
TGF Beta Induces Vitamin D Receptor and Modulates Mitochondrial Activity of Human Pancreatic Cancer Cells

 and
and 
Abstract
Simple Summary
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
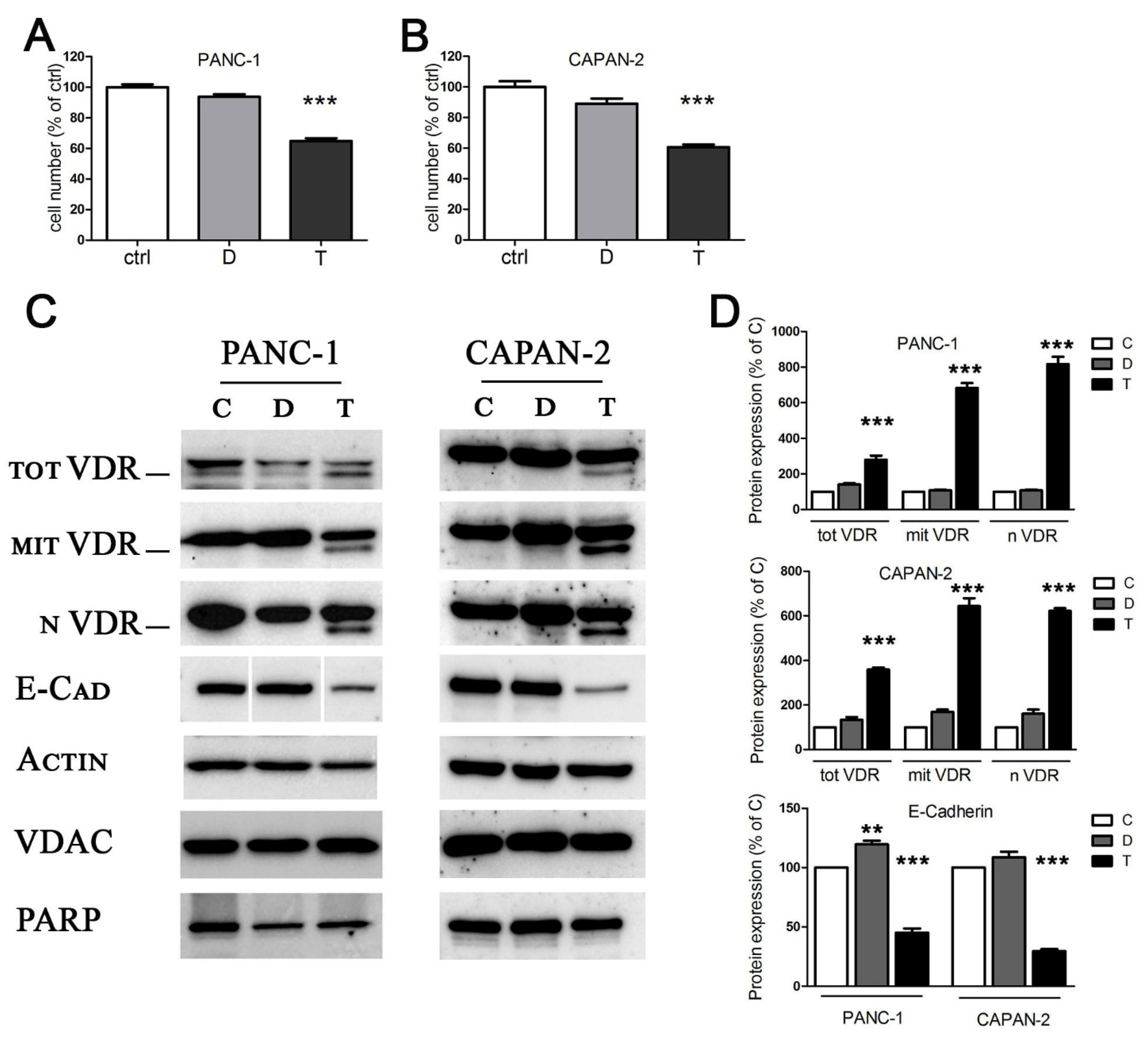
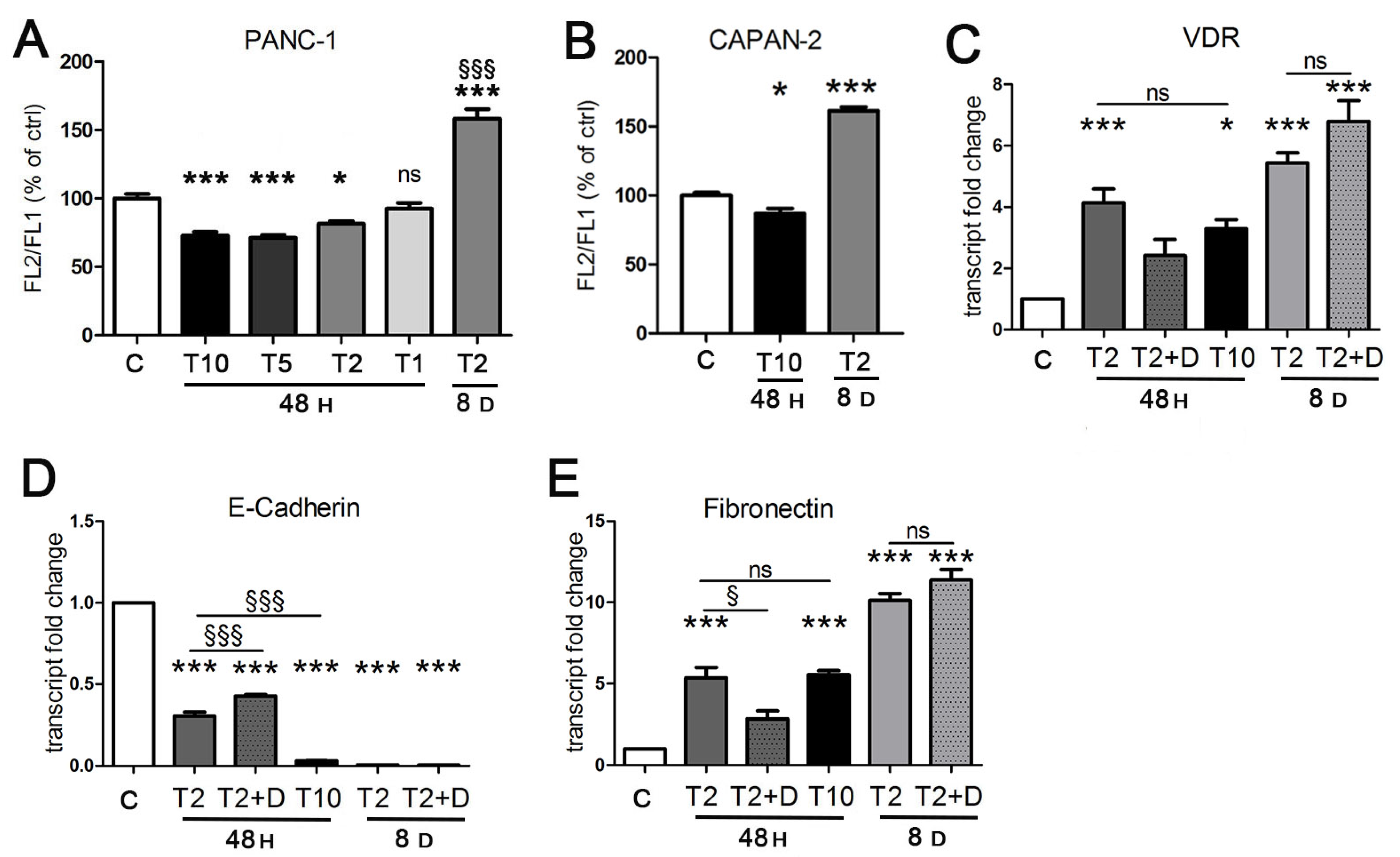
2.1. The Response of Two Human Pancreatic Cancer Models to TGFβ and Vitamin D and the Modulation of Vitamin D Receptor
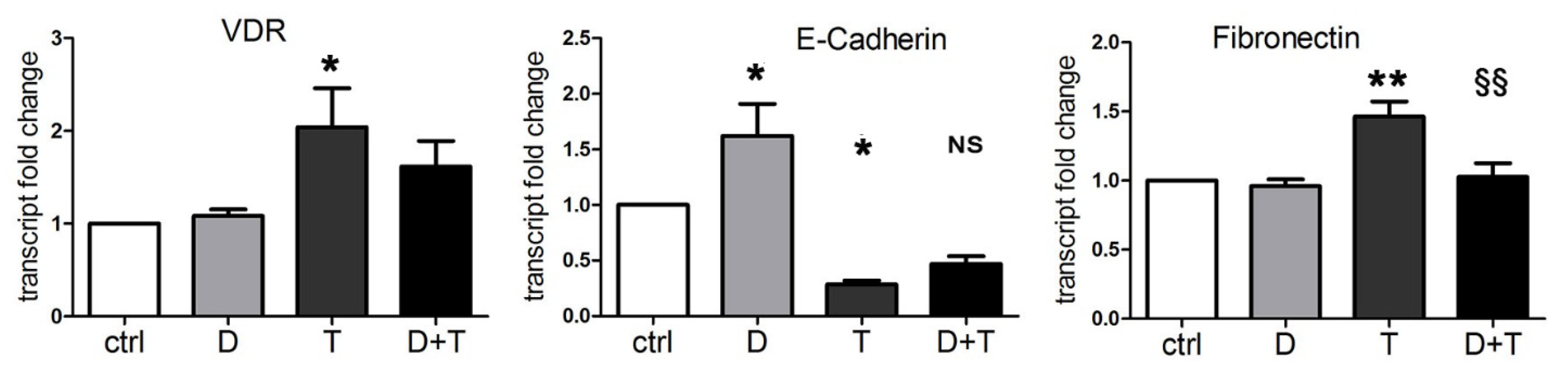
2.2. Vitamin D Opposes the Epithelial–Mesenchymal Transition Induced by TGFβ
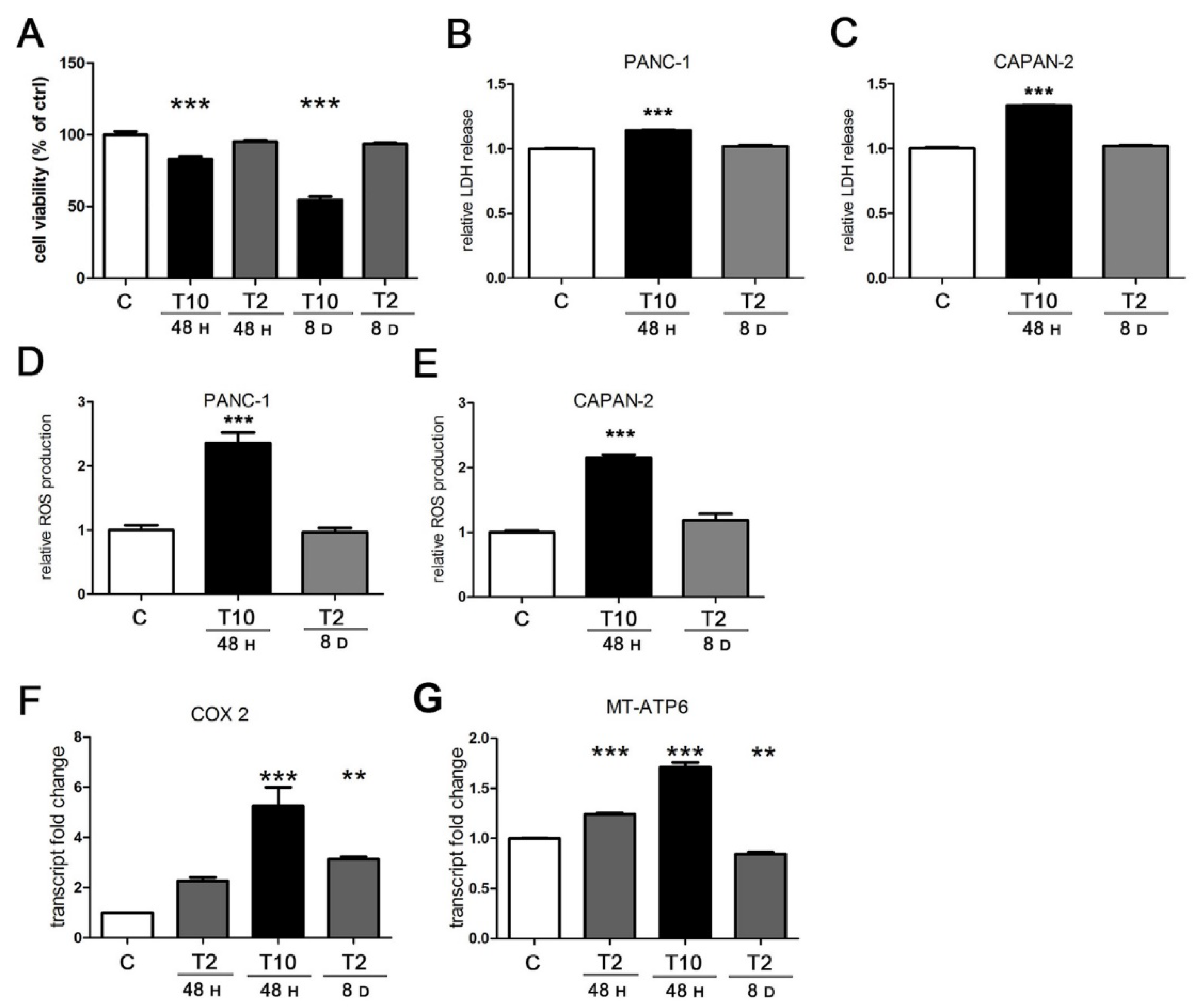
2.3. Characterization of TGFβ Metabolic Effects in Pancreatic Cancer Cells: Dose and Time Dependency
2.4. The Cytotoxic Production of ROS Triggered by TGFβ Is Abated at Low Concentration of the Cytokine
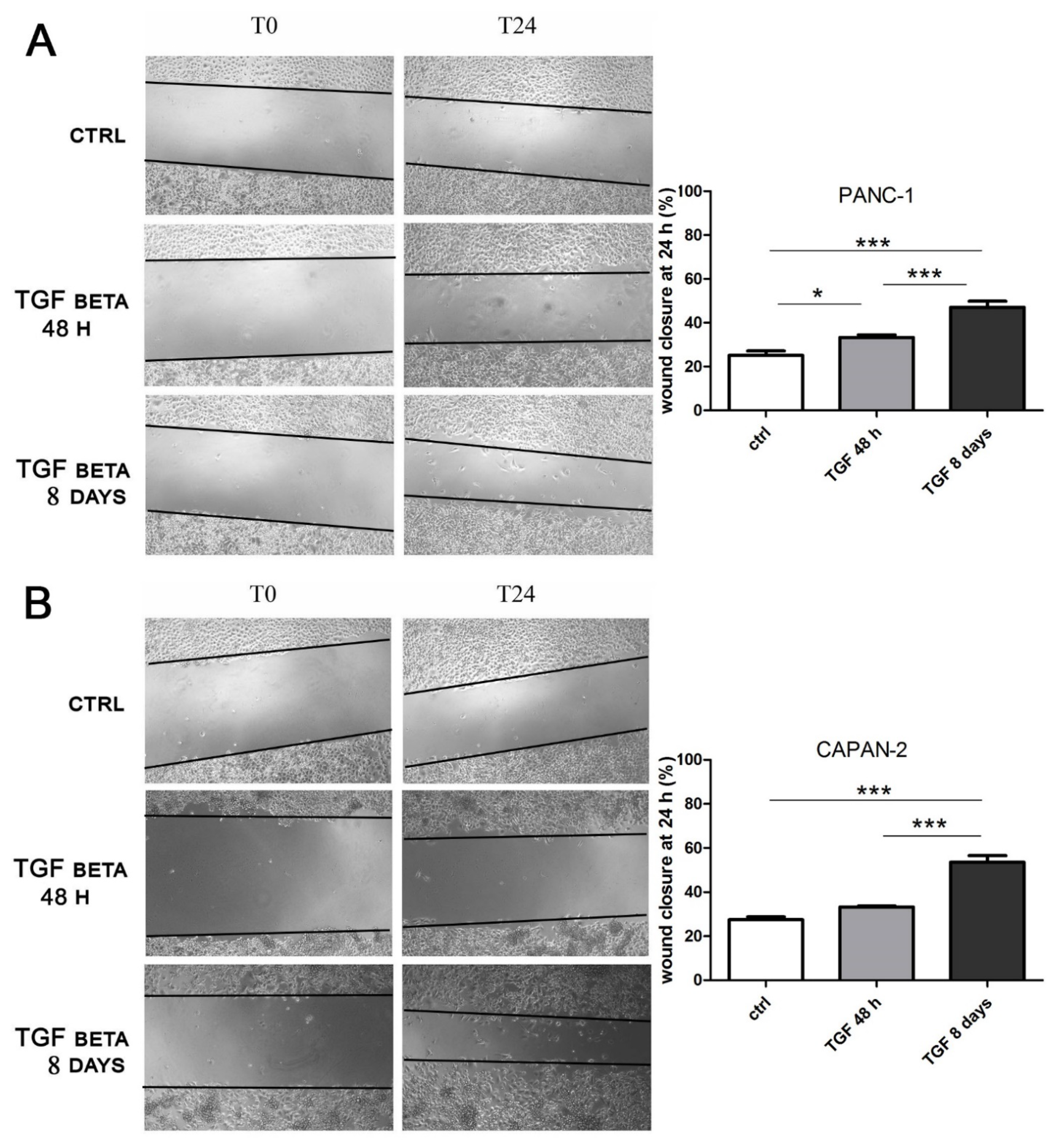
2.5. The Long-Term TGFβ Treatment at Low Doses Promotes Cell Migration
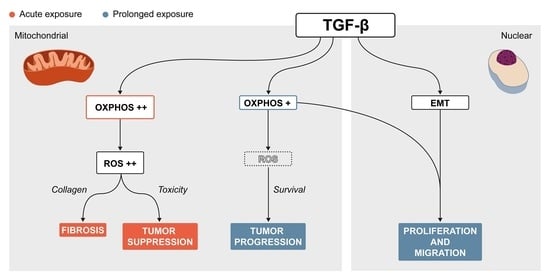
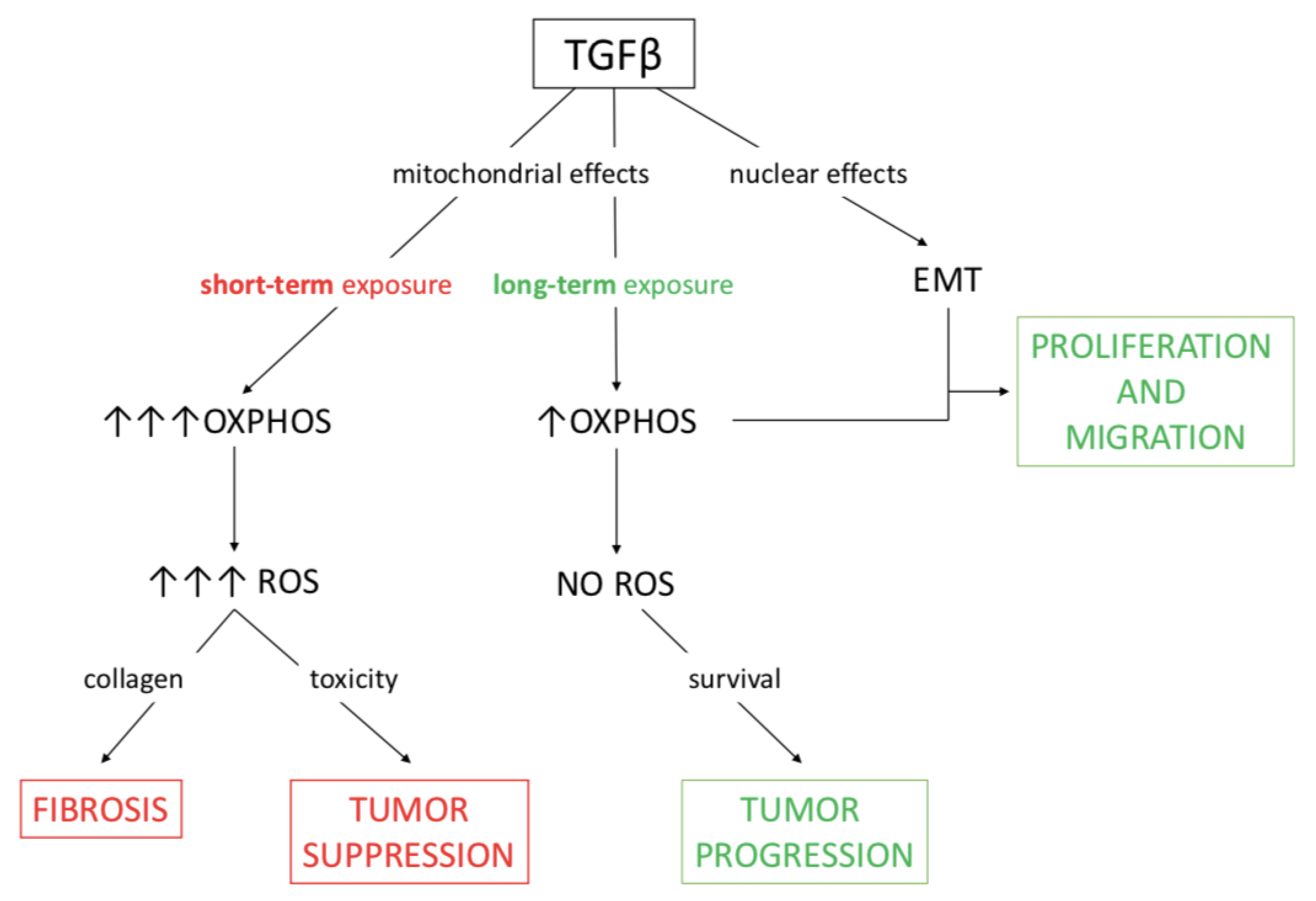
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Treatments
4.2. Cell Proliferation Assay
4.3. Extract Preparation and Western Blotting Analysis
4.4. Real-Time Polymerase Chain Reaction (qRT-PCR)
4.5. Measurement of Mitochondrial Membrane Potential (Δψm)
4.6. Intracellular ROS Production
4.7. Cell Viability Assay
4.8. Toxicity Test (LDH Release)
4.9. Wound Healing Assay
4.10. Band Quantification and Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nolte, M.; Margadant, C. Controlling Immunity and Inflammation through Integrin-Dependent Regulation of TGF-β. Trends Cell Biol. 2020, 30, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-Related Inflammation. Nature 2008, 454, 436–444. [Google Scholar] [CrossRef] [PubMed]
- Seoane, J.; Gomis, R.R. TGF-β Family Signaling in Tumor Suppression and Cancer Progression. Cold Spring Harb. Perspect. Biol. 2017, 9, a022277. [Google Scholar] [CrossRef] [PubMed]
- Akhurst, R.J.; Derynck, R. TGF-β Signaling in Cancer—A Double-Edged Sword. Trends Cell Biol. 2001, 11, S44–S51. [Google Scholar] [CrossRef]
- Padua, D. Roles of TGFβ in Metastasis. Cell Res. 2009, 19, 89–102. [Google Scholar] [CrossRef]
- Kim, S.-J.; Im, Y.-H.; Markowitz, S.D.; Bang, Y.-J. Molecular Mechanisms of Inactivation of TGF-β Receptors during Carcinogenesis. Cytokine Growth Factor Rev. 2000, 11, 159–168. [Google Scholar] [CrossRef]
