
New Heparanase-Inhibiting Triazolo-Thiadiazoles Attenuate Primary Tumor Growth and Metastasis

 , ,
, ,  ,
, 
Abstract
Simple Summary
Abstract
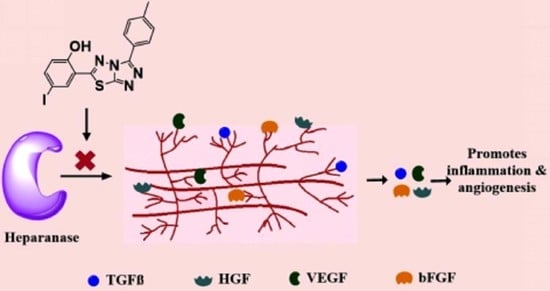
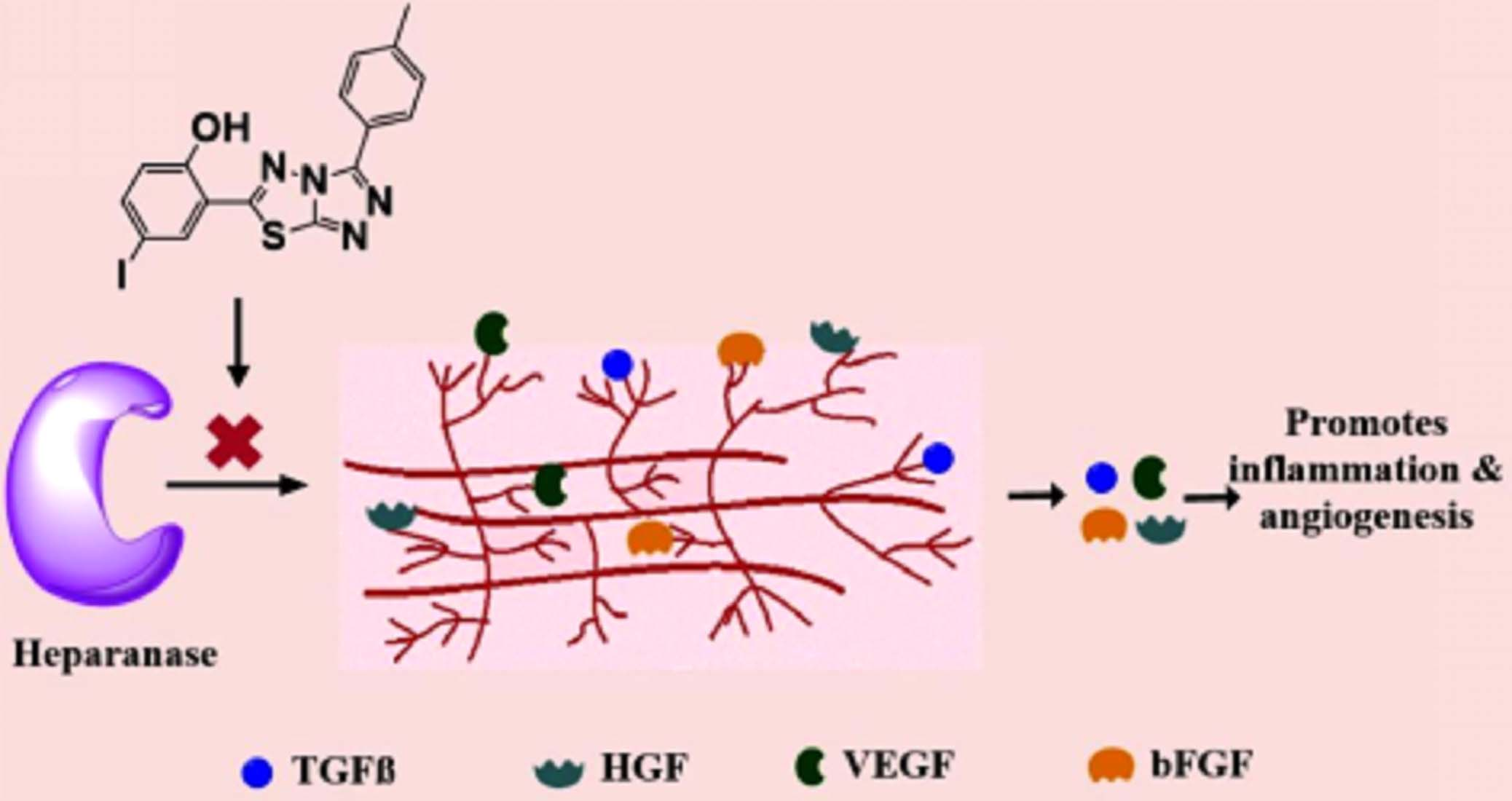
1. Introduction
2. Materials and Methods
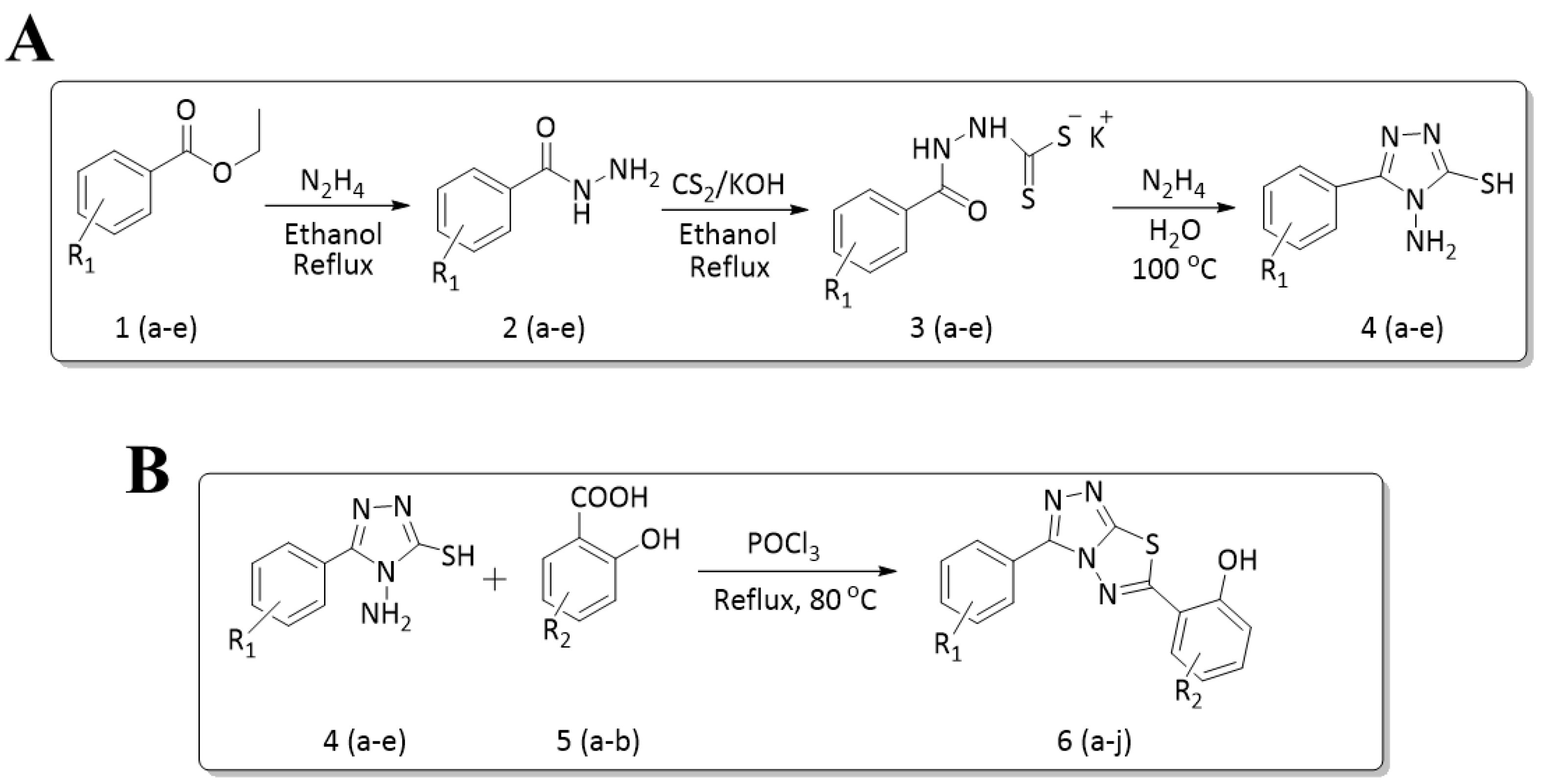
2.1. General Procedure for the Synthesis of 4-amino-5-substituted phenyl-4H-1,2,4-triazole-3-thiol (4a–e)
2.2. General Procedure for the Synthesis of 6-substituted-(3-substituted phenyl-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazole)
2.3. 2,4-diiodo-6-(3-(4-nitrophenyl)-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazol-6-yl)phenol (4-NDI)
2.4. 2-(3-(3-bromophenyl)-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazol-6-yl)-4,6-diiodophenol (3-BDI)
2.5. 2-(3-(3-chlorophenyl)-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazol-6-yl)-4,6-diiodophenol (3-CDI)
2.6. 2,4-diiodo-6-(3-(4-chlorophenyl)-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazol-6-yl)phenol (4-CDI)
2.7. 2,4-diiodo-6-(3-(p-tolyl)-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazol-6-yl)phenol (4-MDI)
2.8. 4-iodo-2-(3-(4-nitrophenyl)-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazol-6-yl)phenol (4-NMI)
2.9. 2-(3-(3-bromophenyl)-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazol-6-yl)-4-iodophenol (3-BMI)
2.10. 2-(3-(3-chlorophenyl)-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazol-6-yl)-4-iodophenol (3-CMI)
2.11. 2-(3-(4-chlorophenyl)-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazol-6-yl)-4-iodophenol (4-CMI)
2.12. 4-iodo-2-(3-(p-tolyl)-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazol-6-yl)phenol (4-MMI)
2.13. Cells and Cell Culture
2.14. Colorimetric Heparanase Assay
2.15. ECM Degradation Heparanase Assay
2.16. Cell Invasion
2.17. Experimental Metastasis
2.18. IVIS Imaging
2.19. MPC-11 Tumor Growth
2.20. Statistics
3. Results
3.1. Synthesis of Title Compounds
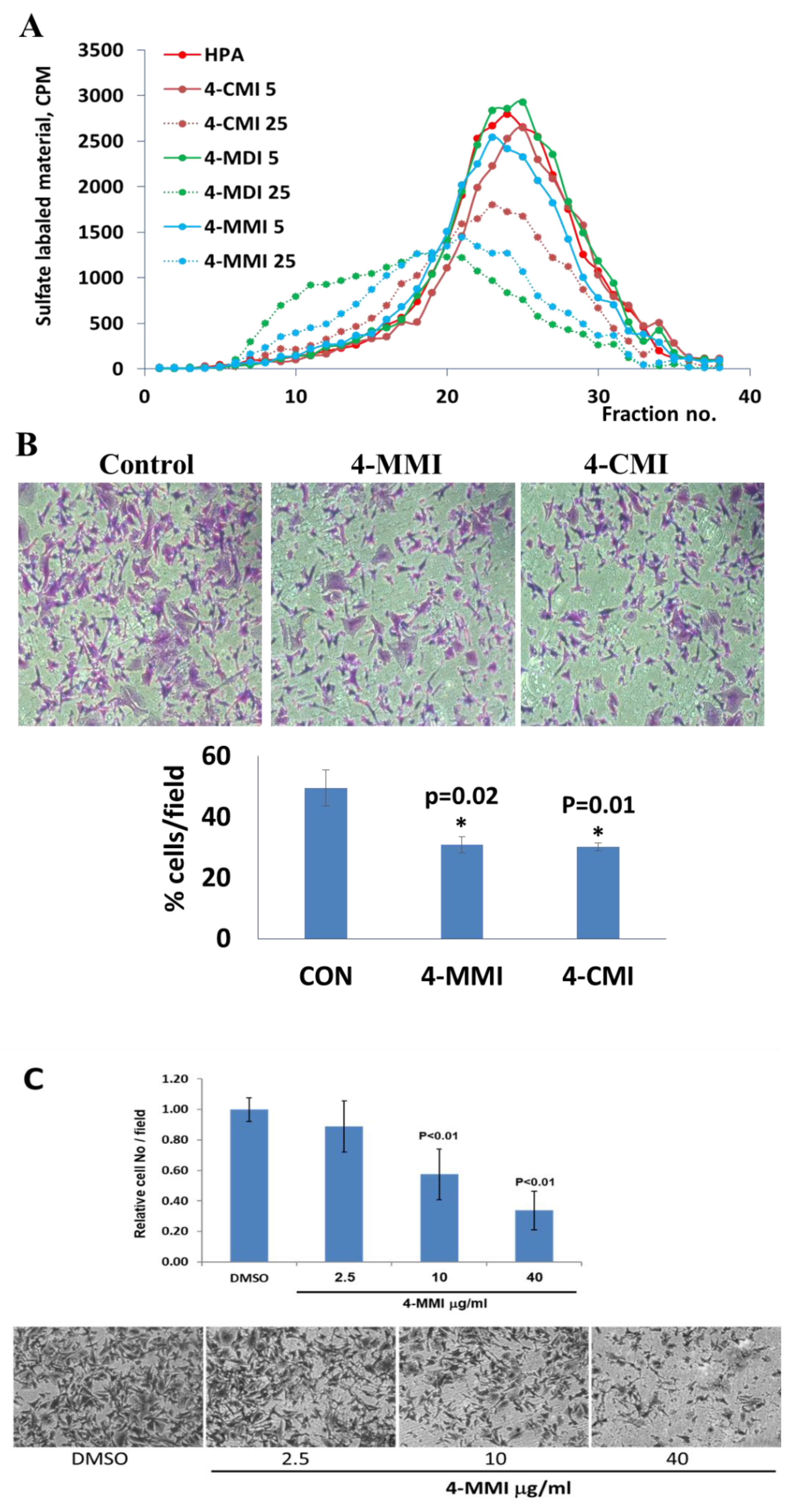
3.2. Newly Synthesized Triazolo–Thiadiazoles Inhibit Heparanase Enzymatic Activity
3.3. Compounds 4-CMI and 4-MMI Inhibit Glioma Cell Invasion
3.4. Compounds 4-CMI and 4-MMI Mitigate Breast Carcinoma Experimental Metastasis
3.5. Compound 4-MMI Reduces Tumor Burden in a Myeloma Model
