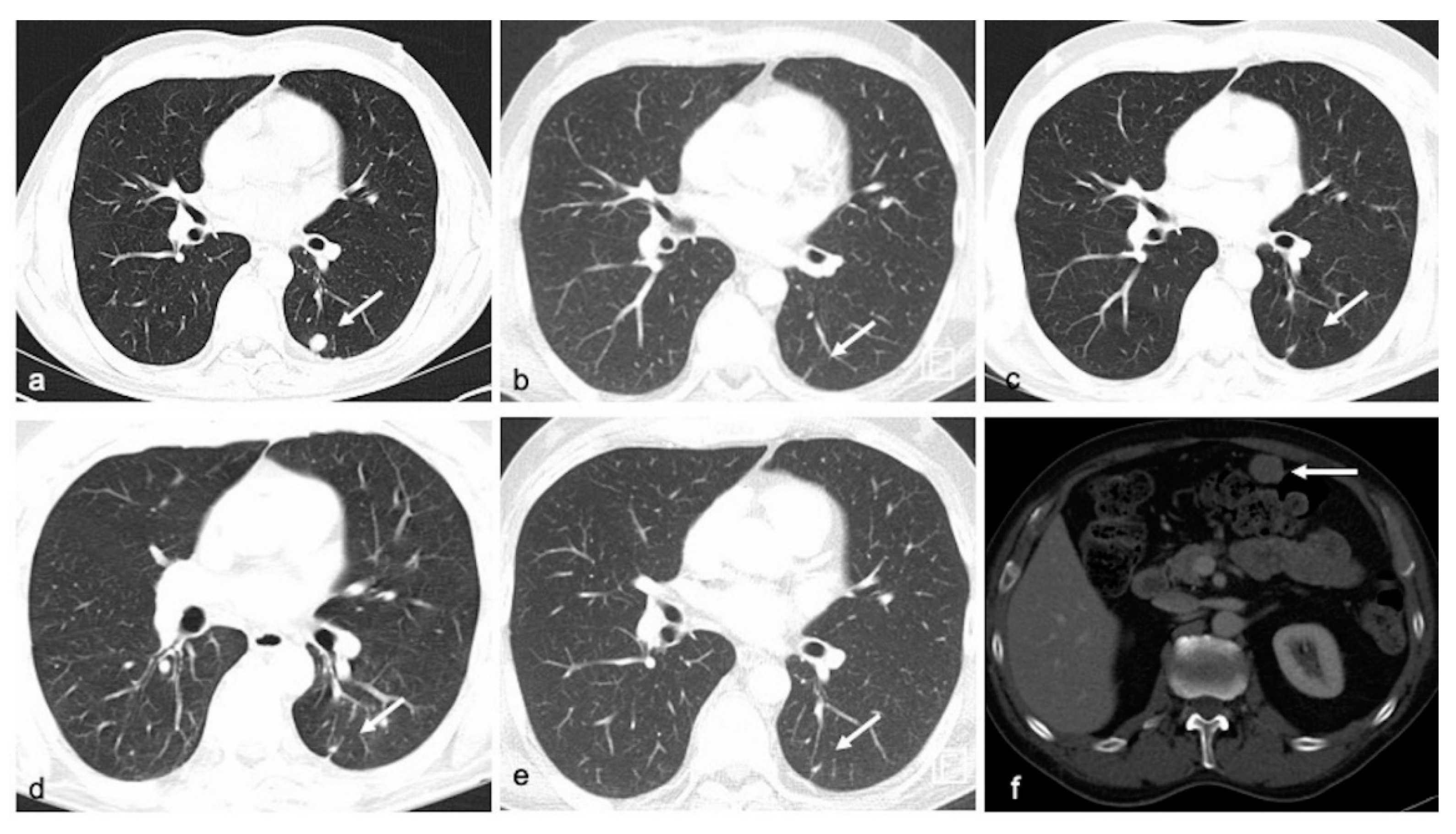
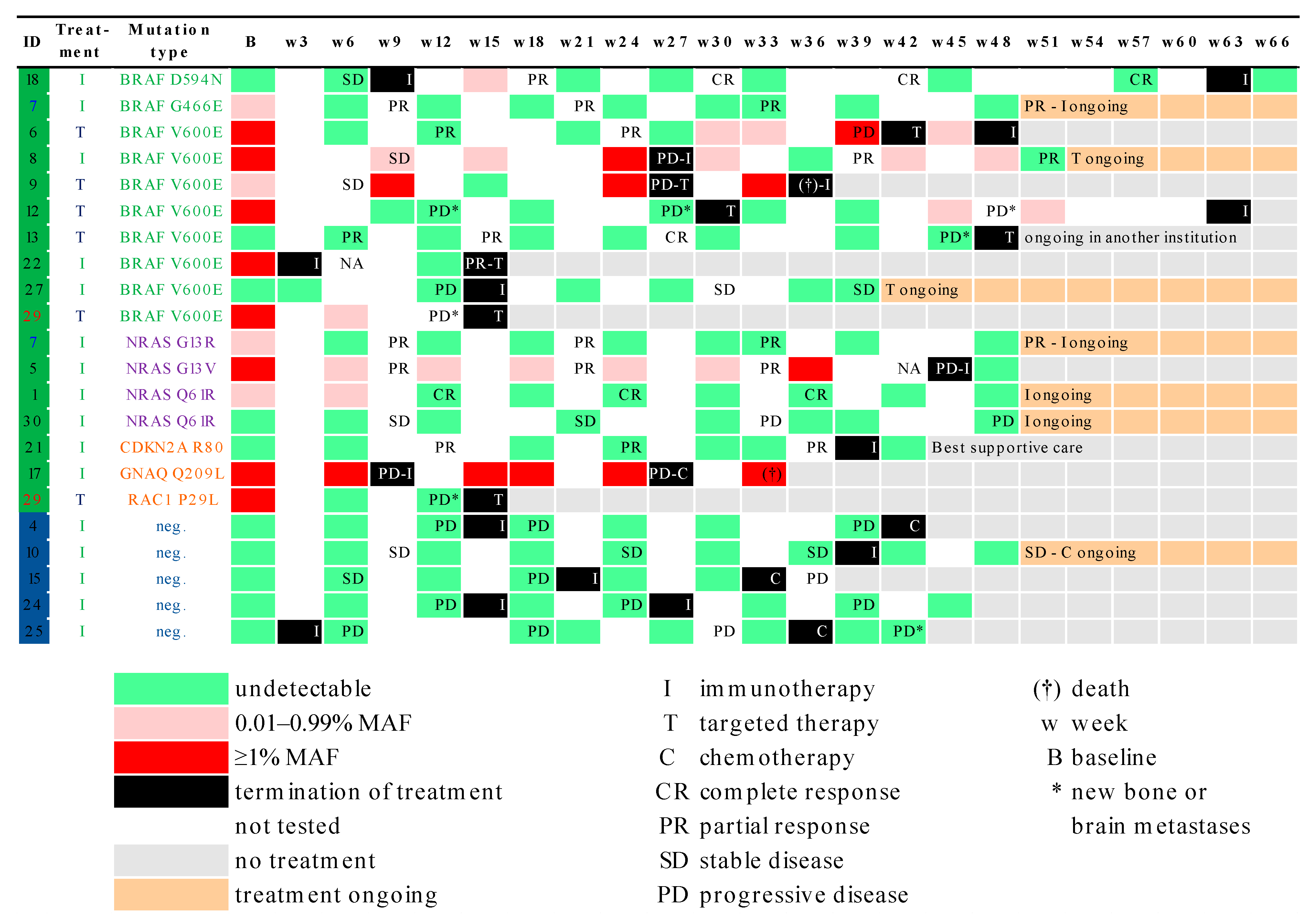
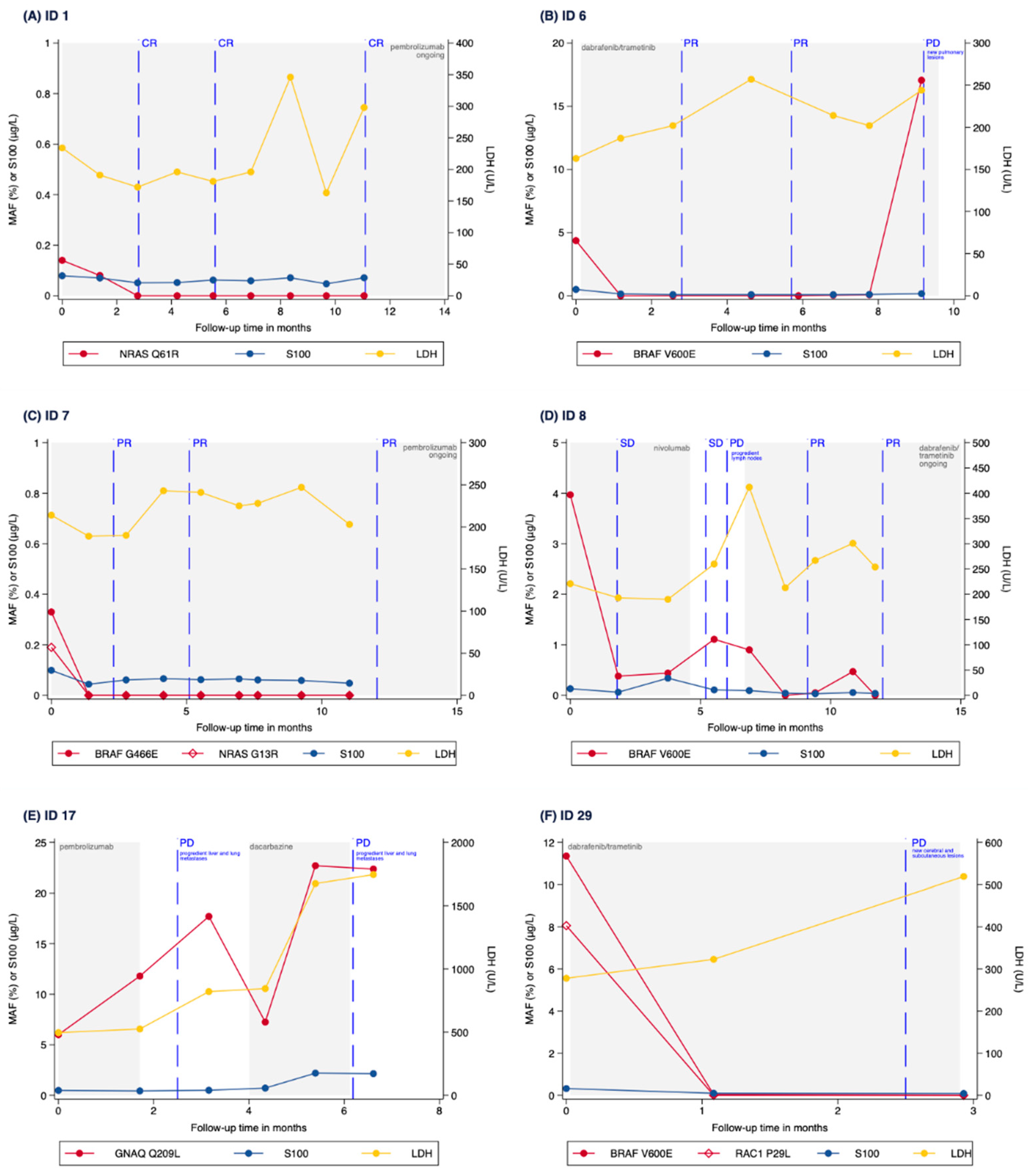
3.3. Correlation of MAF Levels with Radiological Response
3.4. Correlation of MAF Levels with S100, LDH and CRP Levels
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Larkin, J.; Ascierto, P.A.; Dréno, B.; Atkinson, V.; Liszkay, G.; Maio, M.; Mandalà, M.; Demidov, L.; Stroyakovskiy, D.; Thomas, L.; et al. Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. N. Engl. J. Med. 2014, 371, 1867–1876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flaherty, K.T.; Infante, J.R.; Daud, A.; Gonzalez, R.; Kefford, R.F.; Sosman, J.; Hamid, O.; Schuchter, L.; Cebon, J.; Ibrahim, N.; et al. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N. Engl. J. Med. 2012, 367, 1694–1703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Ribas, A.; Puzanov, I.; Dummer, R.; Schadendorf, D.; Hamid, O.; Robert, C.; Hodi, F.S.; Schachter, J.; Pavlick, A.C.; Lewis, K.D.; et al. Pembrolizumab versus investigator-choice chemotherapy for ipilimumab-refractory melanoma (KEYNOTE-002): A randomised, controlled, phase 2 trial. Lancet Oncol. 2015, 16, 908–918. [Google Scholar] [CrossRef] [Green Version]
