
The Role of MacroH2A Histone Variants in Cancer


Abstract
Simple Summary
Abstract
1. Introduction
2. Alteration of H2A Variants in Cancer
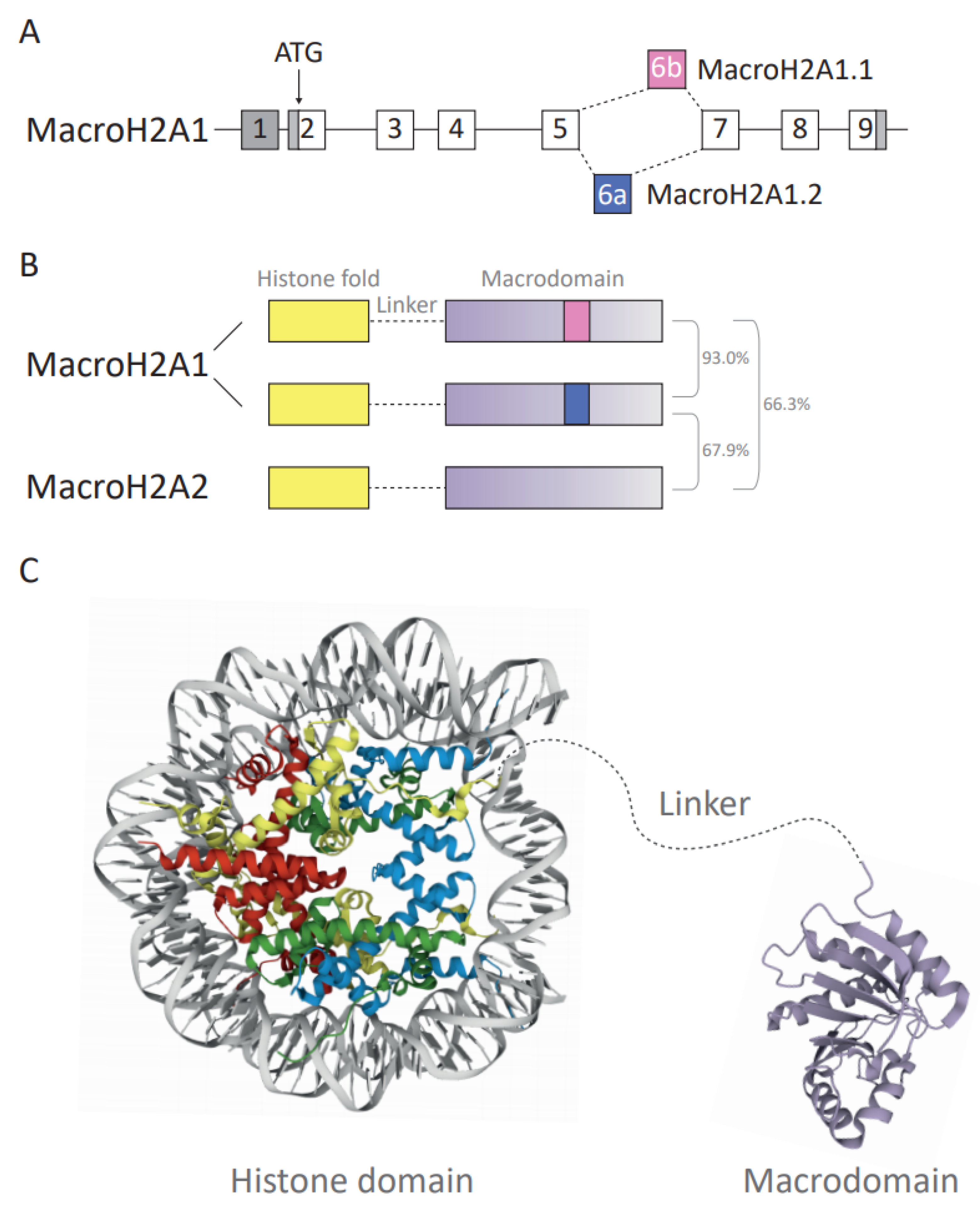
3. MacroH2A Variants: The Three Functional Domains
4. The Ambiguous Role of MacroH2A in Transcriptional Regulation
5. MacroH2A as a Tumor Suppressor
6. MacroH2A as an Oncohistone
7. Conclusions
Author Contributions
Funding
Informed Consent Statement
Conflicts of Interest
References
- Allis, M.-L.C.D.; Jenuwein, T.; Reinberg, D. Epigenetics, 2nd ed.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2015; p. 984. [Google Scholar]
- Stadhouders, R.; Filion, G.J.; Graf, T. Transcription factors and 3D genome conformation in cell-fate decisions. Nature 2019, 569, 345–354. [Google Scholar] [CrossRef]
- Miranda Furtado, C.L.; Dos Santos Luciano, M.C.; Silva Santos, R.D.; Furtado, G.P.; Moraes, M.O.; Pessoa, C. Epidrugs: Targeting epigenetic marks in cancer treatment. Epigenetics 2019, 14, 1164–1176. [Google Scholar] [CrossRef]
- Yoo, C.B.; Jones, P.A. Epigenetic therapy of cancer: Past, present and future. Nat. Rev. Drug Discov. 2006, 5, 37–50. [Google Scholar] [CrossRef]
- Bernt, K.M.; Armstrong, S.A. Targeting epigenetic programs in MLL-rearranged leukemias. Hematol. Am. Soc. Hematol. Educ. Program. 2011, 2011, 354–360. [Google Scholar] [CrossRef] [PubMed]
- Hatzimichael, E.; Lagos, K.; Sim, V.R.; Briasoulis, E.; Crook, T. Epigenetics in diagnosis, prognostic assessment and treatment of cancer: An update. EXCLI J. 2014, 13, 954–976. [Google Scholar] [PubMed]
- Miyamoto, K.; Ushijima, T. Diagnostic and therapeutic applications of epigenetics. Jpn. J. Clin. Oncol. 2005, 35, 293–301. [Google Scholar] [CrossRef] [PubMed]
- Luger, K.; Mader, A.W.; Richmond, R.K.; Sargent, D.F.; Richmond, T.J. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature 1997, 389, 251–260. [Google Scholar] [CrossRef]
- McGinty, R.K.; Tan, S. Nucleosome structure and function. Chem. Rev. 2015, 115, 2255–2273. [Google Scholar] [CrossRef]
- Bednar, J.; Garcia-Saez, I.; Boopathi, R.; Cutter, A.R.; Papai, G.; Reymer, A.; Syed, S.H.; Lone, I.N.; Tonchev, O.; Crucifix, C.; et al. Structure and Dynamics of a 197 bp Nucleosome in Complex with Linker Histone H1. Mol. Cell 2017, 66, 384–397.e8. [Google Scholar] [CrossRef]
- Tessarz, P.; Kouzarides, T. Histone core modifications regulating nucleosome structure and dynamics. Nat. Rev. Mol. Cell Biol. 2014, 15, 703–708. [Google Scholar] [CrossRef]
- Morgan, M.A.J.; Shilatifard, A. Reevaluating the roles of histone-modifying enzymes and their associated chromatin modifications in transcriptional regulation. Nat. Genet. 2020, 52, 1271–1281. [Google Scholar] [CrossRef]
- Buschbeck, M.; Hake, S.B. Variants of core histones and their roles in cell fate decisions, development and cancer. Nat. Rev. Mol. Cell Biol. 2017, 18, 299–314. [Google Scholar] [CrossRef]
- Martire, S.; Banaszynski, L.A. The roles of histone variants in fine-tuning chromatin organization and function. Nat. Rev. Mol. Cell Biol. 2020, 21, 522–541. [Google Scholar] [CrossRef]
- Ghiraldini, F.G.; Filipescu, D.; Bernstein, E. Solid tumours hijack the histone variant network. Nat. Rev. Cancer 2021, 21, 257–275. [Google Scholar] [CrossRef] [PubMed]
- Bonisch, C.; Hake, S.B. Histone H2A variants in nucleosomes and chromatin: More or less stable? Nucleic Acids Res. 2012, 40, 10719–10741. [Google Scholar] [CrossRef] [PubMed]
- Maze, I.; Noh, K.M.; Soshnev, A.A.; Allis, C.D. Every amino acid matters: Essential contributions of histone variants to mammalian development and disease. Nat. Rev. Genet. 2014, 15, 259–271. [Google Scholar] [CrossRef] [PubMed]
- Vardabasso, C.; Hasson, D.; Ratnakumar, K.; Chung, C.Y.; Duarte, L.F.; Bernstein, E. Histone variants: Emerging players in cancer biology. Cell Mol. Life Sci. 2014, 71, 379–404. [Google Scholar] [CrossRef]
- Bernstein, E.; Hake, S.B. The nucleosome: A little variation goes a long way. BioChem. Cell Biol. 2006, 84, 505–517. [Google Scholar] [CrossRef]
- Talbert, P.B.; Henikoff, S. Histone variants—Ancient wrap artists of the epigenome. Nat. Rev. Mol. Cell Biol. 2010, 11, 264–275. [Google Scholar] [CrossRef] [PubMed]
- Lowe, B.R.; Maxham, L.A.; Hamey, J.J.; Wilkins, M.R.; Partridge, J.F. Histone H3 Mutations: An Updated View of Their Role in Chromatin Deregulation and Cancer. Cancers 2019, 11, 660. [Google Scholar] [CrossRef] [PubMed]
- Schwartzentruber, J.; Korshunov, A.; Liu, X.Y.; Jones, D.T.; Pfaff, E.; Jacob, K.; Sturm, D.; Fontebasso, A.M.; Quang, D.A.; Tonjes, M.; et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 2012, 482, 226–231. [Google Scholar] [CrossRef]
