
An Overview on Diffuse Large B-Cell Lymphoma Models: Towards a Functional Genomics Approach

 ,
,
Abstract
Simple Summary
Abstract

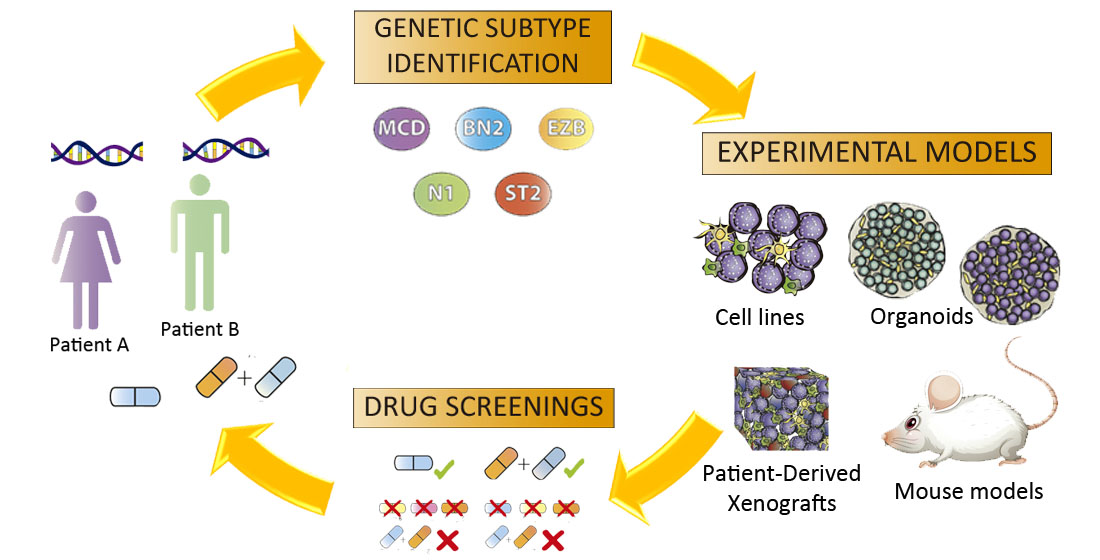
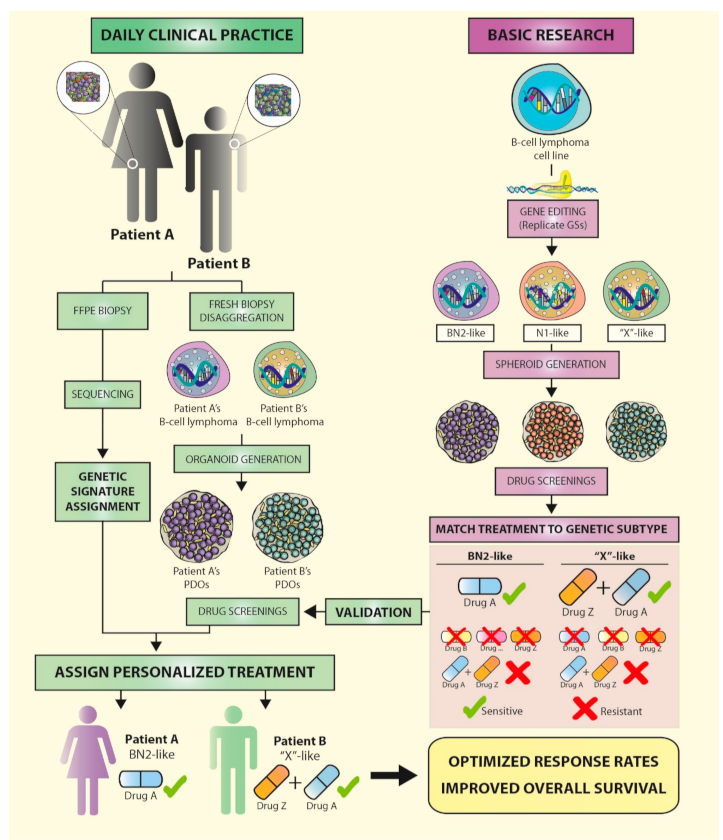
1. Unraveling Aggressive B-Cell Lymphomas: The Elusive Link between Genomics and Personalized Treatment
2. Diffuse Large B-Cell Lymphomas: Clinically Unresolved Genomic Complexity
2.1. Diffuse Large B-Cell Lymphoma: A Traditional Overview
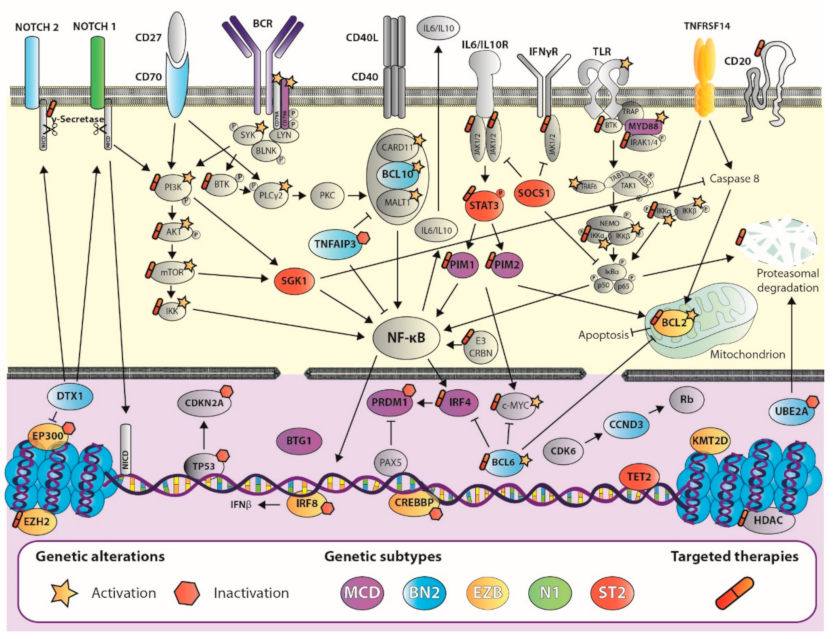
2.2. Advances in DLBCL Genomics: Genetic Signature Helps Design Tailored Therapies
2.3. Friend or Foe: The DLBCL Microenvironment
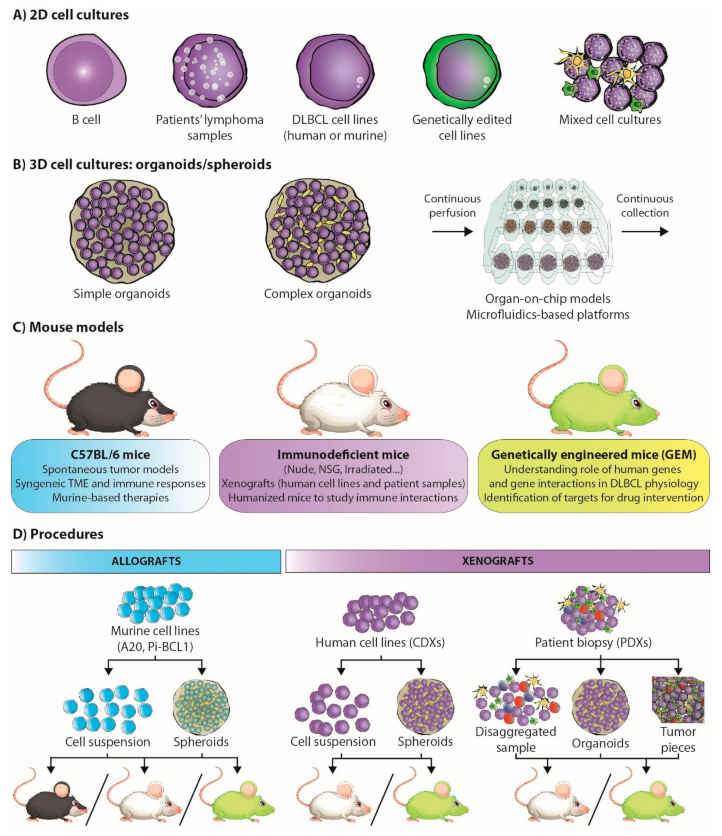
3. Functional Models for B-Cell Lymphoma Research
3.1. Gene Editing of Lymphoma Cells
3.2. The Rise of Drug Screening: Mission Pathways
3.3. Of Mice and Men: Understanding Lymphoma Biology

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strategy | Phenotype/Incidence | Prospective Uses | |
|---|---|---|---|
| Genetically engineered mice | Eμ-Myc | DLBCL (time dependent) [99] | |
| Eμ-BRD2 | DLBCL [91] | ||
| Bcl6 Knock in | GC-DLBCL [92] | ||
| Bcl6/Myc | ABC-DLBCL [92] | ||
| Iµ:HA.BCL6 | 36–62% lymphoma incidence [92] | Combination with conditional Spen and Tnfaip3 knockout or oncogenic Notch2 alleles to model Cluster BN2 | |
| Mb1:Cre;Eµ:Bcl2;Crebbpfl/fl | GC-DLBCL [103] | Combination of the different alleles to generate a sophisticated EZB mouse model | |
| Cγ1Cre/wt;Kmt2dfl/fl;VavP:Bcl2 | 21% incidence GC-DLBCL [104] | ||
| Ezh2cond.p.Y641F/wt;VavP:Bcl2; Cγ1Cre/wt | DLBCL-like lymphoma [105] | ||
| Cd19Cre/wt;Myd88cond.p.L252P/wt;Rosa26LSL.BCL2-IRES-GFP/wt | 85% incidence ABC-DLBCL [106] | Modeling of the MCD cluster by combination of both alleles and a newly generated CD79Bcond.p.Y196H/wt allele, or with the already existing Prdm1fl/fl | |
| Cγ1Cre;Prdm1fl/fl;Rosa26LSL.IKK2ca | 50% incidence IRF4, post-GC DLBCL [107] | ||
| Syngeneic models | Pi-BCL1 (m) iv or ip in BALB/c immunocompetent mice | DLBCL [108,109] | Gene editing Generation of complex organoids prior to inoculation of the cell line |
| A20 (m) iv, intrasplenic, or sc injection in BALB/c immunocompetent mice | DLBCL [110,111,112] | ||
| Xenograft models | SU-DHL-4 (h) iv or sc in SCID immunodeficient mice | DLBCL [113,114] | Gene editing Generation of organoids Inoculation in humanized NOD/SCID mice |
| Transduced HPCs in irradiated mice | GC- and ABC-DLBCL [103,115] | Generation of a-la-carte HPCs reproducing the genetic signatures Inoculation in humanized NOD/SCID mice | |
| PDX iv or sc in immunodeficient mice | 20–30% successful engraftment | Use of humanized mice to study lymphoma physiology and drug responses Personalized medicine | |
| PDO iv or sc in immunodeficient mice | 20–30% successful engraftment | Genetic modification Use of humanized mice to study lymphoma physiology and drug responses Personalized medicine. |
3.4. Back to the Bench: À La Carte-Engineered Spheroids
4. Conclusions
Author Contributions
Funding
Ethics Approval and Consent to Participate
Consent for Publication
Availability of Data and Materials
Acknowledgments
Conflicts of Interest
References
- Bakhshi, T.J.; Georgel, P.T. Genetic and Epigenetic Determinants of Diffuse Large B-Cell Lymphoma. Blood Cancer J. 2020, 10, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Sant, M.; Allemani, C.; Tereanu, C.; De Angelis, R.; Capocaccia, R.; Visser, O.; Marcos-Gragera, R.; Maynadié, M.; Simonetti, A.; Lutz, J.-M.; et al. Incidence of Hematologic Malignancies in Europe by Morphologic Subtype: Results of the HAEMACARE Project. Blood 2010, 116, 3724–3734. [Google Scholar] [CrossRef] [PubMed]
- Howlader, N.; Noone, A.M.; Krapcho, M.; Miller, D.; Brest, A.; Yu, M.; Ruhl, J.; Tatalovich, Z.; Mariotto, A.; Lewis, D.R.; et al. Diffuse Large B-Cell Lymphoma—Cancer Stat Facts. 2019. Available online: https://seer.cancer.gov/statfacts/html/dlbcl.html (accessed on 18 May 2021).