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bonnans, C.; Chou, J.; Werb, Z. Remodelling the extracellular matrix in development and disease. Nat. Rev. Mol. Cell Biol. 2014, 15, 786–801. [Google Scholar] [CrossRef]
- Pozzi, A.; Yurchenco, P.D.; Iozzo, R.V. The nature and biology of basement membranes. Matrix Biol. 2017, 57–58, 1–11. [Google Scholar] [CrossRef]
- Karamanos, N.K.; Piperigkou, Z.; Theocharis, A.D.; Watanabe, H.; Franchi, M.; Baud, S.; Brézillon, S.; Götte, M.; Passi, A.; Vigetti, D.; et al. Proteoglycan Chemical Diversity Drives Multifunctional Cell Regulation and Therapeutics. Chem. Rev. 2018, 118, 9152–9232. [Google Scholar] [CrossRef]
- Kjellén, L.; Lindahl, U. Specificity of glycosaminoglycan-protein interactions. Curr. Opin. Struct. Biol. 2018, 50, 101–108. [Google Scholar] [CrossRef]
- Basappa; Kumar, C.A.; Swamy, S.N.; Sugahara, K.; Rangappa, K.S. Anti-tumor and anti-angiogenic activity of novel hydantoin derivatives: Inhibition of VEGF secretion in liver metastatic osteosarcoma cells. Bioorganic Med. Chem. 2009, 17, 4928–4934. [Google Scholar] [CrossRef]
- Basappa; Murugan, S.; Kavitha, C.V.; Purushothaman, A.; Nevin, K.G.; Sugahara, K.; Rangappa, K.S. A small oxazine compound as an anti-tumor agent: A novel pyranoside mimetic that binds to VEGF, HB-EGF, and TNF-α. Cancer Lett. 2010, 297, 231–243. [Google Scholar] [CrossRef] [PubMed]
- Hari, S.; Swaroop, T.R.; Preetham, H.D.; Mohan, C.D.; Muddegowda, U.; Basappa, S.; Vlodavsky, I.; Sethi, G.; Rangappa, K.S. Synthesis, Cytotoxic and Heparanase Inhibition Studies of 5-oxo-1-arylpyrrolidine-3- carboxamides of Hydrazides and 4-amino-5-aryl-4H-1,2,4-triazole-3-thiol. Curr. Org. Synth. 2020, 17, 243–250. [Google Scholar] [CrossRef] [PubMed]
- Ilan, N.; Bhattacharya, U.; Barash, U.; Boyango, I.; Yanku, Y.; Gross-Cohen, M.; Vlodavsky, I. Heparanase—The Message Comes in Different Flavors. Adv. Exp. Med. Biol. 2020, 1221, 253–283. [Google Scholar] [CrossRef] [PubMed]
- Vlodavsky, I.; Ilan, N.; Sanderson, R.D. Forty Years of Basic and Translational Heparanase Research. Adv. Exp. Med. Biol. 2020, 1221, 3–59. [Google Scholar] [CrossRef]
- Hao, N.-B.; Tang, B.; Wang, G.-Z.; Xie, R.; Hu, C.-J.; Wang, S.-M.; Wu, Y.-Y.; Liu, E.; Xie, X.; Yang, S.-M. Hepatocyte growth factor (HGF) upregulates heparanase expression via the PI3K/Akt/NF-κB signaling pathway for gastric cancer metastasis. Cancer Lett. 2015, 361, 57–66. [Google Scholar] [CrossRef]
