
Blockade of HVEM for Prostate Cancer Immunotherapy in Humanized Mice


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Results
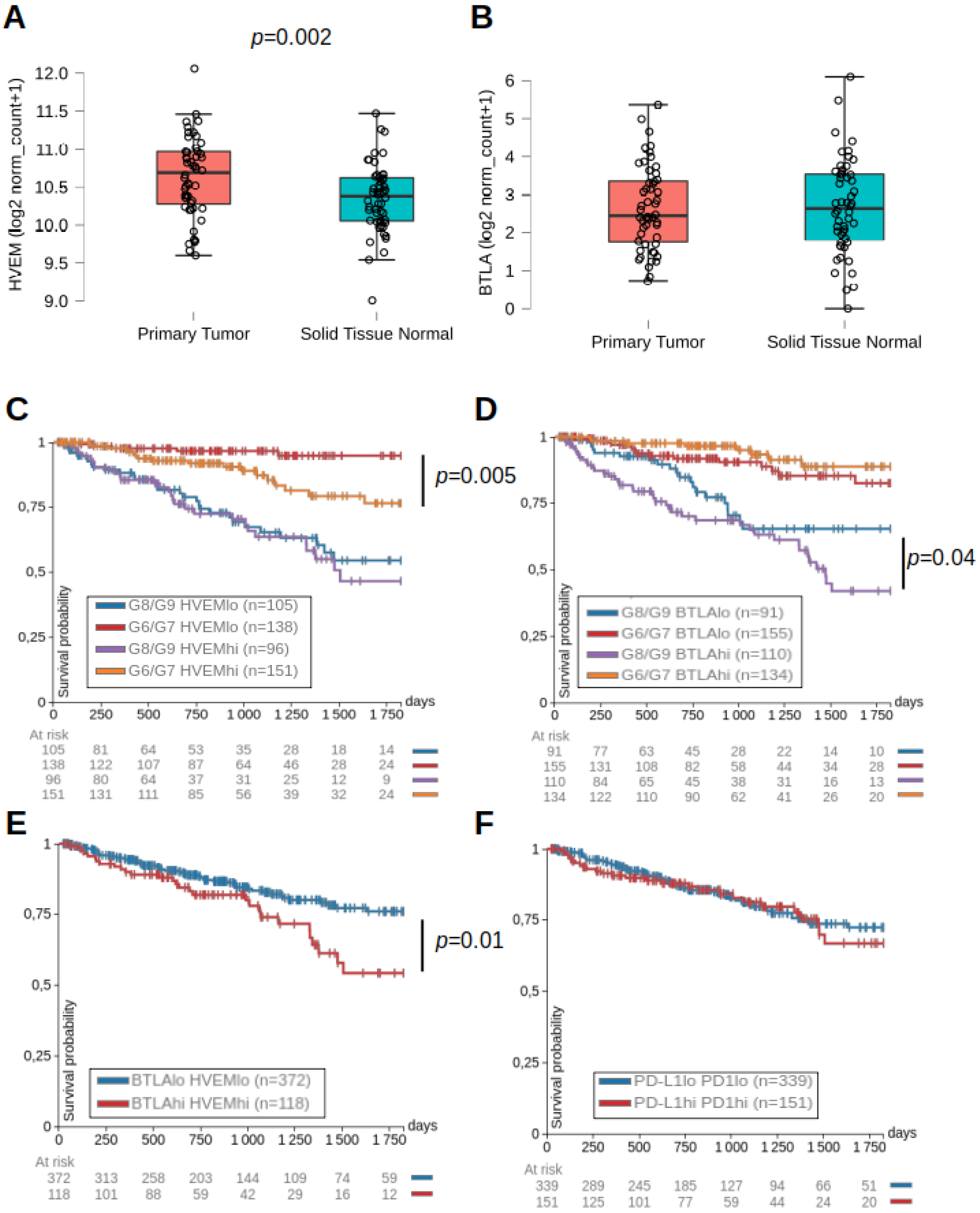
2.1. HVEM and BTLA Were Associated with Lower Progression-Free Intervals in Prostate Cancer Patients
2.2. Targeting HVEM with a mAb Improved Tumor Control in Humanized Mice
2.3. Tumor Control Was Dependent on CD8+ T Cells in Anti-HVEM-Treated Mice
2.4. Treatment with the Anti-HVEM mAb Did Not Increase Graft vs. Host Disease Nor the Number or Proliferation of Human T Cells
2.5. mRNA Enrichment Analysis Showed Increased Activation and Decreased Immuno-Suppression in TILs of Anti-HVEM-Treated Mice
2.6. HVEM Was an Immune Checkpoint during Anti-Tumor T Cell Immune Response in Humanized Mice
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Preparation of Human Peripheral Mononuclear Cells
5.2. Humanized Mice Tumor Model
5.3. Antibodies
5.4. Cell Lines
5.5. Generation of HVEM-Deficient PC3 Clone Using CRISPR-Cas9 RNP Transfection
5.6. Phenotypic Analysis by Flow Cytometry
5.7. NanoString nCounter Expression Assay
5.8. Bioinformatics Analysis
5.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| PBMC | peripheral blood mononuclear cells |
| HVEM | herpes virus entry mediator |
| BTLA | B and T lymphocyte attenuator |
| TILs | tumor-infiltrating leukocytes |
| NSG | NOD.SCID.γc-null |
| ICI | immune checkpoint inhibitors |
| RNP | ribonucleoproteins |
| DEG | differentially expressed genes |
| GVHD | graft vs. host disease |
| IPA | ingenuity pathway analysis |
| GSEA | gene set enrichment analysis |
| FDR | false discovery rate |
| FWER | family-wise error rate |
| PDX | patient-derived xenografts |
| PCa | prostate cancer |
| TCGA | The Cancer Genome Atlas |
| PRAD | prostate adenocarcinomas |
| PFI | progression-free interval |
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Mittal, D.; Gubin, M.M.; Schreiber, R.D.; Smyth, M.J. New insights into cancer immunoediting and its three component phases-elimination, equilibrium and escape. Curr. Opin. Immunol. 2014, 27, 16–25. [Google Scholar] [CrossRef]
- Hargadon, K.M.; Johnson, C.E.; Williams, C.J. Immune checkpoint blockade therapy for cancer: An overview of FDA-approved immune checkpoint inhibitors. Int. Immunopharmacol. 2018, 62, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.J.; Koo, K.C. Current status and future perspectives of checkpoint inhibitor immunotherapy for prostate cancer: A comprehensive review. Int. J. Mol. Sci. 2020, 21, 5484. [Google Scholar] [CrossRef] [PubMed]
