
Biparatopic Protein Nanoparticles for the Precision Therapy of CXCR4+ Cancers

 ,
,  ,
,  , and
, and
Simple Summary
Abstract
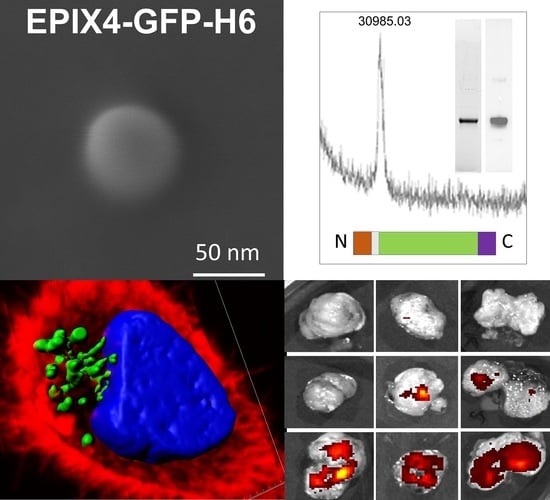
1. Introduction
2. Materials and Methods
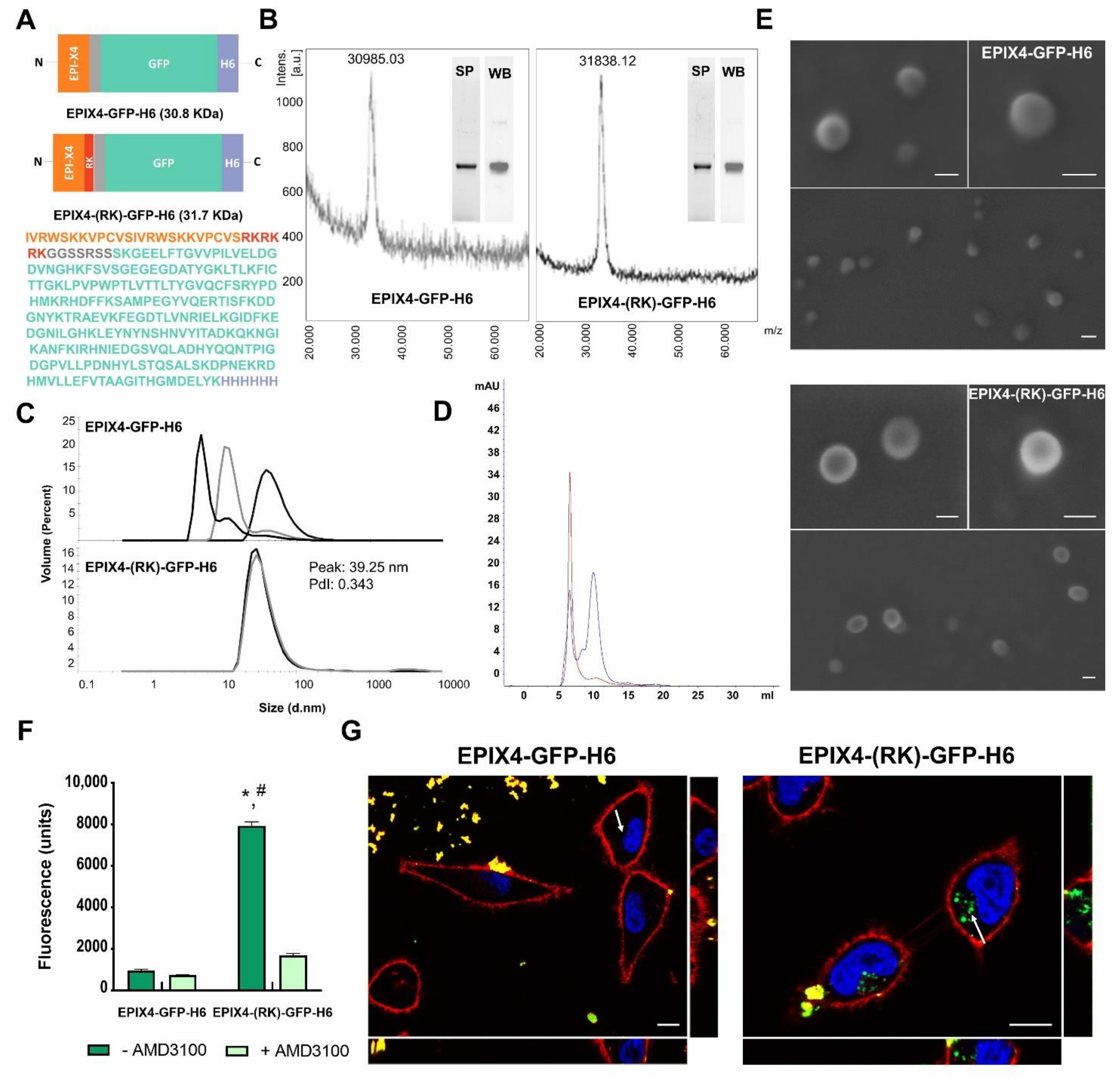
2.1. Protein Design, Production and Purification
2.2. Protein Characterization
2.3. Electron Microscopy
2.4. Cell Culture, Flow Cytometry and Cytotoxicity Assay
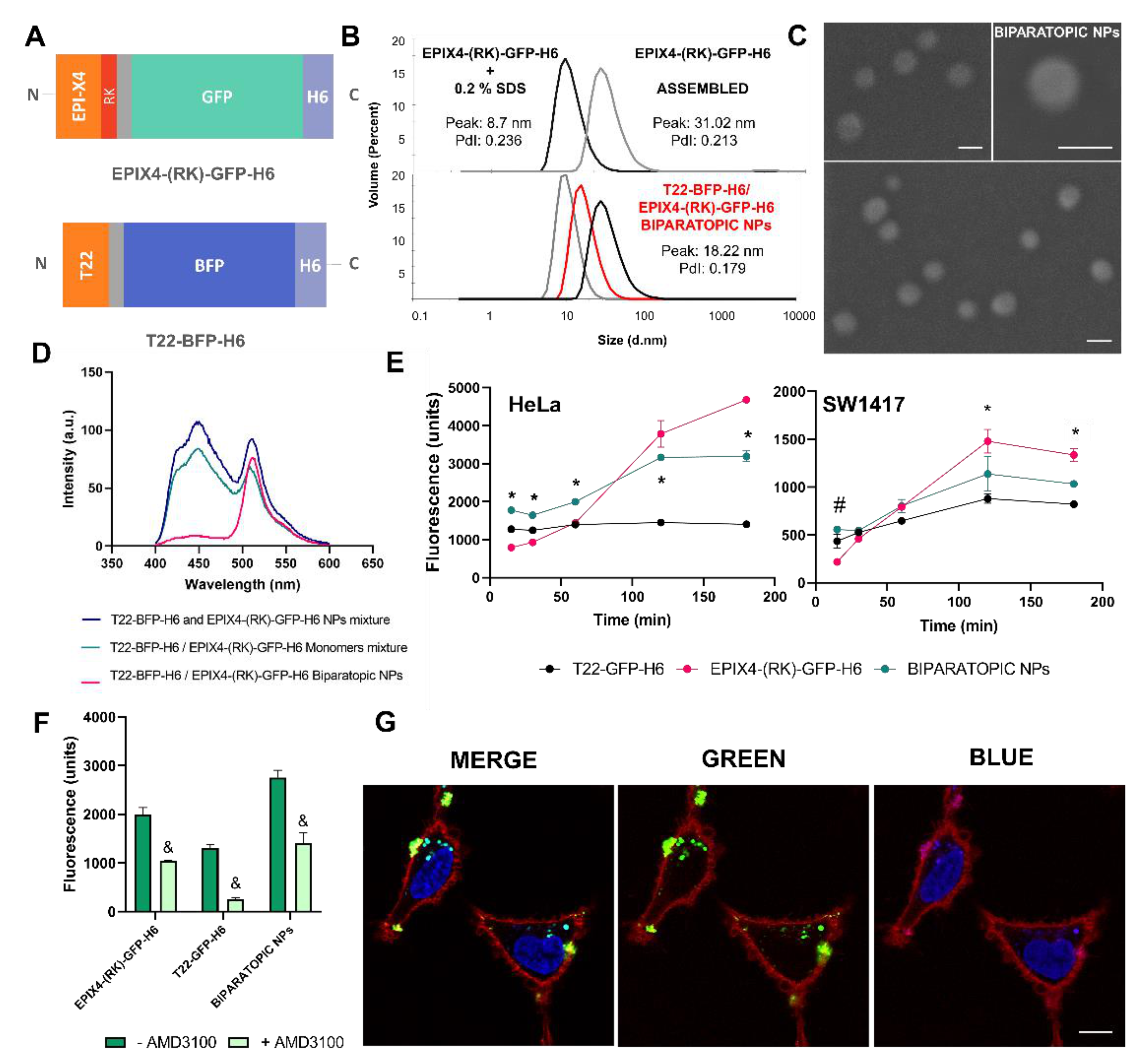
2.5. Production and Characterization of Biparatopic Nanoparticles
2.6. Cytotoxicity Studies
2.7. Analysis of Apoptosis
2.8. Confocal Assay
2.9. Evaluation of EPIX4-(RK)-GFP-H6 and Biparatopic Nanoparticles Biodistribution in a Colorectal Subcutaneous Cancer Mouse
2.10. Histopathology and Detection of Apoptotic Bodies and Mitotic Figures
2.11. Statistical Analyses
3. Results
4. Discussion
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Das, M.; Mohanty, C.; Sahoo, S.K. Ligand-based targeted therapy for cancer tissue. Expert Opin. Drug Deliv. 2009, 6, 285–304. [Google Scholar] [CrossRef] [PubMed]
- Dawidczyk, C.M.; Kim, C.; Park, J.H.; Russell, L.M.; Lee, K.H.; Pomper, M.G.; Searson, P.C. State-of-the-art in design rules for drug delivery platforms: Lessons learned from FDA-approved nanomedicines. J. Control. Release 2014, 187, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Lech, G.; Słotwiński, R.; Słodkowski, M.; Krasnodębski, I.W. Colorectal cancer tumour markers and biomarkers: Recent therapeutic advances. World J. Gastroenterol. 2016, 22, 1745–1755. [Google Scholar] [CrossRef] [PubMed]
- U.S. Food and Drug Administration. Human insulin receives FDA approval. FDA Drug Bull. 1982, 12, 18–19. [Google Scholar]
- Large, D.E.; Sousy, J.R.; Hebert, J.; Auguste, D.T. Advances in Receptor-Mediated, Tumor-Targeted Drug Delivery. Adv. Ther. 2019, 2, 1800091. [Google Scholar] [CrossRef]
- Duncan, R.; Gaspar, R. Nanomedicine(s) under the microscope. Mol. Pharm. 2011, 8, 2101–2141. [Google Scholar] [CrossRef]
- Lee, R.J.; Low, P.S. Delivery of liposomes into cultured KB cells via folate receptor-mediated endocytosis. J. Biol. Chem. 1994, 269, 3198–3204. [Google Scholar] [CrossRef]
- Binkowski, T.A.; Jiang, W.; Roux, B.; Anderson, W.F.; Joachimiak, A. Virtual high-throughput ligand screening. Methods Mol. Biol. 2014, 1140, 251–261. [Google Scholar] [CrossRef]
- Yaginuma, K.; Aoki, W.; Miura, N.; Ohtani, Y.; Aburaya, S.; Kogawa, M.; Nishikawa, Y.; Hosokawa, M.; Takeyama, H.; Ueda, M. High-throughput identification of peptide agonists against GPCRs by co-culture of mammalian reporter cells and peptide-secreting yeast cells using droplet microfluidics. Sci. Rep. 2019, 9, 10920. [Google Scholar] [CrossRef]
- Domanska, U.M.; Kruizinga, R.C.; Nagengast, W.B.; Timmer-Bosscha, H.; Huls, G.; de Vries, E.G.; Walenkamp, A.M. A review on CXCR4/CXCL12 axis in oncology: No place to hide. Eur. J. Cancer 2013, 49, 219–230. [Google Scholar] [CrossRef]
- Zhao, H.; Guo, L.; Zhao, J.; Weng, H.; Zhao, B. CXCR4 over-expression and survival in cancer: A system review and meta-analysis. Oncotarget 2015, 6, 5022–5040. [Google Scholar] [CrossRef]