- Yamamoto, H.; Ishihara, S.; Toda, Y.; Oda, Y. Histone H3.3 mutation in giant cell tumor of bone: An update in pathology. Med. Mol. Morphol. 2020, 53, 1–6. [Google Scholar] [CrossRef]
- Chan, K.M.; Fang, D.; Gan, H.; Hashizume, R.; Yu, C.; Schroeder, M.; Gupta, N.; Mueller, S.; James, C.D.; Jenkins, R.; et al. The histone H3.3K27M mutation in pediatric glioma reprograms H3K27 methylation and gene expression. Genes Dev. 2013, 27, 985–990. [Google Scholar] [CrossRef]
- Fontebasso, A.M.; Liu, X.Y.; Sturm, D.; Jabado, N. Chromatin remodeling defects in pediatric and young adult glioblastoma: A tale of a variant histone 3 tail. Brain Pathol. 2013, 23, 210–216. [Google Scholar] [CrossRef]
- Bender, S.; Tang, Y.; Lindroth, A.M.; Hovestadt, V.; Jones, D.T.; Kool, M.; Zapatka, M.; Northcott, P.A.; Sturm, D.; Wang, W.; et al. Reduced H3K27me3 and DNA hypomethylation are major drivers of gene expression in K27M mutant pediatric high-grade gliomas. Cancer Cell 2013, 24, 660–672. [Google Scholar] [CrossRef] [PubMed]
- Amatori, S.; Tavolaro, S.; Gambardella, S.; Fanelli, M. The dark side of histones: Genomic organization and role of oncohistones in cancer. Clin. Epigenet. 2021, 13, 71. [Google Scholar] [CrossRef] [PubMed]
- Chakravarthy, S.; Gundimella, S.K.; Caron, C.; Perche, P.Y.; Pehrson, J.R.; Khochbin, S.; Luger, K. Structural characterization of the histone variant macroH2A. Mol. Cell Biol. 2005, 25, 7616–7624. [Google Scholar] [CrossRef] [PubMed]
- Barrero, M.J.; Sese, B.; Kuebler, B.; Bilic, J.; Boue, S.; Marti, M.; Izpisua Belmonte, J.C. Macrohistone variants preserve cell identity by preventing the gain of H3K4me2 during reprogramming to pluripotency. Cell Rep. 2013, 3, 1005–1011. [Google Scholar] [CrossRef]
- Pasque, V.; Gillich, A.; Garrett, N.; Gurdon, J.B. Histone variant macroH2A confers resistance to nuclear reprogramming. EMBO J. 2011, 30, 2373–2387. [Google Scholar] [CrossRef] [PubMed]
- Rangasamy, D. Histone variant H2A.Z can serve as a new target for breast cancer therapy. Curr. Med. Chem. 2010, 17, 3155–3161. [Google Scholar] [CrossRef]
- Vardabasso, C.; Gaspar-Maia, A.; Hasson, D.; Punzeler, S.; Valle-Garcia, D.; Straub, T.; Keilhauer, E.C.; Strub, T.; Dong, J.; Panda, T.; et al. Histone Variant H2A.Z.2 Mediates Proliferation and Drug Sensitivity of Malignant Melanoma. Mol. Cell 2015, 59, 75–88. [Google Scholar] [CrossRef]
- Hsu, C.C.; Shi, J.; Yuan, C.; Zhao, D.; Jiang, S.; Lyu, J.; Wang, X.; Li, H.; Wen, H.; Li, W.; et al. Recognition of histone acetylation by the GAS41 YEATS domain promotes H2A.Z deposition in non-small cell lung cancer. Genes Dev. 2018, 32, 58–69. [Google Scholar] [CrossRef]
- Dryhurst, D.; Ausio, J. Histone H2A.Z deregulation in prostate cancer. Cause or effect? Cancer Metastasis Rev. 2014, 33, 429–439. [Google Scholar] [CrossRef] [PubMed]
- Hua, S.; Kallen, C.B.; Dhar, R.; Baquero, M.T.; Mason, C.E.; Russell, B.A.; Shah, P.K.; Liu, J.; Khramtsov, A.; Tretiakova, M.S.; et al. Genomic analysis of estrogen cascade reveals histone variant H2A.Z associated with breast cancer progression. Mol. Syst. Biol. 2008, 4, 188. [Google Scholar] [CrossRef] [PubMed]
- Suto, R.K.; Clarkson, M.J.; Tremethick, D.J.; Luger, K. Crystal structure of a nucleosome core particle containing the variant histone H2A.Z. Nat. Struct. Biol. 2000, 7, 1121–1124. [Google Scholar] [PubMed]
- Yang, H.D.; Kim, P.J.; Eun, J.W.; Shen, Q.; Kim, H.S.; Shin, W.C.; Ahn, Y.M.; Park, W.S.; Lee, J.Y.; Nam, S.W. Oncogenic potential of histone-variant H2A.Z.1 and its regulatory role in cell cycle and epithelial-mesenchymal transition in liver cancer. Oncotarget 2016, 7, 11412–11423. [Google Scholar] [CrossRef]
- Svotelis, A.; Gevry, N.; Grondin, G.; Gaudreau, L. H2A.Z overexpression promotes cellular proliferation of breast cancer cells. Cell Cycle 2010, 9, 364–370. [Google Scholar] [CrossRef]
- Gevry, N.; Hardy, S.; Jacques, P.E.; Laflamme, L.; Svotelis, A.; Robert, F.; Gaudreau, L. Histone H2A.Z is essential for estrogen receptor signaling. Genes Dev. 2009, 23, 1522–1533. [Google Scholar] [CrossRef]
- Gevry, N.; Chan, H.M.; Laflamme, L.; Livingston, D.M.; Gaudreau, L. p21 transcription is regulated by differential localization of histone H2A.Z. Genes Dev. 2007, 21, 1869–1881. [Google Scholar] [CrossRef]
- West, M.H.; Bonner, W.M. Histone 2A, a heteromorphous family of eight protein species. Biochemistry 1980, 19, 3238–3245. [Google Scholar] [CrossRef]
- Rogakou, E.P.; Pilch, D.R.; Orr, A.H.; Ivanova, V.S.; Bonner, W.M. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J. Biol. Chem. 1998, 273, 5858–5868. [Google Scholar] [CrossRef] [PubMed]