- Havel, J.J.; Chowell, D.; Chan, T.A. The evolving landscape of biomarkers for checkpoint inhibitor immunotherapy. Nat. Rev. Cancer 2019, 19, 133. [Google Scholar] [CrossRef]
- Park, Y.-J.; Kuen, D.-S.; Chung, Y. Future prospects of immune checkpoint blockade in cancer: From response prediction to overcoming resistance. Exp. Mol. Med. 2018, 50, 109. [Google Scholar] [CrossRef]
- Pasero, C.; Speiser, D.E.; Derré, L.; Olive, D. The HVEM network: New directions in targeting novel costimulatory/co-inhibitory molecules for cancer therapy. Curr. Opin. Pharmacol. 2012, 12, 478–485. [Google Scholar] [CrossRef]
- Ning, Z.; Liu, K.; Xiong, H. Roles of BTLA in Immunity and Immune Disorders. Front. Immunol. 2021, 12, 990. [Google Scholar] [CrossRef]
- Watanabe, N.; Gavrieli, M.; Sedy, J.R.; Yang, J.; Fallarino, F.; Loftin, S.K.; Hurchla, M.A.; Zimmerman, N.; Sim, J.; Zang, X.; et al. BTLA is a lymphocyte inhibitory receptor with similarities to CTLA-4 and PD-1. Nat. Immunol. 2003, 4, 670–679. [Google Scholar] [CrossRef] [PubMed]
- Cai, G.; Anumanthan, A.; Brown, J.A.; Greenfield, E.A.; Zhu, B.; Freeman, G.J. CD160 inhibits activation of human CD4+ T cells through interaction with herpesvirus entry mediator. Nat. Immunol. 2008, 9, 176–185. [Google Scholar] [CrossRef]
- Tan, C.L.; Peluso, M.J.; Drijvers, J.M.; Mera, C.M.; Grande, S.M.; Brown, K.E.; Godec, J.; Freeman, G.J.; Sharpe, A.H. CD160 Stimulates CD8+ T Cell Responses and Is Required for Optimal Protective Immunity to Listeria monocytogenes. ImmunoHorizons 2018, 2, 238–250. [Google Scholar] [CrossRef]
- Šedý, J.R.; Bjordahl, R.L.; Bekiaris, V.; Macauley, M.G.; Ware, B.C.; Norris, P.S.; Lurain, N.S.; Benedict, C.A.; Ware, C.F. CD160 Activation by Herpesvirus Entry Mediator Augments Inflammatory Cytokine Production and Cytolytic Function by NK Cells. J. Immunol. 2013, 191, 828–836. [Google Scholar] [CrossRef] [PubMed]
- Shaikh, R.B.; Santee, S.; Granger, S.W.; Butrovich, K.; Cheung, T.; Kronenberg, M.; Cheroutre, H.; Ware, C.F. Constitutive Expression of LIGHT on T Cells Leads to Lymphocyte Activation, Inflammation, and Tissue Destruction. J. Immunol. 2001, 167, 6330–6337. [Google Scholar] [CrossRef]
- Harrop, J.A.; McDonnell, P.C.; Brigham-Burke, M.; Lyn, S.D.; Minton, J.; Tan, K.B.; Dede, K.; Spampanato, J.; Silverman, C.; Hensley, P.; et al. Herpesvirus entry mediator ligand (HVEM-L), a novel ligand for HVEM/TR2, stimulates proliferation of T cells and inhibits HT29 cell growth. J. Biol. Chem. 1998, 273, 27548–27556. [Google Scholar] [CrossRef]
