
An Updated Understanding of the Role of YAP in Driving Oncogenic Responses




 and
and 
Abstract
:Simple Summary
Abstract
1. The Hippo Pathway Core
Structural and Functional Differences between YAP and TAZ
2. Extracellular and Intracellular Modulators of YAP
2.1. Mechanotransduction
2.2. Polarity and Tight Junction Proteins
2.3. The Cadherin-Catenin Complex
2.4. Soluble Growth Factors
2.4.1. Growth Factors
2.4.2. Hormones
2.4.3. Metabolic Factors
3. YAP-Dependent Signaling Pathways Impacting Cancer
3.1. YAP Action through the Stages of Cancer Development
3.1.1. Proliferation
3.1.2. Invasion and Migration
3.1.3. YAP as Tumor Suppressor
3.2. YAP at the Intersection of Immune and Tumor Cells
3.2.1. TILs and MDSCs
3.2.2. TAMs
4. Biochemistry and Translational Properties of Currently Known YAP Inhibitors
4.1. Drugs Affecting Nuclear Localization of YAP
4.2. Competitive Inhibition of YAP Binding and Function
4.3. Other Inhibitors
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Chen, Y.; Han, H.; Seo, G.; Vargas, R.E.; Yang, B.; Chuc, K.; Zhao, H.; Wang, W. Systematic Analysis of the Hippo Pathway Organization and Oncogenic Alteration in Evolution. Sci. Rep. 2020, 10, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Justice, R.W.; Zilian, O.; Woods, D.F.; Noll, M.; Bryant, P.J. The Drosophila Tumor Suppressor Gene Warts Encodes a Homolog of Human Myotonic Dystrophy Kinase and Is Required for the Control of Cell Shape and Proliferation. Genes Dev. 1995, 9, 534–546. [Google Scholar] [CrossRef] [Green Version]
- Staley, B.K.; Irvine, K.D. Hippo Signaling in Drosophila: Recent Advances and Insights. Dev. Dyn. 2012, 241, 3–15. [Google Scholar] [CrossRef] [Green Version]
- Xu, T.; Wang, W.; Zhang, S.; Stewart, R.A.; Yu, W. Identifying Tumor Suppressors in Genetic Mosaics: The Drosophila Lats Gene Encodes a Putative Protein Kinase. Development 1995, 121, 1053–1063. [Google Scholar] [CrossRef] [PubMed]
- Piccolo, S.; Dupont, S.; Cordenonsi, M. The Biology of YAP/TAZ: Hippo Signaling and Beyond. Physiol. Rev. 2014, 94, 1287–1312. [Google Scholar] [CrossRef]
- Wada, K.-I.; Itoga, K.; Okano, T.; Yonemura, S.; Sasaki, H. Hippo Pathway Regulation by Cell Morphology and Stress Fibers. Development 2011, 138, 3907–3914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ege, N.; Dowbaj, A.M.; Jiang, M.; Howell, M.; Hooper, S.; Foster, C.; Jenkins, R.P.; Sahai, E. Quantitative Analysis Reveals That Actin and Src-Family Kinases Regulate Nuclear YAP1 and Its Export. Cell Syst. 2018, 6, 692–708. [Google Scholar] [CrossRef] [Green Version]
- Manning, S.A.; Dent, L.G.; Kondo, S.; Zhao, Z.W.; Plachta, N.; Harvey, K.F. Dynamic Fluctuations in Subcellular Localization of the Hippo Pathway Effector Yorkie In Vivo. Curr. Biol. 2018, 28, 1651–1660.e4. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Lu, Y.; Yin, M.-X.; Wang, C.; Wu, W.; Li, J.; Wu, W.; Ge, L.; Hu, L.; Zhao, Y.; et al. Importin A1 Mediates Yorkie Nuclear Import via an N-Terminal Non-Canonical Nuclear Localization Signal. J. Biol. Chem. 2016, 291, 7926–7937. [Google Scholar] [CrossRef] [Green Version]
- Kofler, M.; Speight, P.; Little, D.; Di Ciano-Oliveira, C.; Szászi, K.; Kapus, A. Mediated Nuclear Import and Export of TAZ and the Underlying Molecular Requirements. Nat. Commun. 2018, 9, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Clara, J.A.; Monge, C.; Yang, Y.; Takebe, N. Targeting Signalling Pathways and the Immune Microenvironment of Cancer Stem Cells—A Clinical Update. Nat. Rev. Clin. Oncol. 2020, 17, 204–232. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zhao, B.; Wang, P.; Chen, F.; Dong, Z.; Yang, H.; Guan, K.-L.; Xu, Y. Structural Insights into the YAP and TEAD Complex. Genes Dev. 2010, 24, 235–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reggiani, F.; Gobbi, G.; Ciarrocchi, A.; Sancisi, V. YAP and TAZ Are Not Identical Twins. Trends Biochem. Sci. 2021, 46, 154–168. [Google Scholar] [CrossRef] [PubMed]
- Sudol, M.; Shields, D.C.; Farooq, A. Structures of YAP Protein Domains Reveal Promising Targets for Development of New Cancer Drugs. Semin. Cell Dev. Biol. 2012, 23, 827–833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, R.; Plouffe, S.W.; Guan, K.-L. Determining the Phosphorylation Status of Hippo Components YAP and TAZ Using Phos-Tag. Methods Mol. Biol. 2019, 1893, 281–287. [Google Scholar] [CrossRef] [PubMed]
- Plouffe, S.W.; Lin, K.C.; Moore, J.L.; Tan, F.E.; Ma, S.; Ye, Z.; Qiu, Y.; Ren, B.; Guan, K.-L. The Hippo Pathway Effector Proteins YAP and TAZ Have both Distinct and Overlapping Functions in the Cell. J. Biol. Chem. 2018, 293, 11230–11240. [Google Scholar] [CrossRef] [Green Version]
- Muppala, S.; Raghunathan, V.K.; Jalilian, I.; Thomasy, S.; Murphy, C.J. YAP and TAZ Are Distinct Effectors of Corneal Myofibroblast Transformation. Exp. Eye Res. 2019, 180, 102–109. [Google Scholar] [CrossRef]
- Lebrun, J.-J. The Dual Role of TGFβ in Human Cancer: From Tumor Suppression to Cancer Metastasis. ISRN Mol. Biol. 2012, 2012, 381428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Massagué, J. TGFβ Signalling in Context. Nat. Rev. Mol. Cell Biol. 2012, 13, 616–630. [Google Scholar] [CrossRef]
- Ferrigno, O.; Lallemand, F.; Verrecchia, F.; L’Hoste, S.; Camonis, J.; Atfi, A.; Mauviel, A. Yes-Associated Protein (YAP65) Interacts with Smad7 and Potentiates Its Inhibitory Activity against TGF-Beta/Smad Signaling. Oncogene 2002, 21, 4879–4884. [Google Scholar] [CrossRef] [Green Version]
- Fujii, M.; Toyoda, T.; Nakanishi, H.; Yatabe, Y.; Sato, A.; Matsudaira, Y.; Ito, H.; Murakami, H.; Kondo, Y.; Kondo, E.; et al. TGF-β Synergizes with Defects in the Hippo Pathway to Stimulate Human Malignant Mesothelioma Growth. J. Exp. Med. 2012, 209, 479–494. [Google Scholar] [CrossRef] [Green Version]
- Felley-Bosco, E.; Stahel, R. Hippo/YAP Pathway for Targeted Therapy. Transl. Lung Cancer Res. 2014, 3, 75–83. [Google Scholar] [CrossRef] [Green Version]
- Xu, W.; Zhang, M.; Li, Y.; Wang, Y.; Wang, K.; Chen, Q.; Zhang, R.; Song, W.; Huang, Q.; Zhao, W.; et al. YAP Manipulates Proliferation via PTEN/AKT/MTOR-Mediated Autophagy in Lung Adenocarcinomas. Cancer Cell Int. 2021, 21, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Morin-Kensicki, E.M.; Boone, B.N.; Howell, M.; Stonebraker, J.R.; Teed, J.; Alb, J.G.; Magnuson, T.R.; O’Neal, W.; Milgram, S.L. Defects in Yolk Sac Vasculogenesis, Chorioallantoic Fusion, and Embryonic Axis Elongation in Mice with Targeted Disruption of Yap65. Mol. Cell Biol. 2006, 26, 77–87. [Google Scholar] [CrossRef] [Green Version]
- Finch-Edmondson, M.L.; Strauss, R.P.; Passman, A.M.; Sudol, M.; Yeoh, G.C.; Callus, B.A. TAZ Protein Accumulation Is Negatively Regulated by YAP Abundance in Mammalian Cells. J. Biol. Chem. 2015, 290, 27928–27938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azzolin, L.; Panciera, T.; Soligo, S.; Enzo, E.; Bicciato, S.; Dupont, S.; Bresolin, S.; Frasson, C.; Basso, G.; Guzzardo, V.; et al. YAP/TAZ Incorporation in the β-Catenin Destruction Complex Orchestrates the Wnt Response. Cell 2014, 158, 157–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, M.; Tomlinson, V.; Lara, R.; Holliday, D.; Chelala, C.; Harada, T.; Gangeswaran, R.; Manson-Bishop, C.; Smith, P.; Danovi, S.A.; et al. Yes-Associated Protein (YAP) Functions as a Tumor Suppressor in Breast. Cell Death Differ. 2008, 15, 1752–1759. [Google Scholar] [CrossRef]