- Kundu, S.; Xiong, A.; Spyrou, A.; Wicher, G.; Marinescu, V.D.; Edqvist, P.-H.D.; Zhang, L.; Essand, M.; Dimberg, A.; Smits, A.; et al. Heparanase Promotes Glioma Progression and Is Inversely Correlated with Patient Survival. Mol. Cancer Res. 2016, 14, 1243–1253. [Google Scholar] [CrossRef]
- Ilan, N.; Elkin, M.; Vlodavsky, I. Regulation, function and clinical significance of heparanase in cancer metastasis and angiogenesis. Int. J. Biochem. Cell Biol. 2006, 38, 2018–2039. [Google Scholar] [CrossRef]
- Vlodavsky, I.; Beckhove, P.; Lerner, I.; Pisano, C.; Meirovitz, A.; Ilan, N.; Elkin, M. Significance of Heparanase in Cancer and Inflammation. Cancer Microenviron. 2011, 5, 115–132. [Google Scholar] [CrossRef]
- Mohan, C.D.; Hari, S.; Preetham, H.D.; Rangappa, S.; Barash, U.; Ilan, N.; Nayak, S.C.; Gupta, V.K.; Basappa; Vlodavsky, I.; et al. Targeting Heparanase in Cancer: Inhibition by Synthetic, Chemically Modified, and Natural Compounds. iScience 2019, 15, 360–390. [Google Scholar] [CrossRef] [PubMed]
- Cassinelli, G.; Torri, G.; Naggi, A. Non-Anticoagulant Heparins as Heparanase Inhibitors. Adv. Exp. Med. Biol. 2020, 1221, 493–522. [Google Scholar] [CrossRef]
- Chhabra, M.; Ferro, V. PI-88 and Related Heparan Sulfate Mimetics. Adv. Exp. Med. Biol. 2020, 1221, 473–491. [Google Scholar] [CrossRef] [PubMed]
- Noseda, A.; Barbieri, P. Roneparstat: Development, Preclinical and Clinical Studies. Adv. Exp. Med. Biol. 2020, 1221, 523–538. [Google Scholar] [CrossRef]
- Hammond, E.; Dredge, K. Heparanase Inhibition by Pixatimod (PG545): Basic Aspects and Future Perspectives. Adv. Exp. Med. Biol. 2020, 1221, 539–565. [Google Scholar] [CrossRef] [PubMed]
- Priya, B.; Kumar, C.A.; Swamy, S.N.; Basappa; Naveen, S.; Prasad, J.S.; Rangappa, K. 2-(2-(2-Ethoxybenzoylamino)-4-chlorophenoxy)-N-(2-ethoxybenzoyl)benzamine inhibits EAT cell induced angiogenesis by down regulation of VEGF secretion. Bioorganic Med. Chem. Lett. 2007, 17, 2775–2780. [Google Scholar] [CrossRef] [PubMed]
- Sanderson, R.D.; Sneed, T.B.; Young, L.A.; Sullivan, G.L.; Lander, A.D. Adhesion of B lymphoid (MPC-11) cells to type I collagen is mediated by integral membrane proteoglycan, syndecan. J. Immunol. 1992, 148, 3902–3911. [Google Scholar] [PubMed]