- Schreuer, M.; Jansen, Y.; Planken, S.; Chevolet, I.; Seremet, T.; Kruse, V.; Neyns, B. Combination of dabrafenib plus trametinib for BRAF and MEK inhibitor pretreated patients with advanced BRAFV600-mutant melanoma: An open-label, single arm, dual-centre, phase 2 clinical trial. Lancet Oncol. 2017, 18, 464–472. [Google Scholar] [CrossRef]
- Uhrig, M.; Hassel, J.C.; Schlemmer, H.-P.; Ganten, M.-K. Therapy response assessment in metastatic melanoma patients treated with a braf inhibitor. Adapted choi criteria can reflect early therapy response better than does RECIST. Acad. Radiol. 2013, 20, 423–429. [Google Scholar] [CrossRef]
- Hodi, F.S.; Hwu, W.-J.; Kefford, R.; Weber, J.S.; Daud, A.; Hamid, O.; Patnaik, A.; Ribas, A.; Robert, C.; Gangadhar, T.C.; et al. Evaluation of immune-related response criteria and RECIST v1.1 in patients with advanced melanoma treated with pembrolizumab. J. Clin. Oncol. 2016, 34, 1510–1517. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Network. Genomic Classification of Cutaneous Melanoma The Cancer Genome Atlas Network HHS Public Access. Cell 2015, 161, 1681–1696. [Google Scholar] [CrossRef] [Green Version]