- Rogakou, E.P.; Boon, C.; Redon, C.; Bonner, W.M. Megabase chromatin domains involved in DNA double-strand breaks in vivo. J. Cell Biol. 1999, 146, 905–916. [Google Scholar] [CrossRef] [PubMed]
- Celeste, A.; Difilippantonio, S.; Difilippantonio, M.J.; Fernandez-Capetillo, O.; Pilch, D.R.; Sedelnikova, O.A.; Eckhaus, M.; Ried, T.; Bonner, W.M.; Nussenzweig, A. H2AX haploinsufficiency modifies genomic stability and tumor susceptibility. Cell 2003, 114, 371–383. [Google Scholar] [CrossRef]
- Bassing, C.H.; Suh, H.; Ferguson, D.O.; Chua, K.F.; Manis, J.; Eckersdorff, M.; Gleason, M.; Bronson, R.; Lee, C.; Alt, F.W. Histone H2AX: A dosage-dependent suppressor of oncogenic translocations and tumors. Cell 2003, 114, 359–370. [Google Scholar] [CrossRef]
- Monni, O.; Knuutila, S. 11q deletions in hematological malignancies. Leuk. Lymphoma 2001, 40, 259–266. [Google Scholar] [CrossRef]
- Parikh, R.A.; White, J.S.; Huang, X.; Schoppy, D.W.; Baysal, B.E.; Baskaran, R.; Bakkenist, C.J.; Saunders, W.S.; Hsu, L.C.; Romkes, M.; et al. Loss of distal 11q is associated with DNA repair deficiency and reduced sensitivity to ionizing radiation in head and neck squamous cell carcinoma. Genes Chromosomes Cancer 2007, 46, 761–775. [Google Scholar] [CrossRef]
- Srivastava, N.; Gochhait, S.; Gupta, P.; Bamezai, R.N. Copy number alterations of the H2AFX gene in sporadic breast cancer patients. Cancer Genet. Cytogenet. 2008, 180, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Contrepois, K.; Coudereau, C.; Benayoun, B.A.; Schuler, N.; Roux, P.F.; Bischof, O.; Courbeyrette, R.; Carvalho, C.; Thuret, J.Y.; Ma, Z.; et al. Histone variant H2A.J accumulates in senescent cells and promotes inflammatory gene expression. Nat. Commun. 2017, 8, 14995. [Google Scholar] [CrossRef]
- Cornen, S.; Guille, A.; Adelaide, J.; Addou-Klouche, L.; Finetti, P.; Saade, M.R.; Manai, M.; Carbuccia, N.; Bekhouche, I.; Letessier, A.; et al. Candidate luminal B breast cancer genes identified by genome, gene expression and DNA methylation profiling. PLoS ONE 2014, 9, e81843. [Google Scholar] [CrossRef]
- Colotta, F.; Allavena, P.; Sica, A.; Garlanda, C.; Mantovani, A. Cancer-related inflammation, the seventh hallmark of cancer: Links to genetic instability. Carcinogenesis 2009, 30, 1073–1081. [Google Scholar] [CrossRef]
- Rube, C.E.; Baumert, C.; Schuler, N.; Isermann, A.; Schmal, Z.; Glanemann, M.; Mann, C.; Scherthan, H. Human skin aging is associated with increased expression of the histone variant H2A.J in the epidermis. NPJ Aging Mech. Dis. 2021, 7, 7. [Google Scholar] [CrossRef]
- Chew, G.L.; Bleakley, M.; Bradley, R.K.; Malik, H.S.; Henikoff, S.; Molaro, A.; Sarthy, J. Short H2A histone variants are expressed in cancer. Nat. Commun. 2021, 12, 490. [Google Scholar] [CrossRef] [PubMed]
- Molaro, A.; Young, J.M.; Malik, H.S. Evolutionary origins and diversification of testis-specific short histone H2A variants in mammals. Genome Res. 2018, 28, 460–473. [Google Scholar] [CrossRef]
- Sansoni, V.; Casas-Delucchi, C.S.; Rajan, M.; Schmidt, A.; Bonisch, C.; Thomae, A.W.; Staege, M.S.; Hake, S.B.; Cardoso, M.C.; Imhof, A. The histone variant H2A.Bbd is enriched at sites of DNA synthesis. Nucleic Acids Res. 2014, 42, 6405–6420. [Google Scholar] [CrossRef] [PubMed]
- Soboleva, T.A.; Parker, B.J.; Nekrasov, M.; Hart-Smith, G.; Tay, Y.J.; Tng, W.Q.; Wilkins, M.; Ryan, D.; Tremethick, D.J. A new link between transcriptional initiation and pre-mRNA splicing: The RNA binding histone variant H2A.B. PLoS Genet. 2017, 13, e1006633. [Google Scholar] [CrossRef] [PubMed]
- Pehrson, J.R.; Fried, V.A. MacroH2A, a core histone containing a large nonhistone region. Science 1992, 257, 1398–1400. [Google Scholar] [CrossRef]
- Gaspar-Maia, A.; Qadeer, Z.A.; Hasson, D.; Ratnakumar, K.; Leu, N.A.; Leroy, G.; Liu, S.; Costanzi, C.; Valle-Garcia, D.; Schaniel, C.; et al. MacroH2A histone variants act as a barrier upon reprogramming towards pluripotency. Nat. Commun. 2013, 4, 1565. [Google Scholar] [CrossRef]
- Dardenne, E.; Pierredon, S.; Driouch, K.; Gratadou, L.; Lacroix-Triki, M.; Espinoza, M.P.; Zonta, E.; Germann, S.; Mortada, H.; Villemin, J.P.; et al. Splicing switch of an epigenetic regulator by RNA helicases promotes tumor-cell invasiveness. Nat. Struct. Mol. Biol. 2012, 19, 1139–1146. [Google Scholar] [CrossRef]
- Hodge, D.Q.; Cui, J.; Gamble, M.J.; Guo, W. Histone Variant MacroH2A1 Plays an Isoform-Specific Role in Suppressing Epithelial-Mesenchymal Transition. Sci. Rep. 2018, 8, 841. [Google Scholar] [CrossRef]
- Novikov, L.; Park, J.W.; Chen, H.; Klerman, H.; Jalloh, A.S.; Gamble, M.J. QKI-mediated alternative splicing of the histone variant MacroH2A1 regulates cancer cell proliferation. Mol. Cell Biol. 2011, 31, 4244–4255. [Google Scholar] [CrossRef]