- Alishekevitz, D.; Bril, R.; Loven, D.; Miller, V.; Voloshin, T.; Gingis-Velistki, S.; Fremder, E.; Scherer, S.J.; Shaked, Y. Differential Therapeutic Effects of Anti-VEGF-A Antibody in Different Tumor Models: Implications for Choosing Appropriate Tumor Models for Drug Testing. Mol. Cancer Ther. 2013, 13, 202–213. [Google Scholar] [CrossRef]
- Baburajeev, C.P.; Mohan, C.D.; Rangappa, S.; Mason, D.J.; Fuchs, J.E.; Bender, A.; Barash, U.; Vlodavsky, I.; Basappa; Rangappa, K.S. S. Identification of Novel Class of Triazolo-Thiadiazoles as Potent Inhibitors of Human Heparanase and their Anticancer Activity. BMC Cancer 2017, 17, 235. [Google Scholar] [CrossRef]
- Hammond, E.; Li, C.P.; Ferro, V. Development of a colorimetric assay for heparanase activity suitable for kinetic analysis and inhibitor screening. Anal. Biochem. 2010, 396, 112–116. [Google Scholar] [CrossRef]
- Vlodavsky, I.; Friedmann, Y.; Elkin, M.; Aingorn, H.; Atzmon, R.; Ishai-Michaeli, R.; Bitan, M.; Pappo, O.; Peretz, T.; Michal, I.; et al. Mammalian heparanase: Gene cloning, expression and function in tumor progression and metastasis. Nat. Med. 1999, 5, 793–802. [Google Scholar] [CrossRef] [PubMed]
- Vlodavsky, I. Preparation of Extracellular Matrices Produced by Cultured Corneal Endothelial and PF-HR9 Endodermal Cells. Curr. Protoc. Cell Biol. 1999, 1. [Google Scholar] [CrossRef] [PubMed]
- Vlodavsky, I.; Fuks, Z.; Bar-Ner, M.; Ariav, Y.; Schirrmacher, V. Lymphoma cell-mediated degradation of sulfated proteoglycans in the subendothelial extracellular matrix: Relationship to tumor cell metastasis. Cancer Res. 1983, 43, 2704–2711. [Google Scholar] [PubMed]
- Zetser, A.; Bashenko, Y.; Miao, H.-Q.; Vlodavsky, I.; Ilan, N. Heparanase affects adhesive and tumorigenic potential of human glioma cells. Cancer Res. 2003, 63, 7733–7741. [Google Scholar]
- Barash, U.; Spyrou, A.; Liu, P.; Vlodavsky, E.; Zhu, C.; Luo, J.; Su, D.; Ilan, N.; Forsberg-Nilsson, K.; Vlodavsky, I.; et al. Heparanase promotes glioma progression via enhancing CD24 expression. Int. J. Cancer 2019, 145, 1596–1608. [Google Scholar] [CrossRef] [PubMed]
- Loka, R.S.; Sletten, E.T.; Barash, U.; Vlodavsky, I.; Nguyen, H.M. Specific Inhibition of Heparanase by a Glycopolymer with Well-Defined Sulfation Pattern Prevents Breast Cancer Metastasis in Mice. ACS Appl. Mater. Interfaces 2019, 11, 244–254. [Google Scholar] [CrossRef]