- Siegel, P.M.; Massagué, J. Cytostatic and Apoptotic Actions of TGF-β in Homeostasis and Cancer. Nat. Rev. Cancer 2003, 3, 807–820. [Google Scholar] [CrossRef] [PubMed]
- Siegel, P.M.; Shu, W.; Cardiff, R.D.; Muller, W.J.; Massagué, J. Transforming Growth Factor β Signaling Impairs Neu-Induced Mammary Tumorigenesis While Promoting Pulmonary Metastasis. Proc. Natl. Acad. Sci. USA 2003, 100, 8430–8435. [Google Scholar] [CrossRef]
- Liu, R.-M.; Desai, L.P. Reciprocal Regulation of TGF-β and Reactive Oxygen Species: A Perverse Cycle for Fibrosis. Redox Biol. 2015, 6, 565–577. [Google Scholar] [CrossRef] [PubMed]
- Ricca, C.; Aillon, A.; Viano, M.; Bergandi, L.; Aldieri, E.; Silvagno, F. Vitamin D Inhibits the Epithelial-Mesenchymal Transition by a Negative Feedback Regulation of TGF-β Activity. J. Steroid Biochem. Mol. Biol. 2019, 187, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Roh, Y.S.; Song, J.; Zhang, B.; Liu, C.; Loomba, R.; Seki, E. Transforming growth factor beta signaling in hepatocytes participates in steatohepatitis through regulation of cell death and lipid metabolism in mice. Hepatology 2014, 59, 483–495. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Fang, L.; Tang, X.; Lu, S.; Tamm, M.; Stolz, D.; Roth, M. TGF-β Upregulated Mitochondria Mass through the SMAD2/3 → C/EBPβ → PRMT1 Signal Pathway in Primary Human Lung Fibroblasts. J. Immunol. 2019, 202, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Yadav, H.; Quijano, C.; Kamaraju, A.K.; Gavrilova, O.; Malek, R.; Chen, W.; Zerfas, P.; Zhigang, D.; Wright, E.C.; Stuelten, C.; et al. Protection from Obesity and Diabetes by Blockade of TGF-β/Smad3 Signaling. Cell Metab. 2011, 14, 67–79. [Google Scholar] [CrossRef] [PubMed]
- Dimeloe, S.; Gubser, P.; Loeliger, J.; Frick, C.; Develioglu, L.; Fischer, M.; Marquardsen, F.; Bantug, G.R.; Thommen, D.; Lecoultre, Y.; et al. Tumor-Derived TGF-β Inhibits Mitochondrial Respiration to Suppress IFN-γ Production by Human CD4+ T Cells. Sci. Signal. 2019, 12, eaav3334. [Google Scholar] [CrossRef]
- Yoon, Y.-S.; Lee, J.-H.; Hwang, S.-C.; Choi, K.S.; Yoon, G. TGF Β1 Induces Prolonged Mitochondrial ROS Generation through Decreased Complex IV Activity with Senescent Arrest in Mv1Lu Cells. Oncogene 2005, 24, 1895–1903. [Google Scholar] [CrossRef] [PubMed]