- Cheung, T.C.; Steinberg, M.W.; Oborne, L.M.; Macauley, M.G.; Fukuyama, S.; Sanjo, H.; D’Souza, C.; Norris, P.S.; Pfeffer, K.; Murphy, K.M.; et al. Unconventional ligand activation of herpesvirus entry mediator signals cell survival. Proc. Natl. Acad. Sci. USA 2009, 106, 6244–6249. [Google Scholar] [CrossRef]
- Inoue, T.; Sho, M.; Yasuda, S.; Nishiwada, S.; Nakamura, S.; Ueda, T.; Nishigori, N.; Kawasaki, K.; Obara, S.; Nakamoto, T.; et al. HVEM Expression Contributes to Tumor Progression and Prognosis in Human Colorectal Cancer. Anticancer Res. 2015, 35, 1361–1367. [Google Scholar] [PubMed]
- Malissen, N.; Macagno, N.; Granjeaud, S.; Granier, C.; Moutardier, V.; Gaudy-Marqueste, C.; Habel, N.; Mandavit, M.; Guillot, B.; Pasero, C.; et al. HVEM has a broader expression than PD-L1 and constitutes a negative prognostic marker and potential treatment target for melanoma. Oncoimmunology 2019, 8, e1665976. [Google Scholar] [CrossRef]
- Migita, K.; Sho, M.; Shimada, K.; Yasuda, S.; Yamato, I.; Takayama, T.; Matsumoto, S.; Wakatsuki, K.; Hotta, K.; Tanaka, T.; et al. Significant involvement of herpesvirus entry mediator in human esophageal squamous cell carcinoma. Cancer 2014, 120, 808–817. [Google Scholar] [CrossRef]
- Lan, X.; Li, S.; Gao, H.; Nanding, A.; Quan, L.; Yang, C.; Ding, S.; Xue, Y. Increased BTLA and HVEM in gastric cancer are associated with progression and poor prognosis. OncoTargets Ther. 2017, 10, 919–926. [Google Scholar] [CrossRef] [PubMed]
- Hokuto, D.; Sho, M.; Yamato, I.; Yasuda, S.; Obara, S.; Nomi, T.; Nakajima, Y. Clinical impact of herpesvirus entry mediator expression in human hepatocellular carcinoma. Eur. J. Cancer 2015, 51, 157–165. [Google Scholar] [CrossRef]
- Tsang, J.Y.S.; Chan, K.-W.; Ni, Y.-B.; Hlaing, T.; Hu, J.; Chan, S.-K.; Cheung, S.-Y.; Tse, G.M. Expression and Clinical Significance of Herpes Virus Entry Mediator (HVEM) in Breast Cancer. Ann. Surg. Oncol. 2017, 24, 4042–4050. [Google Scholar] [CrossRef] [PubMed]
- M’Hidi, H.; Thibult, M.L.; Chetaille, B.; Rey, F.; Bouadallah, R.; Nicollas, R.; Olive, D.; Xerri, L. High expression of the inhibitory receptor BTLA in T-follicular helper cells and in B-cell small lymphocytic lymphoma/chronic lymphocytic leukemia. Am. J. Clin. Pathol. 2009, 132, 589–596. [Google Scholar] [CrossRef]
- Wang, Q.; Ye, Y.; Yu, H.; Lin, S.-H.; Tu, H.; Liang, D.; Chang, D.W.; Huang, M.; Wu, X. Immune checkpoint-related serum proteins and genetic variants predict outcomes of localized prostate cancer, a cohort study. Cancer Immunol. Immunother. 2020, 70, 701–712. [Google Scholar] [CrossRef] [PubMed]