- Hayward, N.; Wilmott, J.; Waddell, N.; Johansson, P.A.; Field, M.A.; Nones, K.; Patch, A.-M.; Kakavand, H.; Alexandrov, L.B.; Burke, H.; et al. Whole-genome landscapes of major melanoma subtypes. Nat. Cell Biol. 2017, 545, 175–180. [Google Scholar] [CrossRef]
- Shain, A.H.; Yeh, I.; Kovalyshyn, I.; Sriharan, A.; Talevich, E.; Gagnon, A.; Dummer, R.; North, J.P.; Pincus, L.B.; Ruben, B.S.; et al. The genetic evolution of melanoma from precursor lesions. N. Engl. J. Med. 2015, 373, 1926–1936. [Google Scholar] [CrossRef]
- Rizos, H.; Menzies, A.M.; Pupo, G.M.; Carlino, M.S.; Fung, C.; Hyman, J.; Haydu, L.E.; Mijatov, B.; Becker, T.M.; Boyd, S.C.; et al. BRAF inhibitor resistance mechanisms in metastatic melanoma: Spectrum and clinical impact. Clin. Cancer Res. 2014, 20, 1965–1977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, H.; Hugo, W.; Kong, X.; Hong, A.; Koya, R.C.; Moriceau, G.; Chodon, T.; Guo, R.; Johnson, D.B.; Dahlman, K.B.; et al. Acquired resistance and clonal evolution in melanoma during BRAF inhibitor therapy. Cancer Discov. 2014, 4, 80–93. [Google Scholar] [CrossRef] [Green Version]
- Santiago-Walker, A.; Gagnon, R.; Mazumdar, J.; Casey, M.; Long, G.V.; Schadendorf, D.; Flaherty, K.T.; Kefford, R.; Hauschild, A.; Hwu, P.; et al. Correlation of BRAF mutation status in circulating-free DNA and tumor and association with clinical outcome across four BRAFi and MEKi clinical trials. Clin. Cancer Res. 2016, 22, 567–574. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Long, G.V.; Boyd, S.; Lo, S.; Menzies, A.M.; Tembe, V.; Guminski, A.; Jakrot, V.; Scolyer, R.A.; Mann, G.; et al. Circulating tumour DNA predicts response to anti-PD1 antibodies in metastatic melanoma. Ann. Oncol. 2017, 28, 1130–1136. [Google Scholar] [CrossRef] [PubMed]
- Diefenbach, R.; Lee, J.; Menzies, A.; Carlino, M.; Long, G.; Saw, R.; Howle, J.; Spillane, A.; Scolyer, R.; Kefford, R.; et al. Design and testing of a custom melanoma next generation sequencing panel for analysis of circulating tumor DNA. Cancers 2020, 12, 2228. [Google Scholar] [CrossRef] [PubMed]
- Calapre, L.; Giardina, T.; Robinson, C.; Reid, A.L.; Al-Ogaili, Z.; Pereira, M.R.; McEvoy, A.; Warburton, L.; Hayward, N.; Khattak, M.A.; et al. Locus-specific concordance of genomic alterations between tissue and plasma circulating tumor DNA in metastatic melanoma. Mol. Oncol. 2019, 13, 171–184. [Google Scholar] [CrossRef] [Green Version]
- Ion AmpliSeq HD Technology for Targeted Sequencing. Available online: https://www.thermofisher.com/at/en/home/products-and-services/promotions/life-science/ampliseq-hd.html (accessed on 8 January 2021).