- Negmadjanov, U.; Godic, Z.; Rizvi, F.; Emelyanova, L.; Ross, G.; Richards, J.; Holmuhamedov, E.L.; Jahangir, A. TGF-Β1-Mediated Differentiation of Fibroblasts Is Associated with Increased Mitochondrial Content and Cellular Respiration. PLoS ONE 2015, 10, e0123046. [Google Scholar] [CrossRef] [PubMed]
- Kawanishi, S.; Ohnishi, S.; Ma, N.; Hiraku, Y.; Murata, M. Crosstalk between DNA Damage and Inflammation in the Multiple Steps of Carcinogenesis. Int. J. Mol. Sci. 2017, 18, 1808. [Google Scholar] [CrossRef] [PubMed]
- Snezhkina, A.V.; Kudryavtseva, A.V.; Kardymon, O.L.; Savvateeva, M.V.; Melnikova, N.V.; Krasnov, G.S.; Dmitriev, A.A. ROS Generation and Antioxidant Defense Systems in Normal and Malignant Cells. Oxidative Med. Cell. Longev. 2019, 2019, 6175804. [Google Scholar] [CrossRef] [PubMed]
- Echizen, K.; Oshima, H.; Nakayama, M.; Oshima, M. The Inflammatory Microenvironment That Promotes Gastrointestinal Cancer Development and Invasion. Adv. Biol. Regul. 2018, 68, 39–45. [Google Scholar] [CrossRef]
- Krstić, J.; Trivanović, D.; Mojsilović, S.; Santibanez, J.F. Transforming Growth Factor-Beta and Oxidative Stress Interplay: Implications in Tumorigenesis and Cancer Progression. Oxidative Med. Cell. Longev. 2015, 2015, 654594. [Google Scholar] [CrossRef] [PubMed]
- Abe, Y.; Sakairi, T.; Beeson, C.; Kopp, J.B. TGF-Β1 Stimulates Mitochondrial Oxidative Phosphorylation and Generation of Reactive Oxygen Species in Cultured Mouse Podocytes, Mediated in Part by the MTOR Pathway. Am. J. Physiol. Renal Physiol. 2013, 305, F1477–F1490. [Google Scholar] [CrossRef]
- Yan, F.; Wang, Y.; Wu, X.; Peshavariya, H.M.; Dusting, G.J.; Zhang, M.; Jiang, F. Nox4 and Redox Signaling Mediate TGF-β-Induced Endothelial Cell Apoptosis and Phenotypic Switch. Cell Death Dis. 2014, 5, e1010. [Google Scholar] [CrossRef] [PubMed]
- Morry, J.; Ngamcherdtrakul, W.; Yantasee, W. Oxidative Stress in Cancer and Fibrosis: Opportunity for Therapeutic Intervention with Antioxidant Compounds, Enzymes, and Nanoparticles. Redox Biol. 2017, 11, 240–253. [Google Scholar] [CrossRef] [PubMed]
- Sawada, T.; Kimura, K.; Nishihara, T.; Onoda, N.; Teraoka, H.; Yamashita, Y.; Yamada, N.; Yashiro, M.; Ohira, M.; Hirakawa, K. TGF-Beta1 down-Regulates ICAM-1 Expression and Enhances Liver Metastasis of Pancreatic Cancer. Adv. Med. Sci. 2006, 51, 60–65. [Google Scholar] [PubMed]