- Park, J.-J.; Anand, S.; Zhao, Y.; Matsumura, Y.; Sakoda, Y.; Kuramasu, A.; Strome, S.E.; Chen, L.; Tamada, K. Expression of anti-HVEM single-chain antibody on tumor cells induces tumor-specific immunity with long-term memory. Cancer Immunol. Immunother. 2012, 61, 203–214. [Google Scholar] [CrossRef] [PubMed]
- Han, L.; Wang, W.; Fang, Y.; Feng, Z.; Liao, S.; Li, W.; Li, Y.; Li, C.; Maitituoheti, M.; Dong, H.; et al. Soluble B and T Lymphocyte Attenuator Possesses Antitumor Effects and Facilitates Heat Shock Protein 70 Vaccine-Triggered Antitumor Immunity against a Murine TC-1 Cervical Cancer Model In Vivo. J. Immunol. 2009, 183, 7842–7850. [Google Scholar] [CrossRef] [PubMed]
- Oguro, S.; Ino, Y.; Shimada, K.; Hatanaka, Y.; Matsuno, Y.; Esaki, M.; Nara, S.; Kishi, Y.; Kosuge, T.; Hiraoka, N. Clinical significance of tumor-infiltrating immune cells focusing on BTLA and Cbl-b in patients with gallbladder cancer. Cancer Sci. 2015, 106, 1750–1760. [Google Scholar] [CrossRef]
- Quan, L.; Lan, X.; Meng, Y.; Guo, X.; Guo, Y.; Zhao, L.; Chen, X.; Liu, A. BTLA marks a less cytotoxic T cell subset in diffuse large B-cell lymphoma with high expression of checkpoints. Exp. Hematol. 2018, 60, 47–56. [Google Scholar] [CrossRef]
- Derré, L.; Rivals, J.P.; Jandus, C.; Pastor, S.; Rimoldi, D.; Romero, P.; Michielin, O.; Olive, D.; Speiser, D.E. BTLA mediates inhibition of human tumor-specific CD8+ T cells that can be partially reversed by vaccination. J. Clin. Investig. 2010, 120, 157–167. [Google Scholar] [CrossRef]
- Shultz, L.D.; Schweitzer, P.A.; Christianson, S.W.; Gott, B.; Schweitzer, I.B.; Tennent, B.; McKenna, S.; Mobraaten, L.; Rajan, T.V.; Greiner, D.L. Multiple defects in innate and adaptive immunologic function in NOD/LtSz-scid mice. J. Immunol. 1995, 154, 180–191. [Google Scholar]
- DeRose, Y.S.; Wang, G.; Lin, Y.-C.; Bernard, P.S.; Buys, S.S.; Ebbert, M.T.W.; Factor, R.; Matsen, C.; Milash, B.A.; Nelson, E.; et al. Tumor grafts derived from women with breast cancer authentically reflect tumor pathology, growth, metastasis and disease outcomes. Nat. Med. 2011, 17, 1514–1520. [Google Scholar] [CrossRef]
- Bankert, R.B.; Balu-Iyer, S.V.; Odunsi, K.; Shultz, L.D.; Kelleher, R.J.; Barnas, J.L.; Simpson-Abelson, M.; Parsons, R.; Yokota, S.J. Humanized mouse model of ovarian cancer recapitulates patient solid tumor progression, ascites formation, and metastasis. PLoS ONE 2011, 6, e24420. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Shen, C.; Wei, Z.; Zhang, R.; Wang, Y.; Jiang, L.; Chen, K.; Qiu, S.; Zhang, Y.; Zhang, T.; et al. Patient-derived non-small cell lung cancer xenograft mirrors complex tumor heterogeneity. Cancer Biol. Med. 2021, 18, 184–198. [Google Scholar] [CrossRef]