- Moya, I.M.; Castaldo, S.A.; Van den Mooter, L.; Soheily, S.; Sansores-Garcia, L.; Jacobs, J.; Mannaerts, I.; Xie, J.; Verboven, E.; Hillen, H.; et al. Peritumoral Activation of the Hippo Pathway Effectors YAP and TAZ Suppresses Liver Cancer in Mice. Science 2019, 366, 1029–1034. [Google Scholar] [CrossRef]
- Kaan, H.Y.K.; Chan, S.W.; Tan, S.K.J.; Guo, F.; Lim, C.J.; Hong, W.; Song, H. Crystal Structure of TAZ-TEAD Complex Reveals a Distinct Interaction Mode from That of YAP-TEAD Complex. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef]
- Pobbati, A.V.; Han, X.; Hung, A.W.; Weiguang, S.; Huda, N.; Chen, G.-Y.; Kang, C.; Chia, C.S.B.; Luo, X.; Hong, W.; et al. Targeting the Central Pocket in Human Transcription Factor TEAD as a Potential Cancer Therapeutic Strategy. Structure 2015, 23, 2076–2086. [Google Scholar] [CrossRef] [Green Version]
- Pobbati, A.V.; Rubin, B.P. Protein-Protein Interaction Disruptors of the YAP/TAZ-TEAD Transcriptional Complex. Molecules 2020, 25, 6001. [Google Scholar] [CrossRef] [PubMed]
- Totaro, A.; Panciera, T.; Piccolo, S. YAP/TAZ Upstream Signals and Downstream Responses. Nat. Cell Biol. 2018, 20, 888–899. [Google Scholar] [CrossRef] [PubMed]
- Zou, R.; Xu, Y.; Feng, Y.; Shen, M.; Yuan, F.; Yuan, Y. YAP Nuclear-Cytoplasmic Translocation Is Regulated by Mechanical Signaling, Protein Modification, and Metabolism. Cell Biol. Int. 2020, 44, 1416–1425. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, C.; Lampe, M.; Nillasithanukroh, S.; Han, W.; Lian, X.; Palecek, S.P. Human Pluripotent Stem Cell Culture Density Modulates YAP Signaling. Biotechnol. J. 2016, 11, 662–675. [Google Scholar] [CrossRef] [Green Version]
- Zhao, B.; Wei, X.; Li, W.; Udan, R.S.; Yang, Q.; Kim, J.; Xie, J.; Ikenoue, T.; Yu, J.; Li, L.; et al. Inactivation of YAP Oncoprotein by the Hippo Pathway Is Involved in Cell Contact Inhibition and Tissue Growth Control. Genes Dev. 2007, 21, 2747–2761. [Google Scholar] [CrossRef] [Green Version]
- Das, A.; Fischer, R.S.; Pan, D.; Waterman, C.M. YAP Nuclear Localization in the Absence of Cell-Cell Contact Is Mediated by a Filamentous Actin-Dependent, Myosin II- and Phospho-YAP-Independent Pathway during Extracellular Matrix Mechanosensing. J. Biol. Chem. 2016, 291, 6096–6110. [Google Scholar] [CrossRef] [Green Version]
- Jang, J.-W.; Kim, M.-K.; Bae, S.-C. Reciprocal Regulation of YAP/TAZ by the Hippo Pathway and the Small GTPase Pathway. Small GTPases 2020, 11, 280–288. [Google Scholar] [CrossRef]
- Dupont, S.; Morsut, L.; Aragona, M.; Enzo, E.; Giulitti, S.; Cordenonsi, M.; Zanconato, F.; Le Digabel, J.; Forcato, M.; Bicciato, S.; et al. Role of YAP/TAZ in Mechanotransduction. Nature 2011, 474, 179–183. [Google Scholar] [CrossRef]
- Pavel, M.; Renna, M.; Park, S.J.; Menzies, F.M.; Ricketts, T.; Füllgrabe, J.; Ashkenazi, A.; Frake, R.A.; Lombarte, A.C.; Bento, C.F.; et al. Contact Inhibition Controls Cell Survival and Proliferation via YAP/TAZ-Autophagy Axis. Nat. Commun. 2018, 9, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Mana-Capelli, S.; Paramasivam, M.; Dutta, S.; McCollum, D. Angiomotins Link F-Actin Architecture to Hippo Pathway Signaling. Mol. Biol. Cell 2014, 25, 1676–1685. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Li, L.; Lu, Q.; Wang, L.H.; Liu, C.-Y.; Lei, Q.; Guan, K.-L. Angiomotin Is a Novel Hippo Pathway Component That Inhibits YAP Oncoprotein. Genes Dev. 2011, 25, 51–63. [Google Scholar] [CrossRef] [Green Version]
- Lv, M.; Li, S.; Luo, C.; Zhang, X.; Shen, Y.; Sui, Y.X.; Wang, F.; Wang, X.; Yang, J.; Liu, P.; et al. Angiomotin Promotes Renal Epithelial and Carcinoma Cell Proliferation by Retaining the Nuclear YAP. Oncotarget 2016, 7, 12393–12403. [Google Scholar] [CrossRef] [Green Version]
- Moleirinho, S.; Hoxha, S.; Mandati, V.; Curtale, G.; Troutman, S.; Ehmer, U.; Kissil, J.L. Regulation of Localization and Function of the Transcriptional Co-Activator YAP by Angiomotin. eLife 2017, 6. [Google Scholar] [CrossRef]
- Meng, Z.; Moroishi, T.; Guan, K.-L. Mechanisms of Hippo Pathway Regulation. Genes Dev. 2016, 30, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Meng, Z.; Moroishi, T.; Mottier-Pavie, V.; Plouffe, S.W.; Hansen, C.G.; Hong, A.W.; Park, H.W.; Mo, J.-S.; Lu, W.; Lu, S.; et al. MAP4K Family Kinases Act in Parallel to MST1/2 to Activate LATS1/2 in the Hippo Pathway. Nat. Commun. 2015, 6, 1–13. [Google Scholar] [CrossRef]
- Dobrokhotov, O.; Samsonov, M.; Sokabe, M.; Hirata, H. Mechanoregulation and Pathology of YAP/TAZ via Hippo and Non-Hippo Mechanisms. Clin. Transl. Med. 2018, 7, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Lamar, J.M.; Xiao, Y.; Norton, E.; Jiang, Z.-G.; Gerhard, G.M.; Kooner, S.; Warren, J.S.A.; Hynes, R.O. SRC Tyrosine Kinase Activates the YAP/TAZ Axis and Thereby Drives Tumor Growth and Metastasis. J. Biol. Chem. 2019, 294, 2302–2317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elosegui-Artola, A.; Andreu, I.; Beedle, A.E.M.; Lezamiz, A.; Uroz, M.; Kosmalska, A.J.; Oria, R.; Kechagia, J.Z.; Rico-Lastres, P.; Le Roux, A.-L.; et al. Force Triggers YAP Nuclear Entry by Regulating Transport across Nuclear Pores. Cell 2017, 171, 1397–1410.e14. [Google Scholar] [CrossRef] [PubMed]
- Halaoui, R.; McCaffrey, L. Rewiring Cell Polarity Signaling in Cancer. Oncogene 2015, 34, 939–950. [Google Scholar] [CrossRef] [PubMed]
- Karaman, R.; Halder, G. Cell Junctions in Hippo Signaling. Cold Spring Harb. Perspect. Biol. 2018, 10. [Google Scholar] [CrossRef] [PubMed]
- Su, W.-H.; Mruk, D.D.; Wong, E.W.P.; Lui, W.-Y.; Cheng, C.Y. Polarity Protein Complex Scribble/Lgl/Dlg and Epithelial Cell Barriers. Adv. Exp. Med. Biol. 2012, 763, 149–170. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Li, J.; Li, P.; Wang, Y.; Liang, Z.; Jiang, Y.; Li, J.; Feng, C.; Wang, R.; Chen, H.; et al. Loss of DLG5 Promotes Breast Cancer Malignancy by Inhibiting the Hippo Signaling Pathway. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef]
- Kwan, J.; Sczaniecka, A.; Heidary Arash, E.; Nguyen, L.; Chen, C.-C.; Ratkovic, S.; Klezovitch, O.; Attisano, L.; McNeill, H.; Emili, A.; et al. DLG5 Connects Cell Polarity and Hippo Signaling Protein Networks by Linking PAR-1 with MST1/2. Genes Dev. 2016, 30, 2696–2709. [Google Scholar] [CrossRef] [Green Version]
- Szymaniak, A.D.; Mahoney, J.E.; Cardoso, W.V.; Varelas, X. Crumbs3-Mediated Polarity Directs Airway Epithelial Cell Fate through the Hippo Pathway Effector Yap. Dev. Cell 2015, 34, 283–296. [Google Scholar] [CrossRef] [Green Version]
- Mao, X.; Li, P.; Wang, Y.; Liang, Z.; Liu, J.; Li, J.; Jiang, Y.; Bao, G.; Li, L.; Zhu, B.; et al. CRB3 Regulates Contact Inhibition by Activating the Hippo Pathway in Mammary Epithelial Cells. Cell Death Dis. 2017, 8, e2546. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Yang, N.; Figel, S.A.; Wilson, K.E.; Morrison, C.D.; Gelman, I.H.; Zhang, J. PTPN14 Interacts with and Negatively Regulates the Oncogenic Function of YAP. Oncogene 2013, 32, 1266–1273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, T.; Ludwig, M.Z.; Xu, J.; Fehon, R.G. Kibra and Merlin Activate the Hippo Pathway Spatially Distinct from and Independent of Expanded. Dev. Cell 2017, 40, 478–490.e3. [Google Scholar] [CrossRef] [Green Version]