- Ye, H.; Zhou, Q.; Zheng, S.; Li, G.; Lin, Q.; Wei, L.; Fu, Z.; Zhang, B.; Liu, Y.; Li, Z.; et al. Tumor-Associated Macrophages Promote Progression and the Warburg Effect via CCL18/NF-KB/VCAM-1 Pathway in Pancreatic Ductal Adenocarcinoma. Cell Death Dis. 2018, 9, 453. [Google Scholar] [CrossRef] [PubMed]
- Deer, E.L.; González-Hernández, J.; Coursen, J.D.; Shea, J.E.; Ngatia, J.; Scaife, C.L.; Firpo, M.A.; Mulvihill, S.J. Phenotype and Genotype of Pancreatic Cancer Cell Lines. Pancreas 2010, 39, 425–435. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Dong, M.; Sheng, W.; Liu, Q.; Yu, D.; Dong, Q.; Li, Q.; Wang, J. Expression of Vitamin D Receptor as a Potential Prognostic Factor and Therapeutic Target in Pancreatic Cancer. Histopathology 2015, 67, 386–397. [Google Scholar] [CrossRef]
- Itoigawa, Y.; Harada, N.; Harada, S.; Katsura, Y.; Makino, F.; Ito, J.; Nurwidya, F.; Kato, M.; Takahashi, F.; Atsuta, R.; et al. TWEAK Enhances TGF-β-Induced Epithelial-Mesenchymal Transition in Human Bronchial Epithelial Cells. Respir. Res. 2015, 16, 48. [Google Scholar] [CrossRef] [PubMed]
- Hosper, N.A.; van den Berg, P.P.; de Rond, S.; Popa, E.R.; Wilmer, M.J.; Masereeuw, R.; Bank, R.A. Epithelial-to-Mesenchymal Transition in Fibrosis: Collagen Type I Expression Is Highly Upregulated after EMT, but Does Not Contribute to Collagen Deposition. Exp. Cell Res. 2013, 319, 3000–3009. [Google Scholar] [CrossRef]
- Fischer, K.D.; Agrawal, D.K. Vitamin D Regulating TGF-β Induced Epithelial-Mesenchymal Transition. Respir. Res. 2014, 15, 146. [Google Scholar] [CrossRef] [PubMed]
- Silvagno, F.; Consiglio, M.; Foglizzo, V.; Destefanis, M.; Pescarmona, G. Mitochondrial Translocation of Vitamin D Receptor Is Mediated by the Permeability Transition Pore in Human Keratinocyte Cell Line. PLoS ONE 2013, 8, e54716. [Google Scholar] [CrossRef] [PubMed]
- Consiglio, M.; Destefanis, M.; Morena, D.; Foglizzo, V.; Forneris, M.; Pescarmona, G.; Silvagno, F. The Vitamin D Receptor Inhibits the Respiratory Chain, Contributing to the Metabolic Switch That Is Essential for Cancer Cell Proliferation. PLoS ONE 2014, 9, e115816. [Google Scholar] [CrossRef] [PubMed]