- Sehn, L.H.; Salles, G. Diffuse Large B-Cell Lymphoma. N. Engl. J. Med. 2021. [Google Scholar] [CrossRef]
- Alizadeh, A.A.; Eisen, M.B.; Davis, R.E.; Ma, C.; Lossos, I.S.; Rosenwald, A.; Boldrick, J.C.; Sabet, H.; Tran, T.; Yu, X.; et al. Distinct Types of Diffuse Large B-Cell Lymphoma Identified by Gene Expression Profiling. Nature 2000, 403, 503–511. [Google Scholar] [CrossRef] [PubMed]
- Rosenwald, A.; Wright, G.; Chan, W.C.; Connors, J.M.; Campo, E.; Fisher, R.I.; Gascoyne, R.D.; Muller-Hermelink, H.K.; Smeland, E.B.; Giltnane, J.M.; et al. The Use of Molecular Profiling to Predict Survival after Chemotherapy for Diffuse Large-B-Cell Lymphoma. N. Engl. J. Med. 2002, 346, 1937–1947. [Google Scholar] [CrossRef]
- Jaffe, E.S.; Barr, P.M.; Smith, S.M. Understanding the New WHO Classification of Lymphoid Malignancies: Why It’s Important and How It Will Affect Practice. Am. Soc. Clin. Oncol. Educ. Book 2017, 37, 535–546. [Google Scholar] [CrossRef]
- Campo, E.; Harris, N.L.; Jaffe, E.S.; Pileri, S.A.; Stein, H.; Thiele, J.; Vardiman, J.W. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues; International Agency for Research on Cancer: Lyon, France, 2008; ISBN 978-92-832-4494-3. [Google Scholar]
- Swerdlow, S.H.; Campo, E.; Pileri, S.A.; Harris, N.L.; Stein, H.; Siebert, R.; Advani, R.; Ghielmini, M.; Salles, G.A.; Zelenetz, A.D.; et al. The 2016 Revision of the World Health Organization Classification of Lymphoid Neoplasms. Blood 2016, 127, 2375–2390. [Google Scholar] [CrossRef]
- Basso, K.; Dalla-Favera, R. Germinal Centres and B Cell Lymphomagenesis. Nat. Rev. Immunol. 2015, 15, 172–184. [Google Scholar] [CrossRef]
- Pasqualucci, L.; Trifonov, V.; Fabbri, G.; Ma, J.; Rossi, D.; Chiarenza, A.; Wells, V.A.; Grunn, A.; Messina, M.; Elliot, O.; et al. Analysis of the Coding Genome of Diffuse Large B-Cell Lymphoma. Nat. Genet. 2011, 43, 830. [Google Scholar] [CrossRef]
- Dubois, S.; Viailly, P.-J.; Mareschal, S.; Bohers, E.; Bertrand, P.; Ruminy, P.; Maingonnat, C.; Jais, J.-P.; Peyrouze, P.; Figeac, M.; et al. Next-Generation Sequencing in Diffuse Large B-Cell Lymphoma Highlights Molecular Divergence and Therapeutic Opportunities: A LYSA Study. Clin. Cancer Res. 2016, 22, 2919–2928. [Google Scholar] [CrossRef]
- Mandelbaum, J.; Bhagat, G.; Tang, H.; Mo, T.; Brahmachary, M.; Shen, Q.; Chadburn, A.; Rajewsky, K.; Tarakhovsky, A.; Pasqualucci, L.; et al. BLIMP1 Is a Tumor Suppressor Gene Frequently Disrupted in Activated B Cell-like Diffuse Large B Cell Lymphoma. Cancer Cell 2010, 18, 568–579. [Google Scholar] [CrossRef]
- Morin, R.D.; Mendez-Lago, M.; Mungall, A.J.; Goya, R.; Mungall, K.L.; Corbett, R.D.; Johnson, N.A.; Severson, T.M.; Chiu, R.; Field, M.; et al. Frequent Mutation of Histone-Modifying Genes in Non-Hodgkin Lymphoma. Nature 2011, 476, 298–303. [Google Scholar] [CrossRef]
- Davis, R.E.; Ngo, V.N.; Lenz, G.; Tolar, P.; Young, R.M.; Romesser, P.B.; Kohlhammer, H.; Lamy, L.; Zhao, H.; Yang, Y.; et al. Chronic Active B-Cell-Receptor Signalling in Diffuse Large B-Cell Lymphoma. Nature 2010, 463, 88–92. [Google Scholar] [CrossRef]
- Compagno, M.; Lim, W.K.; Grunn, A.; Nandula, S.V.; Brahmachary, M.; Shen, Q.; Bertoni, F.; Ponzoni, M.; Scandurra, M.; Califano, A.; et al. Mutations of Multiple Genes Cause Deregulation of NF-KappaB in Diffuse Large B-Cell Lymphoma. Nature 2009, 459, 717–721. [Google Scholar] [CrossRef] [PubMed]
- Lohr, J.G.; Stojanov, P.; Lawrence, M.S.; Auclair, D.; Chapuy, B.; Sougnez, C.; Cruz-Gordillo, P.; Knoechel, B.; Asmann, Y.W.; Slager, S.L.; et al. Discovery and Prioritization of Somatic Mutations in Diffuse Large B-Cell Lymphoma (DLBCL) by Whole-Exome Sequencing. Proc. Natl. Acad. Sci. USA 2012, 109, 3879. [Google Scholar] [CrossRef]
- Zhang, J.; Grubor, V.; Love, C.L.; Banerjee, A.; Richards, K.L.; Mieczkowski, P.A.; Dunphy, C.; Choi, W.; Au, W.Y.; Srivastava, G.; et al. Genetic Heterogeneity of Diffuse Large B-Cell Lymphoma. Proc. Natl. Acad. Sci. USA 2013, 110, 1398–1403. [Google Scholar] [CrossRef]
- Novak, A.J.; Asmann, Y.W.; Maurer, M.J.; Wang, C.; Slager, S.L.; Hodge, L.S.; Manske, M.; Price-Troska, T.; Yang, Z.-Z.; Zimmermann, M.T.; et al. Whole-Exome Analysis Reveals Novel Somatic Genomic Alterations Associated with Outcome in Immunochemotherapy-Treated Diffuse Large B-Cell Lymphoma. Blood Cancer J. 2015, 5, e346. [Google Scholar] [CrossRef] [PubMed]
- Karube, K.; Enjuanes, A.; Dlouhy, I.; Jares, P.; Martin-Garcia, D.; Nadeu, F.; Ordóñez, G.R.; Rovira, J.; Clot, G.; Royo, C.; et al. Integrating Genomic Alterations in Diffuse Large B-Cell Lymphoma Identifies New Relevant Pathways and Potential Therapeutic Targets. Leukemia 2018, 32, 675–684. [Google Scholar] [CrossRef]
- Ngo, V.N.; Young, R.M.; Schmitz, R.; Jhavar, S.; Xiao, W.; Lim, K.-H.; Kohlhammer, H.; Xu, W.; Yang, Y.; Zhao, H.; et al. Oncogenically Active MYD88 Mutations in Human Lymphoma. Nature 2011, 470, 115–119. [Google Scholar] [CrossRef] [PubMed]
- Monti, S.; Chapuy, B.; Takeyama, K.; Rodig, S.J.; Hao, Y.; Yeda, K.T.; Inguilizian, H.; Mermel, C.; Currie, T.; Dogan, A.; et al. Integrative Analysis Reveals an Outcome-Associated and Targetable Pattern of P53 and Cell Cycle Deregulation in Diffuse Large B Cell Lymphoma. Cancer Cell 2012, 22, 359–372. [Google Scholar] [CrossRef]
- Jardin, F.; Jais, J.-P.; Molina, T.-J.; Parmentier, F.; Picquenot, J.-M.; Ruminy, P.; Tilly, H.; Bastard, C.; Salles, G.-A.; Feugier, P.; et al. Diffuse Large B-Cell Lymphomas with CDKN2A Deletion Have a Distinct Gene Expression Signature and a Poor Prognosis under R-CHOP Treatment: A GELA Study. Blood 2010, 116, 1092–1104. [Google Scholar] [CrossRef]
- Lenz, G.; Wright, G.; Dave, S.S.; Xiao, W.; Powell, J.; Zhao, H.; Xu, W.; Tan, B.; Goldschmidt, N.; Iqbal, J.; et al. Stromal Gene Signatures in Large-B-Cell Lymphomas. N. Engl. J. Med. 2008, 359, 2313–2323. [Google Scholar] [CrossRef] [PubMed]
- Challa-Malladi, M.; Lieu, Y.K.; Califano, O.; Holmes, A.B.; Bhagat, G.; Murty, V.V.; Dominguez-Sola, D.; Pasqualucci, L.; Dalla-Favera, R. Combined Genetic Inactivation of Β2-Microglobulin and CD58 Reveals Frequent Escape from Immune Recognition in Diffuse Large B Cell Lymphoma. Cancer Cell 2011, 20, 728–740. [Google Scholar] [CrossRef]
- Miao, Y.; Medeiros, L.J.; Li, Y.; Li, J.; Young, K.H. Genetic Alterations and Their Clinical Implications in DLBCL. Nat. Rev. Clin. Oncol. 2019, 16, 634–652. [Google Scholar] [CrossRef] [PubMed]