- Xu, C.; Zheng, L.; Li, D.; Chen, G.; Gu, J.; Chen, J.; Yao, Q. CXCR4 overexpression is correlated with poor prognosis in colorectal cancer. Life Sci. 2018, 208, 333–340. [Google Scholar] [CrossRef]
- Benedicto, A.; Romayor, I.; Arteta, B. CXCR4 receptor blockage reduces the contribution of tumor and stromal cells to the metastatic growth in the liver. Oncol. Rep. 2018, 39, 2022–2030. [Google Scholar] [CrossRef]
- Muz, B.; Azab, F.; Fiala, M.; King, J.; Kohnen, D.; Fogler, W.E.; Smith, T.; Magnani, J.L.; Vij, R.; Azab, A.K. Inhibition of E-Selectin (GMI-1271) or E-selectin together with CXCR4 (GMI-1359) re-sensitizes multiple myeloma to therapy. Blood Cancer J. 2019, 9, 68. [Google Scholar] [CrossRef]
- Murakami, T.; Zhang, T.Y.; Koyanagi, Y.; Tanaka, Y.; Kim, J.; Suzuki, Y.; Minoguchi, S.; Tamamura, H.; Waki, M.; Matsumoto, A.; et al. Inhibitory mechanism of the CXCR4 antagonist T22 against human immunodeficiency virus type 1 infection. J. Virol. 1999, 73, 7489–7496. [Google Scholar] [CrossRef]
- Unzueta, U.; Céspedes, M.V.; Ferrer-Miralles, N.; Casanova, I.; Cedano, J.; Corchero, J.L.; Domingo-Espín, J.; Villaverde, A.; Mangues, R.; Vázquez, E. Intracellular CXCR4+ cell targeting with T22-empowered protein-only nanoparticles. Int. J. Nanomed. 2012, 7, 4533–4544. [Google Scholar] [CrossRef]
- Céspedes, M.V.; Unzueta, U.; Aviñó, A.; Gallardo, A.; Álamo, P.; Sala, R.; Sánchez-Chardi, A.; Casanova, I.; Mangues, M.A.; Lopez-Pousa, A.; et al. Selective depletion of metastatic stem cells as therapy for human colorectal cancer. EMBO Mol. Med. 2018, 10, e8772. [Google Scholar] [CrossRef]
- Céspedes, M.V.; Unzueta, U.; Tatkiewicz, W.; Sánchez-Chardi, A.; Conchillo-Solé, O.; Álamo, P.; Xu, Z.; Casanova, I.; Corchero, J.L.; Pesarrodona, M.; et al. In vivo architectonic stability of fully de novo designed protein-only nanoparticles. ACS Nano 2014, 8, 4166–4176. [Google Scholar] [CrossRef]
- Serna, N.; Cespedes, M.; Sanchez-Garcia, L.; Unzueta, U.; Sala, R.; Sanchez-Chardi, A.; Cortes, F.; Ferrer-Miralles, N.; Mangues, R.; Vazquez, E.; et al. Peptide-Based Nanostructured Materials with Intrinsic Proapoptotic Activities in CXCR4(+) Solid Tumors. Adv. Funct. Mater. 2017, 27, 1700919. [Google Scholar] [CrossRef]
- Díaz, R.; Pallarès, V.; Cano-Garrido, O.; Serna, N.; Sánchez-García, L.; Falgàs, A.; Pesarrodona, M.; Unzueta, U.; Sánchez-Chardi, A.; Sánchez, J.M.; et al. Selective CXCR4. Small 2018, 14, e1800665. [Google Scholar] [CrossRef]
- Sanchez-Garcia, L.; Serna, N.; Alamo, P.; Sala, R.; Cespedes, M.V.; Roldan, M.; Sanchez-Chardi, A.; Unzueta, U.; Casanova, I.; Mangues, R.; et al. Self-assembling toxin-based nanoparticles as self-delivered antitumoral drugs. J. Control. Release 2018, 274, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Diaz, R.; Sanchez-Garcia, L.; Serna, N.; Sanchez-Chardi, A.; Cano-Garrido, O.; Sanchez, J.; Unzueta, U.; Vazquez, E.; Villaverde, A. Engineering a recombinant chlorotoxin as cell-targeted cytotoxic nanoparticles. Sci. China Mater. 2019, 62, 892–898. [Google Scholar] [CrossRef]