- Silvagno, F.; de Vivo, E.; Attanasio, A.; Gallo, V.; Mazzucco, G.; Pescarmona, G. Mitochondrial Localization of Vitamin D Receptor in Human Platelets and Differentiated Megakaryocytes. PLoS ONE 2010, 5, e8670. [Google Scholar] [CrossRef] [PubMed]
- Larriba, M.J.; García de Herreros, A.; Muñoz, A. Vitamin D and the Epithelial to Mesenchymal Transition. Stem Cells Int. 2016, 2016, 6213872. [Google Scholar] [CrossRef] [PubMed]
- Stone, M.L.; Beatty, G.L. Cellular Determinants and Therapeutic Implications of Inflammation in Pancreatic Cancer. Pharmacol. Ther. 2019, 201, 202–213. [Google Scholar] [CrossRef] [PubMed]
- Oglio, R.; Thomasz, L.; Salvarredi, L.; Juvenal, G.; Pisarev, M. Comparative Effects of Transforming Growth Factor Beta Isoforms on Redox Metabolism in Thyroid Cells. Mol. Cell. Endocrinol. 2018, 470, 168–178. [Google Scholar] [CrossRef]
- Hüttemann, M.; Lee, I.; Pecinova, A.; Pecina, P.; Przyklenk, K.; Doan, J.W. Regulation of Oxidative Phosphorylation, the Mitochondrial Membrane Potential, and Their Role in Human Disease. J. Bioenerg. Biomembr. 2008, 40, 445–456. [Google Scholar] [CrossRef]
- Chen, J.; Tang, Z.; Slominski, A.T.; Li, W.; Żmijewski, M.A.; Liu, Y.; Chen, J. Vitamin D and Its Analogs as Anticancer and Anti-Inflammatory Agents. Eur. J. Med. Chem. 2020, 207, 112738. [Google Scholar] [CrossRef]
- Liu, W.; Zhang, L.; Xu, H.-J.; Li, Y.; Hu, C.-M.; Yang, J.-Y.; Sun, M.-Y. The Anti-Inflammatory Effects of Vitamin D in Tumorigenesis. Int. J. Mol. Sci. 2018, 19, 2736. [Google Scholar] [CrossRef]
- Pitarresi, J.R.; Rustgi, A.K. Mechanisms Underlying Metastatic Pancreatic Cancer. Adv. Exp. Med. Biol. 2019, 1164, 3–10. [Google Scholar] [CrossRef]
- Bulle, A.; Lim, K.-H. Beyond Just a Tight Fortress: Contribution of Stroma to Epithelial-Mesenchymal Transition in Pancreatic Cancer. Signal Transduct. Target. Ther. 2020, 5, 249. [Google Scholar] [CrossRef] [PubMed]
- Löhr, M.; Schmidt, C.; Ringel, J.; Kluth, M.; Müller, P.; Nizze, H.; Jesnowski, R. Transforming Growth Factor-β1 Induces Desmoplasia in an Experimental Model of Human Pancreatic Carcinoma. Cancer Res. 2001, 61, 550–555. [Google Scholar] [PubMed]
- Stylianou, A.; Gkretsi, V.; Stylianopoulos, T. Transforming Growth Factor-β Modulates Pancreatic Cancer Associated Fibroblasts Cell Shape, Stiffness and Invasion. Biochim. Biophys. Acta Gen. Subj. 2018, 1862, 1537–1546. [Google Scholar] [CrossRef]