- Pazienza, V.; Panebianco, C.; Rappa, F.; Memoli, D.; Borghesan, M.; Cannito, S.; Oji, A.; Mazza, G.; Tamburrino, D.; Fusai, G.; et al. Histone macroH2A1.2 promotes metabolic health and leanness by inhibiting adipogenesis. Epigenet. Chromatin 2016, 9, 45. [Google Scholar] [CrossRef]
- Hurtado-Bages, S.; Posavec Marjanovic, M.; Valero, V.; Malinverni, R.; Corujo, D.; Bouvet, P.; Lavigne, A.C.; Bystricky, K.; Buschbeck, M. The Histone Variant MacroH2A1 Regulates Key Genes for Myogenic Cell Fusion in a Splice-Isoform Dependent Manner. Cells 2020, 9, 1109. [Google Scholar] [CrossRef]
- Hurtado-Bages, S.; Knobloch, G.; Ladurner, A.G.; Buschbeck, M. The taming of PARP1 and its impact on NAD(+) metabolism. Mol. Metab. 2020, 38, 100950. [Google Scholar] [CrossRef] [PubMed]
- Lo Re, O.; Fusilli, C.; Rappa, F.; Van Haele, M.; Douet, J.; Pindjakova, J.; Rocha, S.W.; Pata, I.; Valcikova, B.; Uldrijan, S.; et al. Induction of cancer cell stemness by depletion of macrohistone H2A1 in hepatocellular carcinoma. Hepatology 2018, 67, 636–650. [Google Scholar] [CrossRef] [PubMed]
- Rappa, F.; Greco, A.; Podrini, C.; Cappello, F.; Foti, M.; Bourgoin, L.; Peyrou, M.; Marino, A.; Scibetta, N.; Williams, R.; et al. Immunopositivity for histone macroH2A1 isoforms marks steatosis-associated hepatocellular carcinoma. PLoS ONE 2013, 8, e54458. [Google Scholar] [CrossRef]
- Borghesan, M.; Fusilli, C.; Rappa, F.; Panebianco, C.; Rizzo, G.; Oben, J.A.; Mazzoccoli, G.; Faulkes, C.; Pata, I.; Agodi, A.; et al. DNA Hypomethylation and Histone Variant macroH2A1 Synergistically Attenuate Chemotherapy-Induced Senescence to Promote Hepatocellular Carcinoma Progression. Cancer Res. 2016, 76, 594–606. [Google Scholar] [CrossRef]
- Ma, X.; Ding, Y.; Zeng, L. The diagnostic and prognostic value of H2AFY in hepatocellular carcinoma. BMC Cancer 2021, 21, 418. [Google Scholar] [CrossRef]
- Kim, J.; Shin, Y.; Lee, S.; Kim, M.; Punj, V.; Lu, J.F.; Shin, H.; Kim, K.; Ulmer, T.S.; Koh, J.; et al. Regulation of Breast Cancer-Induced Osteoclastogenesis by MacroH2A1.2 Involving EZH2-Mediated H3K27me3. Cell Rep. 2018, 24, 224–237. [Google Scholar] [CrossRef]
- Berman, H.; Henrick, K.; Nakamura, H. Announcing the worldwide Protein Data Bank. Nat. Struct. Biol. 2003, 10, 980. [Google Scholar] [CrossRef]
- Drew, H.R.; Wing, R.M.; Takano, T.; Broka, C.; Tanaka, S.; Itakura, K.; Dickerson, R.E. Structure of a B-DNA dodecamer: Conformation and dynamics. Proc. Natl. Acad. Sci. USA 1981, 78, 2179–2183. [Google Scholar] [CrossRef]
- Wu, B.; Davey, G.E.; Nazarov, A.A.; Dyson, P.J.; Davey, C.A. Specific DNA structural attributes modulate platinum anticancer drug site selection and cross-link generation. Nucleic Acids Res. 2011, 39, 8200–8212. [Google Scholar] [CrossRef]
- Costanzi, C.; Pehrson, J.R. MACROH2A2, a new member of the MARCOH2A core histone family. J. Biol. Chem. 2001, 276, 21776–21784. [Google Scholar] [CrossRef]
- Chakravarthy, S.; Luger, K. The histone variant macro-H2A preferentially forms “hybrid nucleosomes”. J. Biol. Chem. 2006, 281, 25522–25531. [Google Scholar] [CrossRef]
- Nusinow, D.A.; Sharp, J.A.; Morris, A.; Salas, S.; Plath, K.; Panning, B. The histone domain of macroH2A1 contains several dispersed elements that are each sufficient to direct enrichment on the inactive X chromosome. J. Mol. Biol. 2007, 371, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, P.D.; Gamble, M.J. MacroH2A1 chromatin specification requires its docking domain and acetylation of H2B lysine 20. Nat. Commun. 2018, 9, 5143. [Google Scholar] [CrossRef]
- Chakravarthy, S.; Patel, A.; Bowman, G.D. The basic linker of macroH2A stabilizes DNA at the entry/exit site of the nucleosome. Nucleic Acids Res. 2012, 40, 8285–8295. [Google Scholar] [CrossRef] [PubMed]
- Muthurajan, U.M.; McBryant, S.J.; Lu, X.; Hansen, J.C.; Luger, K. The linker region of macroH2A promotes self-association of nucleosomal arrays. J. Biol. Chem. 2011, 286, 23852–23864. [Google Scholar] [CrossRef] [PubMed]
- Corujo, D.; Buschbeck, M. Post-Translational Modifications of H2A Histone Variants and Their Role in Cancer. Cancers 2018, 10, 59. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, E.; Muratore-Schroeder, T.L.; Diaz, R.L.; Chow, J.C.; Changolkar, L.N.; Shabanowitz, J.; Heard, E.; Pehrson, J.R.; Hunt, D.F.; Allis, C.D. A phosphorylated subpopulation of the histone variant macroH2A1 is excluded from the inactive X chromosome and enriched during mitosis. Proc. Natl. Acad. Sci. USA 2008, 105, 1533–1538. [Google Scholar] [CrossRef] [PubMed]