- Karube, K.; Campo, E. MYC Alterations in Diffuse Large B-Cell Lymphomas. Semin. Hematol. 2015, 52, 97–106. [Google Scholar] [CrossRef]
- Schmitz, R.; Wright, G.W.; Huang, D.W.; Johnson, C.A.; Phelan, J.D.; Wang, J.Q.; Roulland, S.; Kasbekar, M.; Young, R.M.; Shaffer, A.L.; et al. Genetics and Pathogenesis of Diffuse Large B-Cell Lymphoma. N. Engl. J. Med. 2018, 378, 1396–1407. [Google Scholar] [CrossRef]
- Reddy, A.; Zhang, J.; Davis, N.S.; Moffitt, A.B.; Love, C.L.; Waldrop, A.; Leppa, S.; Pasanen, A.; Meriranta, L.; Karjalainen-Lindsberg, M.-L.; et al. Genetic and Functional Drivers of Diffuse Large B Cell Lymphoma. Cell 2017, 171, 481–494.e15. [Google Scholar] [CrossRef]
- Pasqualucci, L.; Dalla-Favera, R. The Genetic Landscape of Diffuse Large B-Cell Lymphoma. Semin. Hematol. 2015, 52, 67–76. [Google Scholar] [CrossRef]
- Wright, G.W.; Huang, D.W.; Phelan, J.D.; Coulibaly, Z.A.; Roulland, S.; Young, R.M.; Wang, J.Q.; Schmitz, R.; Morin, R.D.; Tang, J.; et al. A Probabilistic Classification Tool for Genetic Subtypes of Diffuse Large B Cell Lymphoma with Therapeutic Implications. Cancer Cell 2020, 37, 551–568.e14. [Google Scholar] [CrossRef]
- Chapuy, B.; Stewart, C.; Dunford, A.J.; Kim, J.; Kamburov, A.; Redd, R.A.; Lawrence, M.S.; Roemer, M.G.M.; Li, A.J.; Ziepert, M.; et al. Molecular Subtypes of Diffuse Large B Cell Lymphoma Are Associated with Distinct Pathogenic Mechanisms and Outcomes. Nat. Med. 2018, 24, 679–690. [Google Scholar] [CrossRef] [PubMed]
- Lacy, S.E.; Barrans, S.L.; Beer, P.A.; Painter, D.; Smith, A.G.; Roman, E.; Cooke, S.L.; Ruiz, C.; Glover, P.; Van Hoppe, S.J.L.; et al. Targeted Sequencing in DLBCL, Molecular Subtypes, and Outcomes: A Haematological Malignancy Research Network Report. Blood 2020, 135, 1759–1771. [Google Scholar] [CrossRef]
- Pedrosa, L.; Fernández-Miranda, I.; Pérez-Callejo, D.; Quero, C.; Rodríguez, M.; Martín-Acosta, P.; Gómez, S.; González-Rincón, J.; Santos, A.; Tarin, C.; et al. Proposal and Validation of a Method to Classify Genetic Subtypes of Diffuse Large B Cell Lymphoma. Sci. Rep. 2021, 11, 1886. [Google Scholar] [CrossRef]
- Runge, H.F.P.; Lacy, S.; Barrans, S.; Beer, P.A.; Painter, D.; Smith, A.; Roman, E.; Burton, C.; Crouch, S.; Tooze, R.; et al. Application of the LymphGen Classification Tool to 928 Clinically and Genetically-Characterised Cases of Diffuse Large B Cell Lymphoma (DLBCL). Br. J. Haematol. 2021, 192, 216–220. [Google Scholar] [CrossRef]
- Young, R.M.; Phelan, J.D.; Wilson, W.H.; Staudt, L.M. Pathogenic B-Cell Receptor Signaling in Lymphoid Malignancies: New Insights to Improve Treatment. Immunol. Rev. 2019, 291, 190–213. [Google Scholar] [CrossRef]
- Young, R.M.; Phelan, J.D.; Shaffer, A.L.; Wright, G.W.; Huang, D.W.; Schmitz, R.; Johnson, C.; Oellerich, T.; Wilson, W.; Staudt, L.M. Taming the Heterogeneity of Aggressive Lymphomas for Precision Therapy. Annu. Rev. Cancer Biol. 2019, 3, 429–455. [Google Scholar] [CrossRef]
- Ciavarella, S.; Vegliante, M.C.; Fabbri, M.; De Summa, S.; Melle, F.; Motta, G.; De Iuliis, V.; Opinto, G.; Enjuanes, A.; Rega, S.; et al. Dissection of DLBCL Microenvironment Provides a Gene Expression-Based Predictor of Survival Applicable to Formalin-Fixed Paraffin-Embedded Tissue. Ann. Oncol. 2018, 29, 2363–2370. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Scott, D.W.; Gascoyne, R.D. The Tumour Microenvironment in B Cell Lymphomas. Nat. Rev. Cancer 2014, 14, 517–534. [Google Scholar] [CrossRef]
- Kotlov, N.; Bagaev, A.; Revuelta, M.V.; Phillip, J.M.; Cacciapuoti, M.T.; Antysheva, Z.; Svekolkin, V.; Tikhonova, E.; Miheecheva, N.; Kuzkina, N.; et al. Clinical and Biological Subtypes of B-Cell Lymphoma Revealed by Microenvironmental Signatures. Cancer Discov. 2021. [Google Scholar] [CrossRef]
- Clozel, T.; Yang, S.; Elstrom, R.L.; Tam, W.; Martin, P.; Kormaksson, M.; Banerjee, S.; Vasanthakumar, A.; Culjkovic, B.; Scott, D.W.; et al. Mechanism-Based Epigenetic Chemosensitization Therapy of Diffuse Large B-Cell Lymphoma. Cancer Discov. 2013, 3, 1002–1019. [Google Scholar] [CrossRef]
- Mulder, T.A.; Wahlin, B.E.; Österborg, A.; Palma, M. Targeting the Immune Microenvironment in Lymphomas of B-Cell Origin: From Biology to Clinical Application. Cancers 2019, 11, 915. [Google Scholar] [CrossRef]
- Gaudelli, N.M.; Komor, A.C.; Rees, H.A.; Packer, M.S.; Badran, A.H.; Bryson, D.I.; Liu, D.R. Programmable Base Editing of A•T to G•C in Genomic DNA without DNA Cleavage. Nature 2017, 551, 464–471. [Google Scholar] [CrossRef]
- Cox, D.B.T.; Platt, R.J.; Zhang, F. Therapeutic Genome Editing: Prospects and Challenges. Nat. Med. 2015, 21, 121–131. [Google Scholar] [CrossRef]
- Caeser, R.; Di Re, M.; Krupka, J.A.; Gao, J.; Lara-Chica, M.; Dias, J.M.L.; Cooke, S.L.; Fenner, R.; Usheva, Z.; Runge, H.F.P.; et al. Genetic Modification of Primary Human B Cells to Model High-Grade Lymphoma. Nat. Commun. 2019, 10, 4543. [Google Scholar] [CrossRef]
- Bai, B.; Myklebust, J.H.; Wälchli, S. Gene Editing in B-Lymphoma Cell Lines Using CRISPR/Cas9 Technology. Methods Mol. Biol. 2020, 2115, 445–454. [Google Scholar] [CrossRef]
- Felce, S.L.; Anderson, A.P.; Maguire, S.; Gascoyne, D.M.; Armstrong, R.N.; Wong, K.K.; Li, D.; Banham, A.H. CRISPR/Cas9-Mediated Foxp1 Silencing Restores Immune Surveillance in an Immunocompetent A20 Lymphoma Model. Front. Oncol. 2020, 10, 448. [Google Scholar] [CrossRef]
- Greiner, V.; Puerto, R.B.; Liu, S.; Herbel, C.; Carmona, E.M.; Goldberg, M.S. CRISPR-Mediated Editing of the B Cell Receptor in Primary Human B Cells. iScience 2019, 12, 369–378. [Google Scholar] [CrossRef]
- Schlager, S.; Salomon, C.; Olt, S.; Albrecht, C.; Ebert, A.; Bergner, O.; Wachter, J.; Trapani, F.; Gerlach, D.; Voss, T.; et al. Inducible Knock-out of BCL6 in Lymphoma Cells Results in Tumor Stasis. Oncotarget 2020, 11, 875–890. [Google Scholar] [CrossRef]
- Nie, M.; Du, L.; Zhang, B.; Ren, W.; Joung, J.; Ye, X.; Fuenzalida, J.A.; Shi, X.; Liu, D.; Wu, K.; et al. Essentiality of CREBBP in EP300 Truncated B-Cell Lymphoma Revealed by Genome-Wide CRISPR-Cas9 Screen. bioRxiv 2019, 746594. [Google Scholar] [CrossRef]
- Eyquem, J.; Mansilla-Soto, J.; Giavridis, T.; van der Stegen, S.J.C.; Hamieh, M.; Cunanan, K.M.; Odak, A.; Gönen, M.; Sadelain, M. Targeting a CAR to the TRAC Locus with CRISPR/Cas9 Enhances Tumour Rejection. Nature 2017, 543, 113–117. [Google Scholar] [CrossRef]