- Quintana, E.; Piskounova, E.; Shackleton, M.; Weinberg, D.; Eskiocak, U.; Fullen, D.R.; Johnson, T.M.; Morrison, S.J. Human melanoma metastasis in NSG mice correlates with clinical outcome in patients. Sci. Transl. Med. 2012, 4, 159ra149. [Google Scholar] [CrossRef] [PubMed]
- Rea, D.; Del Vecchio, V.; Palma, G.; Barbieri, A.; Falco, M.; Luciano, A.; De Biase, D.; Perdonà, S.; Facchini, G.; Arra, C. Mouse Models in Prostate Cancer Translational Research: From Xenograft to PDX. BioMed Res. Int. 2016, 2016. [Google Scholar] [CrossRef]
- De La Rochere, P.; Guil-Luna, S.; Decaudin, D.; Azar, G.; Sidhu, S.S.; Piaggio, E. Humanized Mice for the Study of Immuno-Oncology. Trends Immunol. 2018, 39, 748–763. [Google Scholar] [CrossRef]
- Wang, Y.; Singh, N.K.; Spear, T.T.; Hellman, L.M.; Piepenbrink, K.H.; McMahan, R.H.; Rosen, H.R.; Vander Kooi, C.W.; Nishimura, M.I.; Baker, B.M. How an alloreactive T cell receptor achieves peptide and MHC specificity. Proc. Natl. Acad. Sci. USA 2017, 114, E4792–E4801. [Google Scholar] [CrossRef] [PubMed]
- Felix, N.J.; Allen, P.M. Specificity of T cell alloreactivity. Nat. Rev. Immunol. 2007, 7, 942–953. [Google Scholar] [CrossRef]
- Kearney, C.J.; Vervoort, S.J.; Hogg, S.J.; Ramsbottom, K.M.; Freeman, A.J.; Lalaoui, N.; Pijpers, L.; Michie, J.; Brown, K.K.; Knight, D.A.; et al. Tumor immune evasion arises through loss of TNF sensitivity. Sci. Immunol. 2018, 3, eaar3451. [Google Scholar] [CrossRef]
- Simoni, Y.; Becht, E.; Fehlings, M.; Loh, C.Y.; Koo, S.; Teng, K.W.W.; Yeong, J.P.S.; Nahar, R.; Zhang, T.; Kared, H.; et al. Bystander CD8+ T cells are abundant and phenotypically distinct in human tumour infiltrates. Nature 2018, 557, 575–579. [Google Scholar] [CrossRef]
- Roth, M.D.; Harui, A. Human tumor infiltrating lymphocytes cooperatively regulate prostate tumor growth in a humanized mouse model. J. Immunother. Cancer 2015, 3, 12. [Google Scholar] [CrossRef]
- Jespersen, H.; Lindberg, M.F.; Donia, M.; Söderberg, E.M.V.; Andersen, R.; Keller, U.; Ny, L.; Svane, I.M.; Nilsson, L.M.; Nilsson, J.A. Clinical responses to adoptive T cell transfer can be modeled in an autologous immune-humanized mouse model. Nat. Commun. 2017, 8, 1–10. [Google Scholar] [CrossRef]
- Ali, N.; Flutter, B.; Rodriguez, R.S.; Sharif-Paghaleh, E.; Barber, L.D.; Lombardi, G.; Nestle, F.O. Xenogeneic Graft-versus-Host-Disease in NOD-scid IL- 2Rcnull Mice Display a T-Effector Memory Phenotype. PLoS ONE 2012, 7, e44219. [Google Scholar] [CrossRef]