- Barash, U.; Lapidot, M.; Zohar, Y.; Loomis, C.; Moreira, A.; Feld, S.; Goparaju, C.; Yang, H.; Hammond, E.; Zhang, G.; et al. Involvement of Heparanase in the Pathogenesis of Mesothelioma: Basic Aspects and Clinical Applications. J. Natl. Cancer Inst. 2018, 110, 1102–1114. [Google Scholar] [CrossRef]
- Weissmann, M.; Arvatz, G.; Horowitz, N.; Feld, S.; Naroditsky, I.; Zhang, Y.; Ng, M.; Hammond, E.; Nevo, E.; Vlodavsky, I.; et al. Heparanase-neutralizing antibodies attenuate lymphoma tumor growth and metastasis. Proc. Natl. Acad. Sci. USA 2016, 113, 704–709. [Google Scholar] [CrossRef]
- Barash, U.; Zohar, Y.; Wildbaum, G.; Beider, K.; Nagler, A.; Karin, N.; Ilan, N.; Vlodavsky, I. Heparanase enhances myeloma progression via CXCL10 downregulation. Leukemia 2014, 28, 2178–2187. [Google Scholar] [CrossRef] [PubMed]
- Galli, M.; Chatterjee, M.; Grasso, M.; Specchia, G.; Magen, H.; Einsele, H.; Celeghini, I.; Barbieri, P.; Paoletti, D.; Pace, S.; et al. Phase I study of the heparanase inhibitor roneparstat: An innovative approach for ultiple myeloma therapy. Haematologica 2018, 103, e469–e472. [Google Scholar] [CrossRef] [PubMed]
- Vlodavsky, I.; Singh, P.; Boyango, I.; Gutter-Kapon, L.; Elkin, M.; Sanderson, R.D.; Ilan, N. Heparanase: From basic research to therapeutic applications in cancer and inflammation. Drug Resist. Updates 2016, 29, 54–75. [Google Scholar] [CrossRef]
- Vlodavsky, I.; Gross-Cohen, M.; Weissmann, M.; Ilan, N.; Sanderson, R.D. Opposing Functions of Heparanase-1 and Heparanase-2 in Cancer Progression. Trends Biochem. Sci. 2018, 43, 18–31. [Google Scholar] [CrossRef] [PubMed]
- Giannini, G.; Battistuzzi, G.; Rivara, S. The Control of Heparanase through the Use of Small Molecules. Adv. Exp. Med. Biol. 2020, 1221, 567–603. [Google Scholar] [CrossRef] [PubMed]
- Zcharia, E.; Jia, J.; Zhang, X.; Baraz, L.; Lindahl, U.; Peretz, T.; Vlodavsky, I.; Li, J.-P. Newly Generated Heparanase Knock-Out Mice Unravel Co-Regulation of Heparanase and Matrix Metalloproteinases. PLoS ONE 2009, 4, e5181. [Google Scholar] [CrossRef]
- Pisano, C.; Vlodavsky, I.; Ilan, N.; Zunino, F. The potential of heparanase as a therapeutic target in cancer. Biochem. Pharmacol. 2014, 89, 12–19. [Google Scholar] [CrossRef]