- Barretina, J.; Caponigro, G.; Stransky, N.; Venkatesan, K.; Margolin, A.A.; Kim, S.; Wilson, C.J.; Lehár, J.; Kryukov, G.V.; Sonkin, D.; et al. The Cancer Cell Line Encyclopedia Enables Predictive Modelling of Anticancer Drug Sensitivity. Nature 2012, 483, 603–607. [Google Scholar] [CrossRef]
- Garnett, M.J.; Edelman, E.J.; Heidorn, S.J.; Greenman, C.D.; Dastur, A.; Lau, K.W.; Greninger, P.; Thompson, I.R.; Luo, X.; Soares, J.; et al. Systematic Identification of Genomic Markers of Drug Sensitivity in Cancer Cells. Nature 2012, 483, 570–575. [Google Scholar] [CrossRef] [PubMed]
- Haverty, P.M.; Lin, E.; Tan, J.; Yu, Y.; Lam, B.; Lianoglou, S.; Neve, R.M.; Martin, S.; Settleman, J.; Yauch, R.L.; et al. Reproducible Pharmacogenomic Profiling of Cancer Cell Line Panels. Nature 2016, 533, 333–337. [Google Scholar] [CrossRef] [PubMed]
- Ghandi, M.; Huang, F.W.; Jané-Valbuena, J.; Kryukov, G.V.; Lo, C.C.; McDonald, E.R.; Barretina, J.; Gelfand, E.T.; Bielski, C.M.; Li, H.; et al. Next-Generation Characterization of the Cancer Cell Line Encyclopedia. Nature 2019, 569, 503–508. [Google Scholar] [CrossRef]
- Quentmeier, H.; Pommerenke, C.; Dirks, W.G.; Eberth, S.; Koeppel, M.; MacLeod, R.A.F.; Nagel, S.; Steube, K.; Uphoff, C.C.; Drexler, H.G. The LL-100 Panel: 100 Cell Lines for Blood Cancer Studies. Sci. Rep. 2019, 9, 8218. [Google Scholar] [CrossRef] [PubMed]
- Phelan, J.D.; Young, R.M.; Webster, D.E.; Roulland, S.; Wright, G.W.; Kasbekar, M.; Shaffer, A.L.; Ceribelli, M.; Wang, J.Q.; Schmitz, R.; et al. A Multiprotein Supercomplex Controlling Oncogenic Signalling in Lymphoma. Nature 2018, 560, 387–391. [Google Scholar] [CrossRef]
- Rui, L.; Drennan, A.C.; Ceribelli, M.; Zhu, F.; Wright, G.W.; Huang, D.W.; Xiao, W.; Li, Y.; Grindle, K.M.; Lu, L.; et al. Epigenetic Gene Regulation by Janus Kinase 1 in Diffuse Large B-Cell Lymphoma. Proc. Natl. Acad. Sci. USA 2016, 113, E7260–E7267. [Google Scholar] [CrossRef]
- Solomon, L.A.; Batista, C.R.; DeKoter, R.P. Lenalidomide Modulates Gene Expression in Human ABC-DLBCL Cells by Regulating IKAROS Interaction with an Intronic Control Region of SPIB. Exp. Hematol. 2017, 56, 46–57.e1. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Shaffer, A.L.; Emre, N.C.T.; Ceribelli, M.; Zhang, M.; Wright, G.; Xiao, W.; Powell, J.; Platig, J.; Kohlhammer, H.; et al. Exploiting Synthetic Lethality for the Therapy of ABC Diffuse Large B Cell Lymphoma. Cancer Cell 2012, 21, 723–737. [Google Scholar] [CrossRef]
- Davis, R.E.; Brown, K.D.; Siebenlist, U.; Staudt, L.M. Constitutive Nuclear Factor KappaB Activity Is Required for Survival of Activated B Cell-like Diffuse Large B Cell Lymphoma Cells. J. Exp. Med. 2001, 194, 1861–1874. [Google Scholar] [CrossRef]
- Wilson, W.H.; Young, R.M.; Schmitz, R.; Yang, Y.; Pittaluga, S.; Wright, G.; Lih, C.-J.; Williams, P.M.; Shaffer, A.L.; Gerecitano, J.; et al. Targeting B Cell Receptor Signaling with Ibrutinib in Diffuse Large B Cell Lymphoma. Nat. Med. 2015, 21, 922–926. [Google Scholar] [CrossRef] [PubMed]
- Wen, T.; Wang, J.; Shi, Y.; Qian, H.; Liu, P. Inhibitors Targeting Bruton’s Tyrosine Kinase in Cancers: Drug Development Advances. Leukemia 2021, 35, 312–332. [Google Scholar] [CrossRef]
- Lam, L.T.; Davis, R.E.; Pierce, J.; Hepperle, M.; Xu, Y.; Hottelet, M.; Nong, Y.; Wen, D.; Adams, J.; Dang, L.; et al. Small Molecule Inhibitors of IkappaB Kinase Are Selectively Toxic for Subgroups of Diffuse Large B-Cell Lymphoma Defined by Gene Expression Profiling. Clin. Cancer Res. 2005, 11, 28–40. [Google Scholar] [PubMed]
- Wang, X.; Tan, Y.; Huang, Z.; Huang, N.; Gao, M.; Zhou, F.; Hu, J.; Feng, W. Disrupting Myddosome Assembly in Diffuse Large B-cell Lymphoma Cells Using the MYD88 Dimerization Inhibitor ST2825. Oncol. Rep. 2019, 42, 1755–1766. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.-H.; Kosek, J.; Wang, M.; Heise, C.; Schafer, P.H.; Chopra, R. Lenalidomide Efficacy in Activated B-Cell-like Subtype Diffuse Large B-Cell Lymphoma Is Dependent upon IRF4 and Cereblon Expression. Br. J. Haematol. 2013, 160, 487–502. [Google Scholar] [CrossRef] [PubMed]
- Ramadass, V.; Vaiyapuri, T.; Tergaonkar, V. Small Molecule NF-ΚB Pathway Inhibitors in Clinic. Int. J. Mol. Sci. 2020, 21, 5164. [Google Scholar] [CrossRef]
- Chiappella, A.; Santambrogio, E.; Castellino, A.; Nicolosi, M.; Vitolo, U. Integrating Novel Drugs to Chemoimmunotherapy in Diffuse Large B-Cell Lymphoma. Expert Rev. Hematol. 2017, 10, 697–705. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Li, B.; Xie, B.; Xu, Z.; Chang, G.; Tao, Y.; Zhang, Y.; Chang, S.; Wang, Y.; Yu, D.; et al. DCZ3301, a Novel Cytotoxic Agent, Inhibits Proliferation in Diffuse Large B-Cell Lymphoma via the STAT3 Pathway. Cell Death Dis. 2017, 8, e3111. [Google Scholar] [CrossRef]
- Bojarczuk, K.; Wienand, K.; Ryan, J.A.; Chen, L.; Villalobos-Ortiz, M.; Mandato, E.; Stachura, J.; Letai, A.; Lawton, L.N.; Chapuy, B.; et al. Targeted Inhibition of PI3Kα/δ Is Synergistic with BCL-2 Blockade in Genetically Defined Subtypes of DLBCL. Blood 2019, 133, 70–80. [Google Scholar] [CrossRef]
- Yahiaoui, A.; Meadows, S.A.; Sorensen, R.A.; Cui, Z.-H.; Keegan, K.S.; Brockett, R.; Chen, G.; Quéva, C.; Li, L.; Tannheimer, S.L. PI3Kδ Inhibitor Idelalisib in Combination with BTK Inhibitor ONO/GS-4059 in Diffuse Large B Cell Lymphoma with Acquired Resistance to PI3Kδ and BTK Inhibitors. PLoS ONE 2017, 12, e0171221. [Google Scholar] [CrossRef]
- McCabe, M.T.; Ott, H.M.; Ganji, G.; Korenchuk, S.; Thompson, C.; Van Aller, G.S.; Liu, Y.; Graves, A.P.; Iii, A.D.P.; Diaz, E.; et al. EZH2 Inhibition as a Therapeutic Strategy for Lymphoma with EZH2-Activating Mutations. Nature 2012, 492, 108–112. [Google Scholar] [CrossRef]
- Knutson, S.K.; Kawano, S.; Minoshima, Y.; Warholic, N.M.; Huang, K.-C.; Xiao, Y.; Kadowaki, T.; Uesugi, M.; Kuznetsov, G.; Kumar, N.; et al. Selective Inhibition of EZH2 by EPZ-6438 Leads to Potent Antitumor Activity in EZH2-Mutant Non-Hodgkin Lymphoma. Mol. Cancer Ther. 2014, 13, 842–854. [Google Scholar] [CrossRef] [PubMed]
- Yamagishi, M.; Hori, M.; Fujikawa, D.; Ohsugi, T.; Honma, D.; Adachi, N.; Katano, H.; Hishima, T.; Kobayashi, S.; Nakano, K.; et al. Targeting Excessive EZH1 and EZH2 Activities for Abnormal Histone Methylation and Transcription Network in Malignant Lymphomas. Cell Rep. 2019, 29, 2321–2337.e7. [Google Scholar] [CrossRef]