- Cespedes, M.V.; Cano-Garrido, O.; Alamo, P.; Sala, R.; Gallardo, A.; Serna, N.; Falgas, A.; Volta-Duran, E.; Casanova, I.; Sanchez-Chardi, A.; et al. Engineering Secretory Amyloids for Remote and Highly Selective Destruction of Metastatic Foci. Adv. Mater. 2020, 32, e1907348. [Google Scholar] [CrossRef]
- Serna, N.; Cano-Garrido, O.; Sánchez-García, L.; Pesarrodona, M.; Unzueta, U.; Sánchez-Chardi, A.; Mangues, R.; Vázquez, E.; Villaverde, A. Engineering Protein Venoms as Self-Assembling CXCR4-Targeted Cytotoxic Nanoparticles. Part. Part. Syst. Charact. 2020, 37, 2000040. [Google Scholar] [CrossRef]
- Zirafi, O.; Kim, K.A.; Ständker, L.; Mohr, K.B.; Sauter, D.; Heigele, A.; Kluge, S.F.; Wiercinska, E.; Chudziak, D.; Richter, R.; et al. Discovery and characterization of an endogenous CXCR4 antagonist. Cell Rep. 2015, 11, 737–747. [Google Scholar] [CrossRef] [PubMed]
- Zirafi, O.; Hermann, P.C.; Münch, J. Proteolytic processing of human serum albumin generates EPI-X4, an endogenous antagonist of CXCR4. J. Leukoc. Biol. 2016, 99, 863–868. [Google Scholar] [CrossRef] [PubMed]
- Vyas, S.P.; Sihorkar, V. Endogenous carriers and ligands in non-immunogenic site-specific drug delivery. Adv. Drug Deliv. Rev. 2000, 43, 101–164. [Google Scholar] [CrossRef]
- Serna, N.; Céspedes, M.V.; Saccardo, P.; Xu, Z.; Unzueta, U.; Álamo, P.; Pesarrodona, M.; Sánchez-Chardi, A.; Roldán, M.; Mangues, R.; et al. Rational engineering of single-chain polypeptides into protein-only, BBB-targeted nanoparticles. Nanomedicine 2016, 12, 1241–1251. [Google Scholar] [CrossRef]
- Unzueta, U.; Serna, N.; Sánchez-García, L.; Roldán, M.; Sánchez-Chardi, A.; Mangues, R.; Villaverde, A.; Vázquez, E. Engineering multifunctional protein nanoparticles by in vitro disassembling and reassembling of heterologous building blocks. Nanotechnology 2017, 28, 505102. [Google Scholar] [CrossRef]
- López-Laguna, H.; Unzueta, U.; Conchillo-Solé, O.; Sánchez-Chardi, A.; Pesarrodona, M.; Cano-Garrido, O.; Voltà, E.; Sánchez-García, L.; Serna, N.; Saccardo, P.; et al. Assembly of histidine-rich protein materials controlled through divalent cations. Acta Biomater. 2019, 83, 257–264. [Google Scholar] [CrossRef]
- López-Laguna, H.; Sánchez, J.; Unzueta, U.; Mangues, R.; Vázquez, E.; Villaverde, A. Divalent Cations: A Molecular Glue for Protein Materials. Trends Biochem. Sci. 2020, 45, 992–1003. [Google Scholar] [CrossRef]
- Shen, J.; Wolfram, J.; Ferrari, M.; Shen, H. Taking the vehicle out of drug delivery. Mater. Today 2017, 20, 95–97. [Google Scholar] [CrossRef]
- Kim, J.; Connelly, K.L.; Unterwald, E.M.; Rawls, S.M. Chemokines and cocaine: CXCR4 receptor antagonist AMD3100 attenuates cocaine place preference and locomotor stimulation in rats. Brain Behav. Immun. 2017, 62, 30–34. [Google Scholar] [CrossRef]