- Gumbiner, B.M.; Kim, N.-G. The Hippo-YAP Signaling Pathway and Contact Inhibition of Growth. J. Cell Sci. 2014, 127, 709–717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hulpiau, P.; van Roy, F. Molecular Evolution of the Cadherin Superfamily. Int. J. Biochem. Cell Biol. 2009, 41, 349–369. [Google Scholar] [CrossRef] [PubMed]
- Gumbiner, B.M. Regulation of Cadherin-Mediated Adhesion in Morphogenesis. Nat. Rev. Mol. Cell Biol. 2005, 6, 622–634. [Google Scholar] [CrossRef]
- Clevers, H. Wnt/Beta-Catenin Signaling in Development and Disease. Cell 2006, 127, 469–480. [Google Scholar] [CrossRef] [Green Version]
- Hay, E.D.; Zuk, A. Transformations between Epithelium and Mesenchyme: Normal, Pathological, and Experimentally Induced. Am. J. Kidney Dis. 1995, 26, 678–690. [Google Scholar] [CrossRef]
- Masszi, A.; Fan, L.; Rosivall, L.; McCulloch, C.A.; Rotstein, O.D.; Mucsi, I.; Kapus, A. Integrity of Cell-Cell Contacts Is a Critical Regulator of TGF-Beta 1-Induced Epithelial-to-Myofibroblast Transition: Role for Beta-Catenin. Am. J. Pathol. 2004, 165, 1955–1967. [Google Scholar] [CrossRef]
- Iwano, M.; Plieth, D.; Danoff, T.M.; Xue, C.; Okada, H.; Neilson, E.G. Evidence That Fibroblasts Derive from Epithelium during Tissue Fibrosis. J. Clin. Investig. 2002, 110, 341–350. [Google Scholar] [CrossRef]
- He, T.C.; Sparks, A.B.; Rago, C.; Hermeking, H.; Zawel, L.; da Costa, L.T.; Morin, P.J.; Vogelstein, B.; Kinzler, K.W. Identification of C-MYC as a Target of the APC Pathway. Science 1998, 281, 1509–1512. [Google Scholar] [CrossRef] [PubMed]
- Mendonsa, A.M.; Na, T.-Y.; Gumbiner, B.M. E-Cadherin in Contact Inhibition and Cancer. Oncogene 2018, 37, 4769–4780. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.-G.; Koh, E.; Chen, X.; Gumbiner, B.M. E-Cadherin Mediates Contact Inhibition of Proliferation through Hippo Signaling-Pathway Components. Proc. Natl. Acad. Sci. USA 2011, 108, 11930–11935. [Google Scholar] [CrossRef] [Green Version]
- Giampietro, C.; Disanza, A.; Bravi, L.; Barrios-Rodiles, M.; Corada, M.; Frittoli, E.; Savorani, C.; Lampugnani, M.G.; Boggetti, B.; Niessen, C.; et al. The Actin-Binding Protein EPS8 Binds VE-Cadherin and Modulates YAP Localization and Signaling. J. Cell Biol. 2015, 211, 1177–1192. [Google Scholar] [CrossRef] [Green Version]
- Li, P.; Silvis, M.R.; Honaker, Y.; Lien, W.-H.; Arron, S.T.; Vasioukhin, V. AE-Catenin Inhibits a Src-YAP1 Oncogenic Module That Couples Tyrosine Kinases and the Effector of Hippo Signaling Pathway. Genes Dev. 2016, 30, 798–811. [Google Scholar] [CrossRef] [Green Version]
- Gladden, A.B.; Hebert, A.M.; Schneeberger, E.E.; McClatchey, A.I. The NF2 Tumor Suppressor, Merlin, Regulates Epidermal Development through the Establishment of a Junctional Polarity Complex. Dev. Cell 2010, 19, 727–739. [Google Scholar] [CrossRef] [Green Version]
- Fan, R.; Kim, N.-G.; Gumbiner, B.M. Regulation of Hippo Pathway by Mitogenic Growth Factors via Phosphoinositide 3-Kinase and Phosphoinositide-Dependent Kinase-1. Proc. Natl. Acad. Sci. USA 2013, 110, 2569–2574. [Google Scholar] [CrossRef] [Green Version]
- Straßburger, K.; Tiebe, M.; Pinna, F.; Breuhahn, K.; Teleman, A.A. Insulin/IGF Signaling Drives Cell Proliferation in Part via Yorkie/YAP. Dev. Biol. 2012, 367, 187–196. [Google Scholar] [CrossRef] [Green Version]
- Yu, F.-X.; Zhao, B.; Panupinthu, N.; Jewell, J.L.; Lian, I.; Wang, L.H.; Zhao, J.; Yuan, H.; Tumaneng, K.; Li, H.; et al. Regulation of the Hippo-YAP Pathway by G-Protein-Coupled Receptor Signaling. Cell 2012, 150, 780–791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, X.; Chen, N.; Xu, H.; Zhou, X.; Wang, J.; Fang, X.; Zhang, Y.; Li, Y.; Yang, J.; Wang, X. Regulation of Hippo-YAP Signaling by Insulin-like Growth Factor-1 Receptor in the Tumorigenesis of Diffuse Large B-Cell Lymphoma. J. Hematol. Oncol. 2020, 13, 1–15. [Google Scholar] [CrossRef]
- Smoot, R.L.; Werneburg, N.W.; Sugihara, T.; Hernandez, M.C.; Yang, L.; Mehner, C.; Graham, R.P.; Bronk, S.F.; Truty, M.J.; Gores, G.J. Platelet-Derived Growth Factor Regulates YAP Transcriptional Activity via Src Family Kinase Dependent Tyrosine Phosphorylation. J. Cell Biochem. 2018, 119, 824–836. [Google Scholar] [CrossRef] [PubMed]
- Park, H.W.; Kim, Y.C.; Yu, B.; Moroishi, T.; Mo, J.-S.; Plouffe, S.W.; Meng, Z.; Lin, K.C.; Yu, F.-X.; Alexander, C.M.; et al. Alternative Wnt Signaling Activates YAP/TAZ. Cell 2015, 162, 780–794. [Google Scholar] [CrossRef] [Green Version]
- Enzo, E.; Santinon, G.; Pocaterra, A.; Aragona, M.; Bresolin, S.; Forcato, M.; Grifoni, D.; Pession, A.; Zanconato, F.; Guzzo, G.; et al. Aerobic Glycolysis Tunes YAP/TAZ Transcriptional Activity. EMBO J. 2015, 34, 1349–1370. [Google Scholar] [CrossRef]
- DeRan, M.; Yang, J.; Shen, C.-H.; Peters, E.C.; Fitamant, J.; Chan, P.; Hsieh, M.; Zhu, S.; Asara, J.M.; Zheng, B.; et al. Energy Stress Regulates Hippo-YAP Signaling Involving AMPK-Mediated Regulation of Angiomotin-like 1 Protein. Cell Rep. 2014, 9, 495–503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Xiao, Z.-D.; Li, X.; Aziz, K.E.; Gan, B.; Johnson, R.L.; Chen, J. AMPK Modulates Hippo Pathway Activity to Regulate Energy Homeostasis. Nat. Cell Biol. 2015, 17, 490–499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, C.; Zhu, Y.; Zhang, W.; Liao, Q.; Chen, Y.; Zhao, X.; Guo, Q.; Shen, P.; Zhen, B.; Qian, X.; et al. Regulation of the Hippo-YAP Pathway by Glucose Sensor O-GlcNAcylation. Mol. Cell 2017, 68, 591–604.e5. [Google Scholar] [CrossRef] [PubMed]
- Sorrentino, G.; Ruggeri, N.; Specchia, V.; Cordenonsi, M.; Mano, M.; Dupont, S.; Manfrin, A.; Ingallina, E.; Sommaggio, R.; Piazza, S.; et al. Metabolic Control of YAP and TAZ by the Mevalonate Pathway. Nat. Cell Biol. 2014, 16, 357–366. [Google Scholar] [CrossRef] [PubMed]
- Ma, B.; Chen, Y.; Chen, L.; Cheng, H.; Mu, C.; Li, J.; Gao, R.; Zhou, C.; Cao, L.; Liu, J.; et al. Hypoxia Regulates Hippo Signalling through the SIAH2 Ubiquitin E3 Ligase. Nat. Cell Biol. 2015, 17, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Li, Y.; Ma, Y.; Yang, L.; Wang, T.; Meng, X.; Zong, Z.; Sun, X.; Hua, X.; Li, H. Yes-Associated Protein (YAP) Binds to HIF-1α and Sustains HIF-1α Protein Stability to Promote Hepatocellular Carcinoma Cell Glycolysis under Hypoxic Stress. J. Exp. Clin. Cancer Res. 2018, 37, 1–12. [Google Scholar] [CrossRef]
- Hong, A.W.; Meng, Z.; Yuan, H.-X.; Plouffe, S.W.; Moon, S.; Kim, W.; Jho, E.-H.; Guan, K.-L. Osmotic Stress-Induced Phosphorylation by NLK at Ser128 Activates YAP. EMBO Rep. 2017, 18, 72–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aragona, M.; Panciera, T.; Manfrin, A.; Giulitti, S.; Michielin, F.; Elvassore, N.; Dupont, S.; Piccolo, S. A Mechanical Checkpoint Controls Multicellular Growth through YAP/TAZ Regulation by Actin-Processing Factors. Cell 2013, 154, 1047–1059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, X.; Degese, M.S.; Iglesias-Bartolome, R.; Vaque, J.P.; Molinolo, A.A.; Rodrigues, M.; Zaidi, M.R.; Ksander, B.R.; Merlino, G.; Sodhi, A.; et al. Hippo-Independent Activation of YAP by the GNAQ Uveal Melanoma Oncogene through a Trio-Regulated Rho GTPase Signaling Circuitry. Cancer Cell 2014, 25, 831–845. [Google Scholar] [CrossRef] [Green Version]