- Diefenbach, R.J.; Lee, J.H.; Strbenac, D.; Yang, J.Y.H.; Menzies, A.M.; Carlino, M.S.; Long, G.V.; Spillane, A.J.; Stretch, J.R.; Saw, R.P.M.; et al. Analysis of the whole-exome sequencing of tumor and circulating tumor DNA in metastatic melanoma. Cancers 2019, 11, 1905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bakhoum, S.F.; Cantley, L.C. The Multifaceted role of chromosomal instability in cancer and its microenvironment. Cell 2018, 174, 1347–1360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prasetyanti, P.R.; Medema, J.P. Intra-tumor heterogeneity from a cancer stem cell perspective. Mol. Cancer 2017, 16, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Gray, E.S.; Rizos, H.; Reid, A.L.; Boyd, S.C.; Pereira, M.R.; Lo, J.; Tembe, V.; Freeman, J.; Lee, J.; Scolyer, R.A.; et al. Circulating tumor DNA to monitor treatment response and detect acquired resistance in patients with metastatic melanoma. Oncotarget 2015, 6, 42008–42018. [Google Scholar] [CrossRef] [Green Version]
- Zaretsky, J.M.; Garcia-Diaz, A.; Shin, D.S.; Escuin-Ordinas, H.; Hugo, W.; Hu-Lieskovan, S.; Torrejon, D.Y.; Abril-Rodriguez, G.; Sandoval, S.; Barthly, L.; et al. Mutations associated with acquired resistance to PD-1 blockade in melanoma. N. Engl. J. Med. 2016, 375, 819–829. [Google Scholar] [CrossRef] [PubMed]
- Czarnecka, A.M.; Bartnik, E.; Fiedorowicz, M.; Rutkowski, P. Targeted therapy in melanoma and mechanisms of resistance. Int. J. Mol. Sci. 2020, 21, 4576. [Google Scholar] [CrossRef]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- Eisenhauer, E.A.; Therasse, P.; Bogaerts, J.; Schwartz, L.H.; Sargent, D.; Ford, R.; Dancey, J.; Arbuck, S.; Gwyther, S.; Mooney, M.; et al. New response evaluation criteria in solid tumours: Revised RECIST guideline (version 1.1). Eur. J. Cancer 2009, 45, 228–247. [Google Scholar] [CrossRef] [PubMed]
- Schemper, M.; Smith, T.L. A note on quantifying follow-up in studies of failure time. Control. Clin. Trials 1996, 17, 343–346. [Google Scholar] [CrossRef]
- Diefenbach, R.J.; Lee, J.H.; Rizos, H. Monitoring melanoma using circulating free DNA. Am. J. Clin. Dermatol. 2019, 20, 1–12. [Google Scholar] [CrossRef]