- Serna, N.; Sanchez-Garcia, L.; Sanchez-Chardi, A.; Unzueta, U.; Roldan, M.; Mangues, R.; Vazquez, E.; Villaverde, A. Protein-only, antimicrobial peptide-containing recombinant nanoparticles with inherent built-in antibacterial activity. Acta Biomater. 2017, 60, 256–263. [Google Scholar] [CrossRef]
- Weidle, U.H.; Kontermann, R.E.; Brinkmann, U. Tumor-antigen-binding bispecific antibodies for cancer treatment. Semin. Oncol. 2014, 41, 653–660. [Google Scholar] [CrossRef]
- Kintzing, J.R.; Filsinger Interrante, M.V.; Cochran, J.R. Emerging Strategies for Developing Next-Generation Protein Therapeutics for Cancer Treatment. Trends Pharmacol. Sci. 2016, 37, 993–1008. [Google Scholar] [CrossRef]
- Li, J.Y.; Perry, S.R.; Muniz-Medina, V.; Wang, X.; Wetzel, L.K.; Rebelatto, M.C.; Hinrichs, M.J.; Bezabeh, B.Z.; Fleming, R.L.; Dimasi, N.; et al. A Biparatopic HER2-Targeting Antibody-Drug Conjugate Induces Tumor Regression in Primary Models Refractory to or Ineligible for HER2-Targeted Therapy. Cancer Cell 2016, 29, 117–129. [Google Scholar] [CrossRef]
- Srinivasarao, M.; Low, P.S. Ligand-Targeted Drug Delivery. Chem. Rev. 2017, 117, 12133–12164. [Google Scholar] [CrossRef]
- Alamo, P.; Gallardo, A.; Pavon, M.A.; Casanova, I.; Trias, M.; Mangues, M.A.; Vazquez, E.; Villaverde, A.; Mangues, R.; Cespedes, M.V. Subcutaneous preconditioning increases invasion and metastatic dissemination in mouse colorectal cancer models. Dis. Models Mech. 2014, 7, 387–396. [Google Scholar] [CrossRef]
- Sala, R.; Sanchez-Garcia, L.; Serna, N.; Cespedes, M.V.; Casanova, I.; Roldan, M.; Sanchez-Chardi, A.; Unzueta, U.; Vazquez, E.; Mangues, R.; et al. Collaborative membrane activity and receptor-dependent tumor cell targeting for precise nanoparticle delivery in CXCR4(+) colorectal cancer. Acta Biomater. 2019, 99, 426–432. [Google Scholar] [CrossRef]
- Abraham, M.; Klein, S.; Bulvik, B.; Wald, H.; Weiss, I.D.; Olam, D.; Weiss, L.; Beider, K.; Eizenberg, O.; Wald, O.; et al. The CXCR4 inhibitor BL-8040 induces the apoptosis of AML blasts by downregulating ERK, BCL-2, MCL-1 and cyclin-D1 via altered miR-15a/16-1 expression. Leukemia 2017, 31, 2336–2346. [Google Scholar] [CrossRef]
- Tavor, S.; Weiss, I.; Beider, K.; Wald, H.; Eizenberg, O.; Pereg, Y.; Klapper, L.; Nagler, A.; Peled, A.; Abraham, M. The CXCR4 Antagonist BL-8040 Efficiently Induces Apoptosis and Inhibits The Survival Of AML Cells. Blood 2013, 122, 3939. [Google Scholar] [CrossRef]
- Fogle, W.E.; Flanner, H.; Wolfgang, C.; Smith, J.A.; Thackray, H.M.; Magnani, J.L. Administration of the Dual E-Selectin/CXCR4 Antagonist, GMI-1359, Results in a Unique Profile of Tumor Mobilization from the Bone Marrow and Facilitation of Chemotherapy in a Murine Model of FLT3 ITD AML. Blood 2016, 128, 2826. [Google Scholar] [CrossRef]
- Pallarès, V.; Núñez, Y.; Sánchez-García, L.; Falgàs, A.; Serna, N.; Unzueta, U.; Gallardo, A.; Alba-Castellón, L.; Álamo, P.; Sierra, J.; et al. Antineoplastic effect of a diphtheria toxin-based nanoparticle targeting acute myeloid leukemia cells overexpressing CXCR4. J. Control. Release 2021, 335, 117–129. [Google Scholar] [CrossRef] [PubMed]