- Schlegelmilch, K.; Mohseni, M.; Kirak, O.; Pruszak, J.; Rodriguez, J.R.; Zhou, D.; Kreger, B.T.; Vasioukhin, V.; Avruch, J.; Brummelkamp, T.R.; et al. Yap1 Acts Downstream of α-Catenin to Control Epidermal Proliferation. Cell 2011, 144, 782–795. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Li, W.; Wang, S.; Zhang, P.; Wang, Q.; Xiao, J.; Zhang, C.; Zheng, X.; Xu, X.; Xue, S.; et al. YAP Aggravates Inflammatory Bowel Disease by Regulating M1/M2 Macrophage Polarization and Gut Microbial Homeostasis. Cell Rep. 2019, 27, 1176–1189. [Google Scholar] [CrossRef] [Green Version]
- Meli, V.S.; Atcha, H.; Veerasubramanian, P.K.; Nagalla, R.R.; Luu, T.U.; Chen, E.Y.; Guerrero-Juarez, C.F.; Yamaga, K.; Pandori, W.; Hsieh, J.Y.; et al. YAP-Mediated Mechanotransduction Tunes the Macrophage Inflammatory Response. Sci. Adv. 2020, 6. [Google Scholar] [CrossRef]
- Taniguchi, K.; Wu, L.-W.; Grivennikov, S.I.; de Jong, P.R.; Lian, I.; Yu, F.-X.; Wang, K.; Ho, S.B.; Boland, B.S.; Chang, J.T.; et al. A Gp130-Src-YAP Module Links Inflammation to Epithelial Regeneration. Nature 2015, 519, 57–62. [Google Scholar] [CrossRef] [Green Version]
- Dvorak, H.F. Tumors: Wounds That Do Not Heal. Similarities between Tumor Stroma Generation and Wound Healing. N. Engl. J. Med. 1986, 315, 1650–1659. [Google Scholar] [CrossRef] [PubMed]
- Zanconato, F.; Forcato, M.; Battilana, G.; Azzolin, L.; Quaranta, E.; Bodega, B.; Rosato, A.; Bicciato, S.; Cordenonsi, M.; Piccolo, S. Genome-Wide Association between YAP/TAZ/TEAD and AP-1 at Enhancers Drives Oncogenic Growth. Nat. Cell Biol. 2015, 17, 1218–1227. [Google Scholar] [CrossRef] [PubMed]
- Nicolay, B.N.; Bayarmagnai, B.; Islam, A.B.M.M.K.; Lopez-Bigas, N.; Frolov, M.V. Cooperation between DE2F1 and Yki/Sd Defines a Distinct Transcriptional Program Necessary to Bypass Cell Cycle Exit. Genes Dev. 2011, 25, 323–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizuno, T.; Murakami, H.; Fujii, M.; Ishiguro, F.; Tanaka, I.; Kondo, Y.; Akatsuka, S.; Toyokuni, S.; Yokoi, K.; Osada, H.; et al. YAP Induces Malignant Mesothelioma Cell Proliferation by Upregulating Transcription of Cell Cycle-Promoting Genes. Oncogene 2012, 31, 5117–5122. [Google Scholar] [CrossRef] [Green Version]
- Kapoor, A.; Yao, W.; Ying, H.; Hua, S.; Liewen, A.; Wang, Q.; Zhong, Y.; Wu, C.-J.; Sadanandam, A.; Hu, B.; et al. Yap1 Activation Enables Bypass of Oncogenic Kras Addiction in Pancreatic Cancer. Cell 2014, 158, 185–197. [Google Scholar] [CrossRef] [Green Version]
- Warren, J.S.A.; Xiao, Y.; Lamar, J.M. YAP/TAZ Activation as a Target for Treating Metastatic Cancer. Cancers 2018, 10, 115. [Google Scholar] [CrossRef] [Green Version]
- Overholtzer, M.; Zhang, J.; Smolen, G.A.; Muir, B.; Li, W.; Sgroi, D.C.; Deng, C.-X.; Brugge, J.S.; Haber, D.A. Transforming Properties of YAP, a Candidate Oncogene on the Chromosome 11q22 Amplicon. Proc. Natl. Acad. Sci. USA 2006, 103, 12405–12410. [Google Scholar] [CrossRef] [Green Version]
- Hélias-Rodzewicz, Z.; Pérot, G.; Chibon, F.; Ferreira, C.; Lagarde, P.; Terrier, P.; Coindre, J.-M.; Aurias, A. YAP1 and VGLL3, Encoding Two Cofactors of TEAD Transcription Factors, Are Amplified and Overexpressed in a Subset of Soft Tissue Sarcomas. Genes Chromosomes Cancer 2010, 49, 1161–1171. [Google Scholar] [CrossRef]
- Imoto, I.; Tsuda, H.; Hirasawa, A.; Miura, M.; Sakamoto, M.; Hirohashi, S.; Inazawa, J. Expression of CIAP1, a Target for 11q22 Amplification, Correlates with Resistance of Cervical Cancers to Radiotherapy. Cancer Res. 2002, 62, 4860–4866. [Google Scholar]
- Menzel, M.; Meckbach, D.; Weide, B.; Toussaint, N.C.; Schilbach, K.; Noor, S.; Eigentler, T.; Ikenberg, K.; Busch, C.; Quintanilla-Martinez, L.; et al. In Melanoma, Hippo Signaling Is Affected by Copy Number Alterations and YAP1 Overexpression Impairs Patient Survival. Pigment Cell Melanoma Res. 2014, 27, 671–673. [Google Scholar] [CrossRef]
- Antonescu, C.R.; Le Loarer, F.; Mosquera, J.-M.; Sboner, A.; Zhang, L.; Chen, C.-L.; Chen, H.-W.; Pathan, N.; Krausz, T.; Dickson, B.C.; et al. Novel YAP1-TFE3 Fusion Defines a Distinct Subset of Epithelioid Hemangioendothelioma. Genes Chromosomes Cancer 2013, 52, 775–784. [Google Scholar] [CrossRef] [Green Version]
- Patel, N.R.; Salim, A.A.; Sayeed, H.; Sarabia, S.F.; Hollingsworth, F.; Warren, M.; Jakacky, J.; Tanas, M.; Oliveira, A.M.; Rubin, B.P.; et al. Molecular Characterization of Epithelioid Haemangioendotheliomas Identifies Novel WWTR1-CAMTA1 Fusion Variants. Histopathology 2015, 67, 699–708. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Tang, J.Z.; Vergara, I.A.; Zhang, Y.; Szeto, P.; Yang, L.; Mintoff, C.; Colebatch, A.; McIntosh, L.; Mitchell, K.A.; et al. Somatic Hypermutation of the YAP Oncogene in a Human Cutaneous Melanoma. Mol. Cancer Res. 2019, 17, 1435–1449. [Google Scholar] [CrossRef] [Green Version]
- Thompson, B.J. YAP/TAZ: Drivers of Tumor Growth, Metastasis, and Resistance to Therapy. Bioessays 2020, 42, e1900162. [Google Scholar] [CrossRef] [Green Version]
- Lo Sardo, F.; Strano, S.; Blandino, G. YAP and TAZ in Lung Cancer: Oncogenic Role and Clinical Targeting. Cancers 2018, 10, 137. [Google Scholar] [CrossRef] [Green Version]
- Mohseni, M.; Sun, J.; Lau, A.; Curtis, S.; Goldsmith, J.; Fox, V.L.; Wei, C.; Frazier, M.; Samson, O.; Wong, K.-K.; et al. A Genetic Screen Identifies an LKB1-MARK Signalling Axis Controlling the Hippo-YAP Pathway. Nat. Cell Biol. 2014, 16, 108–117. [Google Scholar] [CrossRef] [PubMed]
- Xie, M.; Wu, X.; Zhang, J.; Zhang, J.; Li, X. Ski Regulates Smads and TAZ Signaling to Suppress Lung Cancer Progression. Mol. Carcinog. 2017, 56, 2178–2189. [Google Scholar] [CrossRef] [PubMed]
- Zanconato, F.; Cordenonsi, M.; Piccolo, S. YAP/TAZ at the Roots of Cancer. Cancer Cell 2016, 29, 783–803. [Google Scholar] [CrossRef] [Green Version]
- Sun, Z.; Xu, R.; Li, X.; Ren, W.; Ou, C.; Wang, Q.; Zhang, H.; Zhang, X.; Ma, J.; Wang, H.; et al. Prognostic Value of Yes-Associated Protein 1 (YAP1) in Various Cancers: A Meta-Analysis. PLoS ONE 2015, 10, e0135119. [Google Scholar] [CrossRef]
- Galli, G.G.; Carrara, M.; Yuan, W.-C.; Valdes-Quezada, C.; Gurung, B.; Pepe-Mooney, B.; Zhang, T.; Geeven, G.; Gray, N.S.; de Laat, W.; et al. YAP Drives Growth by Controlling Transcriptional Pause Release from Dynamic Enhancers. Mol. Cell 2015, 60, 328–337. [Google Scholar] [CrossRef] [Green Version]
- Stein, C.; Bardet, A.F.; Roma, G.; Bergling, S.; Clay, I.; Ruchti, A.; Agarinis, C.; Schmelzle, T.; Bouwmeester, T.; Schübeler, D.; et al. YAP1 Exerts Its Transcriptional Control via TEAD-Mediated Activation of Enhancers. PLoS Genet. 2015, 11, e1005465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jang, W.; Kim, T.; Koo, J.S.; Kim, S.-K.; Lim, D.-S. Mechanical Cue-Induced YAP Instructs Skp2-Dependent Cell Cycle Exit and Oncogenic Signaling. EMBO J. 2017, 36, 2510–2528. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Ji, J.-Y.; Yu, M.; Overholtzer, M.; Smolen, G.A.; Wang, R.; Brugge, J.S.; Dyson, N.J.; Haber, D.A. YAP-Dependent Induction of Amphiregulin Identifies a Non-Cell-Autonomous Component of the Hippo Pathway. Nat. Cell Biol. 2009, 11, 1444–1450. [Google Scholar] [CrossRef]