- Morel, Y.; Schiano de Colella, J.-M.; Harrop, J.; Deen, K.C.; Holmes, S.D.; Wattam, T.A.; Khandekar, S.S.; Truneh, A.; Sweet, R.W.; Gastaut, J.-A.; et al. Reciprocal Expression of the TNF Family Receptor Herpes Virus Entry Mediator and Its Ligand LIGHT on Activated T Cells: LIGHT Down-Regulates Its Own Receptor. J. Immunol. 2000, 165, 4397–4404. [Google Scholar] [CrossRef] [PubMed]
- Murphy, K.M.; Nelson, C.A.; Šedý, J.R. Balancing co-stimulation and inhibition with BTLA and HVEM. Nat. Rev. Immunol. 2006, 6, 671–681. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, Y.; Hokuto, D.; Inoue, T.; Nomi, T.; Yoshikawa, T.; Matsuo, Y.; Koyama, F.; Sho, M. Significance of Herpesvirus Entry Mediator Expression in Human Colorectal Liver Metastasis. Ann. Surg. Oncol. 2019, 26, 3982–3989. [Google Scholar] [CrossRef]
- Compaan, D.M.; Gonzalez, L.C.; Tom, I.; Loyet, K.M.; Eaton, D.; Hymowitz, S.G. Attenuating lymphocyte activity: The crystal structure of the BTLA-HVEM complex. J. Biol. Chem. 2005, 280, 39553–39561. [Google Scholar] [CrossRef]
- Steinberg, M.W.; Cheung, T.C.; Ware, C.F. The signaling networks of the herpesvirus entry mediator (TNFRSF14) in immune regulation. Immunol. Rev. 2011, 244, 169–187. [Google Scholar] [CrossRef]
- Lasaro, M.O.; Sazanovich, M.; Giles-Davis, W.; Mrass, P.; Bunte, R.M.; Sewell, D.A.; Hussain, S.F.; Fu, Y.-X.; Weninger, W.; Paterson, Y.; et al. Active Immunotherapy Combined With Blockade of a Coinhibitory Pathway Achieves Regression of Large Tumor Masses in Cancer-prone Mice. Mol. Ther. 2011, 19, 1727–1736. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Ye, L.; Han, L.; He, Q.; Zhu, J. Knockdown of HVEM, a Lymphocyte Regulator Gene, in Ovarian Cancer Cells Increases Sensitivity to Activated T Cells. Oncol. Res. 2016, 24, 189–196. [Google Scholar] [CrossRef]
- Anderson, A.C.; Joller, N.; Kuchroo, V.K. Lag-3, Tim-3, and TIGIT: Co-inhibitory Receptors with Specialized Functions in Immune Regulation. Immunity 2016, 44, 989–1004. [Google Scholar] [CrossRef]
- De Sousa Linhares, A.; Leitner, J.; Grabmeier-Pfistershammer, K.; Steinberger, P. Not All Immune Checkpoints Are Created Equal. Front. Immunol. 2018, 9, 1909. [Google Scholar] [CrossRef]
- Wang, M.; Yao, L.C.; Cheng, M.; Cai, D.; Martinek, J.; Pan, C.X.; Shi, W.; Ma, A.H.; De Vere White, R.W.; Airhart, S.; et al. Humanized mice in studying efficacy and mechanisms of PD-1-targeted cancer immunotherapy. FASEB J. 2018, 32, 1537–1549. [Google Scholar] [CrossRef]
- Petit, N.Y.; Baillou, C.; Burlion, A.; Dorgham, K.; Levacher, B.; Amiel, C.; Schneider, V.; Lemoine, F.M.; Gorochov, G.; Marodon, G. Gene transfer of two entry inhibitors protects CD4+ T cell from HIV-1 infection in humanized mice. Gene Ther. 2016, 23, 144–150. [Google Scholar] [CrossRef] [PubMed]