- Serna, N.; Álamo, P.; Ramesh, P.; Vinokurova, D.; Sánchez-García, L.; Unzueta, U.; Gallardo, A.; Céspedes, M.V.; Vázquez, E.; Villaverde, A.; et al. Nanostructured toxins for the selective destruction of drug-resistant human CXCR4. J. Control. Release 2020, 320, 96–104. [Google Scholar] [CrossRef] [PubMed]
- López-Laguna, H.; Parladé, E.; Álamo, P.; Sánchez, J.M.; Voltà-Durán, E.; Serna, N.; Sánchez-García, L.; Cano-Garrido, O.; Sánchez-Chardi, A.; Villaverde, A.; et al. In Vitro Fabrication of Microscale Secretory Granules. Adv. Funct. Mater. 2021, 2100914. [Google Scholar] [CrossRef]
- Cespedes, M.V.; Unzueta, U.; Alamo, P.; Gallardo, A.; Sala, R.; Casanova, I.; Pavon, M.A.; Mangues, M.A.; Trias, M.; Lopez-Pousa, A.; et al. Cancer-specific uptake of a liganded protein nanocarrier targeting aggressive CXCR4+ colorectal cancer models. Nanomed. Nanotechnol. Biol. Med. 2016, 12, 1987–1996. [Google Scholar] [CrossRef]
- Falgàs, A.; Pallarès, V.; Unzueta, U.; Céspedes, M.V.; Arroyo-Solera, I.; Moreno, M.J.; Sierra, J.; Gallardo, A.; Mangues, M.A.; Vázquez, E.; et al. A CXCR4-targeted nanocarrier achieves highly selective tumor uptake in diffuse large B-cell lymphoma mouse models. Haematologica 2020, 105, 741–753. [Google Scholar] [CrossRef]
- Xu, Z.; Unzueta, U.; Roldán, M.; Mangues, R.; Sánchez-Chardi, A.; Ferrer-Miralles, N.; Villaverde, A.; Vázquez, E. Formulating tumor-homing peptides as regular nanoparticles enhances receptor-mediated cell penetrability. Mater. Lett. 2015, 154, 140–143. [Google Scholar] [CrossRef][Green Version]
- Álamo, P.; Pallarès, V.; Céspedes, M.; Falgàs, A.; Sanchez, J.; Serna, N.; Sánchez-García, L.; Voltà-Duràn, E.; Morris, G.; Sánchez-Chardi, A.; et al. Fluorescent Dye Labeling Changes the Biodistribution of Tumor-Targeted Nanoparticles. Pharmaceutics 2020, 12, 1004. [Google Scholar] [CrossRef]
- Serna, N.; Carratalá, J.V.; Parladé, E.; Sánchez-Chardi, A.; Aviñó, A.; Unzueta, U.; Mangues, R.; Eritja, R.; Ferrer-Miralles, N.; Vazquez, E.; et al. Developing Protein–Antitumoral Drug Nanoconjugates as Bifunctional Antimicrobial Agents. ACS Appl. Mater. Interfaces 2020, 12, 57746–57756. [Google Scholar] [CrossRef]
- Pallares, V.; Unzueta, U.; Falgas, A.; Sanchez-Garcia, L.; Serna, N.; Gallardo, A.; Morris, G.A.; Alba-Castellon, L.; Alamo, P.; Sierra, J.; et al. An Auristatin nanoconjugate targeting CXCR4+ leukemic cells blocks acute myeloid leukemia dissemination. J. Hematol. Oncol. 2020, 13, 36. [Google Scholar] [CrossRef]
- Volta-Duran, E.; Serna, N.; Sanchez-Garcia, L.; Avino, A.; Sanchez, J.M.; Lopez-Laguna, H.; Cano-Garrido, O.; Casanova, I.; Mangues, R.; Eritja, R.; et al. Design and engineering of tumor-targeted, dual-acting cytotoxic nanoparticles. Acta Biomater. 2021, 119, 312–322. [Google Scholar] [CrossRef]
- Falgas, A.; Pallares, V.; Serna, N.; Sanchez-Garcia, L.; Sierra, J.; Gallardo, A.; Alba-Castellon, L.; Alamo, P.; Unzueta, U.; Villaverde, A.; et al. Selective delivery of T22-PE24-H6 to CXCR4(+) diffuse large B-cell lymphoma cells leads to wide therapeutic index in a disseminated mouse model. Theranostics 2020, 10, 5169–5180. [Google Scholar] [CrossRef]