- Xu, M.Z.; Chan, S.W.; Liu, A.M.; Wong, K.F.; Fan, S.T.; Chen, J.; Poon, R.T.; Zender, L.; Lowe, S.W.; Hong, W.; et al. AXL Receptor Kinase Is a Mediator of YAP-Dependent Oncogenic Functions in Hepatocellular Carcinoma. Oncogene 2011, 30, 1229–1240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gill, M.K.; Christova, T.; Zhang, Y.Y.; Gregorieff, A.; Zhang, L.; Narimatsu, M.; Song, S.; Xiong, S.; Couzens, A.L.; Tong, J.; et al. A Feed Forward Loop Enforces YAP/TAZ Signaling during Tumorigenesis. Nat. Commun. 2018, 9, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Croci, O.; De Fazio, S.; Biagioni, F.; Donato, E.; Caganova, M.; Curti, L.; Doni, M.; Sberna, S.; Aldeghi, D.; Biancotto, C.; et al. Transcriptional Integration of Mitogenic and Mechanical Signals by Myc and YAP. Genes Dev. 2017, 31, 2017–2022. [Google Scholar] [CrossRef] [Green Version]
- Maglic, D.; Schlegelmilch, K.; Dost, A.F.; Panero, R.; Dill, M.T.; Calogero, R.A.; Camargo, F.D. YAP-TEAD Signaling Promotes Basal Cell Carcinoma Development via a c-JUN/AP1 Axis. EMBO J. 2018, 37. [Google Scholar] [CrossRef]
- Hansen, C.G.; Moroishi, T.; Guan, K.-L. YAP and TAZ: A Nexus for Hippo Signaling and Beyond. Trends Cell Biol. 2015, 25, 499–513. [Google Scholar] [CrossRef] [Green Version]
- Hoxha, S.; Shepard, A.; Troutman, S.; Diao, H.; Doherty, J.R.; Janiszewska, M.; Witwicki, R.M.; Pipkin, M.E.; Ja, W.W.; Kareta, M.S.; et al. YAP-Mediated Recruitment of YY1 and EZH2 Represses Transcription of Key Cell-Cycle Regulators. Cancer Res. 2020, 80, 2512–2522. [Google Scholar] [CrossRef]
- Lo Sardo, F.; Pulito, C.; Sacconi, A.; Korita, E.; Sudol, M.; Strano, S.; Blandino, G. YAP/TAZ and EZH2 Synergize to Impair Tumor Suppressor Activity of TGFBR2 in Non-Small Cell Lung Cancer. Cancer Lett. 2021, 500, 51–63. [Google Scholar] [CrossRef] [PubMed]
- Missiroli, S.; Perrone, M.; Genovese, I.; Pinton, P.; Giorgi, C. Cancer Metabolism and Mitochondria: Finding Novel Mechanisms to Fight Tumours. EBioMedicine 2020, 59, 102943. [Google Scholar] [CrossRef] [PubMed]
- Cox, A.G.; Hwang, K.L.; Brown, K.K.; Evason, K.; Beltz, S.; Tsomides, A.; O’Connor, K.; Galli, G.G.; Yimlamai, D.; Chhangawala, S.; et al. Yap Reprograms Glutamine Metabolism to Increase Nucleotide Biosynthesis and Enable Liver Growth. Nat. Cell Biol. 2016, 18, 886–896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santinon, G.; Brian, I.; Pocaterra, A.; Romani, P.; Franzolin, E.; Rampazzo, C.; Bicciato, S.; Dupont, S. DNTP Metabolism Links Mechanical Cues and YAP/TAZ to Cell Growth and Oncogene-Induced Senescence. EMBO J. 2018, 37. [Google Scholar] [CrossRef] [PubMed]
- Cosset, É.; Ilmjärv, S.; Dutoit, V.; Elliott, K.; von Schalscha, T.; Camargo, M.F.; Reiss, A.; Moroishi, T.; Seguin, L.; Gomez, G.; et al. Glut3 Addiction Is a Druggable Vulnerability for a Molecularly Defined Subpopulation of Glioblastoma. Cancer Cell 2017, 32, 856–868.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Qiao, Y.; Wu, Q.; Chen, Y.; Zou, S.; Liu, X.; Zhu, G.; Zhao, Y.; Chen, Y.; Yu, Y.; et al. The Essential Role of YAP O-GlcNAcylation in High-Glucose-Stimulated Liver Tumorigenesis. Nat. Commun. 2017, 8, 1–15. [Google Scholar] [CrossRef]
- Zheng, X.; Han, H.; Liu, G.-P.; Ma, Y.-X.; Pan, R.-L.; Sang, L.-J.; Li, R.-H.; Yang, L.-J.; Marks, J.R.; Wang, W.; et al. LncRNA Wires up Hippo and Hedgehog Signaling to Reprogramme Glucose Metabolism. EMBO J. 2017, 36, 3325–3335. [Google Scholar] [CrossRef]
- Holden, J.K.; Cunningham, C.N. Targeting the Hippo Pathway and Cancer through the TEAD Family of Transcription Factors. Cancers 2018, 10, 81. [Google Scholar] [CrossRef] [Green Version]
- Xie, D.; Nakachi, K.; Wang, H.; Elashoff, R.; Koeffler, H.P. Elevated Levels of Connective Tissue Growth Factor, WISP-1, and CYR61 in Primary Breast Cancers Associated with More Advanced Features. Cancer Res. 2001, 61, 8917–8923. [Google Scholar]
- Zhao, B.; Li, L.; Wang, L.; Wang, C.-Y.; Yu, J.; Guan, K.-L. Cell Detachment Activates the Hippo Pathway via Cytoskeleton Reorganization to Induce Anoikis. Genes Dev. 2012, 26, 54–68. [Google Scholar] [CrossRef] [Green Version]
- Cordenonsi, M.; Zanconato, F.; Azzolin, L.; Forcato, M.; Rosato, A.; Frasson, C.; Inui, M.; Montagner, M.; Parenti, A.R.; Poletti, A.; et al. The Hippo Transducer TAZ Confers Cancer Stem Cell-Related Traits on Breast Cancer Cells. Cell 2011, 147, 759–772. [Google Scholar] [CrossRef]
- Mani, S.A.; Guo, W.; Liao, M.-J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The Epithelial-Mesenchymal Transition Generates Cells with Properties of Stem Cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, B.; Ye, X.; Yu, J.; Li, L.; Li, W.; Li, S.; Yu, J.; Lin, J.D.; Wang, C.-Y.; Chinnaiyan, A.M.; et al. TEAD Mediates YAP-Dependent Gene Induction and Growth Control. Genes Dev. 2008, 22, 1962–1971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, L.; Azzolin, L.; Di Biagio, D.; Zanconato, F.; Battilana, G.; Lucon Xiccato, R.; Aragona, M.; Giulitti, S.; Panciera, T.; Gandin, A.; et al. The SWI/SNF Complex Is a Mechanoregulated Inhibitor of YAP and TAZ. Nature 2018, 563, 265–269. [Google Scholar] [CrossRef]
- Lamar, J.M.; Stern, P.; Liu, H.; Schindler, J.W.; Jiang, Z.-G.; Hynes, R.O. The Hippo Pathway Target, YAP, Promotes Metastasis through Its TEAD-Interaction Domain. Proc. Natl. Acad. Sci. USA 2012, 109, E2441–E2450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiao, Y.; Chen, J.; Lim, Y.B.; Finch-Edmondson, M.L.; Seshachalam, V.P.; Qin, L.; Jiang, T.; Low, B.C.; Singh, H.; Lim, C.T.; et al. YAP Regulates Actin Dynamics through ARHGAP29 and Promotes Metastasis. Cell Rep. 2017, 19, 1495–1502. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.; Zhang, L.; Purohit, V.; Shukla, S.K.; Chen, X.; Yu, F.; Fu, K.; Chen, Y.; Solheim, J.; Singh, P.K.; et al. Active YAP Promotes Pancreatic Cancer Cell Motility, Invasion and Tumorigenesis in a Mitotic Phosphorylation-Dependent Manner through LPAR3. Oncotarget 2015, 6, 36019–36031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diepenbruck, M.; Waldmeier, L.; Ivanek, R.; Berninger, P.; Arnold, P.; van Nimwegen, E.; Christofori, G. Tead2 Expression Levels Control the Subcellular Distribution of Yap and Taz, Zyxin Expression and Epithelial-Mesenchymal Transition. J. Cell Sci. 2014, 127, 1523–1536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ge, L.; Smail, M.; Meng, W.; Shyr, Y.; Ye, F.; Fan, K.-H.; Li, X.; Zhou, H.-M.; Bhowmick, N.A. Yes-Associated Protein Expression in Head and Neck Squamous Cell Carcinoma Nodal Metastasis. PLoS ONE 2011, 6, e27529. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.M.; Jung, W.H.; Koo, J.S. Expression of Yes-Associated Protein (YAP) in Metastatic Breast Cancer. Int. J. Clin. Exp. Pathol. 2015, 8, 11248–11257. [Google Scholar]
- Verfaillie, A.; Imrichova, H.; Atak, Z.K.; Dewaele, M.; Rambow, F.; Hulselmans, G.; Christiaens, V.; Svetlichnyy, D.; Luciani, F.; Van den Mooter, L.; et al. Decoding the Regulatory Landscape of Melanoma Reveals TEADS as Regulators of the Invasive Cell State. Nat. Commun. 2015, 6, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; George, J.; Deb, S.; Degoutin, J.L.; Takano, E.A.; Fox, S.B.; AOCS Study group; Bowtell, D.D.L.; Harvey, K.F. The Hippo Pathway Transcriptional Co-Activator, YAP, Is an Ovarian Cancer Oncogene. Oncogene 2011, 30, 2810–2822. [Google Scholar] [CrossRef] [Green Version]