- Forshew, T.; Murtaza, M.; Parkinson, C.; Gale, D.; Tsui, D.W.Y.; Kaper, F.; Dawson, S.-J.; Piskorz, A.M.; Jimenez-Linan, M.; Bentley, D.; et al. Noninvasive identification and monitoring of cancer mutations by targeted deep sequencing of plasma DNA. Sci. Transl. Med. 2012, 4, 136ra68. [Google Scholar] [CrossRef] [PubMed]
- Kinde, I.; Wu, J.; Papadopoulos, N.; Kinzler, K.W.; Vogelstein, B. Detection and quantification of rare mutations with massively parallel sequencing. Proc. Natl. Acad. Sci. USA 2011, 108, 9530–9535. [Google Scholar] [CrossRef] [Green Version]
- Girotti, M.R.; Gremel, G.; Lee, R.; Galvani, E.; Rothwell, D.; Viros, A.; Mandal, A.K.; Lim, K.H.J.; Saturno, G.; Furney, S.J.; et al. Application of sequencing, liquid biopsies, and patient-derived xenografts for personalized medicine in melanoma. Cancer Discov. 2016, 6, 286–299. [Google Scholar] [CrossRef] [Green Version]
- Bettegowda, C.; Sausen, M.; Leary, R.J.; Kinde, I.; Wang, Y.; Agrawal, N.; Bartlett, B.; Wang, H.; Luber, B.; Alani, R.M.; et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci. Transl. Med. 2014, 6, 224ra24. [Google Scholar] [CrossRef] [Green Version]
- De Mattos-Arruda, L.; Mayor, R.; Ng, C.K.Y.; Weigelt, B.; Martínez-Ricarte, F.; Torrejon, D.; Oliveira, M.; Arias, A.; Raventos, C.; Tang, J.; et al. Cerebrospinal fluid-derived circulating tumour DNA better represents the genomic alterations of brain tumours than plasma. Nat. Commun. 2015, 6, 8839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, S.Q.; Raleigh, J.M.; Callahan, J.; Vergara, I.A.; Ftouni, S.; Hatzimihalis, A.; Colebatch, A.J.; Li, J.; Semple, T.; Doig, K.; et al. Circulating tumor DNA analysis and functional imaging provide complementary approaches for comprehensive disease monitoring in metastatic melanoma. JCO Precis. Oncol. 2017, 1–14. [Google Scholar] [CrossRef]
- Pérez-Guijarro, E.; Yang, H.H.; Araya, R.E.; El Meskini, R.; Michael, H.T.; Vodnala, S.K.; Marie, K.L.; Smith, C.; Chin, S.; Lam, K.; et al. Multimodel preclinical platform predicts clinical response of melanoma to immunotherapy. Nat. Med. 2020, 26, 781–791. [Google Scholar] [CrossRef] [PubMed]