- Douet, J.; Corujo, D.; Malinverni, R.; Renauld, J.; Sansoni, V.; Posavec Marjanovic, M.; Cantarino, N.; Valero, V.; Mongelard, F.; Bouvet, P.; et al. MacroH2A histone variants maintain nuclear organization and heterochromatin architecture. J. Cell Sci. 2017, 130, 1570–1582. [Google Scholar] [CrossRef] [PubMed]
- Kozlowski, M.; Corujo, D.; Hothorn, M.; Guberovic, I.; Mandemaker, I.K.; Blessing, C.; Sporn, J.; Gutierrez-Triana, A.; Smith, R.; Portmann, T.; et al. MacroH2A histone variants limit chromatin plasticity through two distinct mechanisms. EMBO Rep. 2018, 19, e44445. [Google Scholar] [CrossRef]
- Yelagandula, R.; Stroud, H.; Holec, S.; Zhou, K.; Feng, S.; Zhong, X.; Muthurajan, U.M.; Nie, X.; Kawashima, T.; Groth, M.; et al. The histone variant H2A.W defines heterochromatin and promotes chromatin condensation in Arabidopsis. Cell 2014, 158, 98–109. [Google Scholar] [CrossRef]
- Karras, G.I.; Kustatscher, G.; Buhecha, H.R.; Allen, M.D.; Pugieux, C.; Sait, F.; Bycroft, M.; Ladurner, A.G. The macro domain is an ADP-ribose binding module. EMBO J. 2005, 24, 1911–1920. [Google Scholar] [CrossRef] [PubMed]
- Allen, M.D.; Buckle, A.M.; Cordell, S.C.; Lowe, J.; Bycroft, M. The crystal structure of AF1521 a protein from Archaeoglobus fulgidus with homology to the non-histone domain of macroH2A. J. Mol. Biol. 2003, 330, 503–511. [Google Scholar] [CrossRef]
- Rack, J.G.; Perina, D.; Ahel, I. Macrodomains: Structure, Function, Evolution, and Catalytic Activities. Annu. Rev. Biochem. 2016, 85, 431–454. [Google Scholar] [CrossRef] [PubMed]
- Timinszky, G.; Till, S.; Hassa, P.O.; Hothorn, M.; Kustatscher, G.; Nijmeijer, B.; Colombelli, J.; Altmeyer, M.; Stelzer, E.H.; Scheffzek, K.; et al. A macrodomain-containing histone rearranges chromatin upon sensing PARP1 activation. Nat. Struct. Mol. Biol. 2009, 16, 923–929. [Google Scholar] [CrossRef] [PubMed]
- Simonet, N.G.; Thackray, J.K.; Vazquez, B.N.; Ianni, A.; Espinosa-Alcantud, M.; Morales-Sanfrutos, J.; Hurtado-Bages, S.; Sabido, E.; Buschbeck, M.; Tischfield, J.; et al. SirT7 auto-ADP-ribosylation regulates glucose starvation response through mH2A1. Sci. Adv. 2020, 6, eaaz2590. [Google Scholar] [CrossRef] [PubMed]
- Gibson, B.A.; Kraus, W.L. New insights into the molecular and cellular functions of poly(ADP-ribose) and PARPs. Nat. Rev. Mol. Cell Biol. 2012, 13, 411–424. [Google Scholar] [CrossRef] [PubMed]
- Buschbeck, M.; Uribesalgo, I.; Wibowo, I.; Rue, P.; Martin, D.; Gutierrez, A.; Morey, L.; Guigo, R.; Lopez-Schier, H.; Di Croce, L. The histone variant macroH2A is an epigenetic regulator of key developmental genes. Nat. Struct. Mol. Biol. 2009, 16, 1074–1079. [Google Scholar] [CrossRef]
- Kustatscher, G.; Hothorn, M.; Pugieux, C.; Scheffzek, K.; Ladurner, A.G. Splicing regulates NAD metabolite binding to histone macroH2A. Nat. Struct. Mol. Biol. 2005, 12, 624–625. [Google Scholar] [CrossRef]
- Posavec Marjanovic, M.; Hurtado-Bages, S.; Lassi, M.; Valero, V.; Malinverni, R.; Delage, H.; Navarro, M.; Corujo, D.; Guberovic, I.; Douet, J.; et al. MacroH2A1.1 regulates mitochondrial respiration by limiting nuclear NAD(+) consumption. Nat. Struct. Mol. Biol. 2017, 24, 902–910. [Google Scholar] [CrossRef] [PubMed]
- Gamble, M.J.; Frizzell, K.M.; Yang, C.; Krishnakumar, R.; Kraus, W.L. The histone variant macroH2A1 marks repressed autosomal chromatin, but protects a subset of its target genes from silencing. Genes Dev. 2010, 24, 21–32. [Google Scholar] [CrossRef]
- Changolkar, L.N.; Singh, G.; Cui, K.; Berletch, J.B.; Zhao, K.; Disteche, C.M.; Pehrson, J.R. Genome-wide distribution of macroH2A1 histone variants in mouse liver chromatin. Mol. Cell Biol. 2010, 30, 5473–5483. [Google Scholar] [CrossRef] [PubMed]
- Costanzi, C.; Stein, P.; Worrad, D.M.; Schultz, R.M.; Pehrson, J.R. Histone macroH2A1 is concentrated in the inactive X chromosome of female preimplantation mouse embryos. Development 2000, 127, 2283–2289. [Google Scholar] [CrossRef]
- Mietton, F.; Sengupta, A.K.; Molla, A.; Picchi, G.; Barral, S.; Heliot, L.; Grange, T.; Wutz, A.; Dimitrov, S. Weak but uniform enrichment of the histone variant macroH2A1 along the inactive X chromosome. Mol. Cell Biol. 2009, 29, 150–156. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Filipescu, D.; Andrade, J.; Gaspar-Maia, A.; Ueberheide, B.; Bernstein, E. Transcription-associated histone pruning demarcates macroH2A chromatin domains. Nat. Struct. Mol. Biol. 2018, 25, 958–970. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Munoz, I.; Lund, A.H.; van der Stoop, P.; Boutsma, E.; Muijrers, I.; Verhoeven, E.; Nusinow, D.A.; Panning, B.; Marahrens, Y.; van Lohuizen, M. Stable X chromosome inactivation involves the PRC1 Polycomb complex and requires histone MACROH2A1 and the CULLIN3/SPOP ubiquitin E3 ligase. Proc. Natl. Acad. Sci. USA 2005, 102, 7635–7640. [Google Scholar] [CrossRef] [PubMed]