- Cho, K.; Wang, X.; Nie, S.; Chen, Z.G.; Shin, D.M. Therapeutic nanoparticles for drug delivery in cancer. Clin. Cancer Res. 2008, 14, 1310–1316. [Google Scholar] [CrossRef]
- Verdine, G.L.; Walensky, L.D. The challenge of drugging undruggable targets in cancer: Lessons learned from targeting BCL-2 family members. Clin. Cancer Res. 2007, 13, 7264–7270. [Google Scholar] [CrossRef]
- Kumar, V.A.; Wang, B.K.; Kanahara, S.M. Rational design of fiber forming supramolecular structures. Exp. Biol. Med. 2016, 241, 899–908. [Google Scholar] [CrossRef]
- Webber, M.J.; Appel, E.A.; Meijer, E.W.; Langer, R. Supramolecular biomaterials. Nat. Mater. 2016, 15, 13–26. [Google Scholar] [CrossRef]
- Serna, N.; Sanchez-Garcia, L.; Unzueta, U.; Diaz, R.; Vazquez, E.; Mangues, R.; Villaverde, A. Protein-Based Therapeutic Killing for Cancer Therapies. Trends Biotechnol. 2018, 36, 318–335. [Google Scholar] [CrossRef]
- López-Laguna, H.; Sánchez-García, L.; Serna, N.; Voltà-Durán, E.; Sánchez, J.M.; Sánchez-Chardi, A.; Unzueta, U.; Łoś, M.; Villaverde, A.; Vázquez, E. Engineering Protein Nanoparticles Out from Components of the Human Microbiome. Small 2020, 16, e2001885. [Google Scholar] [CrossRef]
- Sánchez, J.M.; López-Laguna, H.; Álamo, P.; Serna, N.; Sánchez-Chardi, A.; Nolan, V.; Cano-Garrido, O.; Casanova, I.; Unzueta, U.; Vazquez, E.; et al. Artificial Inclusion Bodies for Clinical Development. Adv. Sci. 2019, 7, 1902420. [Google Scholar] [CrossRef] [PubMed]
- Poon, K.A.; Flagella, K.; Beyer, J.; Tibbitts, J.; Kaur, S.; Saad, O.; Yi, J.H.; Girish, S.; Dybdal, N.; Reynolds, T. Preclinical safety profile of trastuzumab emtansine (T-DM1): Mechanism of action of its cytotoxic component retained with improved tolerability. Toxicol. Appl. Pharmacol. 2013, 273, 298–313. [Google Scholar] [CrossRef] [PubMed]
- Doronina, S.O.; Toki, B.E.; Torgov, M.Y.; Mendelsohn, B.A.; Cerveny, C.G.; Chace, D.F.; DeBlanc, R.L.; Gearing, R.P.; Bovee, T.D.; Siegall, C.B.; et al. Development of potent monoclonal antibody auristatin conjugates for cancer therapy. Nat. Biotechnol. 2003, 21, 778–784. [Google Scholar] [CrossRef] [PubMed]
- Senter, P.D.; Sievers, E.L. The discovery and development of brentuximab vedotin for use in relapsed Hodgkin lymphoma and systemic anaplastic large cell lymphoma. Nat. Biotechnol. 2012, 30, 631–637. [Google Scholar] [CrossRef]
- Seleci, M.; Seleci, D.A.; Joncyzk, R.; Stahl, F.; Blume, C.; Scheper, T. Smart multifunctional nanoparticles in nanomedicine. BioNanoMaterials 2016, 17, 33–41. [Google Scholar] [CrossRef]
- Glasgow, M.D.; Chougule, M.B. Recent Developments in Active Tumor Targeted Multifunctional Nanoparticles for Combination Chemotherapy in Cancer Treatment and Imaging. J. Biomed. Nanotechnol. 2015, 11, 1859–1898. [Google Scholar] [CrossRef]
- Huang, X.; Shen, J.; Cui, M.; Shen, L.; Luo, X.; Ling, K.; Pei, G.; Jiang, H.; Chen, K. Molecular dynamics simulations on SDF-1alpha: Binding with CXCR4 receptor. Biophys. J. 2003, 84, 171–184. [Google Scholar] [CrossRef]