- Altieri, B.; Grant, W.B.; Della Casa, S.; Orio, F.; Pontecorvi, A.; Colao, A.; Sarno, G.; Muscogiuri, G. Vitamin D and Pancreas: The Role of Sunshine Vitamin in the Pathogenesis of Diabetes Mellitus and Pancreatic Cancer. Crit. Rev. Food Sci. Nutr. 2017, 57, 3472–3488. [Google Scholar] [CrossRef] [PubMed]
- Barreto, S.G.; Neale, R.E. Vitamin D and Pancreatic Cancer. Cancer Lett. 2015, 368, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Quek, L.-E.; Sultani, G.; Turner, N. Epithelial-Mesenchymal Transition Induction Is Associated with Augmented Glucose Uptake and Lactate Production in Pancreatic Ductal Adenocarcinoma. Cancer Metab. 2016, 4, 19. [Google Scholar] [CrossRef]
- Menezes, S.V.; Fouani, L.; Huang, M.L.H.; Geleta, B.; Maleki, S.; Richardson, A.; Richardson, D.R.; Kovacevic, Z. The Metastasis Suppressor, NDRG1, Attenuates Oncogenic TGF-β and NF-ΚB Signaling to Enhance Membrane E-Cadherin Expression in Pancreatic Cancer Cells. Carcinogenesis 2019, 40, 805–818. [Google Scholar] [CrossRef]
- Liu, Q.; Sheng, W.; Dong, M.; Dong, X.; Dong, Q.; Li, F. Gli1 Promotes Transforming Growth Factor-Beta1–and Epidermal Growth Factor-Induced Epithelial to Mesenchymal Transition in Pancreatic Cancer Cells. Surgery 2015, 158, 211–224. [Google Scholar] [CrossRef]
- Meyer-Schaller, N.; Heck, C.; Tiede, S.; Yilmaz, M.; Christofori, G. Foxf2 Plays a Dual Role during Transforming Growth Factor Beta-Induced Epithelial to Mesenchymal Transition by Promoting Apoptosis yet Enabling Cell Junction Dissolution and Migration. Breast Cancer Res. 2018, 20, 118. [Google Scholar] [CrossRef]
- Saxena, M.; Kalathur, R.K.R.; Neutzner, M.; Christofori, G. PyMT-1099, a Versatile Murine Cell Model for EMT in Breast Cancer. Sci. Rep. 2018, 8, 12123. [Google Scholar] [CrossRef]
- Pavan, S.; Meyer-Schaller, N.; Diepenbruck, M.; Kalathur, R.K.R.; Saxena, M.; Christofori, G. A Kinome-Wide High-Content SiRNA Screen Identifies MEK5-ERK5 Signaling as Critical for Breast Cancer Cell EMT and Metastasis. Oncogene 2018, 37, 4197–4213. [Google Scholar] [CrossRef]