- Creppe, C.; Posavec, M.; Douet, J.; Buschbeck, M. MacroH2A in stem cells: A story beyond gene repression. Epigenomics 2012, 4, 221–227. [Google Scholar] [CrossRef]
- Lavigne, M.D.; Vatsellas, G.; Polyzos, A.; Mantouvalou, E.; Sianidis, G.; Maraziotis, I.; Agelopoulos, M.; Thanos, D. Composite macroH2A/NRF-1 Nucleosomes Suppress Noise and Generate Robustness in Gene Expression. Cell Rep. 2015, 11, 1090–1101. [Google Scholar] [CrossRef]
- Agelopoulos, M.; Thanos, D. Epigenetic determination of a cell-specific gene expression program by ATF-2 and the histone variant macroH2A. EMBO J. 2006, 25, 4843–4853. [Google Scholar] [CrossRef]
- Angelov, D.; Molla, A.; Perche, P.Y.; Hans, F.; Cote, J.; Khochbin, S.; Bouvet, P.; Dimitrov, S. The histone variant macroH2A interferes with transcription factor binding and SWI/SNF nucleosome remodeling. Mol. Cell 2003, 11, 1033–1041. [Google Scholar] [CrossRef]
- Chen, H.; Ruiz, P.D.; Novikov, L.; Casill, A.D.; Park, J.W.; Gamble, M.J. MacroH2A1.1 and PARP-1 cooperate to regulate transcription by promoting CBP-mediated H2B acetylation. Nat. Struct. Mol. Biol. 2014, 21, 981–989. [Google Scholar] [CrossRef] [PubMed]
- Dell’Orso, S.; Wang, A.H.; Shih, H.Y.; Saso, K.; Berghella, L.; Gutierrez-Cruz, G.; Ladurner, A.G.; O’Shea, J.J.; Sartorelli, V.; Zare, H. The Histone Variant MacroH2A1.2 Is Necessary for the Activation of Muscle Enhancers and Recruitment of the Transcription Factor Pbx1. Cell Rep. 2016, 14, 1156–1168. [Google Scholar] [CrossRef] [PubMed]
- Chang, E.Y.; Ferreira, H.; Somers, J.; Nusinow, D.A.; Owen-Hughes, T.; Narlikar, G.J. MacroH2A allows ATP-dependent chromatin remodeling by SWI/SNF and ACF complexes but specifically reduces recruitment of SWI/SNF. Biochemistry 2008, 47, 13726–13732. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Lv, P.; Yan, G.; Fan, H.; Cheng, L.; Zhang, F.; Dang, Y.; Wu, H.; Wen, B. MacroH2A1 associates with nuclear lamina and maintains chromatin architecture in mouse liver cells. Sci. Rep. 2015, 5, 17186. [Google Scholar] [CrossRef]
- Kapoor, A.; Goldberg, M.S.; Cumberland, L.K.; Ratnakumar, K.; Segura, M.F.; Emanuel, P.O.; Menendez, S.; Vardabasso, C.; Leroy, G.; Vidal, C.I.; et al. The histone variant macroH2A suppresses melanoma progression through regulation of CDK8. Nature 2010, 468, 1105–1109. [Google Scholar] [CrossRef]
- Sporn, J.C.; Kustatscher, G.; Hothorn, T.; Collado, M.; Serrano, M.; Muley, T.; Schnabel, P.; Ladurner, A.G. Histone macroH2A isoforms predict the risk of lung cancer recurrence. Oncogene 2009, 28, 3423–3428. [Google Scholar] [CrossRef]
- Sporn, J.C.; Jung, B. Differential regulation and predictive potential of MacroH2A1 isoforms in colon cancer. Am. J. Pathol. 2012, 180, 2516–2526. [Google Scholar] [CrossRef]
- Hu, W.H.; Miyai, K.; Sporn, J.C.; Luo, L.; Wang, J.Y.; Cosman, B.; Ramamoorthy, S. Loss of histone variant macroH2A2 expression associates with progression of anal neoplasm. J. Clin. Pathol. 2016, 69, 627–631. [Google Scholar] [CrossRef]
- Zhang, K.; Zhao, H.; Zhang, K.; Hua, C.; Qin, X.; Xu, S. Chromatin-regulating genes are associated with postoperative prognosis and isocitrate dehydrogenase mutation in astrocytoma. Ann. Transl. Med. 2020, 8, 1594. [Google Scholar] [CrossRef] [PubMed]
- Maher, M.; Diesch, J.; Le Pannerer, M.M.; Buschbeck, M. Epigenetics in a Spectrum of Myeloid Diseases and Its Exploitation for Therapy. Cancers 2021, 13, 1746. [Google Scholar] [CrossRef]
- Bereshchenko, O.; Lo Re, O.; Nikulenkov, F.; Flamini, S.; Kotaskova, J.; Mazza, T.; Le Pannerer, M.M.; Buschbeck, M.; Giallongo, C.; Palumbo, G.; et al. Deficiency and haploinsufficiency of histone macroH2A1.1 in mice recapitulate hematopoietic defects of human myelodysplastic syndrome. Clin. Epigenet. 2019, 11, 121. [Google Scholar] [CrossRef] [PubMed]
- Yip, B.H.; Steeples, V.; Repapi, E.; Armstrong, R.N.; Llorian, M.; Roy, S.; Shaw, J.; Dolatshad, H.; Taylor, S.; Verma, A.; et al. The U2AF1S34F mutation induces lineage-specific splicing alterations in myelodysplastic syndromes. J. Clin. Invest. 2017, 127, 2206–2221. [Google Scholar] [CrossRef] [PubMed]
- Danan-Gotthold, M.; Golan-Gerstl, R.; Eisenberg, E.; Meir, K.; Karni, R.; Levanon, E.Y. Identification of recurrent regulated alternative splicing events across human solid tumors. Nucleic Acids Res. 2015, 43, 5130–5144. [Google Scholar] [CrossRef]