- Vlug, E.J.; van de Ven, R.A.H.; Vermeulen, J.F.; Bult, P.; van Diest, P.J.; Derksen, P.W.B. Nuclear Localization of the Transcriptional Coactivator YAP Is Associated with Invasive Lobular Breast Cancer. Cell Oncol. 2013, 36, 375–384. [Google Scholar] [CrossRef] [Green Version]
- Cao, L.; Sun, P.-L.; Yao, M.; Jia, M.; Gao, H. Expression of YES-Associated Protein (YAP) and Its Clinical Significance in Breast Cancer Tissues. Hum. Pathol. 2017, 68, 166–174. [Google Scholar] [CrossRef]
- Hu, X.; Jia, Y.; Yu, J.; Chen, J.; Fu, Q. Loss of YAP Protein in Prostate Cancer Is Associated with Gleason Score Increase. Tumori 2015, 101, 189–193. [Google Scholar] [CrossRef]
- Yu, S.-J.; Hu, J.-Y.; Kuang, X.-Y.; Luo, J.-M.; Hou, Y.-F.; Di, G.-H.; Wu, J.; Shen, Z.-Z.; Song, H.-Y.; Shao, Z.-M. MicroRNA-200a Promotes Anoikis Resistance and Metastasis by Targeting YAP1 in Human Breast Cancer. Clin. Cancer Res. 2013, 19, 1389–1399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, T.; Bondar, T.; Zhou, X.; Zhang, C.; He, H.; Medzhitov, R. Two-Signal Requirement for Growth-Promoting Function of Yap in Hepatocytes. eLife 2015, 4. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Abdelrahman, A.; Vollmar, B.; Zechner, D. The Ambivalent Function of YAP in Apoptosis and Cancer. Int. J. Mol. Sci. 2018, 19, 3770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barry, E.R.; Morikawa, T.; Butler, B.L.; Shrestha, K.; de la Rosa, R.; Yan, K.S.; Fuchs, C.S.; Magness, S.T.; Smits, R.; Ogino, S.; et al. Restriction of Intestinal Stem Cell Expansion and the Regenerative Response by YAP. Nature 2013, 493, 106–110. [Google Scholar] [CrossRef]
- Allegra, A.; Pioggia, G.; Innao, V.; Musolino, C.; Gangemi, S. New Insights into YES-Associated Protein Signaling Pathways in Hematological Malignancies: Diagnostic and Therapeutic Challenges. Cancers 2021, 13, 1981. [Google Scholar] [CrossRef] [PubMed]
- Cottini, F.; Hideshima, T.; Xu, C.; Sattler, M.; Dori, M.; Agnelli, L.; ten Hacken, E.; Bertilaccio, M.T.; Antonini, E.; Neri, A.; et al. Rescue of Hippo Coactivator YAP1 Triggers DNA Damage-Induced Apoptosis in Hematological Cancers. Nat. Med. 2014, 20, 599–606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maruyama, J.; Inami, K.; Michishita, F.; Jiang, X.; Iwasa, H.; Nakagawa, K.; Ishigami-Yuasa, M.; Kagechika, H.; Miyamura, N.; Hirayama, J.; et al. Novel YAP1 Activator, Identified by Transcription-Based Functional Screen, Limits Multiple Myeloma Growth. Mol. Cancer Res. 2018, 16, 197–211. [Google Scholar] [CrossRef] [Green Version]
- Fan, S.; Price, T.; Huang, W.; Plue, M.; Warren, J.; Sundaramoorthy, P.; Paul, B.; Feinberg, D.; MacIver, N.; Chao, N.; et al. PINK1-Dependent Mitophagy Regulates the Migration and Homing of Multiple Myeloma Cells via the MOB1B-Mediated Hippo-YAP/TAZ Pathway. Adv. Sci. 2020, 7, 1900860. [Google Scholar] [CrossRef] [Green Version]
- Pan, Z.; Tian, Y.; Cao, C.; Niu, G. The Emerging Role of YAP/TAZ in Tumor Immunity. Mol. Cancer Res. 2019, 17, 1777–1786. [Google Scholar] [CrossRef] [Green Version]
- Nishimura, H.; Agata, Y.; Kawasaki, A.; Sato, M.; Imamura, S.; Minato, N.; Yagita, H.; Nakano, T.; Honjo, T. Developmentally Regulated Expression of the PD-1 Protein on the Surface of Double-Negative (CD4-CD8-) Thymocytes. Int. Immunol. 1996, 8, 773–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dijkstra, K.K.; Cattaneo, C.M.; Weeber, F.; Chalabi, M.; van de Haar, J.; Fanchi, L.F.; Slagter, M.; van der Velden, D.L.; Kaing, S.; Kelderman, S.; et al. Generation of Tumor-Reactive T Cells by Co-Culture of Peripheral Blood Lymphocytes and Tumor Organoids. Cell 2018, 174, 1586–1598.e12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flecken, T.; Schmidt, N.; Hild, S.; Gostick, E.; Drognitz, O.; Zeiser, R.; Schemmer, P.; Bruns, H.; Eiermann, T.; Price, D.A.; et al. Immunodominance and Functional Alterations of Tumor-Associated Antigen-Specific CD8+ T-Cell Responses in Hepatocellular Carcinoma. Hepatology 2014, 59, 1415–1426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakaguchi, S.; Sakaguchi, N.; Asano, M.; Itoh, M.; Toda, M. Immunologic Self-Tolerance Maintained by Activated T Cells Expressing IL-2 Receptor Alpha-Chains (CD25). Breakdown of a Single Mechanism of Self-Tolerance Causes Various Autoimmune Diseases. J. Immunol. 1995, 155, 1151–1164. [Google Scholar]
- Suh, J.-H.; Won, K.Y.; Kim, G.Y.; Bae, G.E.; Lim, S.-J.; Sung, J.-Y.; Park, Y.-K.; Kim, Y.W.; Lee, J. Expression of Tumoral FOXP3 in Gastric Adenocarcinoma Is Associated with Favorable Clinicopathological Variables and Related with Hippo Pathway. Int. J. Clin. Exp. Pathol. 2015, 8, 14608–14618. [Google Scholar]
- Murakami, S.; Shahbazian, D.; Surana, R.; Zhang, W.; Chen, H.; Graham, G.T.; White, S.M.; Weiner, L.M.; Yi, C. Yes-Associated Protein Mediates Immune Reprogramming in Pancreatic Ductal Adenocarcinoma. Oncogene 2017, 36, 1232–1244. [Google Scholar] [CrossRef] [Green Version]
- Moroishi, T.; Hayashi, T.; Pan, W.-W.; Fujita, Y.; Holt, M.V.; Qin, J.; Carson, D.A.; Guan, K.-L. The Hippo Pathway Kinases LATS1/2 Suppress Cancer Immunity. Cell 2016, 167, 1525–1539.e17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shou, D.; Wen, L.; Song, Z.; Yin, J.; Sun, Q.; Gong, W. Suppressive Role of Myeloid-Derived Suppressor Cells (MDSCs) in the Microenvironment of Breast Cancer and Targeted Immunotherapies. Oncotarget 2016, 7, 64505–64511. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Guo, J.; Weng, L.; Tang, W.; Jin, S.; Ma, W. Myeloid-Derived Suppressor Cells-New and Exciting Players in Lung Cancer. J. Hematol. Oncol. 2020, 13, 10. [Google Scholar] [CrossRef] [PubMed]
- Sanaei, M.-J.; Salimzadeh, L.; Bagheri, N. Crosstalk between Myeloid-Derived Suppressor Cells and the Immune System in Prostate Cancer: MDSCs and Immune System in Prostate Cancer. J. Leukoc. Biol. 2020, 107, 43–56. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A. The Growing Diversity and Spectrum of Action of Myeloid-Derived Suppressor Cells. Eur. J. Immunol. 2010, 40, 3317–3320. [Google Scholar] [CrossRef]
- Wang, G.; Lu, X.; Dey, P.; Deng, P.; Wu, C.C.; Jiang, S.; Fang, Z.; Zhao, K.; Konaparthi, R.; Hua, S.; et al. Targeting YAP-Dependent MDSC Infiltration Impairs Tumor Progression. Cancer Discov. 2016, 6, 80–95. [Google Scholar] [CrossRef] [Green Version]
- Sarkar, S.; Bristow, C.A.; Dey, P.; Rai, K.; Perets, R.; Ramirez-Cardenas, A.; Malasi, S.; Huang-Hobbs, E.; Haemmerle, M.; Wu, S.Y.; et al. PRKCI Promotes Immune Suppression in Ovarian Cancer. Genes Dev. 2017, 31, 1109–1121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Justilien, V.; Brennan, K.I.; Jamieson, L.; Murray, N.R.; Fields, A.P. PKCι Regulates Nuclear YAP1 Localization and Ovarian Cancer Tumorigenesis. Oncogene 2017, 36, 534–545. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Rong, L.; Zhao, X.; Li, X.; Liu, X.; Deng, J.; Wu, H.; Xu, X.; Erben, U.; Wu, P.; et al. TNF Signaling Drives Myeloid-Derived Suppressor Cell Accumulation. J. Clin. Investig. 2012, 122, 4094–4104. [Google Scholar] [CrossRef]
- Yang, R.; Cai, T.-T.; Wu, X.-J.; Liu, Y.-N.; He, J.; Zhang, X.-S.; Ma, G.; Li, J. Tumour YAP1 and PTEN Expression Correlates with Tumour-Associated Myeloid Suppressor Cell Expansion and Reduced Survival in Colorectal Cancer. Immunology 2018, 155, 263–272. [Google Scholar] [CrossRef]