- Camicia, R.; Winkler, H.C.; Hassa, P.O. Novel Drug Targets for Personalized Precision Medicine in Relapsed/Refractory Diffuse Large B-Cell Lymphoma: A Comprehensive Review. Mol. Cancer 2015, 14, 207. [Google Scholar] [CrossRef] [PubMed]
- Cycon, K.A.; Mulvaney, K.; Rimsza, L.M.; Persky, D.; Murphy, S.P. Histone Deacetylase Inhibitors Activate CIITA and MHC Class II Antigen Expression in Diffuse Large B-Cell Lymphoma. Immunology 2013, 140, 259–272. [Google Scholar] [CrossRef] [PubMed]
- Deng, S.; Hu, Q.; Zhang, H.; Yang, F.; Peng, C.; Huang, C. HDAC3 Inhibition Upregulates PD-L1 Expression in B-Cell Lymphomas and Augments the Efficacy of Anti–PD-L1 Therapy. Mol. Cancer Ther. 2019, 18, 900–908. [Google Scholar] [CrossRef]
- Wang, X.; Waschke, B.C.; Woolaver, R.A.; Chen, S.M.Y.; Chen, Z.; Wang, J.H. HDAC Inhibitors Overcome Immunotherapy Resistance in B-Cell Lymphoma. Protein Cell 2020, 11, 472–482. [Google Scholar] [CrossRef] [PubMed]
- Tohda, S.; Sato, T.; Kogoshi, H.; Fu, L.; Sakano, S.; Nara, N. Establishment of a Novel B-Cell Lymphoma Cell Line with Suppressed Growth by Gamma-Secretase Inhibitors. Leuk. Res. 2006, 30, 1385–1390. [Google Scholar] [CrossRef]
- Takimoto-Shimomura, T.; Tsukamoto, T.; Maegawa, S.; Fujibayashi, Y.; Matsumura-Kimoto, Y.; Mizuno, Y.; Chinen, Y.; Shimura, Y.; Mizutani, S.; Horiike, S.; et al. Dual Targeting of Bromodomain-Containing 4 by AZD5153 and BCL2 by AZD4320 against B-Cell Lymphomas Concomitantly Overexpressing c-MYC and BCL2. Investig. New Drugs 2019, 37, 210–222. [Google Scholar] [CrossRef]
- Li, W.; Gupta, S.K.; Han, W.; Kundson, R.A.; Nelson, S.; Knutson, D.; Greipp, P.T.; Elsawa, S.F.; Sotomayor, E.M.; Gupta, M. Targeting MYC Activity in Double-Hit Lymphoma with MYC and BCL2 and/or BCL6 Rearrangements with Epigenetic Bromodomain Inhibitors. J. Hematol. Oncol. 2019, 12, 73. [Google Scholar] [CrossRef]
- Cerchietti, L.C.; Yang, S.N.; Shaknovich, R.; Hatzi, K.; Polo, J.M.; Chadburn, A.; Dowdy, S.F.; Melnick, A. A Peptomimetic Inhibitor of BCL6 with Potent Antilymphoma Effects in Vitro and in Vivo. Blood 2009, 113, 3397–3405. [Google Scholar] [CrossRef]
- Polo, J.M.; Dell’Oso, T.; Ranuncolo, S.M.; Cerchietti, L.; Beck, D.; Da Silva, G.F.; Prive, G.G.; Licht, J.D.; Melnick, A. Specific Peptide Interference Reveals BCL6 Transcriptional and Oncogenic Mechanisms in B-Cell Lymphoma Cells. Nat. Med. 2004, 10, 1329–1335. [Google Scholar] [CrossRef] [PubMed]
- Cerchietti, L.C.; Ghetu, A.F.; Zhu, X.; Da Silva, G.F.; Zhong, S.; Matthews, M.; Bunting, K.L.; Polo, J.M.; Farès, C.; Arrowsmith, C.H.; et al. A Small-Molecule Inhibitor of BCL6 Kills DLBCL Cells In Vitro and In Vivo. Cancer Cell 2010, 17, 400–411. [Google Scholar] [CrossRef]
- Sandberg, R.; Ernberg, I. Assessment of Tumor Characteristic Gene Expression in Cell Lines Using a Tissue Similarity Index (TSI). Proc. Natl. Acad. Sci. USA 2005, 102, 2052–2057. [Google Scholar] [CrossRef] [PubMed]
- Mullard, A. Can You Trust Your Cancer Cell Lines? Nat. Rev. Drug Discov. 2018, 17, 613. [Google Scholar] [CrossRef]
- Kohnken, R.; Porcu, P.; Mishra, A. Overview of the Use of Murine Models in Leukemia and Lymphoma Research. Front. Oncol. 2017, 7, 22. [Google Scholar] [CrossRef]
- Perlman, R.L. Mouse Models of Human Disease: An Evolutionary Perspective. Evol. Med. Public Health 2016, 2016, 170–176. [Google Scholar] [CrossRef]
- Harris, A.W.; Pinkert, C.A.; Crawford, M.; Langdon, W.Y.; Brinster, R.L.; Adams, J.M. The E Mu-Myc Transgenic Mouse. A Model for High-Incidence Spontaneous Lymphoma and Leukemia of Early B Cells. J. Exp. Med. 1988, 167, 353–371. [Google Scholar] [CrossRef] [PubMed]
- Greenwald, R.J.; Tumang, J.R.; Sinha, A.; Currier, N.; Cardiff, R.D.; Rothstein, T.L.; Faller, D.V.; Denis, G.V. E Mu-BRD2 Transgenic Mice Develop B-Cell Lymphoma and Leukemia. Blood 2004, 103, 1475–1484. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Cattoretti, G.; Pasqualucci, L.; Ballon, G.; Tam, W.; Nandula, S.V.; Shen, Q.; Mo, T.; Murty, V.V.; Dalla-Favera, R. Deregulated BCL6 Expression Recapitulates the Pathogenesis of Human Diffuse Large B Cell Lymphomas in Mice. Cancer Cell 2005, 7, 445–455. [Google Scholar] [CrossRef]
- Cho, S.-Y.; Kang, W.; Han, J.Y.; Min, S.; Kang, J.; Lee, A.; Kwon, J.Y.; Lee, C.; Park, H. An Integrative Approach to Precision Cancer Medicine Using Patient-Derived Xenografts. Mol. Cells 2016, 39, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Abdelrasoul, H.; Werner, M.; Setz, C.S.; Okkenhaug, K.; Jumaa, H. PI3K Induces B-Cell Development and Regulates B Cell Identity. Sci. Rep. 2018, 8, 1327. [Google Scholar] [CrossRef]
- Béguelin, W.; Teater, M.; Meydan, C.; Hoehn, K.B.; Phillip, J.M.; Soshnev, A.A.; Venturutti, L.; Rivas, M.A.; Calvo-Fernández, M.T.; Gutierrez, J.; et al. Mutant EZH2 Induces a Pre-Malignant Lymphoma Niche by Reprogramming the Immune Response. Cancer Cell 2020, 37, 655–673.e11. [Google Scholar] [CrossRef]
- Brescia, P.; Schneider, C.; Holmes, A.B.; Shen, Q.; Hussein, S.; Pasqualucci, L.; Basso, K.; Dalla-Favera, R. MEF2B Instructs Germinal Center Development and Acts as an Oncogene in B Cell Lymphomagenesis. Cancer Cell 2018, 34, 453–465.e9. [Google Scholar] [CrossRef] [PubMed]
- Dominguez, P.M.; Ghamlouch, H.; Rosikiewicz, W.; Kumar, P.; Béguelin, W.; Fontán, L.; Rivas, M.A.; Pawlikowska, P.; Armand, M.; Mouly, E.; et al. TET2 Deficiency Causes Germinal Center Hyperplasia, Impairs Plasma Cell Differentiation, and Promotes B-Cell Lymphomagenesis. Cancer Discov. 2018, 8, 1632–1653. [Google Scholar] [CrossRef]
- Zhang, J.; Vlasevska, S.; Wells, V.A.; Nataraj, S.; Holmes, A.B.; Duval, R.; Meyer, S.N.; Mo, T.; Basso, K.; Brindle, P.K.; et al. The CREBBP Acetyltransferase Is a Haploinsufficient Tumor Suppressor in B-Cell Lymphoma. Cancer Discov. 2017, 7, 322–337. [Google Scholar] [CrossRef]
- Mori, S.; Rempel, R.E.; Chang, J.T.; Yao, G.; Lagoo, A.S.; Potti, A.; Bild, A.; Nevins, J.R. Utilization of Pathway Signatures to Reveal Distinct Types of B Lymphoma in the Emicro-Myc Model and Human Diffuse Large B-Cell Lymphoma. Cancer Res. 2008, 68, 8525–8534. [Google Scholar] [CrossRef] [PubMed]