- Katsuno, Y.; Meyer, D.S.; Zhang, Z.; Shokat, K.M.; Akhurst, R.J.; Miyazono, K.; Derynck, R. Chronic TGF-β Exposure Drives Stabilized EMT, Tumor Stemness, and Cancer Drug Resistance with Vulnerability to Bitopic MTOR Inhibition. Sci. Signal. 2019, 12, eaau8544. [Google Scholar] [CrossRef]
- Schlegel, N.C.; von Planta, A.; Widmer, D.S.; Dummer, R.; Christofori, G. PI3K Signalling Is Required for a TGFβ-Induced Epithelial-Mesenchymal-like Transition (EMT-like) in Human Melanoma Cells. Exp. Dermatol. 2015, 24, 22–28. [Google Scholar] [CrossRef]
- Zhao, J.; Zhang, J.; Yu, M.; Xie, Y.; Huang, Y.; Wolff, D.W.; Abel, P.W.; Tu, Y. Mitochondrial Dynamics Regulates Migration and Invasion of Breast Cancer Cells. Oncogene 2013, 32, 4814–4824. [Google Scholar] [CrossRef]
- Zhou, H.; Zhang, B.; Zheng, J.; Yu, M.; Zhou, T.; Zhao, K.; Jia, Y.; Gao, X.; Chen, C.; Wei, T. The Inhibition of Migration and Invasion of Cancer Cells by Graphene via the Impairment of Mitochondrial Respiration. Biomaterials 2014, 35, 1597–1607. [Google Scholar] [CrossRef] [PubMed]
- Cunniff, B.; McKenzie, A.J.; Heintz, N.H.; Howe, A.K. AMPK Activity Regulates Trafficking of Mitochondria to the Leading Edge during Cell Migration and Matrix Invasion. Mol. Biol. Cell 2016, 27, 2662–2674. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, W.; Zhang, T.; Zhou, Q.; Liu, J.; Liu, Y.; Kong, D.; Yu, W.; Liu, R.; Hai, C. TGF-Β1 Induces Epithelial-to-Mesenchymal Transition via Inhibiting Mitochondrial Functions in A549 Cells. Free Radic. Res. 2018, 52, 1432–1444. [Google Scholar] [CrossRef] [PubMed]
- Schwörer, S.; Berisa, M.; Violante, S.; Qin, W.; Zhu, J.; Hendrickson, R.C.; Cross, J.R.; Thompson, C.B. Proline Biosynthesis Is a Vent for TGFβ-Induced Mitochondrial Redox Stress. EMBO J. 2020, 39, e103334. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Bermúdez, A.; Laza-Briviesca, R.; Vicente-Blanco, R.J.; García-Grande, A.; Coronado, M.J.; Laine-Menéndez, S.; Alfaro, C.; Sanchez, J.C.; Franco, F.; Calvo, V.; et al. Cancer-Associated Fibroblasts Modify Lung Cancer Metabolism Involving ROS and TGF-β Signaling. Free Radic. Biol. Med. 2019, 130, 163–173. [Google Scholar] [CrossRef]
- Son, J.; Lyssiotis, C.A.; Ying, H.; Wang, X.; Hua, S.; Ligorio, M.; Perera, R.M.; Ferrone, C.R.; Mullarky, E.; Shyh-Chang, N.; et al. Glutamine Supports Pancreatic Cancer Growth through a KRAS-Regulated Metabolic Pathway. Nature 2013, 496, 101–105. [Google Scholar] [CrossRef]