- Marie, K.L.; Sassano, A.; Yang, H.H.; Michalowski, A.M.; Michael, H.T.; Guo, T.; Tsai, Y.C.; Weissman, A.M.; Lee, M.P.; Jenkins, L.M.; et al. Melanoblast transcriptome analysis reveals pathways promoting melanoma metastasis. Nat. Commun. 2020, 11, 1–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
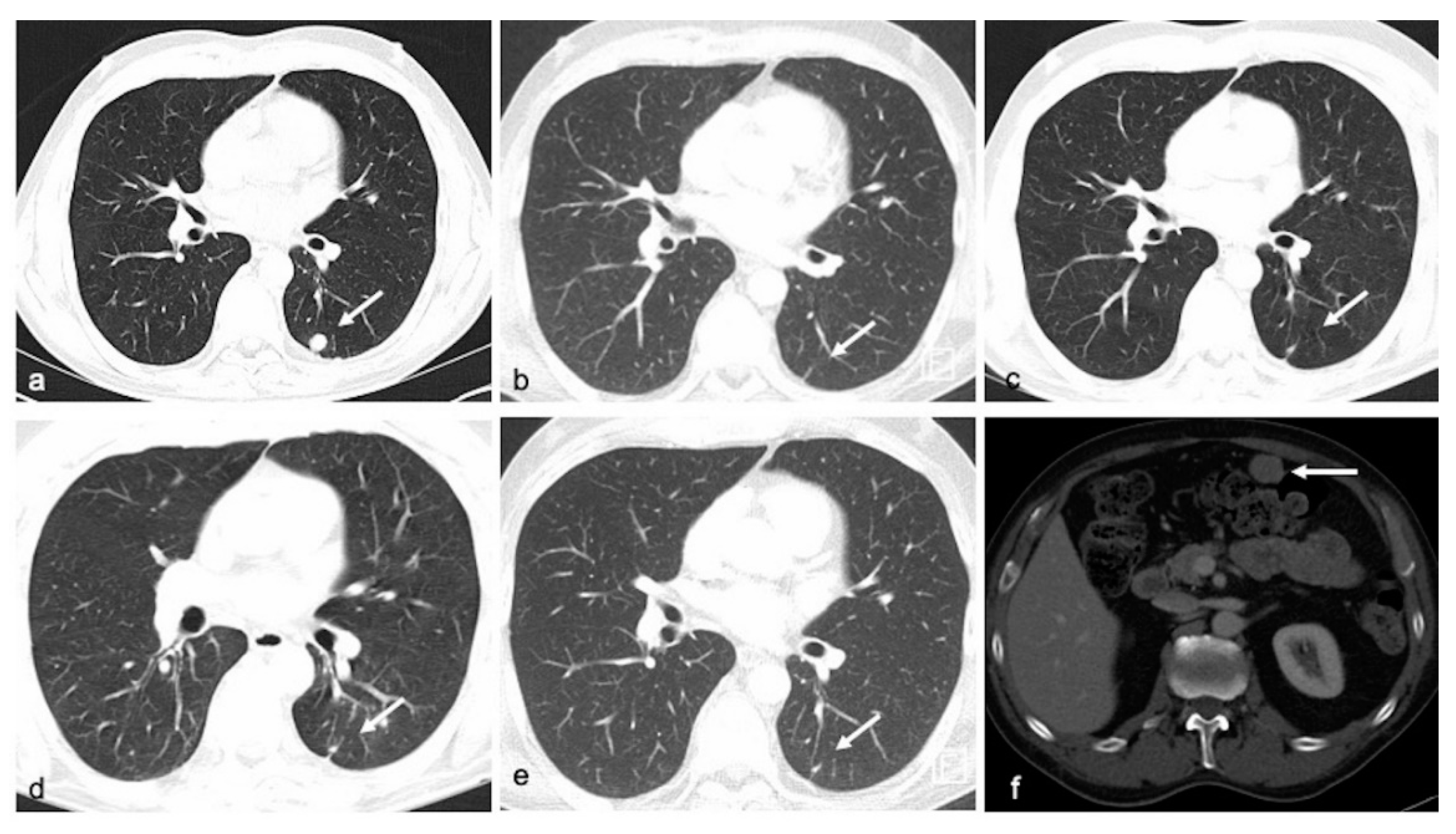{kind=link}
| Subcategories | Baseline ctDNA Low * MAF < 1% (n = 8) | Baseline ctDNA High MAF ≥ 1% (n = 7) | Tissue/Plasma Neg. (n = 5) | Total (n = 20) |
|---|---|---|---|---|
| Age | 70 (67–78) | 70 (59–76) | 70 (68–75) | 71 (67–76) |
| Gender | ||||
| Male | 5 (62.5) | 5 (71.4) | 3 (60) | 13 (65) |
| Female | 3 (37.5) | 2 (28.6) | 2 (40) | 7 (35) |
| ECOG | ||||
| 0 | 7 (87.5) | 3 (42.8) | 4 (80) | 14 (70) |
| 1 | 1 (12.5) | 2 (28.6) | 0 (0) | 3 (15) |
| 2 | 0 (0) | 1 (14.3) | 0 (0) | 1 (5) |
| Unknown | 0 (0) | 1 (14.3) | 1 (20) | 2 (10) |
| Melanoma Type | ||||
| Cutaneous melanoma | 8 (100) | 4 (57.1) | 3 (60) | 15 (75) |
| Uveal melanoma | 0 (0) | 0 (0) | 1 (10) | 1 (5) |
| Mucosal melanoma | 0 (0) | 1 (14.3) | 0 (0) | 1 (5) |
| Unknown primary | 0 (0) | 2 (28.6) | 1 (20) | 3 (15) |
| Tissue localization | ||||
| Primary | 4 (50) | 4 (57.1) | 2 (40) | 10 (50) |
| Metastasis | 4 (50) | 3 (42.9) | 2 (40) | 9 (45) |
| Missing | 0 (0) | 0 (0) | 1 (20) | 1 (5) |
| Tissue mutation | ||||
| BRAF | 5 (62.5) | 5 (71.4) | 0 (0) | 10 (50) |
| NRAS | 2 (25) | 1(14.3) | 0 (0) | 3 (15) |
| CDKN2A | 1 (12.5) | 0 (0) | 0 (0) | 1 (5) |
| GNAQ | 0 (0) | 1 (14.3) | 0 (0) | 1 (5) |
| neg. | 0 (0) | 0 (0) | 5 (100) | 5 (25) |
| AJCC tumor stage | ||||
| IIIC/IIID | 1 (12.5) | 1 (14.3) | 0 (0) | 2 (10) |
| IV | 7 (87.5) | 6 (85.7) | 5 (100) | 18 (90) |
| pT | ||||
| T0 | 1 (12.5) | 0 (0) | 0 (0) | 1 (5) |
| pT1 | 0 (0) | 1 (14.3) | 0 (0) | 1 (5) |
| pT2 | 3 (37.5) | 2 (28.6) | 0 (0) | 5 (25) |
| pT3 | 1 (12.5) | 1 (14.3) | 2 (40) | 4 (20) |
| pT4 | 3 (37.5) | 1 (14.3) | 1 (20) | 5 (25) |
| pTX/Missing | 0 (0) | 2 (28.6) | 2 (40) | 4 (20) |
| pN | ||||
| N0 | 4 (50) | 2 (28.6) | 1 (20) | 7 (35) |
| N1 | 1 (12.5) | 2 (28.6) | 3 (60) | 6 (30) |
| N2 | 3 (37.5) | 0 (0) | 1 (20) | 4 (20) |
| N3 | 0 (0) | 3 (42.8) | 0 (0) | 3 (15) |
| M stage | ||||
| M0 | 1 (12.5) | 1 (14.3) | 0 (0) | 2 (10) |
| M1a | 0 (0) | 1 (14.3) | 0 (0) | 1 (5) |
| M1b | 5 (62.5) | 1 (14.3) | 2 (40) | 8 (40) |
| M1c | 2 (25) | 2 (28.6) | 3 (60) | 7 (35) |