- Creppe, C.; Janich, P.; Cantarino, N.; Noguera, M.; Valero, V.; Musulen, E.; Douet, J.; Posavec, M.; Martin-Caballero, J.; Sumoy, L.; et al. MacroH2A1 regulates the balance between self-renewal and differentiation commitment in embryonic and adult stem cells. Mol. Cell Biol. 2012, 32, 1442–1452. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.M.; Heo, K.; Choi, J.; Kim, K.; An, W. The histone variant MacroH2A regulates Ca(2+) influx through TRPC3 and TRPC6 channels. Oncogenesis 2013, 2, e77. [Google Scholar] [CrossRef]
- Vieira-Silva, T.S.; Monteiro-Reis, S.; Barros-Silva, D.; Ramalho-Carvalho, J.; Graca, I.; Carneiro, I.; Martins, A.T.; Oliveira, J.; Antunes, L.; Hurtado-Bages, S.; et al. Histone variant MacroH2A1 is downregulated in prostate cancer and influences malignant cell phenotype. Cancer Cell Int. 2019, 19, 112. [Google Scholar] [CrossRef]
- Lei, S.; Long, J.; Li, J. MacroH2A suppresses the proliferation of the B16 melanoma cell line. Mol. Med. Rep. 2014, 10, 1845–1850. [Google Scholar] [CrossRef][Green Version]
- Yang, P.; Yin, K.; Zhong, D.; Liao, Q.; Li, K. Inhibition of osteosarcoma cell progression by MacroH2A via the downregulation of cyclin D and cyclindependent kinase genes. Mol. Med. Rep. 2015, 11, 1905–1910. [Google Scholar] [CrossRef]
- Xu, D.; Li, C.F.; Zhang, X.; Gong, Z.; Chan, C.H.; Lee, S.W.; Jin, G.; Rezaeian, A.H.; Han, F.; Wang, J.; et al. Skp2-macroH2A1-CDK8 axis orchestrates G2/M transition and tumorigenesis. Nat. Commun. 2015, 6, 6641. [Google Scholar] [CrossRef]
- Solum, E.; Hansen, T.V.; Aesoy, R.; Herfindal, L. New CDK8 inhibitors as potential anti-leukemic agents—Design, synthesis and biological evaluation. Bioorg. Med. Chem. 2020, 28, 115461. [Google Scholar] [CrossRef]
- Park, S.J.; Shim, J.W.; Park, H.S.; Eum, D.Y.; Park, M.T.; Mi Yi, J.; Choi, S.H.; Kim, S.D.; Son, T.G.; Lu, W.; et al. MacroH2A1 downregulation enhances the stem-like properties of bladder cancer cells by transactivation of Lin28B. Oncogene 2016, 35, 1292–1301. [Google Scholar] [CrossRef]
- Kim, J.M.; Shin, Y.; Lee, S.; Kim, M.Y.; Punj, V.; Shin, H.I.; Kim, K.; Koh, J.M.; Jeong, D.; An, W. MacroH2A1.2 inhibits prostate cancer-induced osteoclastogenesis through cooperation with HP1alpha and H1.2. Oncogene 2018, 37, 5749–5765. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Tillo, E.; de Barrios, O.; Valls, E.; Darling, D.S.; Castells, A.; Postigo, A. ZEB1 and TCF4 reciprocally modulate their transcriptional activities to regulate Wnt target gene expression. Oncogene 2015, 34, 5760–5770. [Google Scholar] [CrossRef] [PubMed]
- de Barrios, O.; Gyorffy, B.; Fernandez-Acenero, M.J.; Sanchez-Tillo, E.; Sanchez-Moral, L.; Siles, L.; Esteve-Arenys, A.; Roue, G.; Casal, J.I.; Darling, D.S.; et al. ZEB1-induced tumourigenesis requires senescence inhibition via activation of DKK1/mutant p53/Mdm2/CtBP and repression of macroH2A1. Gut 2017, 66, 666–682. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Sun, C.; Tran, A.D.; Chin, P.J.; Ruiz, P.D.; Wang, K.; Gibbons, R.J.; Gamble, M.J.; Liu, Y.; Oberdoerffer, P. The macroH2A1.2 histone variant links ATRX loss to alternative telomere lengthening. Nat. Struct. Mol. Biol. 2019, 26, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Sebastian, R.; Hosogane, E.K.; Sun, E.G.; Tran, A.D.; Reinhold, W.C.; Burkett, S.; Sturgill, D.M.; Gudla, P.R.; Pommier, Y.; Aladjem, M.I.; et al. Epigenetic Regulation of DNA Repair Pathway Choice by MacroH2A1 Splice Variants Ensures Genome Stability. Mol. Cell 2020, 79, 836–845.e7. [Google Scholar] [CrossRef]
- Leung, J.W.; Makharashvili, N.; Agarwal, P.; Chiu, L.Y.; Pourpre, R.; Cammarata, M.B.; Cannon, J.R.; Sherker, A.; Durocher, D.; Brodbelt, J.S.; et al. ZMYM3 regulates BRCA1 localization at damaged chromatin to promote DNA repair. Genes Dev. 2017, 31, 260–274. [Google Scholar] [CrossRef]
- Ramamoorthy, M.; Smith, S. Loss of ATRX Suppresses Resolution of Telomere Cohesion to Control Recombination in ALT Cancer Cells. Cancer Cell 2015, 28, 357–369. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Xu, Y.; Gursoy-Yuzugullu, O.; Price, B.D. The histone variant macroH2A1.1 is recruited to DSBs through a mechanism involving PARP1. FEBS Lett. 2012, 586, 3920–3925. [Google Scholar] [CrossRef]
- Ferrand, J.; Rondinelli, B.; Polo, S.E. Histone Variants: Guardians of Genome Integrity. Cells 2020, 9, 2424. [Google Scholar] [CrossRef]
- Nacev, B.A.; Feng, L.; Bagert, J.D.; Lemiesz, A.E.; Gao, J.; Soshnev, A.A.; Kundra, R.; Schultz, N.; Muir, T.W.; Allis, C.D. The expanding landscape of ‘oncohistone’ mutations in human cancers. Nature 2019, 567, 473–478. [Google Scholar] [CrossRef]
- Lavigne, A.C.; Castells, M.; Mermet, J.; Kocanova, S.; Dalvai, M.; Bystricky, K. Increased macroH2A1.1 expression correlates with poor survival of triple-negative breast cancer patients. PLoS ONE 2014, 9, e98930. [Google Scholar] [CrossRef]