- Dong, Y.; Sun, Q.; Zhang, X. PD-1 and Its Ligands Are Important Immune Checkpoints in Cancer. Oncotarget 2017, 8, 2171–2186. [Google Scholar] [CrossRef] [Green Version]
- Keir, M.E.; Butte, M.J.; Freeman, G.J.; Sharpe, A.H. PD-1 and Its Ligands in Tolerance and Immunity. Annu. Rev. Immunol. 2008, 26, 677–704. [Google Scholar] [CrossRef] [Green Version]
- Wei, S.C.; Duffy, C.R.; Allison, J.P. Fundamental Mechanisms of Immune Checkpoint Blockade Therapy. Cancer Discov. 2018, 8, 1069–1086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ribas, A.; Wolchok, J.D. Cancer Immunotherapy Using Checkpoint Blockade. Science 2018, 359, 1350–1355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, P.-C.; Miao, J.; Wang, Y.-C.; Zhang, W.-Q.; Yang, Y.-L.; Wang, C.-W.; Yang, C.-T.; Huang, Z.; You, J.; Xu, Z.; et al. Inhibition of Yes-Associated Protein down-Regulates PD-L1 (CD274) Expression in Human Malignant Pleural Mesothelioma. J. Cell Mol. Med. 2018, 22, 3139–3148. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.H.; Kim, C.G.; Kim, S.-K.; Shin, S.J.; Choe, E.A.; Park, S.-H.; Shin, E.-C.; Kim, J. YAP-Induced PD-L1 Expression Drives Immune Evasion in BRAFi-Resistant Melanoma. Cancer Immunol. Res. 2018, 6, 255–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miao, J.; Hsu, P.-C.; Yang, Y.-L.; Xu, Z.; Dai, Y.; Wang, Y.; Chan, G.; Huang, Z.; Hu, B.; Li, H.; et al. YAP Regulates PD-L1 Expression in Human NSCLC Cells. Oncotarget 2017, 8, 114576–114587. [Google Scholar] [CrossRef] [Green Version]
- He, J.; Hu, Y.; Hu, M.; Li, B. Development of PD-1/PD-L1 Pathway in Tumor Immune Microenvironment and Treatment for Non-Small Cell Lung Cancer. Sci. Rep. 2015, 5, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Mancuso, M.R.; Neal, J.W. Novel Systemic Therapy against Malignant Pleural Mesothelioma. Transl. Lung Cancer Res. 2017, 6, 295–314. [Google Scholar] [CrossRef] [Green Version]
- Zanconato, F.; Cordenonsi, M.; Piccolo, S. YAP and TAZ: A Signalling Hub of the Tumour Microenvironment. Nat. Rev. Cancer 2019, 19, 454–464. [Google Scholar] [CrossRef]
- Bingle, L.; Brown, N.J.; Lewis, C.E. The Role of Tumour-Associated Macrophages in Tumour Progression: Implications for New Anticancer Therapies. J. Pathol. 2002, 196, 254–265. [Google Scholar] [CrossRef]
- Long, K.B.; Collier, A.I.; Beatty, G.L. Macrophages: Key Orchestrators of a Tumor Microenvironment Defined by Therapeutic Resistance. Mol. Immunol. 2019, 110, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Pollard, J.W. Tumour-Educated Macrophages Promote Tumour Progression and Metastasis. Nat. Rev. Cancer 2004, 4, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Li, Y.; Kim, S.M.; Bossuyt, W.; Liu, P.; Qiu, Q.; Wang, Y.; Halder, G.; Finegold, M.J.; Lee, J.-S.; et al. Hippo Signaling Is a Potent in Vivo Growth and Tumor Suppressor Pathway in the Mammalian Liver. Proc. Natl. Acad. Sci. USA 2010, 107, 1437–1442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, G.; Dhar, D.; Nakagawa, H.; Font-Burgada, J.; Ogata, H.; Jiang, Y.; Shalapour, S.; Seki, E.; Yost, S.E.; Jepsen, K.; et al. Identification of Liver Cancer Progenitors Whose Malignant Progression Depends on Autocrine IL-6 Signaling. Cell 2013, 155, 384–396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, X.; Zhao, Y.; Yan, H.; Yang, Y.; Shen, S.; Dai, X.; Ji, X.; Ji, F.; Gong, X.-G.; Li, L.; et al. Single Tumor-Initiating Cells Evade Immune Clearance by Recruiting Type II Macrophages. Genes Dev. 2017, 31, 247–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, W.; Khan, S.K.; Liu, Y.; Xu, R.; Park, O.; He, Y.; Cha, B.; Gao, B.; Yang, Y. Hepatic Hippo Signaling Inhibits Protumoural Microenvironment to Suppress Hepatocellular Carcinoma. Gut 2018, 67, 1692–1703. [Google Scholar] [CrossRef]
- Huang, Y.-J.; Yang, C.-K.; Wei, P.-L.; Huynh, T.-T.; Whang-Peng, J.; Meng, T.-C.; Hsiao, M.; Tzeng, Y.-M.; Wu, A.T.; Yen, Y. Ovatodiolide Suppresses Colon Tumorigenesis and Prevents Polarization of M2 Tumor-Associated Macrophages through YAP Oncogenic Pathways. J. Hematol. Oncol. 2017, 10, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, C.D.K.; Yi, C. YAP/TAZ Signaling and Resistance to Cancer Therapy. Trends Cancer 2019, 5, 283–296. [Google Scholar] [CrossRef]
- Zhao, W.; Liu, H.; Wang, J.; Wang, M.; Shao, R. Cyclizing-Berberine A35 Induces G2/M Arrest and Apoptosis by Activating YAP Phosphorylation (Ser127). J. Exp. Clin. Cancer Res. 2018, 37, 1–12. [Google Scholar] [CrossRef]
- Su, D.; Lin, Z. Dichloroacetate Attenuates the Stemness of Hepatocellular Carcinoma Cells via Promoting Nucleus-Cytoplasm Translocation of YAP. Environ. Toxicol. 2021, 36, 975–983. [Google Scholar] [CrossRef]
- Jin, D.; Wu, Y.; Shao, C.; Gao, Y.; Wang, D.; Guo, J. Norcantharidin Reverses Cisplatin Resistance and Inhibits the Epithelial Mesenchymal Transition of Human Non-small Lung Cancer Cells by Regulating the YAP Pathway. Oncol. Rep. 2021, 45, 609–620. [Google Scholar] [CrossRef]
- Oku, Y.; Nishiya, N.; Shito, T.; Yamamoto, R.; Yamamoto, Y.; Oyama, C.; Uehara, Y. Small Molecules Inhibiting the Nuclear Localization of YAP/TAZ for Chemotherapeutics and Chemosensitizers against Breast Cancers. FEBS Open Bio 2015, 5, 542–549. [Google Scholar] [CrossRef] [Green Version]
- Shao, D.D.; Xue, W.; Krall, E.B.; Bhutkar, A.; Piccioni, F.; Wang, X.; Schinzel, A.C.; Sood, S.; Rosenbluh, J.; Kim, J.W.; et al. KRAS and YAP1 Converge to Regulate EMT and Tumor Survival. Cell 2014, 158, 171–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitajima, S.; Asahina, H.; Chen, T.; Guo, S.; Quiceno, L.G.; Cavanaugh, J.D.; Merlino, A.A.; Tange, S.; Terai, H.; Kim, J.W.; et al. Overcoming Resistance to Dual Innate Immune and MEK Inhibition Downstream of KRAS. Cancer Cell 2018, 34, 439–452.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu-Chittenden, Y.; Huang, B.; Shim, J.S.; Chen, Q.; Lee, S.-J.; Anders, R.A.; Liu, J.O.; Pan, D. Genetic and Pharmacological Disruption of the TEAD-YAP Complex Suppresses the Oncogenic Activity of YAP. Genes Dev. 2012, 26, 1300–1305. [Google Scholar] [CrossRef] [Green Version]
- Wei, H.; Wang, F.; Wang, Y.; Li, T.; Xiu, P.; Zhong, J.; Sun, X.; Li, J. Verteporfin Suppresses Cell Survival, Angiogenesis and Vasculogenic Mimicry of Pancreatic Ductal Adenocarcinoma via Disrupting the YAP-TEAD Complex. Cancer Sci. 2017, 108, 478–487. [Google Scholar] [CrossRef]
- Vigneswaran, K.; Boyd, N.H.; Oh, S.-Y.; Lallani, S.; Boucher, A.; Neill, S.G.; Olson, J.J.; Read, R.D. YAP/TAZ Transcriptional Coactivators Create Therapeutic Vulnerability to Verteporfin in EGFR-Mutant Glioblastoma. Clin. Cancer Res. 2021, 27, 1553–1569. [Google Scholar] [CrossRef]
- Wei, C.; Li, X. Determination of the Migration Effect and Molecular Docking of Verteporfin in Different Subtypes of Breast Cancer Cells. Mol. Med. Rep. 2020, 22, 3955–3961. [Google Scholar] [CrossRef]
- Morice, S.; Mullard, M.; Brion, R.; Dupuy, M.; Renault, S.; Tesfaye, R.; Brounais-Le Royer, B.; Ory, B.; Redini, F.; Verrecchia, F. The YAP/TEAD Axis as a New Therapeutic Target in Osteosarcoma: Effect of Verteporfin and CA3 on Primary Tumor Growth. Cancers 2020, 12, 3847. [Google Scholar] [CrossRef]
- Dasari, V.R.; Mazack, V.; Feng, W.; Nash, J.; Carey, D.J.; Gogoi, R. Verteporfin Exhibits YAP-Independent Anti-Proliferative and Cytotoxic Effects in Endometrial Cancer Cells. Oncotarget 2017, 8, 28628–28640. [Google Scholar] [CrossRef] [Green Version]