- Lefebure, M.; Tothill, R.W.; Kruse, E.; Hawkins, E.D.; Shortt, J.; Matthews, G.M.; Gregory, G.P.; Martin, B.P.; Kelly, M.J.; Todorovski, I.; et al. Genomic Characterisation of Eμ-Myc Mouse Lymphomas Identifies Bcor as a Myc Co-Operative Tumour-Suppressor Gene. Nat. Commun. 2017, 8, 14581. [Google Scholar] [CrossRef]
- Lucas, F.; Rogers, K.A.; Harrington, B.K.; Pan, A.; Yu, L.; Breitbach, J.; Bundschuh, R.; Goettl, V.M.; Hing, Z.A.; Kanga, P.; et al. Eμ-TCL1xMyc: A Novel Mouse Model for Concurrent CLL and B-Cell Lymphoma. Clin. Cancer Res. 2019, 25, 6260–6273. [Google Scholar] [CrossRef]
- Young, R.M.; Hardy, I.R.; Clarke, R.L.; Lundy, N.; Pine, P.; Turner, B.C.; Potter, T.A.; Refaeli, Y. Mouse Models of Non-Hodgkin Lymphoma Reveal Syk as an Important Therapeutic Target. Blood 2009, 113, 2508–2516. [Google Scholar] [CrossRef]
- García-Ramírez, I.; Tadros, S.; González-Herrero, I.; Martín-Lorenzo, A.; Rodríguez-Hernández, G.; Moore, D.; Ruiz-Roca, L.; Blanco, O.; Alonso-López, D.; Rivas, J.D.L.; et al. Crebbp Loss Cooperates with Bcl2 Overexpression to Promote Lymphoma in Mice. Blood 2017, 129, 2645–2656. [Google Scholar] [CrossRef]
- Zhang, J.; Dominguez-Sola, D.; Hussein, S.; Lee, J.-E.; Holmes, A.B.; Bansal, M.; Vlasevska, S.; Mo, T.; Tang, H.; Basso, K.; et al. Disruption of KMT2D Perturbs Germinal Center B Cell Development and Promotes Lymphomagenesis. Nat. Med. 2015, 21, 1190–1198. [Google Scholar] [CrossRef]
- Béguelin, W.; Popovic, R.; Teater, M.; Jiang, Y.; Bunting, K.L.; Rosen, M.; Shen, H.; Yang, S.N.; Wang, L.; Ezponda, T.; et al. EZH2 Is Required for Germinal Center Formation and Somatic EZH2 Mutations Promote Lymphoid Transformation. Cancer Cell 2013, 23, 677–692. [Google Scholar] [CrossRef]
- Knittel, G.; Liedgens, P.; Korovkina, D.; Seeger, J.M.; Al-Baldawi, Y.; Al-Maarri, M.; Fritz, C.; Vlantis, K.; Bezhanova, S.; Scheel, A.H.; et al. B-Cell-Specific Conditional Expression of Myd88p.L252P Leads to the Development of Diffuse Large B-Cell Lymphoma in Mice. Blood 2016, 127, 2732–2741. [Google Scholar] [CrossRef]
- Calado, D.P.; Zhang, B.; Srinivasan, L.; Sasaki, Y.; Seagal, J.; Unitt, C.; Rodig, S.; Kutok, J.; Tarakhovsky, A.; Schmidt-Supprian, M.; et al. Constitutive Canonical NF-ΚB Activation Cooperates with Disruption of BLIMP1 in the Pathogenesis of Activated B-Cell like Diffuse Large B-Cell Lymphoma. Cancer Cell 2010, 18, 580–589. [Google Scholar] [CrossRef]
- Illidge, T.; Honeychurch, J.; Howatt, W.; Ross, F.; Wilkins, B.; Cragg, M. A New in Vivo and in Vitro B Cell Lymphoma Model, Pi-BCL1. Cancer Biother. Radiopharm. 2000, 15, 571–580. [Google Scholar] [CrossRef]
- Timmerman, J.M.; Caspar, C.B.; Lambert, S.L.; Syrengelas, A.D.; Levy, R. Idiotype-Encoding Recombinant Adenoviruses Provide Protective Immunity against Murine B-Cell Lymphomas. Blood 2001, 97, 1370–1377. [Google Scholar] [CrossRef] [PubMed]
- Chaise, C.; Itti, E.; Petegnief, Y.; Wirquin, E.; Copie-Bergman, C.; Farcet, J.-P.; Delfau-Larue, M.-H.; Meignan, M.; Talbot, J.-N.; Molinier-Frenkel, V. [F-18]-Fluoro-2-Deoxy-D: -Glucose Positron Emission Tomography as a Tool for Early Detection of Immunotherapy Response in a Murine B Cell Lymphoma Model. Cancer Immunol. Immunother. 2007, 56, 1163–1171. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Palmieri, C.; Falcone, C.; Iaccino, E.; Tuccillo, F.M.; Gaspari, M.; Trimboli, F.; De Laurentiis, A.; Luberto, L.; Pontoriero, M.; Pisano, A.; et al. In Vivo Targeting and Growth Inhibition of the A20 Murine B-Cell Lymphoma by an Idiotype-Specific Peptide Binder. Blood 2010, 116, 226–238. [Google Scholar] [CrossRef] [PubMed]
- Curti, A.; Pandolfi, S.; Valzasina, B.; Aluigi, M.; Isidori, A.; Ferri, E.; Salvestrini, V.; Bonanno, G.; Rutella, S.; Durelli, I.; et al. Modulation of Tryptophan Catabolism by Human Leukemic Cells Results in the Conversion of CD25- into CD25+ T Regulatory Cells. Blood 2007, 109, 2871–2877. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.-S.; Chen, X.-Y.; Li, W.-P.; Yang, Y.; Song, Z.-L. Establishing SCID Mouse Models of B-Cell Non-Hodgkin’s Lymphoma. Ai Zheng 2009, 28, 181–183. [Google Scholar]
- Ackler, S.; Xiao, Y.; Mitten, M.J.; Foster, K.; Oleksijew, A.; Refici, M.; Schlessinger, S.; Wang, B.; Chemburkar, S.R.; Bauch, J.; et al. ABT-263 and Rapamycin Act Cooperatively to Kill Lymphoma Cells In Vitro and In Vivo. Mol. Cancer Ther. 2008, 7, 3265–3274. [Google Scholar] [CrossRef]
- Jiang, Y.; Ortega-Molina, A.; Geng, H.; Ying, H.-Y.; Hatzi, K.; Parsa, S.; McNally, D.; Wang, L.; Doane, A.S.; Agirre, X.; et al. CREBBP Inactivation Promotes the Development of HDAC3-Dependent Lymphomas. Cancer Discov. 2017, 7, 38–53. [Google Scholar] [CrossRef]
- Flümann, R.; Nieper, P.; Reinhardt, H.C.; Knittel, G. New Murine Models of Aggressive Lymphoma. Leuk. Lymphoma 2020, 61, 788–798. [Google Scholar] [CrossRef] [PubMed]
- Yabe, D.; Fukuda, H.; Aoki, M.; Yamada, S.; Takebayashi, S.; Shinkura, R.; Yamamoto, N.; Honjo, T. Generation of a Conditional Knockout Allele for Mammalian Spen Protein Mint/SHARP. Genesis 2007, 45, 300–306. [Google Scholar] [CrossRef] [PubMed]
- Hampel, F.; Ehrenberg, S.; Hojer, C.; Draeseke, A.; Marschall-Schröter, G.; Kühn, R.; Mack, B.; Gires, O.; Vahl, C.J.; Schmidt-Supprian, M.; et al. CD19-Independent Instruction of Murine Marginal Zone B-Cell Development by Constitutive Notch2 Signaling. Blood 2011, 118, 6321–6331. [Google Scholar] [CrossRef] [PubMed]
- Vereecke, L.; Sze, M.; Mc Guire, C.; Rogiers, B.; Chu, Y.; Schmidt-Supprian, M.; Pasparakis, M.; Beyaert, R.; van Loo, G. Enterocyte-Specific A20 Deficiency Sensitizes to Tumor Necrosis Factor-Induced Toxicity and Experimental Colitis. J. Exp. Med. 2010, 207, 1513–1523. [Google Scholar] [CrossRef]
- Ortega-Molina, A.; Boss, I.W.; Canela, A.; Pan, H.; Jiang, Y.; Zhao, C.; Jiang, M.; Hu, D.; Agirre, X.; Niesvizky, I.; et al. The Histone Lysine Methyltransferase KMT2D Sustains a Gene Expression Program That Represses B Cell Lymphoma Development. Nat. Med. 2015, 21, 1199–1208. [Google Scholar] [CrossRef]
- Ennishi, D.; Takata, K.; Béguelin, W.; Duns, G.; Mottok, A.; Farinha, P.; Bashashati, A.; Saberi, S.; Boyle, M.; Meissner, B.; et al. Molecular and Genetic Characterization of MHC Deficiency Identifies EZH2 as Therapeutic Target for Enhancing Immune Recognition. Cancer Discov. 2019, 9, 546–563. [Google Scholar] [CrossRef]