- Petit, N.Y.; Lambert-Niclot, S.; Marcelin, A.-G.; Garcia, S.; Marodon, G. HIV Replication Is Not Controlled by CD8+ T Cells during the Acute Phase of the Infection in Humanized Mice. PLoS ONE 2015, 10, e0138420. [Google Scholar] [CrossRef]
- Schmitz, J.E.; Simon, M.A.; Kuroda, M.J.; Lifton, M.A.; Ollert, M.W.; Vogel, C.W.; Racz, P.; Tenner-Racz, K.; Scallon, B.J.; Dalesandro, M.; et al. A nonhuman primate model for the selective elimination of CD8+ lymphocytes using a mouse-human chimeric monoclonal antibody. Am. J. Pathol. 1999, 154, 1923–1932. [Google Scholar] [CrossRef]
- Gertner-Dardenne, J.; Fauriat, C.; Orlanducci, F.; Thibult, M.L.; Pastor, S.; Fitzgibbon, J.; Bouabdallah, R.; Xerri, L.; Olive, D. The co-receptor BTLA negatively regulates human Vg9Vd2 T cell proliferation: A potential way of immune escape for lymphoma cells. Blood 2013, 122, 922–931. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Bailey, A.; Kuleshov, M.V.; Clarke, D.J.B.; Evangelista, J.E.; Jenkins, S.L.; Lachmann, A.; Wojciechowicz, M.L.; Kropiwnicki, E.; Jagodnik, K.M.; et al. Gene Set Knowledge Discovery with Enrichr. Curr. Protoc. 2021, 1, e90. [Google Scholar] [CrossRef]
- Subramanian, A.; Kuehn, H.; Gould, J.; Tamayo, P.; Mesirov, J.P. GSEA-P: A desktop application for gene set enrichment analysis. Bioinformatics 2007, 23, 3251–3253. [Google Scholar] [CrossRef]
- Goldman, M.J.; Craft, B.; Hastie, M.; Repečka, K.; McDade, F.; Kamath, A.; Banerjee, A.; Luo, Y.; Rogers, D.; Brooks, A.N.; et al. Visualizing and interpreting cancer genomics data via the Xena platform. Nat. Biotechnol. 2020, 38, 675–678. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aubert, N.; Brunel, S.; Olive, D.; Marodon, G. Blockade of HVEM for Prostate Cancer Immunotherapy in Humanized Mice. Cancers 2021, 13, 3009. https://doi.org/10.3390/cancers13123009
Aubert N, Brunel S, Olive D, Marodon G. Blockade of HVEM for Prostate Cancer Immunotherapy in Humanized Mice. Cancers. 2021; 13(12):3009. https://doi.org/10.3390/cancers13123009
Chicago/Turabian StyleAubert, Nicolas, Simon Brunel, Daniel Olive, and Gilles Marodon. 2021. "Blockade of HVEM for Prostate Cancer Immunotherapy in Humanized Mice" Cancers 13, no. 12: 3009. https://doi.org/10.3390/cancers13123009
APA StyleAubert, N., Brunel, S., Olive, D., & Marodon, G. (2021). Blockade of HVEM for Prostate Cancer Immunotherapy in Humanized Mice. Cancers, 13(12), 3009. https://doi.org/10.3390/cancers13123009