- Eales, K.L.; Wilkinson, E.A.; Cruickshank, G.; Tucker, J.H.R.; Tennant, D.A. Verteporfin Selectively Kills Hypoxic Glioma Cells through Iron-Binding and Increased Production of Reactive Oxygen Species. Sci. Rep. 2018, 8, 1–12. [Google Scholar] [CrossRef]
- Lui, J.W.; Xiao, S.; Ogomori, K.; Hammarstedt, J.E.; Little, E.C.; Lang, D. The Efficiency of Verteporfin as a Therapeutic Option in Pre-Clinical Models of Melanoma. J. Cancer 2019, 10, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Zanconato, F.; Battilana, G.; Forcato, M.; Filippi, L.; Azzolin, L.; Manfrin, A.; Quaranta, E.; Di Biagio, D.; Sigismondo, G.; Guzzardo, V.; et al. Transcriptional Addiction in Cancer Cells Is Mediated by YAP/TAZ through BRD4. Nat. Med. 2018, 24, 1599–1610. [Google Scholar] [CrossRef]
- Zhang, Y.; Shen, H.; Withers, H.G.; Yang, N.; Denson, K.E.; Mussell, A.L.; Truskinovsky, A.; Fan, Q.; Gelman, I.H.; Frangou, C.; et al. VGLL4 Selectively Represses YAP-Dependent Gene Induction and Tumorigenic Phenotypes in Breast Cancer. Sci. Rep. 2017, 7, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Jiao, S.; Wang, H.; Shi, Z.; Dong, A.; Zhang, W.; Song, X.; He, F.; Wang, Y.; Zhang, Z.; Wang, W.; et al. A Peptide Mimicking VGLL4 Function Acts as a YAP Antagonist Therapy against Gastric Cancer. Cancer Cell 2014, 25, 166–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, S.; Xie, M.; Scott, A.W.; Jin, J.; Ma, L.; Dong, X.; Skinner, H.D.; Johnson, R.L.; Ding, S.; Ajani, J.A. A Novel YAP1 Inhibitor Targets CSC-Enriched Radiation-Resistant Cells and Exerts Strong Antitumor Activity in Esophageal Adenocarcinoma. Mol. Cancer Ther. 2018, 17, 443–454. [Google Scholar] [CrossRef] [Green Version]
- Qi, L.; Shi, C.; Li, J.; Xu, S.; Han, Y.; Li, J.; Zhang, L. Yes-Associated Protein Promotes Cell Migration via Activating Wiskott-Aldrich Syndrome Protein Family Member 1 in Oral Squamous Cell Carcinoma. J. Oral Pathol. Med. 2019, 48, 290–298. [Google Scholar] [CrossRef]
- Zhao, B.; Xie, J.; Zhou, X.; Zhang, L.; Cheng, X.; Liang, C. YAP Activation in Melanoma Contributes to Anoikis Resistance and Metastasis. Exp. Biol. Med. 2021, 246, 888–896. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Wang, X.; Fang, L.; Lan, C.; Zheng, X.; Wang, Y.; Zhang, Y.; Han, X.; Liu, S.; Cheng, K.; et al. A Combinatorial Strategy Using YAP and Pan-RAF Inhibitors for Treating KRAS-Mutant Pancreatic Cancer. Cancer Lett. 2017, 402, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.-Y.; Zhuang, L.-H.; Hu, Y.; Zhou, Y.-L.; Lin, W.-K.; Wang, D.-D.; Wan, Z.-Q.; Chang, L.-L.; Chen, Y.; Ying, M.-D.; et al. Inactivation of Hypoxia-Induced YAP by Statins Overcomes Hypoxic Resistance Tosorafenib in Hepatocellular Carcinoma Cells. Sci. Rep. 2016, 6, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, T.-F.; Tseng, Y.-C.; Nguyen, P.A.; Li, Y.-C.; Ho, C.-C.; Wu, C.-W. Enhanced YAP Expression Leads to EGFR TKI Resistance in Lung Adenocarcinomas. Sci. Rep. 2018, 8, 1–8. [Google Scholar] [CrossRef]
- Sun, J.; Wang, X.; Tang, B.; Liu, H.; Zhang, M.; Wang, Y.; Ping, F.; Ding, J.; Shen, A.; Geng, M. A Tightly Controlled Src-YAP Signaling Axis Determines Therapeutic Response to Dasatinib in Renal Cell Carcinoma. Theranostics 2018, 8, 3256–3267. [Google Scholar] [CrossRef] [Green Version]
- Zhou, L.; Wang, Q.; Zhang, H.; Li, Y.; Xie, S.; Xu, M. YAP Inhibition by Nuciferine via AMPK-Mediated Downregulation of HMGCR Sensitizes Pancreatic Cancer Cells to Gemcitabine. Biomolecules 2019, 9, 620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiorica, F.; Colella, M.; Taibi, R.; Bonetti, A.; Giuliani, J.; Perrone, M.S.; Missiroli, S.; Giorgi, C. Glioblastoma: Prognostic Factors and Predictive Response to Radio and Chemotherapy. Curr. Med. Chem. 2020, 27, 2814–2825. [Google Scholar] [CrossRef]
- Akervall, J.; Nandalur, S.; Zhang, J.; Qian, C.-N.; Goldstein, N.; Gyllerup, P.; Gardinger, Y.; Alm, J.; Lorenc, K.; Nilsson, K.; et al. A Novel Panel of Biomarkers Predicts Radioresistance in Patients with Squamous Cell Carcinoma of the Head and Neck. Eur. J. Cancer 2014, 50, 570–581. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-L, A.; Squatrito, M.; Northcott, P.; Awan, A.; Holland, E.C.; Taylor, M.D.; Nahlé, Z.; Kenney, A.M. Oncogenic YAP Promotes Radioresistance and Genomic Instability in Medulloblastoma through IGF2-Mediated Akt Activation. Oncogene 2012, 31, 1923–1937. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.; Chen, Y.; Wang, X.; Mu, X. Role of Hippo/YAP Signaling in Irradiation-Induced Glioma Cell Apoptosis. Cancer Manag. Res. 2019, 11, 7577–7585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrade, D.; Mehta, M.; Griffith, J.; Panneerselvam, J.; Srivastava, A.; Kim, T.-D.; Janknecht, R.; Herman, T.; Ramesh, R.; Munshi, A. YAP1 Inhibition Radiosensitizes Triple Negative Breast Cancer Cells by Targeting the DNA Damage Response and Cell Survival Pathways. Oncotarget 2017, 8, 98495–98508. [Google Scholar] [CrossRef] [Green Version]
- Gopal, U.; Mowery, Y.; Young, K.; Pizzo, S.V. Targeting Cell Surface GRP78 Enhances Pancreatic Cancer Radiosensitivity through YAP/TAZ Protein Signaling. J. Biol. Chem. 2019, 294, 13939–13952. [Google Scholar] [CrossRef]
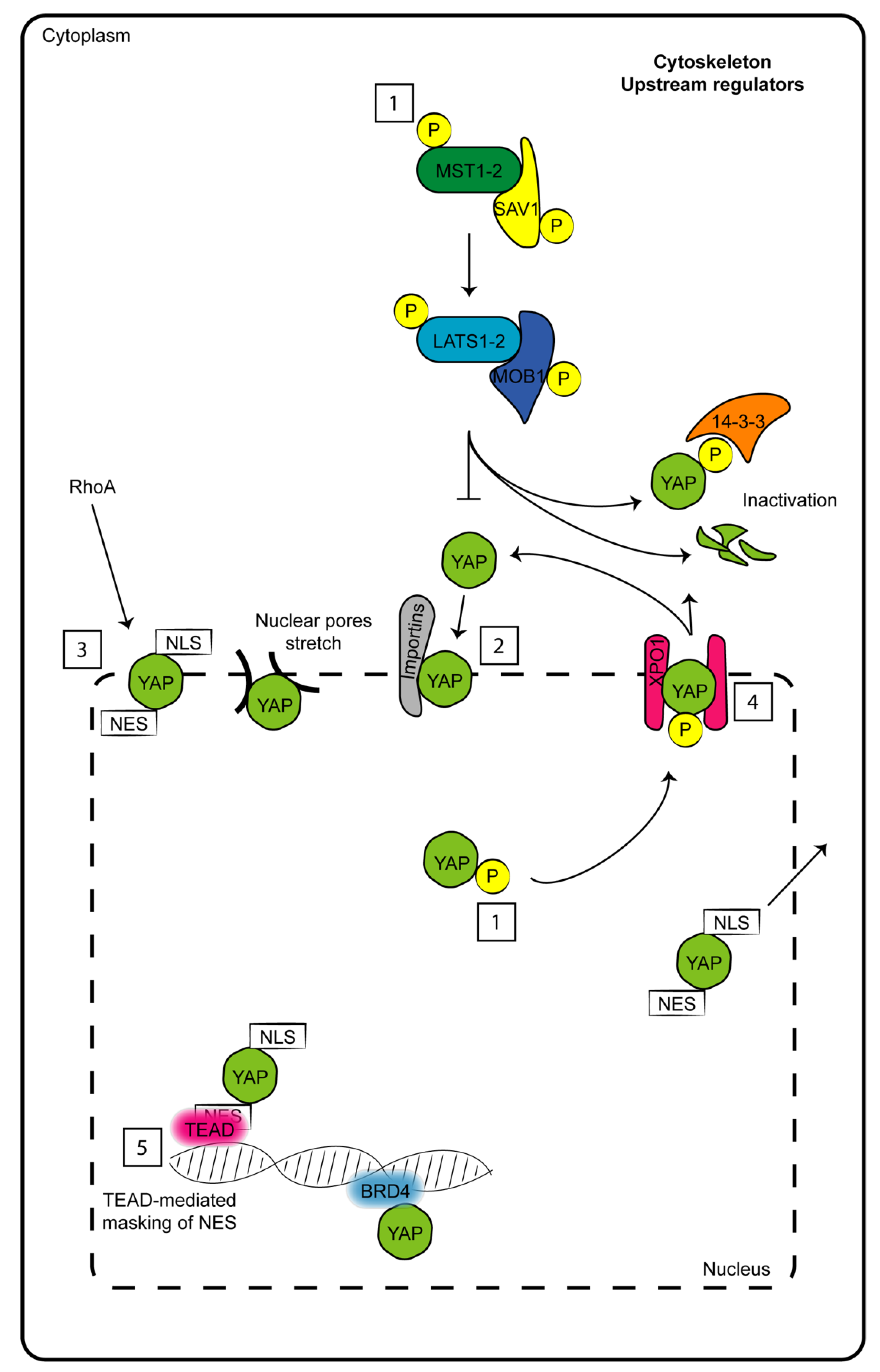



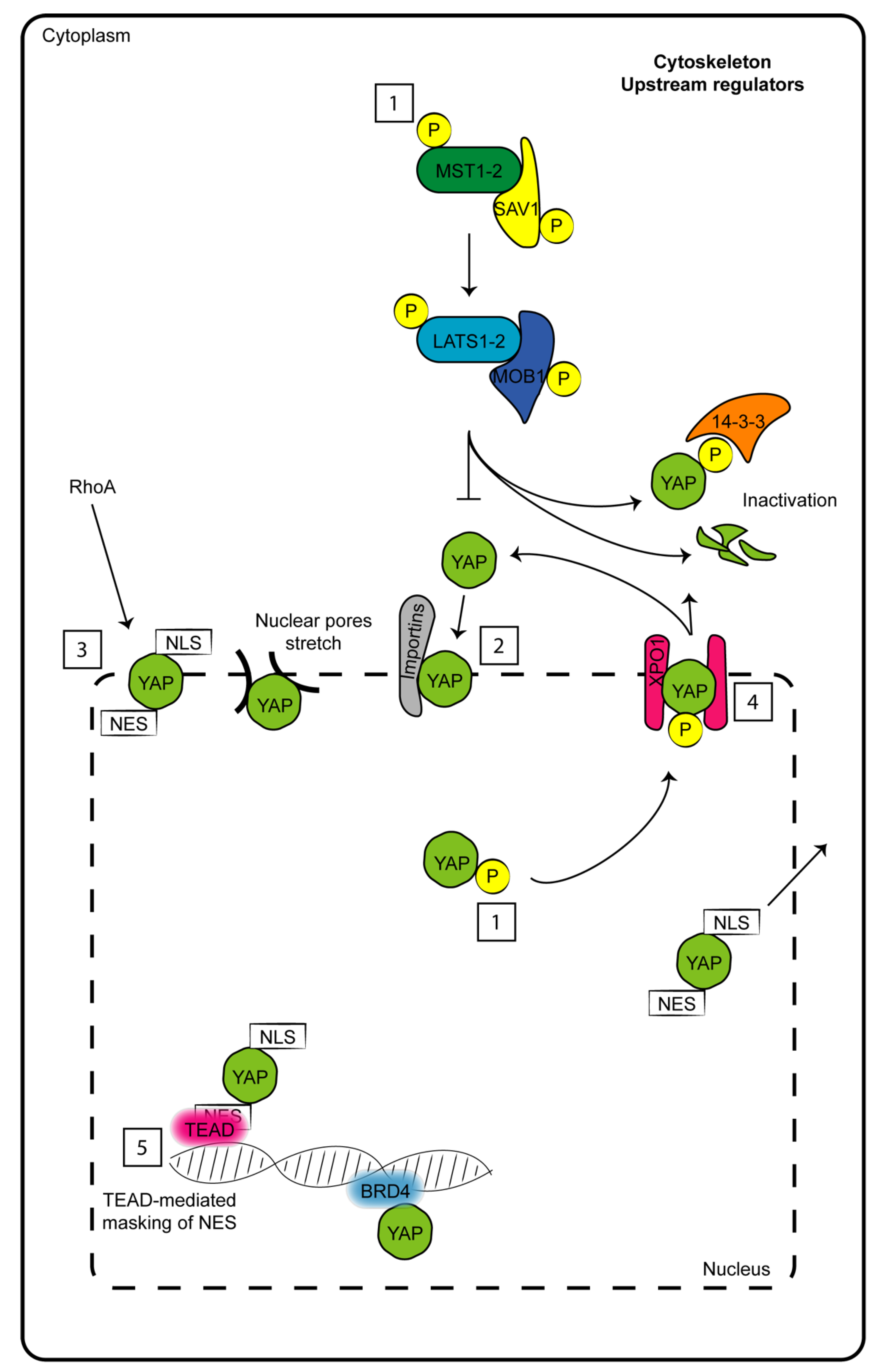{kind=link}
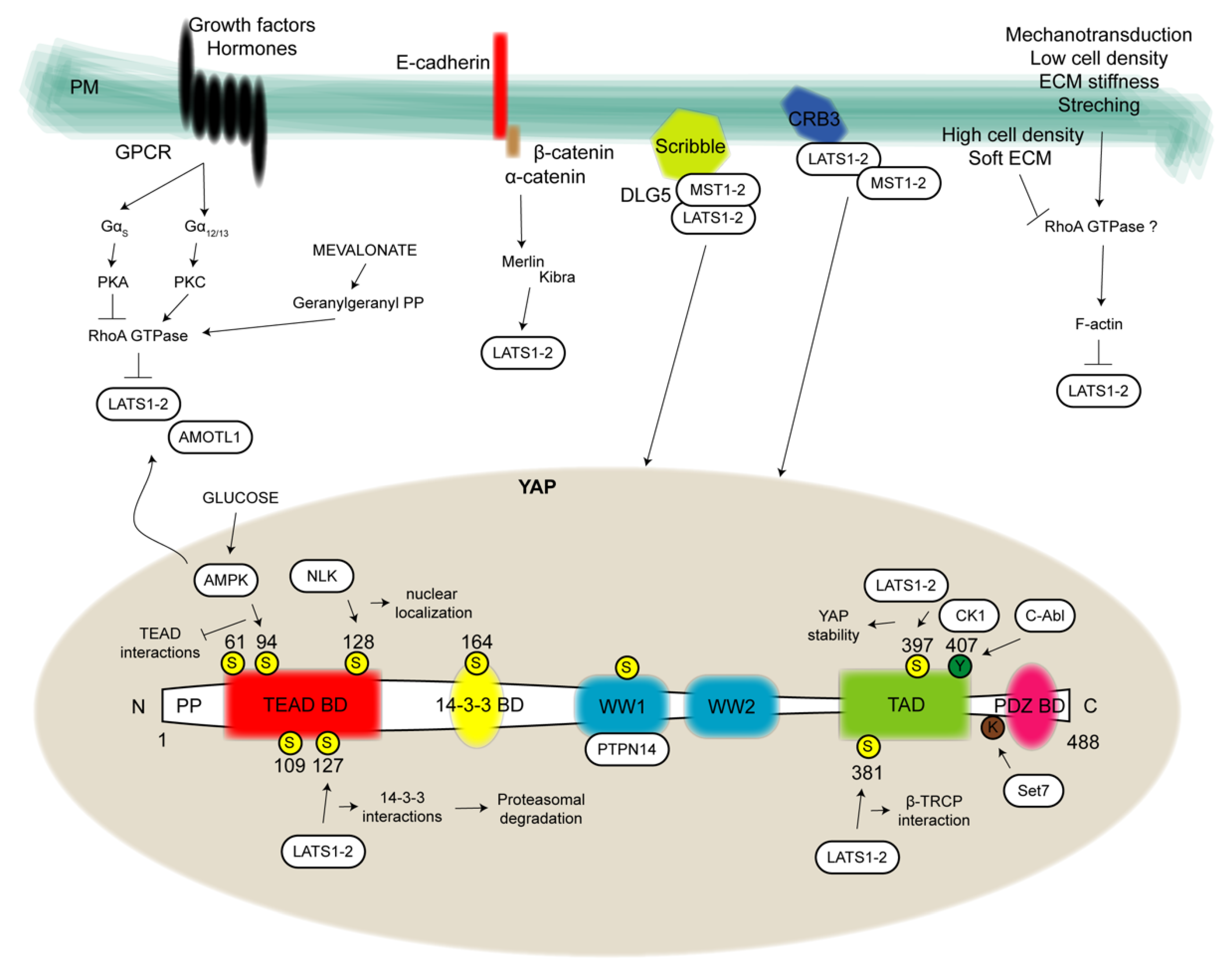{kind=link}
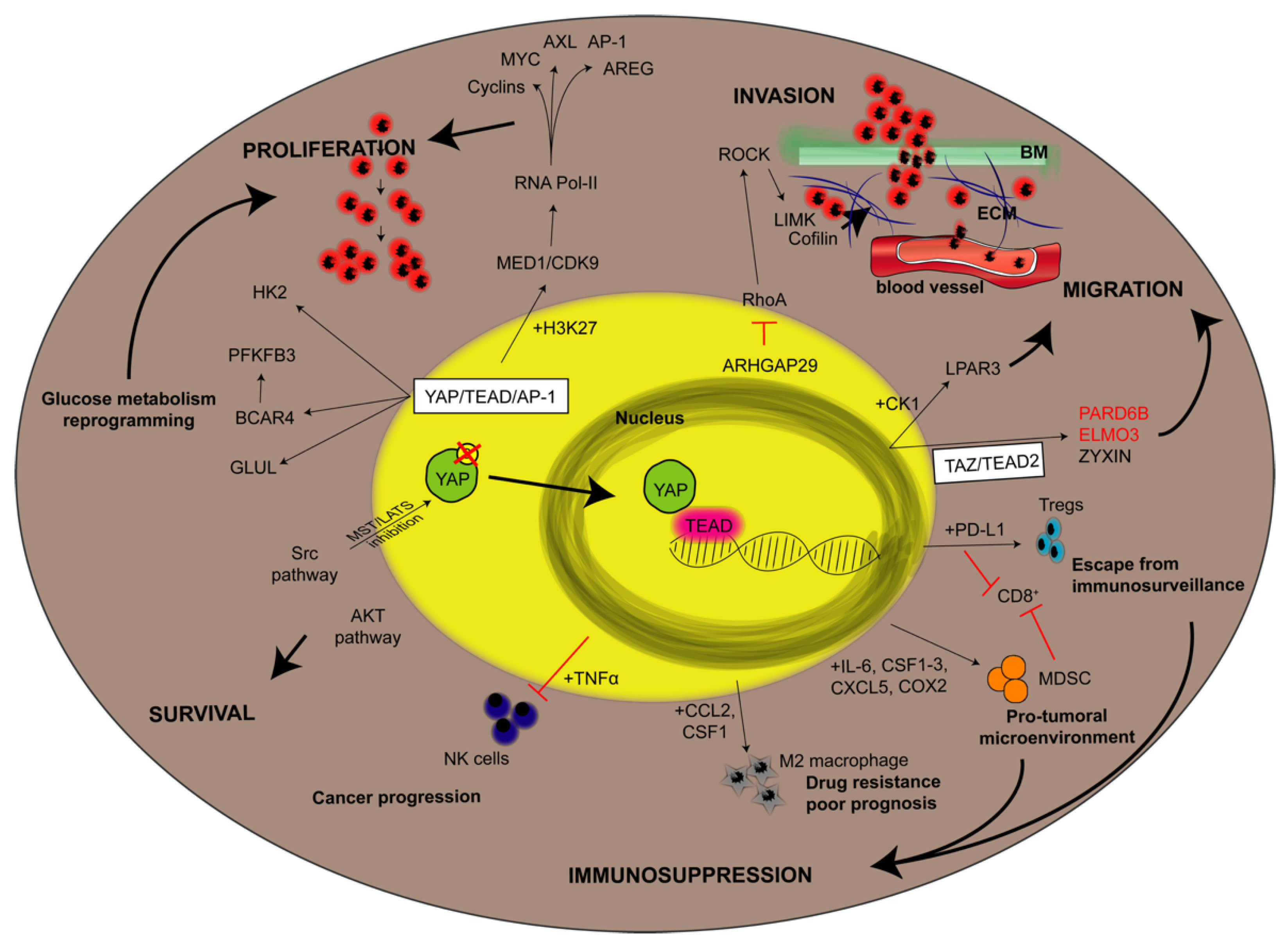{kind=link}
| Drug | Mode of Action | Preclinical (P)/Clinical (C) Use |
|---|---|---|
| A35 | Lower YAP nuclear accumulation | P |
| DCA | Lower YAP nuclear accumulation | P/C |
| NCTD | Lower YAP nuclear accumulation | P |
| Pazopanib | Lower YAP nuclear accumulation | P/C |
| Dasatinib | Lower YAP nuclear accumulation | P/C |
| Blocking Src kinase signaling | ||
| Statins | Lower YAP nuclear accumulation | P/C |
| VP | Competitive binding to YAP | P/C |
| BET inhibitors (JQ1) | Competitive binding to YAP | P/C |
| VGLL4 | Competitive binding to YAP | P |
| Super TD4 | Competitive binding to YAP | P |
| CA3 | Competitive binding to YAP | P |
| VP+LY3009120 | Blocking AKT signaling | P/C |
| Nuciferine | Blocking AMPK signaling | P |
| C38+RT | YAP downregulation | P/C |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morciano, G.; Vezzani, B.; Missiroli, S.; Boncompagni, C.; Pinton, P.; Giorgi, C. An Updated Understanding of the Role of YAP in Driving Oncogenic Responses. Cancers 2021, 13, 3100. https://doi.org/10.3390/cancers13123100
Morciano G, Vezzani B, Missiroli S, Boncompagni C, Pinton P, Giorgi C. An Updated Understanding of the Role of YAP in Driving Oncogenic Responses. Cancers. 2021; 13(12):3100. https://doi.org/10.3390/cancers13123100
Chicago/Turabian StyleMorciano, Giampaolo, Bianca Vezzani, Sonia Missiroli, Caterina Boncompagni, Paolo Pinton, and Carlotta Giorgi. 2021. "An Updated Understanding of the Role of YAP in Driving Oncogenic Responses" Cancers 13, no. 12: 3100. https://doi.org/10.3390/cancers13123100
APA StyleMorciano, G., Vezzani, B., Missiroli, S., Boncompagni, C., Pinton, P., & Giorgi, C. (2021). An Updated Understanding of the Role of YAP in Driving Oncogenic Responses. Cancers, 13(12), 3100. https://doi.org/10.3390/cancers13123100