- Barzily-Rokni, M.; Friedman, N.; Ron-Bigger, S.; Isaac, S.; Michlin, D.; Eden, A. Synergism between DNA methylation and macroH2A1 occupancy in epigenetic silencing of the tumor suppressor gene p16(CDKN2A). Nucleic Acids Res. 2011, 39, 1326–1335. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.H.; Sahoo, D.; Arvanitis, C.; Bradon, N.; Dill, D.L.; Felsher, D.W. Combined analysis of murine and human microarrays and ChIP analysis reveals genes associated with the ability of MYC to maintain tumorigenesis. PLoS Genet. 2008, 4, e1000090. [Google Scholar] [CrossRef] [PubMed]
- Giallongo, S.; Di Rosa, M.; Caltabiano, R.; Longhitano, L.; Reibaldi, M.; Distefano, A.; Lo Re, O.; Amorini, A.M.; Puzzo, L.; Salvatorelli, L.; et al. Loss of macroH2A1 decreases mitochondrial metabolism and reduces the aggressiveness of uveal melanoma cells. Aging 2020, 12, 9745–9760. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.H.; van Riggelen, J.; Yetil, A.; Fan, A.C.; Bachireddy, P.; Felsher, D.W. Cellular senescence is an important mechanism of tumor regression upon c-Myc inactivation. Proc. Natl. Acad. Sci. USA 2007, 104, 13028–13033. [Google Scholar] [CrossRef]
- Guccione, E.; Martinato, F.; Finocchiaro, G.; Luzi, L.; Tizzoni, L.; Dall’ Olio, V.; Zardo, G.; Nervi, C.; Bernard, L.; Amati, B. Myc-binding-site recognition in the human genome is determined by chromatin context. Nat. Cell Biol. 2006, 8, 764–770. [Google Scholar] [CrossRef]
- Podrini, C.; Koffas, A.; Chokshi, S.; Vinciguerra, M.; Lelliott, C.J.; White, J.K.; Adissu, H.A.; Williams, R.; Greco, A. MacroH2A1 isoforms are associated with epigenetic markers for activation of lipogenic genes in fat-induced steatosis. FASEB J. 2015, 29, 1676–1687. [Google Scholar] [CrossRef]
- Li, X.; Kuang, J.; Shen, Y.; Majer, M.M.; Nelson, C.C.; Parsawar, K.; Heichman, K.A.; Kuwada, S.K. The atypical histone macroH2A1.2 interacts with HER-2 protein in cancer cells. J. Biol. Chem. 2012, 287, 23171–23183. [Google Scholar] [CrossRef]
- Jayavelu, A.K.; Schnoder, T.M.; Perner, F.; Herzog, C.; Meiler, A.; Krishnamoorthy, G.; Huber, N.; Mohr, J.; Edelmann-Stephan, B.; Austin, R.; et al. Splicing factor YBX1 mediates persistence of JAK2-mutated neoplasms. Nature 2020, 588, 157–163. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Canonical Histone | Histone Variants |
|---|---|
| H2A | H2A.X, H2A.B, H2A.Z.1, H2A.Z.2, H2A.Z.2.2, H2A.J, macroH2A1.1, macroH2A1.2, macroH2A2 |
| H2B | H2BE, H2BW, TH2B |
| H3 | H3.3, H3.Y.1, H3.Y.2, H3.4, H3.5, CENP-A |
| H4 | H4G |
| Types of Cancer | Backgrounds | MacroH2A | Potential Mechanisms | References |
|---|---|---|---|---|
| Anal cancer | MacroH2A2 | - | [110] | |
| Bladder cancer | MacroH2A1 | Repression of calcium channel genes | [117] | |
| MacroH2A1.1 | Repression of Lin28B | [123] | ||
| Breast cancer | Triple-negative | MacroH2A1.1 | EMT involvement | [134] |
| MacroH2A1.2 | Promoting HER-2 signaling | [141] | ||
| Osteoclastogenesis | MacroH2A1.2 | Repression of LOX gene | [69] | |
| Colon cancer | MacroH2A1.1 | Promoting differentiation | [109] | |
| MacroH2A1 | Repression of p16 | [135] | ||
| Hepatocellular carcinoma | Steatosis-associated | MacroH2A1 | Metabolic implications | [66] |
| MacroH2A1 | Limiting drug response | [67] | ||
| MacroH2A1 | Reducing cancer stemness | [65] | ||
| MacroH2A1 | Loss of heterochromatin compaction | [81] | ||
| Lung cancer | MacroH2A1.1 | Senescence association | [108] | |
| MacroH2A1.1 | Metabolic implications | [61] | ||
| Melanoma | Cutaneous | MacroH2A1/2 | Regulation of CDK8 genes | [107,119] |
| Uveal | MacroH2A1 | Potential metabolic implications | [137] | |
| Myelodysplasia | UA2F1 mutation | MacroH2A1 | - | [114] |
| MacroH2A1.1 | Gene regulation of ribosomal biology | [113] | ||
| Prostate cancer | Osteoclastogenesis | MacroH2A1 | Regulation in LTβ | [124] |
| Teratoma | MacroH2A1 | Promoting differentiation | [116] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hsu, C.-J.; Meers, O.; Buschbeck, M.; Heidel, F.H. The Role of MacroH2A Histone Variants in Cancer. Cancers 2021, 13, 3003. https://doi.org/10.3390/cancers13123003
Hsu C-J, Meers O, Buschbeck M, Heidel FH. The Role of MacroH2A Histone Variants in Cancer. Cancers. 2021; 13(12):3003. https://doi.org/10.3390/cancers13123003
Chicago/Turabian StyleHsu, Chen-Jen, Oliver Meers, Marcus Buschbeck, and Florian H. Heidel. 2021. "The Role of MacroH2A Histone Variants in Cancer" Cancers 13, no. 12: 3003. https://doi.org/10.3390/cancers13123003
APA StyleHsu, C.-J., Meers, O., Buschbeck, M., & Heidel, F. H. (2021). The Role of MacroH2A Histone Variants in Cancer. Cancers, 13(12), 3003. https://doi.org/10.3390/cancers13123003