| M1d | 0 (0) | 2 (28.6) | 0 (0) | 2 (10) |
| Number of metastatic sites | ||||
| 1 | 3 (37.5) | 2 (28.6) | 2 (40) | 7 (35) |
| 2 | 4 (50) | 3 (42.8) | 1 (20) | 8 (40) |
| ≥3 | 1 (12.5) | 2 (28.6) | 2 (40) | 5 (25) |
| S100 | ||||
| ≤1× ULN | 8 (100) | 0 (0) | 3 (60) | 11 (55) |
| >1× ULN | 0 (0) | 7 (100) | 2 (40) | 9 (45) |
| LDH | ||||
| ≤1× ULN | 7 (87.5) | 3 (42.9) | 0 (0) | 10 (50) |
| >1× ULN | 1 (12.5) | 4 (57.1) | 5 (100) | 10 (50) |
| Treatment type | ||||
| Targeted therapy | 2 (25) | 3 (42.9) | 0 (0) | 5 (25) |
| Immunotherapy | 6 (75) | 4 (57.1) | 5 (100) | 15 (75) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Berger, M.; Thueringer, A.; Franz, D.; Dandachi, N.; Talakić, E.; Richtig, G.; Richtig, E.; Rohrer, P.M.; Koch, L.; Wolf, I.H.; et al. Circulating Tumor DNA as a Marker for Treatment Response in Metastatic Melanoma Patients Using Next-Generation Sequencing—A Prospective Feasibility Study. Cancers 2021, 13, 3101. https://doi.org/10.3390/cancers13123101
Berger M, Thueringer A, Franz D, Dandachi N, Talakić E, Richtig G, Richtig E, Rohrer PM, Koch L, Wolf IH, et al. Circulating Tumor DNA as a Marker for Treatment Response in Metastatic Melanoma Patients Using Next-Generation Sequencing—A Prospective Feasibility Study. Cancers. 2021; 13(12):3101. https://doi.org/10.3390/cancers13123101
Chicago/Turabian StyleBerger, Marina, Andrea Thueringer, Doritt Franz, Nadia Dandachi, Emina Talakić, Georg Richtig, Erika Richtig, Peter Michael Rohrer, Lukas Koch, Ingrid Hildegard Wolf, and et al. 2021. "Circulating Tumor DNA as a Marker for Treatment Response in Metastatic Melanoma Patients Using Next-Generation Sequencing—A Prospective Feasibility Study" Cancers 13, no. 12: 3101. https://doi.org/10.3390/cancers13123101
APA StyleBerger, M., Thueringer, A., Franz, D., Dandachi, N., Talakić, E., Richtig, G., Richtig, E., Rohrer, P. M., Koch, L., Wolf, I. H., Koch, C., Rainer, B. M., Koeller, M., Pichler, M., Gerritsmann, H., Kashofer, K., & Aigelsreiter, A. (2021). Circulating Tumor DNA as a Marker for Treatment Response in Metastatic Melanoma Patients Using Next-Generation Sequencing—A Prospective Feasibility Study. Cancers, 13(12), 3101. https://doi.org/10.3390/cancers13123101