- Rivara, S.; Milazzo, F.M.; Giannini, G. Heparanase: A rainbow pharmacological target associated to multiple pathologies including rare diseases. Future Med. Chem. 2016, 8, 647–680. [Google Scholar] [CrossRef]
- Vlodavsky, I.; Ilan, N.; Naggi, A.; Casu, B. Heparanase: Structure, Biological Functions, and Inhibition by Heparin-Derived Mimetics of Heparan Sulfate. Curr. Pharm. Des. 2007, 13, 2057–2073. [Google Scholar] [CrossRef]
- Messore, A.; Madia, V.N.; Pescatori, L.; Saccoliti, F.; Tudino, V.; De Leo, A.; Bortolami, M.; De Vita, D.; Scipione, L.; Pepi, F.; et al. Novel Symmetrical Benzazolyl Derivatives Endowed with Potent Anti-Heparanase Activity. J. Med. Chem. 2018, 61, 10834–10859. [Google Scholar] [CrossRef]
- Simizu, S.; Ishida, K.; Osada, H. Heparanase as a molecular target of cancer chemotherapy. Cancer Sci. 2004, 95, 553–558. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.-J.; Miao, H.-Q.; Pan, W.; Navarro, E.C.; Tonra, J.R.; Mitelman, S.; Camara, M.M.; Deevi, D.S.; Kiselyov, A.S.; Kussie, P.; et al. N-(4-{[4-(1H-Benzoimidazol-2-yl)-arylamino]-methyl}-phenyl)-benzamide derivatives as small molecule heparanase inhibitors. Bioorganic Med. Chem. Lett. 2006, 16, 404–408. [Google Scholar] [CrossRef]
- Pan, W.; Miao, H.-Q.; Xu, Y.-J.; Navarro, E.C.; Tonra, J.R.; Corcoran, E.; Lahiji, A.; Kussie, P.; Kiselyov, A.S.; Wong, W.C.; et al. 1-[4-(1H-Benzoimidazol-2-yl)-phenyl]-3-[4-(1H-benzoimidazol-2-yl)-phenyl]-urea derivatives as small molecule heparanase inhibitors. Bioorganic Med. Chem. Lett. 2006, 16, 409–412. [Google Scholar] [CrossRef]
- Torre, L.A.; Bray, F.; Siegel, R.L.; Ferlay, J.; Lortet-Tieulent, J.; Jemal, A. Global cancer statistics, 2012: Global Cancer Statistics, 2012. CA Cancer J. Clin. 2015, 65, 87–108. [Google Scholar] [CrossRef] [PubMed]
- Marino, S.; Roodman, G.D. Multiple Myeloma and Bone: The Fatal Interaction. Cold Spring Harb. Perspect. Med. 2018, 8, a031286. [Google Scholar] [CrossRef]
- Purushothaman, A.; Sanderson, R.D. Heparanase: A Dynamic Promoter of Myeloma Progression. Adv. Exp. Med. Biol. 2020, 1221, 331–349. [Google Scholar] [CrossRef]
- Wu, L.; Davies, G.J. An Overview of the Structure, Mechanism and Specificity of Human Heparanase. Adv. Exp. Med. Biol. 2020, 1221, 139–167. [Google Scholar] [CrossRef]