- Flümann, R.; Rehkämper, T.; Nieper, P.; Pfeiffer, P.; Holzem, A.; Klein, S.; Bhatia, S.; Kochanek, M.; Kisis, I.; Pelzer, B.W.; et al. An Autochthonous Mouse Model of Myd88- and BCL2-Driven Diffuse Large B-Cell Lymphoma Reveals Actionable Molecular Vulnerabilities. Blood Cancer Discov. 2021, 2, 70–91. [Google Scholar] [CrossRef]
- Heckl, D.; Kowalczyk, M.S.; Yudovich, D.; Belizaire, R.; Puram, R.V.; McConkey, M.E.; Thielke, A.; Aster, J.C.; Regev, A.; Ebert, B.L. Generation of Mouse Models of Myeloid Malignancy with Combinatorial Genetic Lesions Using CRISPR-Cas9 Genome Editing. Nat. Biotechnol. 2014, 32, 941–946. [Google Scholar] [CrossRef] [PubMed]
- Wefers, B.; Bashir, S.; Rossius, J.; Wurst, W.; Kühn, R. Gene Editing in Mouse Zygotes Using the CRISPR/Cas9 System. Methods 2017, 121–122, 55–67. [Google Scholar] [CrossRef]
- Turaj, A.H.; Cox, K.L.; Penfold, C.A.; French, R.R.; Mockridge, C.I.; Willoughby, J.E.; Tutt, A.L.; Griffiths, J.; Johnson, P.W.M.; Glennie, M.J.; et al. Augmentation of CD134 (OX40)-Dependent NK Anti-Tumour Activity Is Dependent on Antibody Cross-Linking. Sci. Rep. 2018, 8, 2278. [Google Scholar] [CrossRef] [PubMed]
- Day, C.-P.; Merlino, G.; Van Dyke, T. Preclinical Mouse Cancer Models: A Maze of Opportunities and Challenges. Cell 2015, 163, 39–53. [Google Scholar] [CrossRef] [PubMed]
- Di Rosso, M.E.; Sterle, H.A.; Cremaschi, G.A.; Genaro, A.M. Beneficial Effect of Fluoxetine and Sertraline on Chronic Stress-Induced Tumor Growth and Cell Dissemination in a Mouse Model of Lymphoma: Crucial Role of Antitumor Immunity. Front. Immunol. 2018, 9, 1341. [Google Scholar] [CrossRef] [PubMed]
- Sagiv-Barfi, I.; Kohrt, H.E.; Burckhardt, L.; Czerwinski, D.K.; Levy, R. Ibrutinib Enhances the Antitumor Immune Response Induced by Intratumoral Injection of a TLR9 Ligand in Mouse Lymphoma. Blood 2015, 125, 2079–2086. [Google Scholar] [CrossRef]
- Passineau, M.J.; Siegal, G.P.; Everts, M.; Pereboev, A.; Jhala, D.; Wang, M.; Zhu, Z.B.; Park, S.K.; Curiel, D.T.; Nelson, G.M. The Natural History of a Novel, Systemic, Disseminated Model of Syngeneic Mouse B-Cell Lymphoma. Leuk. Lymphoma 2005, 46, 1627–1638. [Google Scholar] [CrossRef]
- Talmadge, J.E.; Singh, R.K.; Fidler, I.J.; Raz, A. Murine Models to Evaluate Novel and Conventional Therapeutic Strategies for Cancer. Am. J. Pathol. 2007, 170, 793–804. [Google Scholar] [CrossRef]
- Chapuy, B.; Cheng, H.; Watahiki, A.; Ducar, M.D.; Tan, Y.; Chen, L.; Roemer, M.G.M.; Ouyang, J.; Christie, A.L.; Zhang, L.; et al. Diffuse Large B-Cell Lymphoma Patient-Derived Xenograft Models Capture the Molecular and Biological Heterogeneity of the Disease. Blood 2016, 127, 2203–2213. [Google Scholar] [CrossRef]
- DiFranco, K.M.; Johnson-Farley, N.; Bertino, J.R.; Elson, D.; Vega, B.A.; Belinka, B.A.; Kachlany, S.C. LFA-1-Targeting Leukotoxin (LtxA.; Leukothera®) Causes Lymphoma Tumor Regression in a Humanized Mouse Model and Requires Caspase-8 and Fas to Kill Malignant Lymphocytes. Leuk. Res. 2015, 39, 649–656. [Google Scholar] [CrossRef]
- Hao, Y.; Chapuy, B.; Monti, S.; Sun, H.H.; Rodig, S.J.; Shipp, M.A. Selective JAK2 Inhibition Specifically Decreases Hodgkin Lymphoma and Mediastinal Large B-Cell Lymphoma Growth in Vitro and in Vivo. Clin. Cancer Res. 2014, 20, 2674–2683. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Nomie, K.; Zhang, H.; Bell, T.; Pham, L.; Kadri, S.; Segal, J.; Li, S.; Zhou, S.; Santos, D.; et al. B-Cell Lymphoma Patient-Derived Xenograft Models Enable Drug Discovery and Are a Platform for Personalized Therapy. Clin. Cancer Res. 2017, 23, 4212–4223. [Google Scholar] [CrossRef] [PubMed]
- Vidal-Crespo, A.; Matas-Céspedes, A.; Rodriguez, V.; Rossi, C.; Valero, J.G.; Serrat, N.; Sanjuan-Pla, A.; Menéndez, P.; Roué, G.; López-Guillermo, A.; et al. Daratumumab Displays in Vitro and In Vivo Anti-Tumor Activity in Models of B-Cell Non-Hodgkin Lymphoma and Improves Responses to Standard Chemo-Immunotherapy Regimens. Haematologica 2020, 105, 1032–1041. [Google Scholar] [CrossRef]
- Kopetz, S.; Lemos, R.; Powis, G. The Promise of Patient-Derived Xenografts: The Best Laid Plans of Mice and Men. Clin. Cancer Res. 2012, 18, 5160–5162. [Google Scholar] [CrossRef]
- Walsh, N.; Kenney, L.; Jangalwe, S.; Aryee, K.-E.; Greiner, D.L.; Brehm, M.A.; Shultz, L.D. Humanized Mouse Models of Clinical Disease. Annu. Rev. Pathol. 2017, 12, 187–215. [Google Scholar] [CrossRef] [PubMed]
- Zitvogel, L.; Pitt, J.M.; Daillère, R.; Smyth, M.J.; Kroemer, G. Mouse Models in Oncoimmunology. Nat. Rev. Cancer 2016, 16, 759–773. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.-D.; Tsai, M.-H.; Romero-Masters, J.C.; Ranheim, E.A.; Huebner, S.M.; Bristol, J.A.; Delecluse, H.-J.; Kenney, S.C. Latent Membrane Protein 1 (LMP1) and LMP2A Collaborate To Promote Epstein-Barr Virus-Induced B Cell Lymphomas in a Cord Blood-Humanized Mouse Model but Are Not Essential. J. Virol. 2017, 91. [Google Scholar] [CrossRef]
- Romero-Masters, J.C.; Ohashi, M.; Djavadian, R.; Eichelberg, M.R.; Hayes, M.; Bristol, J.A.; Ma, S.; Ranheim, E.A.; Gumperz, J.; Johannsen, E.C.; et al. An EBNA3C-Deleted Epstein-Barr Virus (EBV) Mutant Causes B-Cell Lymphomas with Delayed Onset in a Cord Blood-Humanized Mouse Model. PLoS Pathog. 2018, 14, e1007221. [Google Scholar] [CrossRef]
- Alizadeh, A.A.; Gentles, A.J.; Alencar, A.J.; Liu, C.L.; Kohrt, H.E.; Houot, R.; Goldstein, M.J.; Zhao, S.; Natkunam, Y.; Advani, R.H.; et al. Prediction of Survival in Diffuse Large B-Cell Lymphoma Based on the Expression of 2 Genes Reflecting Tumor and Microenvironment. Blood 2011, 118, 1350–1358. [Google Scholar] [CrossRef]
- Gentles, A.J.; Newman, A.M.; Liu, C.L.; Bratman, S.V.; Feng, W.; Kim, D.; Nair, V.S.; Xu, Y.; Khuong, A.; Hoang, C.D.; et al. The Prognostic Landscape of Genes and Infiltrating Immune Cells across Human Cancers. Nat. Med. 2015, 21, 938–945. [Google Scholar] [CrossRef]
- Pizzi, M.; Boi, M.; Bertoni, F.; Inghirami, G. Emerging Therapies Provide New Opportunities to Reshape the Multifaceted Interactions between the Immune System and Lymphoma Cells. Leukemia 2016, 30, 1805–1815. [Google Scholar] [CrossRef] [PubMed]