- Kalyanaraman, B.; Darley-Usmar, V.; Davies, K.J.A.; Dennery, P.A.; Forman, H.J.; Grisham, M.B.; Mann, G.E.; Moore, K.; Roberts, L.J.; Ischiropoulos, H. Measuring Reactive Oxygen and Nitrogen Species with Fluorescent Probes: Challenges and Limitations. Free Radic. Biol. Med. 2012, 52, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Mosmann, T. Rapid Colorimetric Assay for Cellular Growth and Survival: Application to Proliferation and Cytotoxicity Assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Ricca, C.; Aillon, A.; Bergandi, L.; Alotto, D.; Castagnoli, C.; Silvagno, F. Vitamin D Receptor Is Necessary for Mitochondrial Function and Cell Health. Int. J. Mol. Sci. 2018, 19, 1672. [Google Scholar] [CrossRef] [PubMed]
- Loughlin, D.T.; Artlett, C.M. Modification of Collagen by 3-Deoxyglucosone Alters Wound Healing through Differential Regulation of P38 MAP Kinase. PLoS ONE 2011, 6, e18676. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, M.A.; Freitas, J.P.; Mazher Hussain, S.; Glazer, E.S. TGF-β Inhibitors in Metastatic Pancreatic Ductal Adenocarcinoma. J. Gastrointest. Cancer 2019, 50, 207–213. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fiz, C.; Apprato, G.; Ricca, C.; Aillon, A.; Bergandi, L.; Silvagno, F. TGF Beta Induces Vitamin D Receptor and Modulates Mitochondrial Activity of Human Pancreatic Cancer Cells. Cancers 2021, 13, 2932. https://doi.org/10.3390/cancers13122932
Fiz C, Apprato G, Ricca C, Aillon A, Bergandi L, Silvagno F. TGF Beta Induces Vitamin D Receptor and Modulates Mitochondrial Activity of Human Pancreatic Cancer Cells. Cancers. 2021; 13(12):2932. https://doi.org/10.3390/cancers13122932
Chicago/Turabian StyleFiz, Camilla, Giulia Apprato, Chiara Ricca, Alessia Aillon, Loredana Bergandi, and Francesca Silvagno. 2021. "TGF Beta Induces Vitamin D Receptor and Modulates Mitochondrial Activity of Human Pancreatic Cancer Cells" Cancers 13, no. 12: 2932. https://doi.org/10.3390/cancers13122932
APA StyleFiz, C., Apprato, G., Ricca, C., Aillon, A., Bergandi, L., & Silvagno, F. (2021). TGF Beta Induces Vitamin D Receptor and Modulates Mitochondrial Activity of Human Pancreatic Cancer Cells. Cancers, 13(12), 2932. https://doi.org/10.3390/cancers13122932