- Wu, L.; Viola, C.M.; Brzozowski, A.M.; Davies, G.J. Structural characterization of human heparanase reveals insights into substrate recognition. Nat. Struct. Mol. Biol. 2015, 22, 1016–1022. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Jiang, J.; Jin, Y.; Kallemeijn, W.W.; Kuo, C.-L.; Artola, M.; Dai, W.; Van Elk, C.; van Eijk, M.; Van Der Marel, G.A.; et al. Activity-based probes for functional interrogation of retaining β-glucuronidases. Nat. Chem. Biol. 2017, 13, 867–873. [Google Scholar] [CrossRef]
- Khamaysi, I.; Hamo-Giladi, D.B.; Abassi, Z. Heparanase in Acute Pancreatitis. Adv. Exp. Med. Biol. 2020, 1221, 703–719. [Google Scholar] [CrossRef]
- Lerner, I.; Hermano, E.; Zcharia, E.; Rodkin, D.; Bulvik, R.; Doviner, V.; Rubinstein, A.M.; Ishai-Michaeli, R.; Atzmon, R.; Sherman, Y.; et al. Heparanase powers a chronic inflammatory circuit that promotes colitis-associated tumorigenesis in mice. J. Clin. Investig. 2011, 121, 1709–1721. [Google Scholar] [CrossRef]
- Masola, V.; Gambaro, G.; Onisto, M. Impact of Heparanse on Organ Fibrosis. Adv. Exp. Med. Biol. 2020, 1221, 669–684. [Google Scholar] [CrossRef]
- Abassi, Z.; Goligorsky, M.S. Heparanase in Acute Kidney Injury. Adv. Exp. Med. Biol. 2020, 1221, 685–702. [Google Scholar] [CrossRef]
- Van Der Vlag, J.; Buijsers, B. Heparanase in Kidney Disease. Adv. Exp. Med. Biol. 2020, 1221, 647–667. [Google Scholar] [CrossRef]
- Rabelink, T.; Berg, B.M.V.D.; Garsen, M.; Wang, G.; Elkin, M.; Van Der Vlag, J. Heparanase: Roles in cell survival, extracellular matrix remodelling and the development of kidney disease. Nat. Rev. Nephrol. 2017, 13, 201–212. [Google Scholar] [CrossRef]
- Gil, N.; Goldberg, R.; Neuman, T.; Garsen, M.; Zcharia, E.; Rubinstein, A.M.; Van Kuppevelt, T.; Meirovitz, A.; Pisano, C.; Li, J.-P.; et al. Heparanase Is Essential for the Development of Diabetic Nephropathy in Mice. Diabetes 2011, 61, 208–216. [Google Scholar] [CrossRef] [PubMed]
- Simeonovic, C.J.; Popp, S.K.; Brown, D.J.; Li, F.-J.; Lafferty, A.R.A.; Freeman, C.; Parish, C.R. Heparanase and Type 1 Diabetes. Adv. Exp. Med. Biol. 2020, 1221, 607–630. [Google Scholar] [CrossRef] [PubMed]
- Shi, C.; Wang, C.; Wang, H.; Yang, C.; Cai, F.; Zeng, F.; Cheng, F.; Liu, Y.; Zhou, T.; Deng, B.; et al. The Potential of Low Molecular Weight Heparin to Mitigate Cytokine Storm in Severe COVID-19 Patients: A Retrospective Cohort Study. Clin. Transl. Sci. 2020, 13, 1087–1095. [Google Scholar] [CrossRef]