- Kapałczyńska, M.; Kolenda, T.; Przybyła, W.; Zajączkowska, M.; Teresiak, A.; Filas, V.; Ibbs, M.; Bliźniak, R.; Łuczewski, Ł.; Lamperska, K. 2D and 3D Cell Culture—A Comparison of Different Types of Cancer Cell Cultures. Arch. Med. Sci. 2018, 14, 910–919. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.W.; Ho, W.J.; Wu, B.M. The Role of the 3D Environment in Hypoxia-Induced Drug and Apoptosis Resistance. Anticancer Res. 2011, 31, 3237–3245. [Google Scholar]
- Katt, M.E.; Placone, A.L.; Wong, A.D.; Xu, Z.S.; Searson, P.C. In Vitro Tumor Models: Advantages, Disadvantages, Variables, and Selecting the Right Platform. Front. Bioeng. Biotechnol. 2016, 4, 12. [Google Scholar] [CrossRef] [PubMed]
- Esch, E.W.; Bahinski, A.; Huh, D. Organs-on-Chips at the Frontiers of Drug Discovery. Nat. Rev. Drug Discov. 2015, 14, 248–260. [Google Scholar] [CrossRef] [PubMed]
- Brassard, J.A.; Lutolf, M.P. Engineering Stem Cell Self-Organization to Build Better Organoids. Cell Stem Cell 2019, 24, 860–876. [Google Scholar] [CrossRef]
- Weiswald, L.-B.; Bellet, D.; Dangles-Marie, V. Spherical Cancer Models in Tumor Biology. Neoplasia 2015, 17, 1–15. [Google Scholar] [CrossRef]
- Han, K.; Pierce, S.E.; Li, A.; Spees, K.; Anderson, G.R.; Seoane, J.A.; Lo, Y.-H.; Dubreuil, M.; Olivas, M.; Kamber, R.A.; et al. CRISPR Screens in Cancer Spheroids Identify 3D Growth-Specific Vulnerabilities. Nature 2020, 580, 136–141. [Google Scholar] [CrossRef]
- Sabhachandani, P.; Sarkar, S.; Mckenney, S.; Ravi, D.; Evens, A.M.; Konry, T. Microfluidic Assembly of Hydrogel-Based Immunogenic Tumor Spheroids for Evaluation of Anticancer Therapies and Biomarker Release. J. Control. Release 2019, 295, 21–30. [Google Scholar] [CrossRef]
- Tian, Y.F.; Ahn, H.; Schneider, R.S.; Yang, S.N.; Roman-Gonzalez, L.; Melnick, A.M.; Cerchietti, L.; Singh, A. Integrin-Specific Hydrogels as Adaptable Tumor Organoids for Malignant B and T Cells. Biomaterials 2015, 73, 110–119. [Google Scholar] [CrossRef] [PubMed]
- Rosenthal, N.; Brown, S. The Mouse Ascending: Perspectives for Human-Disease Models. Nat. Cell Biol. 2007, 9, 993–999. [Google Scholar] [CrossRef]
- Mellman, I.; Coukos, G.; Dranoff, G. Cancer Immunotherapy Comes of Age. Nature 2011, 480, 480–489. [Google Scholar] [CrossRef] [PubMed]
- Bar-Ephraim, Y.E.; Kretzschmar, K.; Clevers, H. Organoids in Immunological Research. Nat. Rev. Immunol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Clevers, H.C. Organoids: Avatars for Personalized Medicine. Keio J. Med. 2019, 68, 95. [Google Scholar] [CrossRef] [PubMed]
- Aboulkheyr Es, H.; Montazeri, L.; Aref, A.R.; Vosough, M.; Baharvand, H. Personalized Cancer Medicine: An Organoid Approach. Trends Biotechnol. 2018, 36, 358–371. [Google Scholar] [CrossRef]
- Béguelin, W.; Rivas, M.A.; Calvo Fernández, M.T.; Teater, M.; Purwada, A.; Redmond, D.; Shen, H.; Challman, M.F.; Elemento, O.; Singh, A.; et al. EZH2 Enables Germinal Centre Formation through Epigenetic Silencing of CDKN1A and an Rb-E2F1 Feedback Loop. Nat. Commun. 2017, 8, 877. [Google Scholar] [CrossRef]
- Scholze, H.; Stephenson, R.E.; Reynolds, R.; Shah, S.; Puri, R.; Butler, S.D.; Trujillo-Alonso, V.; Teater, M.R.; van Besien, H.; Gibbs-Curtis, D.; et al. Combined EZH2 and Bcl-2 Inhibitors as Precision Therapy for Genetically Defined DLBCL Subtypes. Blood Adv. 2020, 4, 5226–5231. [Google Scholar] [CrossRef]
- Fontan Gabas, L.; Goldstein, R.L.; Casalena, G.; Durant, M.; Teater, M.R.; Wilson, J.; Phillip, J.M.; Xia, M.; Shah, S.; Us, I.; et al. Identification of MALT1 Feedback Mechanisms Enables Rational Design of Potent Anti-Lymphoma Regimens for ABC-DLBCL. Blood 2020. [Google Scholar] [CrossRef]
- Mannino, R.G.; Pradhan, P.; Roy, K.; Lam, W.A. 3D In Vitro Microvascular Model-Based Lymphoma Model. Methods Cell Biol. 2018, 146, 149–158. [Google Scholar] [CrossRef] [PubMed]
- Junttila, M.R.; de Sauvage, F.J. Influence of Tumour Micro-Environment Heterogeneity on Therapeutic Response. Nature 2013, 501, 346–354. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, T. Organoids for Drug Discovery and Personalized Medicine. Annu. Rev. Pharmacol. Toxicol. 2019, 59, 447–462. [Google Scholar] [CrossRef] [PubMed]



| Wright and Schmitz [28,31] | Chapuy [32] | Lacy [33] | Pedrosa [34] | COO and Patient Outcome |
|---|---|---|---|---|
| MCD | C5 | MYD88 | MCD2-S | ABC |
| MYD88L265P, CD79B, PIM1, CDKN2A | MYD88L265P, CD79B, PIM1, PRMD1, ETV6 | MYD88L265P, CD79B, PIM1, ETV6, CDKN2A | MYD88, CD79B, PIM1, PRDM1, PIM2, BTG1 | Bad prognosis |
| BN2 | C1 | NOTCH2 | BN22-S | ABC and GCB |
| NOTCH2, BCL6 transl., SPEN, TNFAIP3, CD70, UBE2A, BCL10, CCND3 | NOTCH2, BCL6 transl., SPEN, TNFAIP3, CD70, BCL10 | NOTCH2, BCL10, TNFAIP3, CD70, SPEN, CCND3 | NOTCH2, BCL6 transl., TNFAIP3, CD70, BCL10, UBE2A, CCND3, DTX1 | Variable prognosis |
| EZB | C3 | BCL2 | EZB2-S | GCB |
| EZH2, BCL2 transl., CREBBP, EP300, IRF8, MEF2B, GNA13, KMT2D, REL amp. | EZH2, BCL2 *, CREBBP, EP300, IRF8, MEF2B, GNA13, KMT2D | EZH2, BCL2, CREBBP, IRF8, MEF2B, KMT2D | EZH2, BCL2 *, CREBBP, TNFRSF14, IRF8, KMT2D, EP300 | Generally favorable prognosis |
| N1 | NEC | N12-S | Mostly ABC | |
| NOTCH1 | NOTCH1, REL amp., TP53 | NOTCH1 | Bad prognosis | |
| A53 | C2 | NEC | Mostly ABC | |
| TP53, aneuploidy | TP53, REL, CDKN2A loss, aneuploidy | NOTCH1, REL amp., TP53 | Bad prognosis | |
| ST2 | C4 | SOCS1/SGK1–TET2/SGK1 | ST22-S | Mostly GCB |
| SOCS1, TET2, SGK1 | SGK1, HIST1H1E, NFKBIE, BRAF, CD83 | SOCS1, TET2, SGK1 | SOCS1, TET2, SGK1, STAT3 | Favorable prognosis |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yanguas-Casás, N.; Pedrosa, L.; Fernández-Miranda, I.; Sánchez-Beato, M. An Overview on Diffuse Large B-Cell Lymphoma Models: Towards a Functional Genomics Approach. Cancers 2021, 13, 2893. https://doi.org/10.3390/cancers13122893
Yanguas-Casás N, Pedrosa L, Fernández-Miranda I, Sánchez-Beato M. An Overview on Diffuse Large B-Cell Lymphoma Models: Towards a Functional Genomics Approach. Cancers. 2021; 13(12):2893. https://doi.org/10.3390/cancers13122893
Chicago/Turabian StyleYanguas-Casás, Natalia, Lucía Pedrosa, Ismael Fernández-Miranda, and Margarita Sánchez-Beato. 2021. "An Overview on Diffuse Large B-Cell Lymphoma Models: Towards a Functional Genomics Approach" Cancers 13, no. 12: 2893. https://doi.org/10.3390/cancers13122893
APA StyleYanguas-Casás, N., Pedrosa, L., Fernández-Miranda, I., & Sánchez-Beato, M. (2021). An Overview on Diffuse Large B-Cell Lymphoma Models: Towards a Functional Genomics Approach. Cancers, 13(12), 2893. https://doi.org/10.3390/cancers13122893