- Crump, M.P.; Gong, J.H.; Loetscher, P.; Rajarathnam, K.; Amara, A.; Arenzana-Seisdedos, F.; Virelizier, J.L.; Baggiolini, M.; Sykes, B.D.; Clark-Lewis, I. Solution structure and basis for functional activity of stromal cell-derived factor-1; dissociation of CXCR4 activation from binding and inhibition of HIV-1. EMBO J. 1997, 16, 6996–7007. [Google Scholar] [CrossRef]
- Sakaida, H.; Hori, T.; Yonezawa, A.; Sato, A.; Isaka, Y.; Yoshie, O.; Hattori, T.; Uchiyama, T. T-tropic human immunodeficiency virus type 1 (HIV-1)-derived V3 loop peptides directly bind to CXCR-4 and inhibit T-tropic HIV-1 infection. J. Virol. 1998, 72, 9763–9770. [Google Scholar] [CrossRef]
- Kledal, T.N.; Rosenkilde, M.M.; Coulin, F.; Simmons, G.; Johnsen, A.H.; Alouani, S.; Power, C.A.; Lüttichau, H.R.; Gerstoft, J.; Clapham, P.R.; et al. A broad-spectrum chemokine antagonist encoded by Kaposi’s sarcoma-associated herpesvirus. Science 1997, 277, 1656–1659. [Google Scholar] [CrossRef]
- Liang, X. CXCR4, inhibitors and mechanisms of action. Chem. Biol. Drug Des. 2008, 72, 97–110. [Google Scholar] [CrossRef]
- Vazquez, E.; Roldán, M.; Diez-Gil, C.; Unzueta, U.; Domingo-Espín, J.; Cedano, J.; Conchillo, O.; Ratera, I.; Veciana, J.; Daura, X.; et al. Protein nanodisk assembling and intracellular trafficking powered by an arginine-rich (R9) peptide. Nanomedicine 2010, 5, 259–268. [Google Scholar] [CrossRef]
- Sánchez-García, L.; Sala, R.; Serna, N.; Álamo, P.; Parladé, E.; Alba-Castellón, L.; Voltà-Durán, E.; Sánchez-Chardi, A.; Unzueta, U.; Vázquez, E.; et al. A refined cocktailing of pro-apoptotic nanoparticles boosts anti-tumor activity. Acta Biomater. 2020, 113, 584–596. [Google Scholar] [CrossRef]
- Kapałczyńska, M.; Kolenda, T.; Przybyła, W.; Zajączkowska, M.; Teresiak, A.; Filas, V.; Ibbs, M.; Bliźniak, R.; Łuczewski, Ł.; Lamperska, K. 2D and 3D cell cultures—A comparison of different types of cancer cell cultures. Arch. Med. Sci. 2018, 14, 910–919. [Google Scholar] [CrossRef]
- Engler, A.J.; Sen, S.; Sweeney, H.L.; Discher, D.E. Matrix elasticity directs stem cell lineage specification. Cell 2006, 126, 677–689. [Google Scholar] [CrossRef]
- Busillo, J.M.; Benovic, J.L. Regulation of CXCR4 signaling. Biochim. Biophys. Acta 2007, 1768, 952–963. [Google Scholar] [CrossRef]
- Wang, S.H.; Lin, S.Y. Tumor dormancy: Potential therapeutic target in tumor recurrence and metastasis prevention. Exp. Hematol. Oncol. 2013, 2, 29. [Google Scholar] [CrossRef]
- Hartmann, T.N.; Burger, J.A.; Glodek, A.; Fujii, N.; Burger, M. CXCR4 chemokine receptor and integrin signaling co-operate in mediating adhesion and chemoresistance in small cell lung cancer (SCLC) cells. Oncogene 2005, 24, 4462–4471. [Google Scholar] [CrossRef]
- Larochelle, A.; Krouse, A.; Metzger, M.; Orlic, D.; Donahue, R.E.; Fricker, S.; Bridger, G.; Dunbar, C.E.; Hematti, P. AMD3100 mobilizes hematopoietic stem cells with long-term repopulating capacity in nonhuman primates. Blood 2006, 107, 3772–3778. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