- Buijsers, B.; Yanginlar, C.; Maciej-Hulme, M.L.; de Mast, Q.; van der Vlag, J. Beneficial non-anticoagulant mechanisms underlying heparin treatment of COVID-19 patients. EBioMedicine 2020, 59, 102969. [Google Scholar] [CrossRef] [PubMed]
- Martino, C.; Kellman, B.P.; Sandoval, D.R.; Clausen, T.M.; Marotz, C.A.; Song, S.J.; Wandro, S.; Zaramela, L.S.; Salido Benítez, R.A.; Zhu, Q.; et al. Bacterial modification of the host glycosaminoglycan heparan sulfate modulates SARS-CoV-2 infectivity. bioRxiv 2020. [Google Scholar] [CrossRef]
- Agelidis, A.; Shukla, D. Heparanase, Heparan Sulfate and Viral Infection. Adv. Exp. Med. Biol. 2020, 1221, 759–770. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Triazole Amino Thiol Derivatives | Acid | Products |
 4a |  5a |  4-NDI |
 4b |  5a |  3-BDI |
 4c |  5a |  3-CDI |
 4d |  5a |  4-CDI |
 4e |  5a |  4-MDI |
 4a |  5b |  4-NMI |
 4b |  5b |  3-BMI |
 4c |  5b |  3-CMI |
 4d |  5b |  4-CMI |
 4e |  5b |  4-MMI |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barash, U.; Rangappa, S.; Mohan, C.D.; Vishwanath, D.; Boyango, I.; Basappa, B.; Vlodavsky, I.; Rangappa, K.S. New Heparanase-Inhibiting Triazolo-Thiadiazoles Attenuate Primary Tumor Growth and Metastasis. Cancers 2021, 13, 2959. https://doi.org/10.3390/cancers13122959
Barash U, Rangappa S, Mohan CD, Vishwanath D, Boyango I, Basappa B, Vlodavsky I, Rangappa KS. New Heparanase-Inhibiting Triazolo-Thiadiazoles Attenuate Primary Tumor Growth and Metastasis. Cancers. 2021; 13(12):2959. https://doi.org/10.3390/cancers13122959
Chicago/Turabian StyleBarash, Uri, Shobith Rangappa, Chakrabhavi Dhananjaya Mohan, Divakar Vishwanath, Ilanit Boyango, Basappa Basappa, Israel Vlodavsky, and Kanchugarakoppal S. Rangappa. 2021. "New Heparanase-Inhibiting Triazolo-Thiadiazoles Attenuate Primary Tumor Growth and Metastasis" Cancers 13, no. 12: 2959. https://doi.org/10.3390/cancers13122959
APA StyleBarash, U., Rangappa, S., Mohan, C. D., Vishwanath, D., Boyango, I., Basappa, B., Vlodavsky, I., & Rangappa, K. S. (2021). New Heparanase-Inhibiting Triazolo-Thiadiazoles Attenuate Primary Tumor Growth and Metastasis. Cancers, 13(12), 2959. https://doi.org/10.3390/cancers13122959