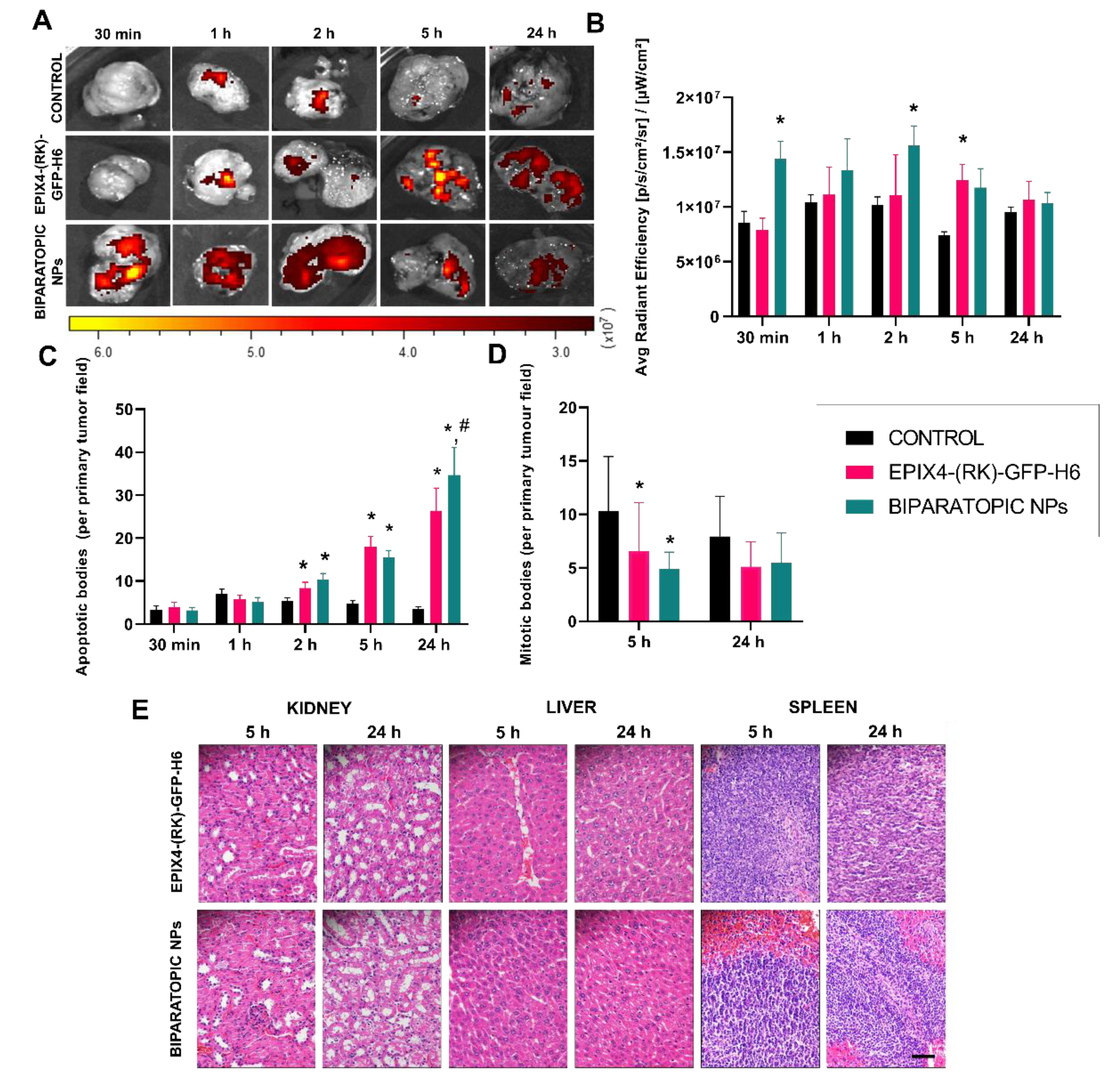{kind=link}
| LIVER | KIDNEY | SPLEEN | LUNG | BONE MARROW | BRAIN | |
|---|---|---|---|---|---|---|
| 0.5 h | ||||||
| BUFFER | 4.59 ± 0.9 | 3.24 ± 0.1 a,b | 3.14 ± 0.4 | 2.5 ± 0.1 | 4.0 ± 0.2 | 3.0 ± 0.2 |
| EPIX4-(RK)-GFP-H6 | 7.14 ± 0.6 | 7.14 ± 0.7 a | 4.2 ± 0.5 | 5.0 ± 0.5 | 5.1 ± 0.4 | 4.1 ± 0.1 |
| BIPARATOPIC NPs | 7.02 ± 0.5 | 7.02 ± 0.8 b | 4.5 ± 0.4 | 3.8 ± 0.3 | 4.3 ± 0.6 | 3.4 ± 0.4 |
| 1 h | ||||||
| BUFFER | 4.35 ± 0.0 | 3.62 ± 0.7 | 2.7 ± 0.0 | 3.0 ± 0.0 | 4.1 ± 0.0 | 3.6 ± 0.0 |
| EPIX4-(RK)-GFP-H6 | 4.93 ± 0.3 | 4.93 ± 0.4 | 3.6 ± 0.2 | 3.3 ± 0.2 | 5.0 ± 0.1 | 3.9 ± 0.1 |
| BIPARATOPIC NPs | 4.73 ± 0.2 | 4.73 ± 1.1 | 3.3 ± 0.2 | 2.7 ± 0.5 | 4.1 ± 0.9 | 3.4 ± 0.2 |
| 2 h | ||||||
| BUFFER | 4.57 ± 0.2 | 3.73 ± 0.3 | 3.4 ± 0.2 | 2.9 ± 0.3 | 5.2 ± 0.04 | 3.4 ± 0.4 |
| EPIX4-(RK)-GFP-H6 | 4.46 ± 0.1 | 4.46 ± 0.4 | 4.4 ± 0.3 | 3.5 ± 0.5 | 5.3 ± 0.9 | 4.3 ± 1.0 |
| BIPARATOPIC NPs | 4.64 ± 0.4 | 4.64 ± 0.5 | 3.7 ± 0.2 | 2.8 ± 0.1 | 4.1 ± 0.2 | 4.0 ± 0.3 |
| 5 h | ||||||
| BUFFER | 4.78 ± 0.3 | 4.3 ± 0.4 | 3.5 ± 0.3 | 2.7 ± 0.3 | 4.5 ± 0.1 | 3.7 ± 0.2 |
| EPIX4-(RK)-GFP-H6 | 4.53 ± 0.2 | 4.53 ± 2.2 | 4.3 ± 0.2 | 2.9 ± 0.1 | 4.3 ± 0.2 | 4.7 ± 0.9 |
| BIPARATOPIC NPs | 3.93 ± 0.3 | 3.93 ± 1.9 | 3.9 ± 0.5 | 2.5 ± 0.1 | 4.5 ± 0.3 | 3.6 ± 0.2 |
| 24 h | ||||||
| BUFFER | 4.48 ± 1.3 | 4.48 ± 1.8 | 4.4 ± 0.4 | 2.6 ± 0.2 | 5.1 ± 0.4 | 3.5 ± 0.2 |
| EPIX4-(RK)-GFP-H6 | 5.36 ± 0.2 | 5.36 ± 2.2 | 4.5 ± 0.4 | 2.6 ± 0.1 | 4.8 ± 0.1 | 5.3 ± 0.8 |
| BIPARATOPIC NPs | 4.38 ± 2.0 | 4.38 ± 2.2 | 4.0 ± 0.2 | 2.8 ± 0.1 | 4.6 ± 0.3 | 4.8 ± 0.6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cano-Garrido, O.; Álamo, P.; Sánchez-García, L.; Falgàs, A.; Sánchez-Chardi, A.; Serna, N.; Parladé, E.; Unzueta, U.; Roldán, M.; Voltà-Durán, E.; et al. Biparatopic Protein Nanoparticles for the Precision Therapy of CXCR4+ Cancers. Cancers 2021, 13, 2929. https://doi.org/10.3390/cancers13122929
Cano-Garrido O, Álamo P, Sánchez-García L, Falgàs A, Sánchez-Chardi A, Serna N, Parladé E, Unzueta U, Roldán M, Voltà-Durán E, et al. Biparatopic Protein Nanoparticles for the Precision Therapy of CXCR4+ Cancers. Cancers. 2021; 13(12):2929. https://doi.org/10.3390/cancers13122929
Chicago/Turabian StyleCano-Garrido, Olivia, Patricia Álamo, Laura Sánchez-García, Aïda Falgàs, Alejandro Sánchez-Chardi, Naroa Serna, Eloi Parladé, Ugutz Unzueta, Mònica Roldán, Eric Voltà-Durán, and et al. 2021. "Biparatopic Protein Nanoparticles for the Precision Therapy of CXCR4+ Cancers" Cancers 13, no. 12: 2929. https://doi.org/10.3390/cancers13122929
APA StyleCano-Garrido, O., Álamo, P., Sánchez-García, L., Falgàs, A., Sánchez-Chardi, A., Serna, N., Parladé, E., Unzueta, U., Roldán, M., Voltà-Durán, E., Casanova, I., Villaverde, A., Mangues, R., & Vázquez, E. (2021). Biparatopic Protein Nanoparticles for the Precision Therapy of CXCR4+ Cancers. Cancers, 13(12), 2929. https://doi.org/10.3390/cancers13122929